
Mechanism, reactivity, and selectivity in a palladium-catalyzed organosilicon-based cross coupling reaction†
Qian
Wang
,
Hui-Mei
Jiang
,
Shuping
Zhuo
 * and
Li-Ping
Xu
*
* and
Li-Ping
Xu
*
School of Chemistry and Chemical Engineering, Shandong University of Technology, Zibo, 255000, China. E-mail: xulp@sdut.edu.cn; zhuosp_academic@yahoo.com
First published on 11th November 2021
Abstract
A comprehensive density functional theory (DFT) study has been performed to elucidate the mechanistic details of the Pd-catalyzed strain-release organosilicon based cross coupling reaction of four- and five-membered silacycles. The reaction is proposed to proceed through Ar–Br oxidative addition (OA), Br-to-tBuO ligand exchange (LE), transmetalation, and reductive elimination (RE) steps, and among which the transmetalation is the rate-determining step. Mechanistically, after ligand exchange, the reaction would lead to two distinct pathways: C–O RE to form the C–O coupling product or transmetalation to provide the C–C coupling product, in which the former is often found to be favorable in many other transformations. The experimentally observed uncommon C–C coupling selectivity over the C–O RE in the title reaction is elucidated by an unusual “thermodynamic control scenario”, that is, although the C–O RE has a slightly lower barrier than the transmetalation, however, the product of the transmetalation is much more stable compared to that of the C–O RE. The computational results from different density functional methods and the experimental details verified our findings. The steric effect in the rate-determining step accounts for the contrasting reactivity with different nucleophiles in the reactions with silacyclopentane. The amine base was found to facilitate the transmetalation process by forming a penta-coordinated silacycle. We predict that the −NMe2 containing base would further promote the reaction by decreasing the activation barrier.
Introduction
Organic compounds containing silicon atoms may exhibit different chemical and physicochemical properties compared to all carbon systems as silicon and carbon atoms are different in many aspects such as covalent radius and electronegativity.1–4 In fact, like metal elements, the chemistry and coordination properties of silicon are affected by the availability of its empty 3d orbitals, which make it facile to form hypervalent 5- and 6-coordinated complexes. Numerous organosilicon compounds are shown to have wide applications in pharmaceuticals, materials science and electronic or optoelectronic devices.5,6 Therefore, growing attention has been paid to the synthesis and transformation of organosilicon compounds.7–12 Silacycles containing small ring structures (e.g., silacyclobutane, SCB) are highly reactive molecules because of the inherent ring strain and Lewis acidity.13–15 The transition metal-catalyzed ring-opening and ring expansion process through the silicon–carbon bond cleavage has been revealed by chemists for the unique reactivity of silacycles.16,17 In this context, both silacyclobutanes and benzosilacyclobutenes were important as well as air-stable four-membered silacycles that have been widely used in a variety of chemical transformations.18For instance, highly efficient insertion-type cycloaddition of silacyclobutanes with alkynes has been well established. Hayashi and coworkers have disclosed an intermolecular palladium-catalyzed ring-expansion cycloaddition of silacyclobutanes with activated alkynes to furnish 1-sila-2-cyclohexenes with high enantioselectivity (Scheme 1a).19 The intramolecular ring-expansion transformation of silacyclobutanes has also seen its rapid progress in recent years. He and coworkers demonstrated that silacyclobutane could act as a new C–H silylation reagent with the aid of Rh/TMS-segphos, which might undergo Si–C bond cleavage and C–H bond activation (Scheme 1b).20
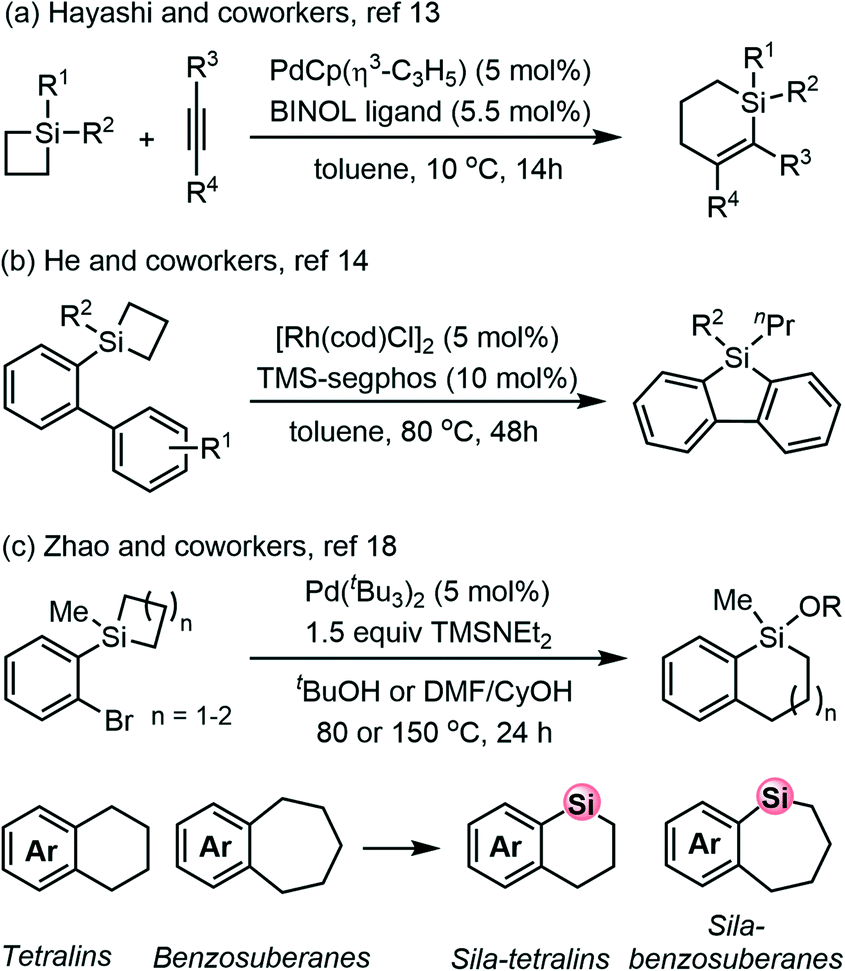 | ||
| Scheme 1 Selected examples for chemical transformations of silacyclobutanes and benzosilacyclobutenes. | ||
Recently, Zhao and coworkers described a Pd-catalyzed alcohol-promoted strain-release intramolecular cross-coupling of silacyclobutane derivatives as an unprecedented ring-expansion method for preparing diverse sila-tetralins and sila-benzosuberanes,21–23 which have potential applications in marketed drugs and natural products (Scheme 1c).24 Notably, this strategy compromises the ring-opening from the benzosilacycles and the intramolecular Hiyama–Denmark cross-coupling25,26 by incorporating aryl halide into the adjacent position.
