
Photocatalytic valorization of furfural to value-added chemicals via mesoporous carbon nitride: a possibility through a metal-free pathway†
Deepak K.
Chauhan
a,
Venugopala R.
Battula
a,
Arkaprabha
Giri
b,
Abhijit
Patra
 b and
Kamalakannan
Kailasam
*a
b and
Kamalakannan
Kailasam
*a
aAdvanced Functional Nanomaterials, Energy and Environment Unit, Institute of Nano Science and Technology (INST), Knowledge city, Sector-81 Manauli, SAS Nagar, 140306 Mohali, Punjab, India. E-mail: kamal@inst.ac.in; kkamal17@gmail.com
bDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhauri, Bhopal 462066, Madhya Pradesh, India
First published on 17th November 2021
Abstract
Strategizing the exploitation of renewable solar light could undoubtedly provide new insight into the field of biomass valorization. Therefore, for the first time, we reported a heterogeneous photocatalytic oxidation route of renewable furfural (FUR) to produce industrial feedstocks maleic anhydride (MAN) and 5-hydroxy-2(5H)-furanone (HFO) under simulated solar light (AM 1.5G) using molecular oxygen (O2) as a terminal oxidant and mesoporous graphitic carbon nitride (SGCN) as a photocatalyst. SGCN showed an excellent photoconversion (>95%) of FUR with 42% and 33% selectivity to MAN and HFO, respectively. Moreover, an excellent selectivity towards MAN (66%) under natural sunlight indicates a pioneering route for the sustainable production of MAN. In addition, the underlying mechanistic route of the FUR photo-oxidation was investigated via various experiments including scavenger studies, substrate studies, and electron spin resonance (ESR) studies which constructively proved the pivotal role of singlet oxygen (1O2) and holes (h+) in FUR photo-oxidation.
1. Introduction
Voluminous biomass on earth provides a tremendous renewable feedstock for upgrading biomass derivatives into fuels and value-added chemicals.1,2 A sister chemical to 5-hydroxymethylfurfural (HMF),3i.e., C5 furfural (FUR), is an indispensable biomass-derived platform chemical. As compared to HMF, FUR has been considered an advantageous starting material for biomass conversion into a wide range of upgraded products.4–6 FUR oxidation could provide vital feedstock production of maleic anhydride (MAN),7 maleic acid (MA),8 succinic acid (SA),9 and furanones10 which have shown great importance in industry and are widely utilized for large scale production of pharmaceuticals, lubricant additives, unsaturated polyester resins, vinyl copolymers, plasticizers and so on.Previous reports demonstrated different strategies including the gas-phase oxidation,11,12 chemical oxidation,13,14 and electro-oxidation10,15 reactions of FUR into MAN, furanones, and other acid derivatives.16,17 Even though adequate driving potential is required for FUR oxidation into MAN and furanones, these strategies may suffer from some limitations comprising the use of elevated temperature,11 corrosive or expensive oxidants,14,18 (electro)catalysts (metals, metal oxides, and metal chalcogenides),10,15 and a noble metal (Pt)19 in the electro-oxidation process.
Photocatalysis has received worldwide attention due to its economic, safe, clean, and renewable characteristic properties.20,21 It embodies to accelerate an otherwise kinetically/thermal hindered reaction under light illumination.22,23 Moreover, photocatalytic driven pathways rule out most of the aforementioned limitations. In recent years, polymeric carbon nitride (commonly called g-C3N4)24 has turned out to be a hot choice as a photocatalyst because of its high thermal and chemical photostability and ease-of-synthesis procedure compared to metal oxides or chalcogenides.25 In recent studies, emphasis has been given to the utilization of the potential of carbon nitride-based semiconductors in organic photo-redox transformations under visible light irradiation.26–28 However, photo-oxidation of FUR into MAN and furanones over g-C3N4 is still challenging and in its infancy. Therefore, rigorous scientific endeavors have to be carried out to achieve sufficient insights towards FUR photo-oxidation in the presence of g-C3N4 as a semiconductor. Furthermore, the importance of the FUR photo-oxidation could be demonstrated by the feasible production of industrially value-added products, offering inexpensive and highly sustainable pathways as compared to the conventional thermal and electrochemical pathways. Probably, a wide range of value added products as shown in Scheme 1 can be produced from FUR photo-oxidation by changing the reaction parameters such as the choice of solvents, catalysts, and the oxidant environment.