Although the field of organosilicon cross coupling chemistry is prosperous and those methods provide efficient means of obtaining useful organosilicon compounds to date, the mechanistic studies remain in their infancy.27–31 For the recent work by Zhao and coworkers (Scheme 1c),24 the reaction mechanism, controlling factors, and the roles of the alcohol and the base remain unclear. More importantly, it is known that one of the key challenges in this transformation is the competitive cross-coupling of aryl halides with oxygen-atom containing nucleophiles, which is the major reaction pathway in other transformations.32–36 Thus, it is of great interest to understand how this reaction exactly inhibits the problematic C–O cross-coupling. A thorough understanding of the reaction mechanism would undoubtedly enable us to broaden this chemistry, develop more efficient transition-metal catalyzed ring-opening and ring expansion transformations of silacycles, and provide more elegant routes for functional organosilicon materials. Therefore, herein we performed a systematic density functional theory study on the Pd-catalyzed alcohol-assisted ring expansion to benzosilacycle through a strain-release silicon-based cross coupling reaction.
Computational details
All geometry optimizations were performed using the Gaussian09 (ref. 37) suite of program at the B3LYP-D3(BJ)/[6-31G(d,p) + Lanl2dz (Pd)] level of theory with the Hay–Wadt effective core potential38,39 for Pd. The B3LYP density functional40–42 with Grimme's empirical dispersion–correction (D3)43 was used for better describing the dispersion interaction in this system. The frequency calculations were performed at the same level of theory as the optimization to verify each stationary point to be intermediate or transition state (with zero or one imaginary frequency, respectively) and to provide thermal correction to the final energetics. Intrinsic reaction coordinate (IRC) calculations were performed for key TSs to identify their true nature. Solvent effects were incorporated in the geometry optimizations and frequency calculations with the SMD44 solvent model (with hexanol as the solvent). 3D geometries were drawn using CYLView software.45Results and discussion
1. The catalytic cycle
Based on the experiments,24 we have proposed the possible reaction pathways (Scheme 2). The Pd(0) catalyst would undergo the C–Br or C–Si oxidative addition (OA) to form A or B, followed by C–Si or C–Br OA to form the Pd(IV) species C. After Br−-to-tBuO− ligand exchange (LE), D would proceed via reductive elimination to generate E or F. A second RE would provide the final product and regenerate the Pd(0) active species (path I, Scheme 2). Alternatively, from the C–Br OA adduct complex A, the Br−-to-tBuO− LE would occur to generate G, followed by the transmetalation (TM) to produce F. Finally, the RE would release the product and the active species (path II, Scheme 2). It should be noted that from G, the competing C–O RE could also take place to form the C–O coupling product H, and is a potential side pathway of the reaction (path III, Scheme 2). | ||
| Scheme 2 The proposed catalytic cycle. | ||
2. Reaction with silacyclobutane: oxidative addition and ligand exchange
Firstly, we investigated the C–Br oxidative addition reaction pathway with two bulky phosphine ligands coordinating to palladium. From the catalyst Pd(PtBu3)2, the coordination of the silacyclobutane substrate (1) is endergonic by 21.5 kcal mol−1 to form intermediate 2 (Fig. 1). Interestingly, the coordination leads to an automatic detachment of one of the phosphine ligands from the palladium center (the longer Pd–P bond distance is 5.299 Å, see Fig. 2 for the geometry details), which is likely because of the large steric repulsion between the silacyclobutane substrate and the PtBu3 ligand. The oxidative addition of Ar–Br to Pd(0) occurs through transition state 3-ts to obtain 4, and has a free energy barrier of 24.4 kcal mol−1 and is endergonic by 1.0 kcal mol−1 with respect to the catalyst, Pd(PtBu3)2. Since one of the PtBu3 ligands only binds loosely to the palladium center in this pathway (Fig. 1, red), we considered a ligand exchange process in which substrate 1 replaces one PtBu3 ligand to generate the monophosphine complex 5 (Fig. 1). Pleasingly, calculation shows that the formation of 5 is endergonic by 16.5 kcal mol−1, which is 5.0 kcal mol−1 more stable than that of the biphosphine counterpart, 2. This is possibly due to the entropy gain associated with one PtBu3 ligand release.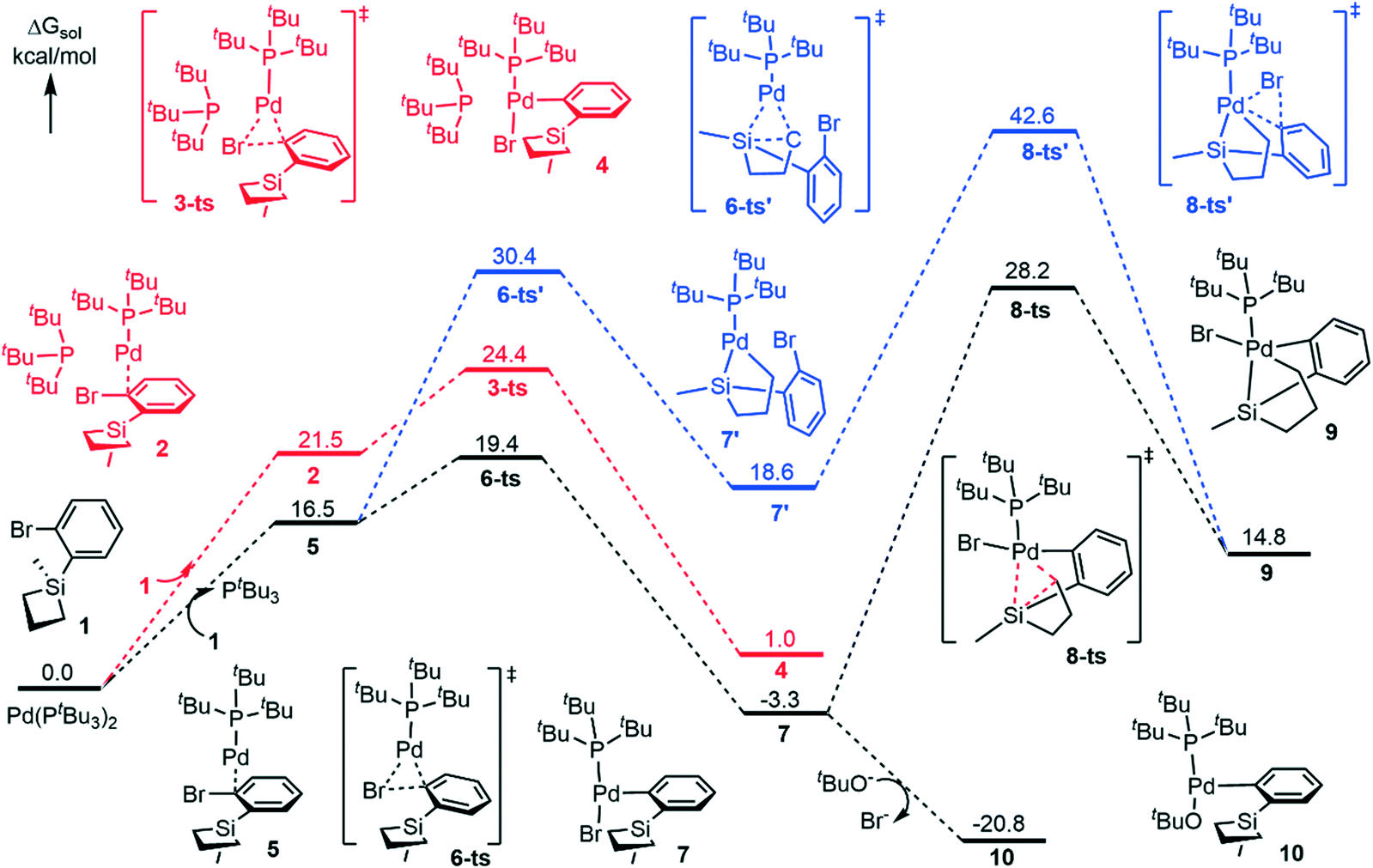 | ||
| Fig. 1 Free energy profile for the oxidative addition and ligand exchange steps. | ||
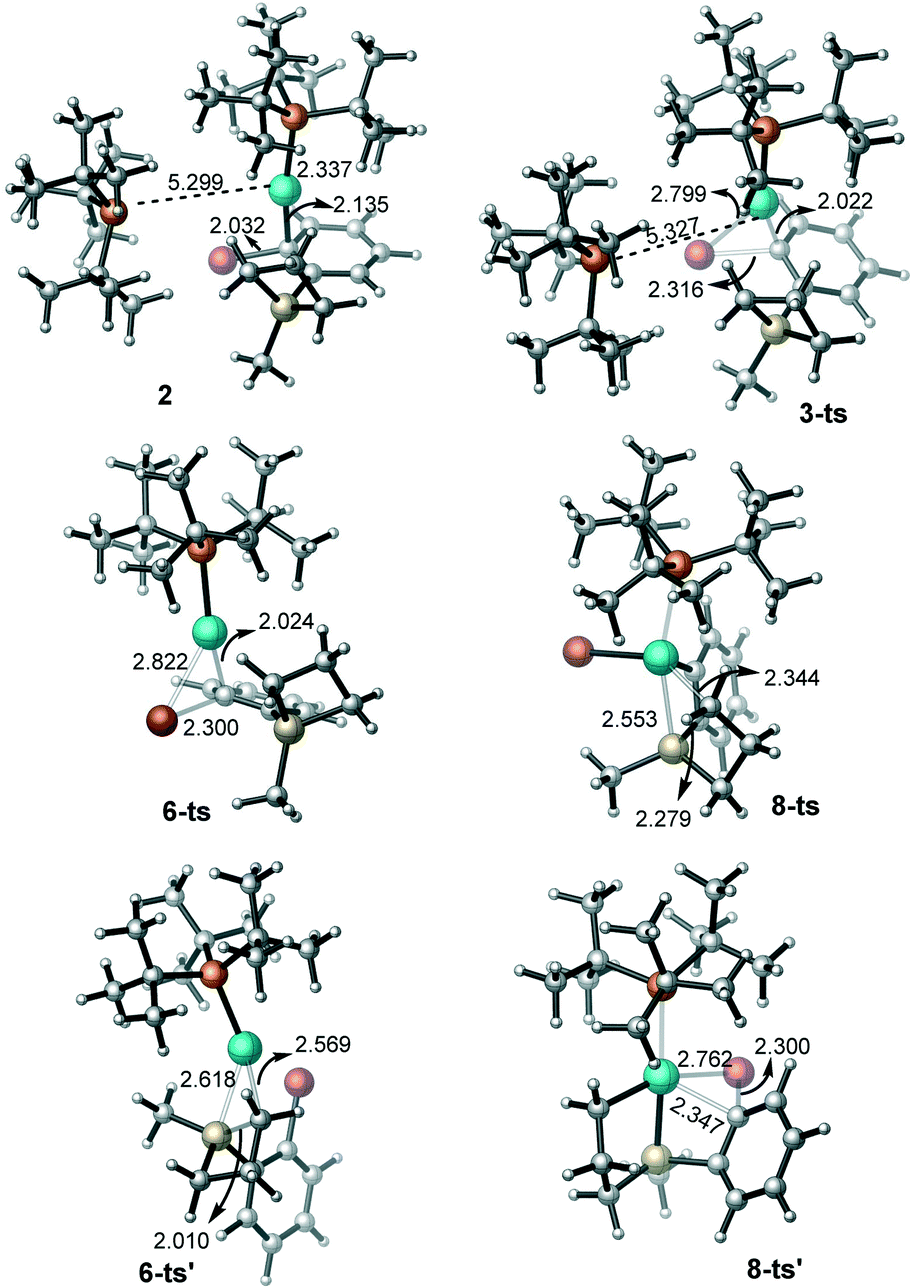 | ||
| Fig. 2 The geometries for the key intermediates and transition states in the oxidative addition steps (bond distances are given in Å). | ||
Following that, the reaction could proceed either through C–Br oxidative addition (Fig. 1, black) or C–Si oxidative addition (Fig. 1, blue). We will first describe the C–Br oxidative addition pathway. This step has a free energy barrier of 19.4 kcal mol−1via6-ts to provide 7. By comparison with the biphospine pathway (Fig. 1, red), the monophosphine pathway (Fig. 1, black) has a lower free energy barrier and the formation of 7 is exergonic by 3.3 kcal mol−1 with respect to Pd(PtBu3)2.
Therefore, our calculation suggests that the monophosphine pathway should be more likely. It is noteworthy that even for the diphosphine pathway, there is only one ligand binding effectively to palladium, indicating the strong steric dependence of this type of phosphine ligand. This is consistent with previous findings by Schoenebeck,46 Maseras,47,48 Harvey,49 and Hartwig.50,51 From 7, the C–Si ring-opening oxidative addition could occur (at 8-ts) to form the Pd(IV) species 9. This step has an activation free energy barrier of 31.5 kcal mol−1 relative to 7. The formation of 9 is endergonic by 14.8 kcal mol−1 with respect to Pd(PtBu3)2.
Another possible monophosphine pathway, starting firstly with the C–Si OA process, requires a 30.4 kcal mol−1 activation barrier (at 6-ts′) to generate the C–Si OA product 7′ (Fig. 1, blue). The following C–Br OA takes place at 8-ts′ to converge to the Pd(IV) species 9. The second OA step demands a dramatically high free energy barrier of 42.6 kcal mol−1 with respect to the starting catalyst. Thus, in these two monophosphine pathways, the mechanism of C–Br OA occurring firstly is relatively more favorable (Fig. 1, black pathway).
Since the Pd(IV) pathways (see path I in Scheme 2 and Fig. 1) have high activation barriers (at least 31.5 kcal mol−1) and are endergonic, we sought to investigate the ligand exchange from the Pd(II) species (7) and the following processes thereafter (see path II in Scheme 2). Gratifyingly, from 7, the ligand exchange between the nucleophile tBuO anion and the Br anion is exergonic by 17.5 kcal mol−1, leading to a thermodynamically stable (PtBu3)Pd(Ar)(OtBu) intermediate 10 (Fig. 1).