 | ||
| Scheme 1 Schematic illustration showing the photo-oxidized valuable products derived from FUR (biomass feedstock). | ||
To the best of our knowledge, no report has shown the photo-oxidation of FUR into valuable MAN and furanones with mesoporous carbon nitride. Progress has been made in photo-oxidation of FUR to furanones where a homogeneous catalysis route was employed only during the overall synthesis of surfactants or drugs in the presence of dyes or photosensitizers.29,30
However, such homogeneous hazardous photosensitizers and their recyclability severely limited the photocatalytic application processes to a further extent. Therefore, we report herein the most efficient and easiest ways for photo-oxidation of FUR into valuable MAN and furanone (5-hydroxy-2(5H)-furanone, i.e., HFO) with the help of heterogeneous mesoporous carbon nitride (SGCN) as a visible light photocatalyst. The state-of-the-art SGCN photocatalyst has been compared with the existing thermally driven catalysts along with Rose Bengal as the only reported photocatalyst for FUR oxidation (Table S2, ESI†), showing the superiority of SGCN to the existing photo(catalysts). FUR could be photo-oxidized with high conversion (>95%) under ambient O2 conditions and using a solar simulator in the presence of an acetonitrile medium (ACN). In addition, aside from the major FUR photo-oxidized products, i.e., MAN and HFO, a minor amount of succinic anhydride (SAN) and cis-beta-formylacrylic acid (β-FAA) could also be co-produced during the reaction.
Notably, the mechanistic investigation of FUR photo-oxidation into MAN and HFO has been proven by control and scavenging studies, followed by the typical electron spin resonance (ESR) technique. Overall, this first successful heterogeneous photocatalytic FUR biomass transformation into value-added products displayed the most efficient and sustainable protocol. However, aiming towards excellent selectivity to oxidized value-added products could still be the most challenging task under light irradiation as presented in this study. Therefore, considerable attention must be given to strategizing not only the FUR oxidation but also the oxidation of biomass-based furan derivatives towards high selectivity products under solar light irradiation in the near future.
2. Experimental section
2.1. Materials
Cyanamide (CA), tetraethoxy orthosilicate (TEOS), ammonium hydrogen difluoride (NH4HF2), furoic acid and furan were purchased from Sigma Aldrich, India. HCl (35%, Fisher Scientific) and ethanol (99.99%, analytical grade, supplier Changshu Hongsheng Fine Chemicals) were purchased and used during the synthesis procedure. Furfural (FUR), p-benzoquinone (BQ), sodium azide (NaN3), potassium iodide (KI), 1,5-dihydroxynaphthalene (1,5-DHN), 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were purchased from TCI Chemicals, India. All the chemicals were used without further purification.2.2. Synthesis procedure of SGCN
The synthesis procedure of mesoporous carbon nitride (SGCN) was adapted according to the previous literature.31 Firstly, CA and TEOS were taken at a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and dissolved in 0.01 M aqueous HCl (4 g) and ethanol (4 g). The pH of the resultant mixture was maintained to 2 by adding HCl dropwise. After that, the solution mixture was stirred for 30 min and the solvent was evaporated by rotary evaporation and subsequently dried at 80 °C for 24 h. The obtained solid was further calcined at 550 °C under an inert atmosphere with a ramping rate of 5 °C min−1 for 4 h to obtain the mesoporous carbon nitride–silica composite. The composite was washed with 4 M ammonium bifluoride (NH4HF2) solution for 48 h to remove the silica followed by washing with water and ethanol several times to obtain pure mesoporous carbon nitride (SGCN). The obtained SGCN was dried in an oven overnight at 120 °C.
:1 molar ratio and dissolved in 0.01 M aqueous HCl (4 g) and ethanol (4 g). The pH of the resultant mixture was maintained to 2 by adding HCl dropwise. After that, the solution mixture was stirred for 30 min and the solvent was evaporated by rotary evaporation and subsequently dried at 80 °C for 24 h. The obtained solid was further calcined at 550 °C under an inert atmosphere with a ramping rate of 5 °C min−1 for 4 h to obtain the mesoporous carbon nitride–silica composite. The composite was washed with 4 M ammonium bifluoride (NH4HF2) solution for 48 h to remove the silica followed by washing with water and ethanol several times to obtain pure mesoporous carbon nitride (SGCN). The obtained SGCN was dried in an oven overnight at 120 °C.