3. Competing transmetalation and C–O reductive elimination
After ligand exchange, the generated intermediate 10 could undergo either transmetalation (see path II, Scheme 2), which involves the silacyclobutane ring-opening and the Pd–C and Si–O bond formation, or direct C–O reductive elimination (see path III, Scheme 2). It should be noted that the C–O RE product has not been observed in the experiment.24 As seen in Fig. 3a, the transmetalation pathway has a free energy barrier of 23.0 kcal mol−1 associated with 11-ts and is highly exergonic by 27.2 kcal mol−1 to obtain the palladacycle complex 12 with respect to intermediate 10. By comparison, the competing C–O reductive elimination occurring through 13-ts bears a slightly lower barrier of 20.0 kcal mol−1, which is 3.0 kcal mol−1 lower than that of the transmetalation pathway. However, the C–O RE pathway is much less exergonic (by only 10.0 kcal mol−1) to generate the ether product 14. Therefore, the results indicate that the C–O RE pathway is kinetically favorable while the transmetalation pathway is thermodynamically advantageous. Because no C–O coupling product was observed in the reported experiment,24 this interesting phenomenon led us to hypothesize a “thermodynamic control” situation of the reaction.52–56 That is, although the formation of ether through C–O reductive elimination is slightly faster (lower barrier), this can be considered as a reversible pathway albeit with a slightly high reverse barrier and the reaction would lead to the palladacycle complex eventually because the latter is a much more thermodynamically stable product. To the best of our knowledge, this is the first time that the thermodynamic-control-situation is proposed as a likely mechanism in the silacycle-based cross coupling reactions.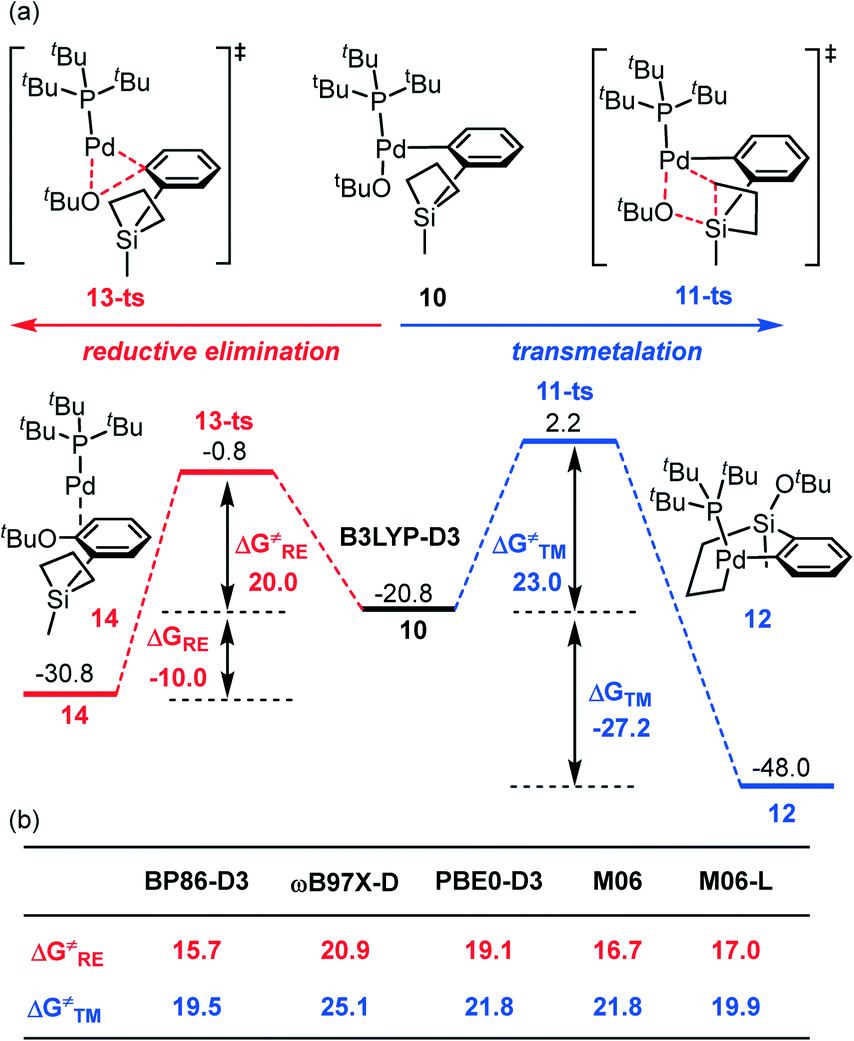 | ||
| Fig. 3 (a) Energy profiles for the competing transmetalation and the C–O reductive elimination (3D geometries for the key intermediates and transition states are shown in the ESI†); (b) the validation with different density functional methods. | ||
It should be also noted that the finding about the kinetically more accessible C–O reductive elimination is consistent with many experiments. For instance, in the palladium-catalyzed cross-coupling of aryl halide with O or N nucleophiles, this often exists as a common reaction pathway.57–61 The reason why in this palladium-catalyzed silacycle-based cross coupling reaction the C–O coupling has been inhibited is because the transmetalation product is much more stable to drive the reaction to proceed through a “thermodynamic control scenario” and to lead to the C–C coupling direction. More importantly, our “thermodynamic control” conclusion finds support from the utilized reaction conditions in which a reasonably long reaction time (24 h) and high temperature (80 °C) are critical for the efficiency of this transformation.24
To obtain unambiguous results from the computation, we have investigated the competing transition states with other density functional theory methods, including BP86-D3,62–64 ωB97X-D,65 PBE0-D3,66 M06,67 and M06-L68 (Fig. 3b). All the results demonstrate that the transmetalation has a higher barrier (ranging from 2.7 to 5.1 kcal mol−1) than the C–O reductive elimination and therefore validates the “thermodynamic control” hypothesis in the palladium-catalyzed silacycle-based cross-coupling reaction.
4. C–C reductive elimination
The following C–C reductive elimination would generate the C–C coupling product. This step bears a small free energy barrier of 6.9 kcal mol−1 associated with 15-ts and is highly exergonic by 35.1 kcal mol−1 to produce 16 (Fig. 4). Finally, a second equivalent of silacyclobutane substrate (1) coordination would release the product (Prod) and regenerate the reactive species facilely.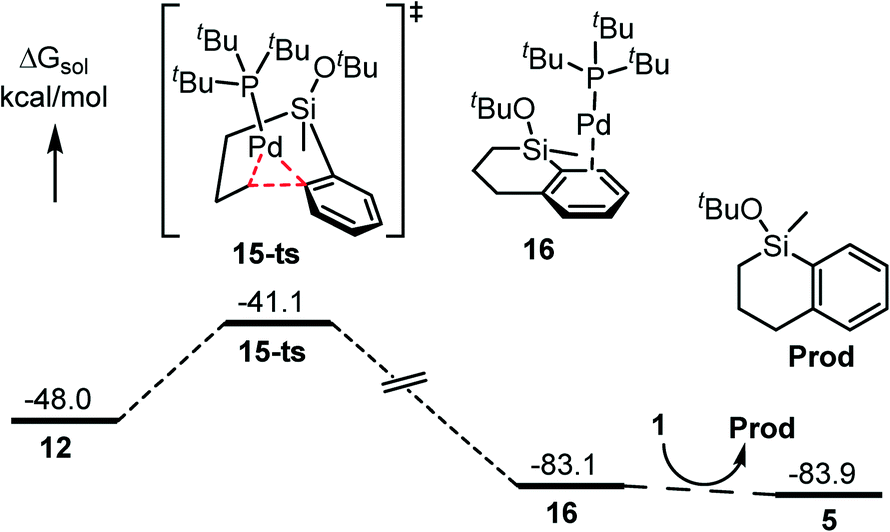 | ||
| Fig. 4 Energy profile for the reductive elimination and product release step (energies are relative to Pd(PtBu3)2). | ||
5. Reaction with silacyclopentane and the solvent effect
In the experiment, when silacyclopentane was used as the substrate, the cross coupling product was observed only after changing the nucleophile from tBuOH to CyOH.24 Thus, it is of much interest to investigate the origin of the distinct reactivities with different nucleophiles. Since the rate-determining step is transmetalation, we calculated the pathways associated with transmetalation with both tBuO− (from tBuOH) and CyO− (from CyOH). As can be seen from Fig. 5, with silacyclopentane, the transmetalation step with tBuO− bears a dramatically high activation barrier of 37.2 kcal mol−1 (red pathway, via18-ts), which is not practical under the reaction conditions. On the other hand, with the CyO− nucleophile, the activation barrier decreases to 31.9 kcal mol−1 (blue pathway, via21-ts) which is 5.3 kcal mol−1 more favorable and is more feasible considering the utilized conditions (e.g., 150 °C, 24 h). Our calculation is consistent with the experiment that the reaction of silacyclopentane would occur only by changing tBuOH to CyOH. | ||
| Fig. 5 Transmetalation step for silacyclopentane with tBuO− (red) and CyO− (blue) as the nucleophiles (ΔG is given in kcal mol−1). | ||
Notably, with silacyclopentane, the transmetalation barriers (with tBuO− or CyO−) are higher than those with the silacyclobutane, which can be attributed to the lower reactivity induced by the less-ring-constrain of the five-membered silacyclopentane compared to the four-membered silacyclobutane.