2.3. Analytical techniques
The diffraction pattern of the material was determined by powder XRD using a Bruker D8 Advance diffractometer equipped with a scintillation counter detector, with a Cu-Kα radiation (λ = 0.15418 nm) source operating at 40 kV and 40 mA. The diffuse reflectance spectrum of the solid sample was recorded on a UV-visible spectrophotometer, Shimadzu UV-2600, which was operated in a solid-state mode with barium sulfate as a reference. The BET surface area and pore size distribution were estimated by the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods on an Autosorb iQ3 instrument (Quantachrome). Transmission electron microscopy (TEM) analysis was carried out using a JEOL operated at an accelerating voltage of 200 kV. Analyses of various functional groups and different vibrational spectra were conducted by Fourier transform infrared-attenuated total reflection (FTIR-ATR) spectroscopy by using a Bruker VERTEX70 instrument. X-ray photoelectron spectroscopy (XPS) was further carried out to realize the chemical states and elemental compositions of the sample by using a PHI 5000 Versa Prob II instrument. Electrochemical analysis was performed in a standard three-electrode system, using a platinum electrode, Ag/AgCl (in saturated KCl) electrode, and SGCN coated glassy carbon electrode as counter, reference and working electrodes, respectively. The measurement was recorded via a Metrohm Autolab (M204 multichannel potentiostat galvanostat). Electron spin resonance (ESR) analysis was carried out to estimate the production of reactive oxygen species (ROS) by using a Bruker EMX Microx spectrometer at a frequency of 100 kHz and a modulation amplitude of 0.2 G. After the reaction, an estimation of the gaseous products COx was done by gas chromatography (Perkin Elmer Clarus 680) and the analysis of the liquid samples such as the conversion of furfural and the selectivity to the products was performed using a Shimadzu gas chromatograph-mass spectrometer (GC-MS) equipped with an SH-Rxi-5Sil MS column.2.4. General reaction procedure
Photocatalytic oxidation of biomass-derived FUR was performed in a homemade glass round bottom flask under a solar simulator (AM 1.5), 100 mW cm−2. In a typical reaction, a certain amount of catalyst was taken in 5 ml acetonitrile (HPLC grade) containing 0.11 mmol of substrate i.e., FUR. Further, the solution was purged with O2 at atmospheric pressure and kept under the solar simulator (AM 1.5), 100 mW cm−2. After the reaction, the gaseous products COx were measured and quantified by gas chromatography (Perkin Elmer Clarus 680). The liquid mixture was simply separated from the catalyst by centrifugation and analyzed using a Shimadzu gas chromatograph-mass spectrometer (GC-MS). Meanwhile, the suspended catalyst was collected and washed with copious amounts of water–ethanol solution. After that, it was dried at 120 °C and further characterized for subsequent cycles.3. Results and discussion
3.1. Characterization
The XRD pattern of SGCN (Fig. 1a) shows an intense peak at 27.4° arising from the (002) planes, which reveals the graphite-like stacking of carbon nitride layers separated at an interlayer distance of 0.326 nm. The diffuse reflectance UV-vis spectrum of SGCN is shown in Fig. 1b. Besides the onset absorption of SGCN at 450 nm which can be assigned to the electronic transition from N 2p to C 2p orbitals, the extended absorption shoulder band from 450 to 600 nm indicated the presence of carbon impurities in SGCN which might arise from the residual ethanol during the thermal condensation process.31,32 Moreover, the band gap of SGCN was calculated from the Tauc plot to be 2.6 eV as shown in Fig. 1c. The surface area and porosity of SGCN were estimated by the nitrogen (N2) adsorption/desorption experiment. The N2 adsorption/desorption isotherm of SGCN (Fig. 1d) is a type-IV isotherm, which is typical for mesoporous materials. The measured specific surface area (SSA) for SGCN was 186 m2 g−1. Moreover, the corresponding pore volume of ca. 0.43 cm3 g−1 and pore radius of 2–3 nm of SGCN were calculated via the BJH (Barrett–Joyner–Halenda) method (Fig. 1d: inset). Transmission electron microscopy (TEM) analysis was performed to examine the surface morphology of SGCN. The morphology of SGCN depicted a layered structure with porosity as shown in Fig. S1a.†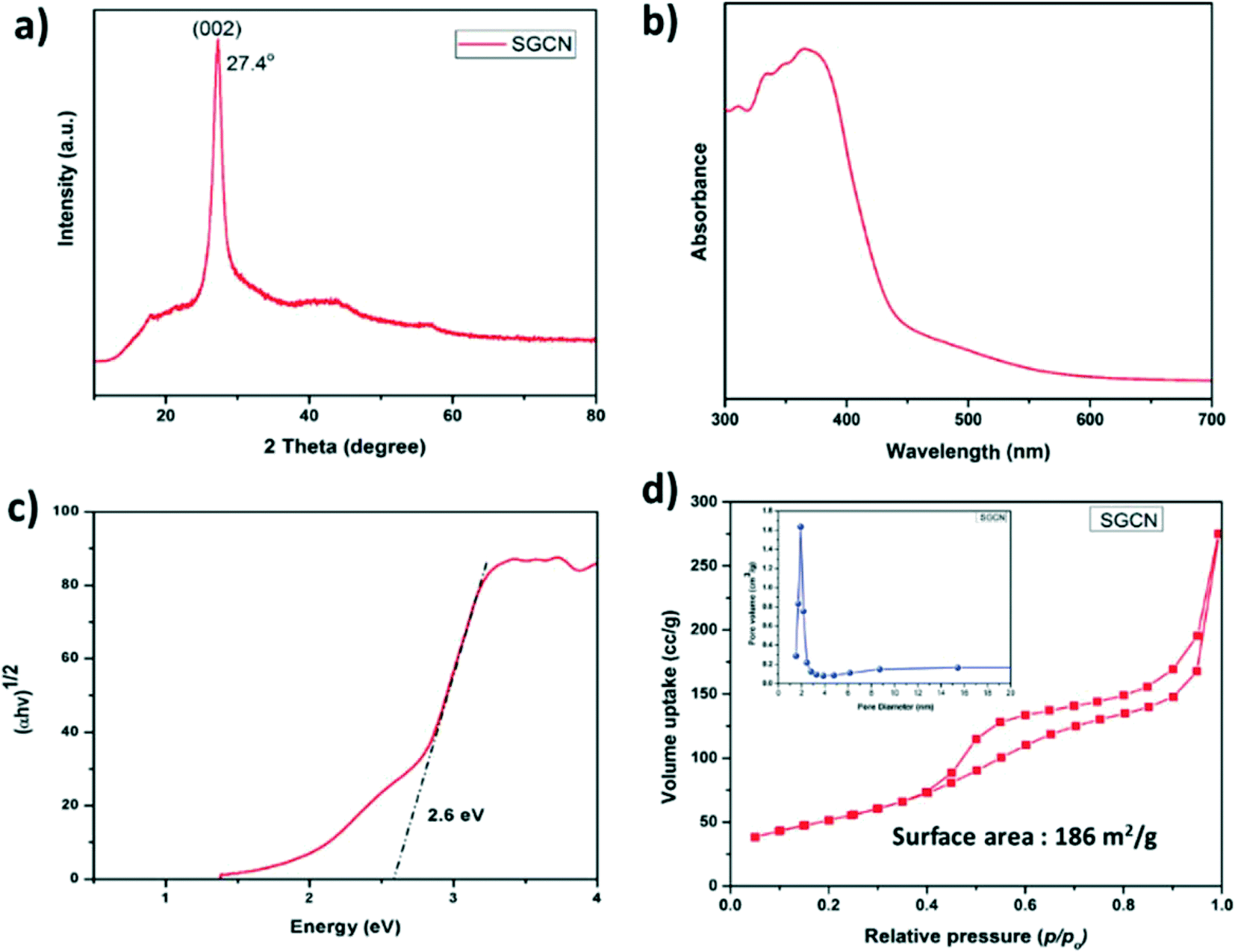 | ||
| Fig. 1 (a) XRD pattern of SGCN, (b) DRUV-vis spectrum of SGCN, (c) Tauc plot of SGCN, and (d) N2 physisorption isotherm of SGCN (inset: pore size distribution graph of SGCN). | ||
Fourier transform infrared (FT-IR) spectroscopy was further employed to confirm the functional groups and various molecular vibrations present in SGCN. As shown in Fig. S1b,† the absorption bands located at 810 and 1200–1650 cm−1 are the characteristic breathing-vibration of tri-s-triazine and the typical stretching modes of the C–N heterocycle bonds, respectively, suggesting the presence of the tri-s-triazine-based moiety in the SGCN matrix. The broad bands at around 3400 cm−1 are associated with the stretching modes of N–H bonds in the aromatic moieties.
X-ray photoelectron spectroscopy (XPS) measurements were applied to confirm the surface chemical compositions of SGCN and the results are shown in Table S1.† The full survey scan of SGCN (Fig. 2a) demonstrated that apart from C 1s and N 1s signals, an O 1s signal was also observed, showing a small number of adsorbed oxygen molecules over the SGCN surface. The C 1s spectrum (Fig. 2b) is comprised of three components: an intense peak at 288.6 eV corresponding to the sp2-hybridised carbon (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N) of the tri-s-triazine-based structure, a second one at 284.6 eV due to the existence of adventitious carbon (C–C), and a third one at 286.7 eV assigned to the surface C–NH2 moieties of the SGCN. The high resolution N 1s spectrum (Fig. 2c) could be divided into four peaks. The signal occurring at 399.5 eV indicates the sp2-hybridized nitrogen atom linked with a carbon atom (C–CN) and the peak at 400.9 eV is attributed to the presence of the N–(C)3 functional group in the carbon nitride core, i.e., the tri-s-triazine motif, further validating the polymerization of melamine. The N 1s peak centered at 401.8 eV signifies the existence of amino functional groups carrying a hydrogen atom (C–N–H). A weak N 1s peak at 405.1 eV is also observed which basically originates from π-excitation.33,34 The peak at 532.4 eV in the high resolution O 1s spectrum could be assigned to the surface adsorbed H2O over SGCN (Fig. 2d).35
C–N) of the tri-s-triazine-based structure, a second one at 284.6 eV due to the existence of adventitious carbon (C–C), and a third one at 286.7 eV assigned to the surface C–NH2 moieties of the SGCN. The high resolution N 1s spectrum (Fig. 2c) could be divided into four peaks. The signal occurring at 399.5 eV indicates the sp2-hybridized nitrogen atom linked with a carbon atom (C–CN) and the peak at 400.9 eV is attributed to the presence of the N–(C)3 functional group in the carbon nitride core, i.e., the