To better understand the origin of the distinct reactivity with the two nucleophiles, we have performed the distortion interaction analysis (Fig. 6a).69,70 It is seen that the larger distortion of both the sub part and the cat part in 18-ts mainly causes the high activation barrier. By the analysis of the transition state structures (Fig. 6b), it is seen that the H1–H3, H2–H3, and H4–H5 bond distances are 2.102, 2.087, and 2.109 Å in 18-ts, which are much shorter than those in 21-ts (2.142, 2.424, and 2.253 Å, respectively), indicating that the tBu group has larger steric repulsion with both the silacyclopentane and the phosphine ligand compared to that of the Cy group and therefore would induce larger distortions. This is also consistent with the steric A value of tBu (>4) and Cy (2.15).71 Thus, the steric effect and the steric-effect-induced-larger-distortion in the rate-determining transmetalation transition states account for the contrasting reactivity with different nucleophiles.
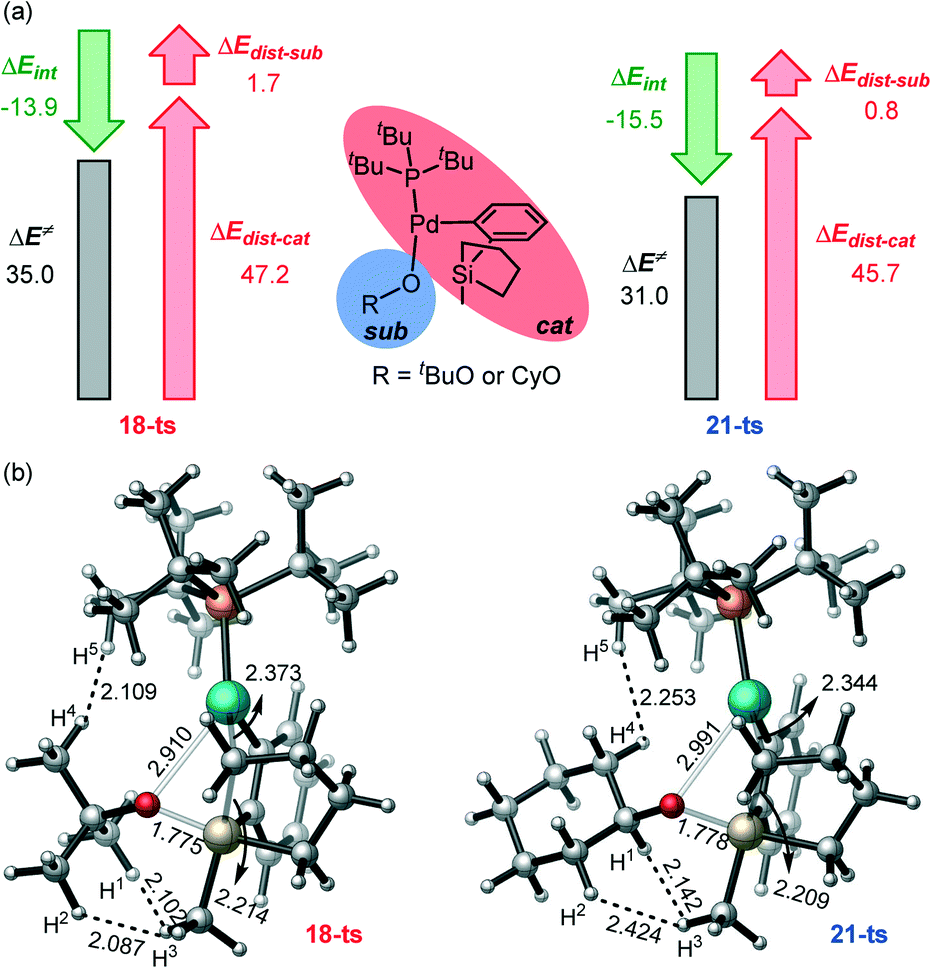 | ||
| Fig. 6 (a) The distortion–interaction analysis of the transmetalation transition states with tBuO− and CyO− as nucleophiles; (b) the 3D geometries of the transition states 18-ts and 21-ts (bond distances are given in Å). | ||
6. Proof of the “strain release Lewis acidity” and elucidation of the role of the base
The four-coordinated silacyclobutane has strong Lewis acidity and it would favorably interact with a nucleophile to generate a penta-coordinated species, which is known as “strain release Lewis acidity”.72,73 Moreover, as shown from previous studies,74,75 the polarization of the Si–C bond would promote the transmetalation process. Therefore, we investigated the activation of the silacycle by coordinating to an additional base anion (X−, Fig. 7). In this scenario, we studied both the transmetalation and the competing reductive elimination pathways from the penta-coordinated silicon species. | ||
| Fig. 7 (a) The possible transmetalation and reductive elimination pathways with additional base; (b) the transmetalation and reductive elimination barriers with different bases. | ||
Compared to that without coordinating a base, the addition of OtBu leads to a slightly unstable (by 3.9 kcal mol−1) intermediate 10–OtBu compared to 10. The following transmetalation and reductive elimination have free energy barriers of 23.0 and 20.4 kcal mol−1 relative to 10–OtBu, and the overall barriers are 26.9 and 24.3 kcal mol−1, which are unfavorable compared to that without OtBu coordination (23.0 and 20.0 kcal mol−1, respectively, Fig. 7). This is likely due to the large steric repulsion between the tert-butyl group of the base and the P(tBu)3 ligand as well as the Pd-bound OtBu group.