tri-s-triazine motif, further validating the polymerization of melamine. The N 1s peak centered at 401.8 eV signifies the existence of amino functional groups carrying a hydrogen atom (C–N–H). A weak N 1s peak at 405.1 eV is also observed which basically originates from π-excitation.33,34 The peak at 532.4 eV in the high resolution O 1s spectrum could be assigned to the surface adsorbed H2O over SGCN (Fig. 2d).35
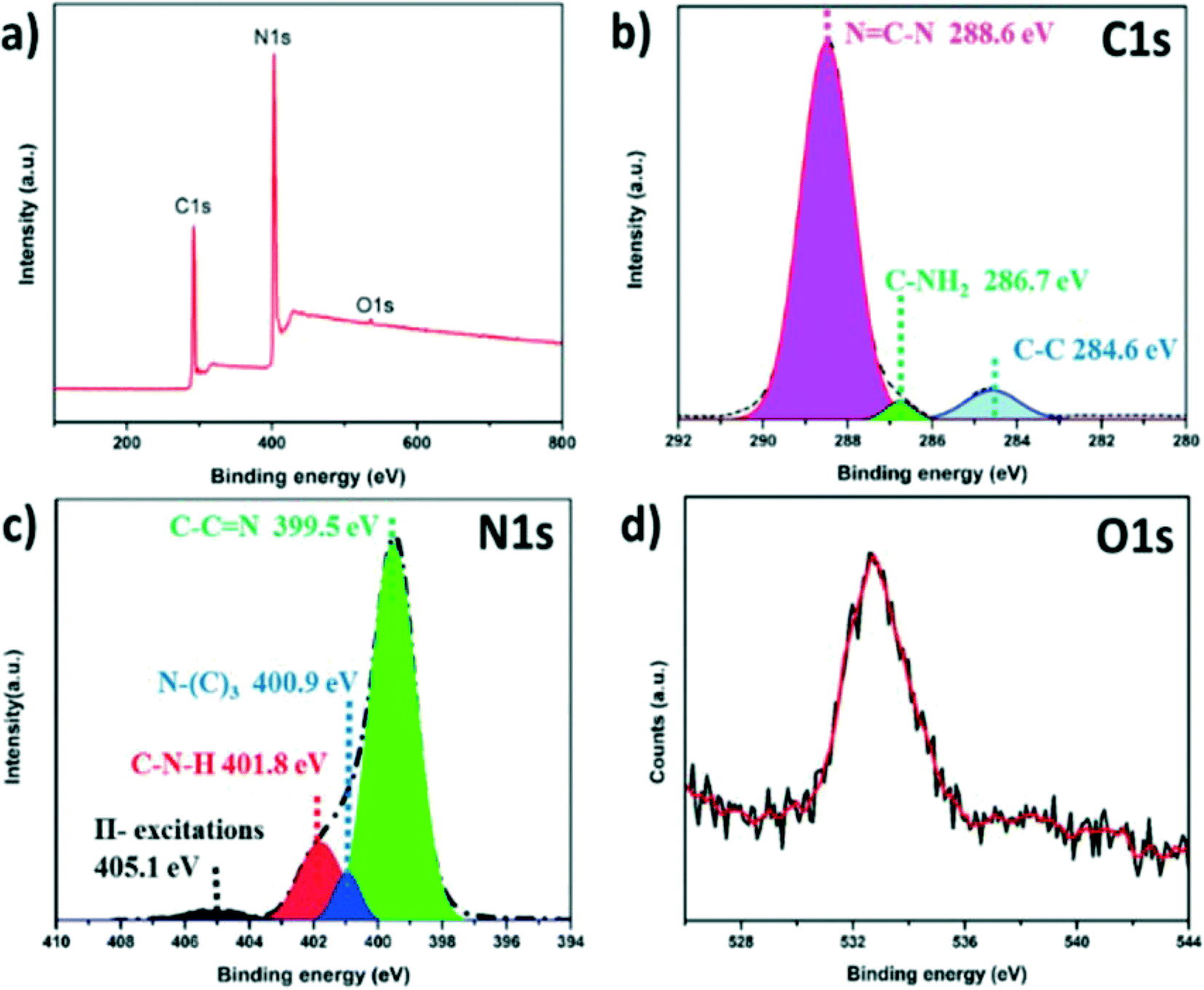 | ||
| Fig. 2 (a) XPS survey scan of SGCN, (b) C 1s spectrum of SGCN, (c) N 1s spectrum of SGCN, and (d) O 1s spectrum of SGCN. | ||
Furthermore, the band potentials of the conduction band minimum (CBM) and the valence band maximum (VBM) of SGCN were found to be integral parameters to uphold the photo-oxidation of FUR. The CBM of SGCN was estimated from the flat-band potential (Vfb), which was analyzed through the Mott–Schottky plot shown in Fig. 3a. The obtained positive slope confirms that SGCN is a typical n-type semiconductor and the Vfb of SGCN is determined to be −0.9 V (vs. Ag/AgCl). This value could further be converted against the NHE by using the following equation (ENHE = EAg/AgCl + 0.197), which was found to be −0.7 V (vs. NHE at pH 7). Basically, the Vfb for most n-type semiconductors resides 0.1 V below the CBM.36 On that account, the CBM of SGCN was found to be −0.8 V (vs. NHE at pH 7). Moreover, Fig. 3b shows the XPS valence band spectrum of SGCN. Of note, the VB potential which is the contact potential difference between the samples and the analyzer could be extrapolated from the XPS valence band spectrum. Therefore, the VBM of SGCN was estimated to be +1.8 V (vs. NHE at pH 7). The values of the CBM and VBM with respect to the NHE were rationally highlighted in the band energy diagram as shown in Fig. 3c. The validation of the obtained CBM and VBM was further done using the following equation: Eg = VBM − CBM with the help of Eg (band gap obtained from the Tauc plot).