Conversely, binding of NEt2 (from TMSNEt2) leads to a more thermodynamically stable (by 2.0 kcal mol−1) complex, 10-NEt2. Compared with that of OtBu, NEt2 has a stronger nucleophicility and relatively smaller steric bulkiness; therefore, the coordination of the NEt2 ligand is consistent with the “strain release Lewis acidity” rule and thus makes the adduct more favorable. Indeed, the four-coordinated silacyclobutane is in a tetrahedron geometry (10, Scheme 3), in which the expected and calculated C–Si–C angles are 109.5° and 78.5° respectively, indicating that this is a highly strained complex. In contrast, after binding with the fifth ligand, NEt2, the penta-coordinated complex changes to a trigonal bipyramidal geometry (10-NEt2, Scheme 3). Thus, the expected C–Si–C angle decreases to 90°, and the actual angle is calculated to be 70.7°. The remarkable decrease in the difference between the expected and actual angles in 10-NEt2 (19.3°) compared to that in 10 (31.0°) implies the release of the strain.
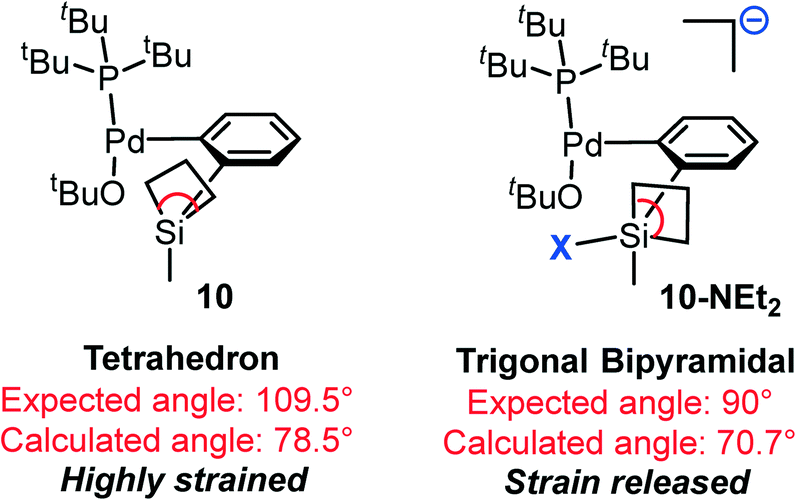 | ||
| Scheme 3 Schematic representation of the angle “strain release” concept by coordinating a fifth ligand. | ||
From 10-NEt2, the subsequent transmetalation and reductive elimination bear barriers of 21.4 and 18.3 kcal mol−1 with respect to 10-NEt2 (Fig. 7). In other words, the binding of NEt2 decreases both the transmetalation and reductive elimination barriers. On the one hand, the binding of the NEt2 anion increases both the electron density of the substrate and the steric hindrance around the Pd coordination sphere, thus promoting the reductive elimination process. On the other hand, this binding activates the silacycle, which is indicated by the much longer Si–C bond distance in 11-NEt2-ts (2.484 Å, Fig. 8) than in 11-ts (2.220 Å), and facilitates the transmetalation event. It should be mentioned that regardless of tetravalent or pentavalent (with binding of different bases) silicon, the free energy barrier difference between ΔG‡TM and ΔG‡RE remain nearly the same (without base: 3.0 kcal mol−1; with OtBu: 2.6 kcal mol−1; with NEt2: 3.1 kcal mol−1), indicating an inherent kinetic preference of the C–O reductive elimination compared to the transmetalation.
 | ||
| Fig. 8 Geometries for the key transition states 11-ts and 11-NEt2-ts (bond distances are given in Å). | ||
Furthermore, we have predicted that for the amine base with an even smaller steric bulkiness, NMe2, the free energy barrier for the transmetalation would further be lowered down (to 20.3 kcal mol−1, Fig. 7), providing insights on enhancing the reaction efficiency by utilizing other kinds of organic bases. Thus, our calculations show that the base (i.e., NEt2 anion) is essential to bind to Si to stabilize the silacycles (via “strain release”) and to lower the activation barriers for both the transmetalation and reductive elimination.
Conclusions
In conclusion, we have performed a comprehensive theoretical study on the mechanism of the palladium-catalyzed strain-release organosilicon-based cross-coupling reaction. We find that:(1) the reaction pathway with one bulky phosphine ligand is favorable than that with two. The most favorable pathway proceeds via Ar–Br oxidative addition, Br-to-tBuO ligand exchange, transmetalation, and reductive elimination steps (path II, Scheme 2).
(2) The chemical selectivity in favor of the C–C cross-coupling rather than the competing C–O reductive elimination is ascribed to the thermodynamic control feature of the two pathways, i.e., although the transmetalation leading to C–C cross-coupling has a slightly higher barrier, the corresponding product is much more stable than the C–O RE product.
(3) The steric effect between the nucleophiles and the ligand as well as the substrate accounts for the distinct reactivity in the reaction with silacyclopentane.
(4) The base plays vital roles in decreasing barriers for both the transmetalation and reductive elimination steps and enhancing reactivities through binding to the silicon center. The angle strain release concept with four- and five-coordinated silicon compounds has been demonstrated. Furthermore, −NMe2 is predicted to be a better base in the palladium-catalyzed strain-release organosilicon-based cross-coupling reaction.
The findings from this study will highlight the “thermodynamic control scenario” in the understanding of reactivity and selectivity in palladium-catalyzed organosilicon-based cross coupling reactions and shed light on the development of more efficient transition-metal catalyzed ring-opening and ring expansion transformations of silacycles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (NSFC 21702126 and 21801155).Notes and references
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, 81st edn, 2000, ISBN 0-8493-0481-4 Search PubMed.
- R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Polyferrocenylsilanes: Synthesis, Properties, and Applications, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC.
- E. Remond, C. Martin, J. Martinez and F. Cavelier, Silicon-Containing Amino Acids: Synthetic Aspects, Conformational Studies, and Applications to Bioactive Peptides, Chem. Rev., 2016, 116, 11654–11684 CrossRef CAS.
- T. A. Su, H. Li, R. S. Klausen, N. T. Kim, M. Neupane, J. L. Leighton, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Silane and Germane Molecular Electronics, Acc. Chem. Res., 2017, 50, 1088–1095 CrossRef CAS PubMed.
- R. Ramesh and D. S. Reddy, Quest for Novel Chemical Entities through Incorporation of Silicon in Drug Scaffolds, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed.
- S. Patai and Z. Rappoport, The Chemistry of Organic Silicon Compounds, J. Wiley and Sons, New York, 1989, p. 1015 Search PubMed.
- Y. Qin, L. Li, J.-Y. Liang, K. Li and D. Zhao, Silacyclization through Palladium-catalyzed Intermolecular Silicon-based C(sp2)–C(sp3) Cross-coupling, Chem. Sci., 2021, 12, 14224–14229 RSC.
- X.-C. Wang, H.-R. Wang, X. Xu and D. Zhao, Ring Expansion to 8-Membered Silacycles through Formal Cross-Dimerization of 5-Membered Palladacycles with Silacyclobutanes, Eur. J. Org. Chem., 2021, 21, 3039–3042 CrossRef.
- J. Zhang, N. Yan, C.-W. Ju and D. Zhao, Nickel(0)-Catalyzed Asymmetric Ring Expansion Toward Enantioenriched Silicon-Stereogenic Benzosiloles, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202111025.
- W.-T. Zhao, F. Gao and D. Zhao, Intermolecular σ-Bond Cross-Exchange Reaction between Cyclopropenones and (Benzo)silacyclobutanes: Straightforward Access towards Sila(benzo)cycloheptenones, Angew. Chem., Int. Ed., 2018, 57, 6329–6332 CrossRef CAS PubMed.
- H. Chen, Y. Chen, X. Tang, S. Liu, R. Wang, T. Hu, L. Gao and Z. Song, Rhodium-Catalyzed Reaction of Silacyclobutanes with Unactivated Alkynes to Afford Silacyclohexenes, Angew. Chem., Int. Ed., 2019, 58, 4695–4699 CrossRef CAS PubMed.
- X. Wang and D. Zhao, Palladium-Catalyzed[4+2] Annulation of Cyclopropenes with Benzosilacyclobutanes, Wuji Huaxue Xuebao, 2020, 40, 1080–1081 CAS.
- S. E. Denmark, B. D. Griedel, D. M. Coe and M. E. Schnute, Chemistry of Enoxysilacyclobutanes: Highly Selective Uncatalyzed Aldol Additions, J. Am. Chem. Soc., 1994, 116, 7026–7043 CrossRef CAS.
- J. W. A. Kinnaird, P. Y. Ng, K. Kubota, X. Wang and J. L. Leighton, Strained Silacycles in Organic Synthesis: A New Reagent for the Enantioselective Allylation of Aldehyde, J. Am. Chem. Soc., 2002, 124, 7920–7921 CrossRef CAS.
- X. Zhang, K. N. Houk and J. L. Leighton, Origins of Stereoselectivity in Strain–Release Allylations, Angew. Chem., Int. Ed., 2005, 44, 938–941 CrossRef CAS PubMed.
- G. Raabe and J. Michl, Multiple Bonding to Silicon, Chem. Rev., 1985, 85, 419–509 CrossRef CAS.
- K. Tanino, M. Takahashi, Y. Tomata, H. Tokura, T. Uehara, T. Narabu and M. Miyashita, Total Synthesis of Solanoeclepin A, Nat. Chem., 2011, 3, 484–488 CrossRef CAS.
- Q.-C. Mu, J. Chen, C.-G. Xia and L.-W. Xu, Synthesis of Silacyclobutanes and Their Catalytic Transformations Enabled by Transition-metal Complexes, Coord. Chem. Rev., 2018, 374, 93–113 CrossRef CAS.
- R. Shintani, K. Moriya and T. Hayashi, Palladium-Catalyzed Desymmetrization of Silacyclobutanes with Alkynes: Enantioselective Synthesis of Silicon-Stereogenic 1-Sila-2-cyclohexenes and Mechanistic Considerations, Org. Lett., 2012, 14, 2902–2905 CrossRef CAS.
- Q.-W. Zhang, K. An, L.-C. Liu, S. Guo, C. Jiang, H. Guo and W. He, Rhodium-Catalyzed Intramolecular C−H Silylation by Silacyclobutanes, Angew. Chem., Int. Ed., 2016, 55, 6319–6323 CrossRef CAS PubMed.
- N. Hirone, H. Sanjiki, R. Tanaka, T. Hata and H. Urabe, Acceleration of the Substitution of Silanes with Grignard Reagents by Using either LiCl or YCl3/MeLi, Angew. Chem., Int. Ed., 2010, 49, 7762–7764 CrossRef CAS.
- K. Ouyang, Y. Liang and Z. Xi, Construction of Benzosiloles, Six- and Eight-Membered Silacyclic Skeletons, via a Pd-Catalyzed Intramolecular Mizoroki–Heck Reaction of Vinylsilanes, Org. Lett., 2012, 14, 4572–4575 CrossRef CAS.
- K. Takamoto, S. Yoshioka, H. Fujioka and M. Arisawa, Palladium-Catalyzed Seven-Membered Silacycle Construction: 1,7-Enyne Hydroxycyclization to Give a Benzosilepine Skeleton, Org. Lett., 2018, 20, 1773–1776 CrossRef CAS PubMed.
- Y. Qin, J.-L. Han, C.-W. Ju and D. Zhao, Ring Expansion to 6-, 7-, and 8-Membered Benzosilacycles through Strain-Release Silicon-Based Cross-Coupling, Angew. Chem., Int. Ed., 2020, 59, 8481–8485 CrossRef CAS.
- Y. Hatanaka and T. Hiyama, Cross-coupling of Organosilanes with Organic Halides Mediated by a Palladium Catalyst and Tris(diethylamino)sulfonium Difluorotrimethylsilicate, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS.
- S. E. Denmark and C. S. Regens, Palladium-Catalyzed Cross-Coupling Reactions of Organosilanols and their Salts, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS PubMed.
- L. Zhang, K. An, Y. Wang, Y.-D. Wu, X. Zhang, Z.-X. Yu and W. He, A Combined Computational and Experimental Study of Rh-Catalyzed C–H Silylation with Silacyclobutanes: Insights Leading to a More Efficient Catalyst System, J. Am. Chem. Soc., 2021, 143, 3571–3582 CrossRef CAS.
- H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Palladium-Catalysed Cross-Coupling of Organosilicon Reagents, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
- L. Zhang and D.-C. Fang, Catalytic C–H Activation/C–C Coupling Reaction: DFT Studies on the Mechanism, Solvent Effect, and Role of Additive, J. Org. Chem., 2013, 78, 2405–2412 CrossRef CAS.
- C. P. Johnston, T. H. West, R. E. Dooley, M. Reid, A. B. Jones, E. J. King, A. G. Leach and G. C. Lloyd-Jones, Anion-Initiated Trifluoromethylation by TMSCF3: Deconvolution of the Siliconate–Carbanion Dichotomy by Stopped-Flow NMR/IR, J. Am. Chem. Soc., 2018, 140, 11112–11124 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, F. Liu, G. He, J. Zhang, K. N. Houk, A. B. Smith III and Y. Liang, The Role of CuI in the Siloxane-mediated Pd-catalyzed Cross-coupling Reactions of Aryl Iodides with Aryl Lithium Reagents, Chin. Chem. Lett., 2021, 32, 441–444 CrossRef CAS PubMed.
- G. Mann and J. F. Hartwig, Palladium Alkoxides: Potential Intermediacy in Catalytic Amination, Reductive Elimination of Ethers, and Catalytic Etheration. Comments on Alcohol Elimination from Ir(III), J. Am. Chem. Soc., 1996, 118, 13109–13110 CrossRef CAS.
- M. Palucki, J. P. Wolfe and S. L. Buchwald, Palladium-Catalyzed Intermolecular Carbon−Oxygen Bond Formation: A New Synthesis of Aryl Ethers, J. Am. Chem. Soc., 1997, 119, 3395–3396 CrossRef CAS.
- A. V. Vorogushin, X. Huang and S. L. Buchwald, Use of Tunable Ligands Allows for Intermolecular Pd-Catalyzed C−O Bond Formation, J. Am. Chem. Soc., 2005, 127, 8146–8149 CrossRef CAS PubMed.
- S. Gowrisankar, A. G. Sergeev, P. Anbarasan, A. Spannenberg, H. Neumann and M. Beller, A General and Efficient Catalyst for Palladium-Catalyzed C−O Coupling Reactions of Aryl Halides with Primary Alcohols, J. Am. Chem. Soc., 2010, 132, 11592–11598 CrossRef CAS PubMed.
- P. E. Maligres, J. Li, S. W. Krska, J. D. Schreier and I. T. Raheem, C–O Cross–Coupling of Activated Aryl and Heteroaryl Halides with Aliphatic Alcohols, Angew. Chem., Int. Ed., 2012, 51, 9071–9074 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2009 (see ESI for full citation) Search PubMed.
- P. J. Hay and W. R. Wadt, Ab initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, Ab initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- A. D. Becke, Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, A new mixing of Hartree–Fock and Local Density-Functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009 (http://www.cylview.org) Search PubMed.
- F. Proutiere, E. Lyngvi, M. Aufiero, I. A. Sanhueza and F. Schoenebeck, Combining the Reactivity Properties of PCy3 and PtBu3 into a Single Ligand, P(iPr)(tBu)2. Reaction via Mono- or Bisphosphine Palladium(0) Centers and Palladium(I) Dimer Formation, Organometallics, 2014, 33, 6879–6884 CrossRef CAS.
- K. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, Oxidative Additions of Aryl Halides to Palladium Proceed through the Monoligated Complex, ChemCatChem, 2013, 5, 3604–3609 CrossRef CAS.
- I. Funes-Ardoiz, D. J. Nelson and F. Maseras, Halide Abstraction Competes with Oxidative Addition in the Reactions of Aryl Halides with [Ni(PMenPh(3−n))4], Chem. – Eur. J., 2017, 23, 16728–16733 CrossRef CAS.
- C. L. McMullin, N. Fey and J. N. Harvey, Computed Ligand Effects on the Oxidative Addition of Phenyl Halides to Phosphine Supported Palladium(0) Catalysts, Dalton Trans., 2014, 43, 13545–13556 RSC.
- F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, Autocatalytic Oxidative Addition of PhBr to Pd(PtBu3)2 via Pd(PtBu3)2(H)(Br), J. Am. Chem. Soc., 2008, 130, 5842–5843 CrossRef.
- F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, Effect of Ligand Steric Properties and Halide Identity on the Mechanism for Oxidative Addition of Haloarenes to Trialkylphosphine Pd(0) Complexes, J. Am. Chem. Soc., 2009, 131, 8141–8154 CrossRef CAS PubMed.
- P. Sykes and P. P. Hall, A Guidebook to Mechanism in Organic Chemistry, 6th edn, 1986, ISBN 0–582–44695–3 Search PubMed.
- R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, The Importance of Kinetic and Thermodynamic Control when Assessing Mechanisms of Carboxylate-Assisted C–H Activation, J. Am. Chem. Soc., 2019, 141, 8896–8906 CrossRef CAS PubMed.
- S. R. Marsden, L. Mestrom, D. G. G. McMillan and U. Hanefeld, Thermodynamically and Kinetically Controlled Reactions in Biocatalysis – from Concepts to Perspectives, ChemCatChem, 2019, 12, 426–437 CrossRef.
- J. K. Lam, H. V. Pham, K. N. Houk and C. D. Vanderwal, Computation and Experiment Reveal That the Ring-Rearrangement Metathesis of Himbert Cycloadducts Can Be Subject to Kinetic or Thermodynamic Control, J. Am. Chem. Soc., 2013, 135, 17585–17594 CrossRef CAS PubMed.
- L. Omann, B. Pudasaini, E. Irran, H. F. T. Klare, M.-H. Baik and M. Oestreich, Thermodynamic versus Kinetic Control in Substituent Redistribution Reactions of Silylium Ions Steered by the Counteranion, Chem. Sci., 2018, 9, 5600–5607 RSC.
- G. Mann and J. F. Hartwig, Palladium Alkoxides: Potential Intermediacy in Catalytic Amination, Reductive Elimination of Ethers, and Catalytic Etheration. Comments on Alcohol Elimination from Ir(III), J. Am. Chem. Soc., 1996, 118, 13109–13110 CrossRef CAS.
- M. Palucki, J. P. Wolfe and S. L. Buchwald, Palladium-Catalyzed Intermolecular Carbon−Oxygen Bond Formation: A New Synthesis of Aryl Ethers, J. Am. Chem. Soc., 1997, 119, 3395–3396 CrossRef CAS.
- A. V. Vorogushin, X. Huang and S. L. Buchwald, Use of Tunable Ligands Allows for Intermolecular Pd-Catalyzed C−O Bond Formation, J. Am. Chem. Soc., 2005, 127, 8146–8149 CrossRef CAS PubMed.
- S. Gowrisankar, A. G. Sergeev, P. Anbarasan, A. Spannenberg, H. Neumann and M. Beller, A General and Efficient Catalyst for Palladium-Catalyzed C−O Coupling Reactions of Aryl Halides with Primary Alcohols, J. Am. Chem. Soc., 2010, 132, 11592–11598 CrossRef CAS.
- P. E. Maligres, J. Li, S. W. Krska, J. D. Schreier and I. T. Raheem, C–O Cross-Coupling of Activated Aryl and Heteroaryl Halides with Aliphatic Alcohols, Angew. Chem., Int. Ed., 2012, 51, 9071–9074 CrossRef CAS.
- A. D. Becke, Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Density-functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- J.-D. Chai and M. Head-Gordon, Systematic Optimization of Long-range Corrected Hybrid Density Functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, A New Local Density Functional for Main-group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef.
- D. H. Ess and K. N. Houk, Distortion/interaction Energy Control of 1,3-dipolar Cycloaddition Reactivity, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS PubMed.
- L.-P. Xu, J. B. Roque, R. Sarpong and D. G. Musaev, Reactivity and Selectivity Controlling Factors in the Pd/Dialkylbiarylphosphine-Catalyzed C–C Cleavage/Cross-Coupling of an N-Fused Bicyclo α-Hydroxy-β-Lactam, J. Am. Chem. Soc., 2020, 142, 21140–21152 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley-Interscience, New York, 1st edn, 1994 Search PubMed.
- S. E. Denmark, R. T. Jacobs, G. Dai-Ho and S. Wilson, Synthesis, Structure, and Reactivity of An Organogermanium Lewis Acid, Organometallics, 1990, 9, 3015–3019 CrossRef CAS.
- X. Zhang, K. N. Houk and J. L. Leighton, Origins of Stereoselectivity in Strain-Release Allylations, Angew. Chem., Int. Ed., 2005, 44, 938–941 CrossRef CAS PubMed.
- S. E. Denmark and R. F. Sweis, Design and Implementation of New, Silicon-Based, Cross-Coupling Reactions: Importance of Silicon−Oxygen Bonds, Acc. Chem. Res., 2002, 35, 835–846 CrossRef CAS.
- S. A. Tymonko, R. C. Smith, A. Ambrosi and S. E. Denmark, Mechanistic Significance of the Si–O–Pd Bond in the Palladium-Catalyzed Cross-Coupling Reactions of Alkenylsilanolates, J. Am. Chem. Soc., 2015, 137, 6192–6199 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Energetics and Cartesian coordinates for all reported structures. See DOI: 10.1039/d1cy01890f |
| This journal is © The Royal Society of Chemistry 2022 |