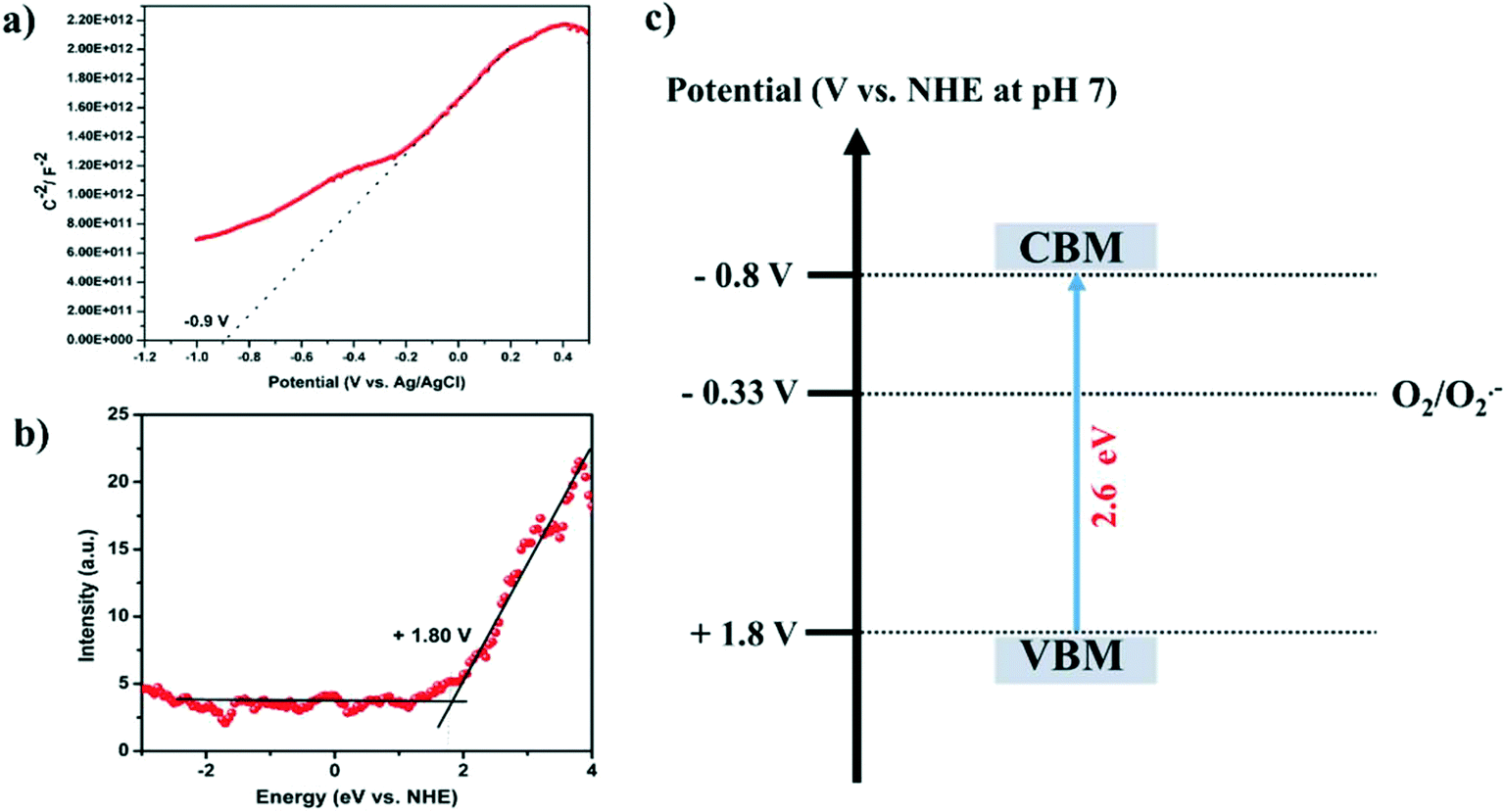 | ||
| Fig. 3 (a) Mott–Schottky plot of SGCN, (b) valence band spectrum of SGCN, and (c) band energy diagram of SGCN. | ||
3.2. Photocatalytic experiments
The photocatalytic activity of the SGCN for photo-oxidation of FUR in ACN was evaluated under simulated solar light irradiation (AM 1.5G). Apparently, the large negative CBM of SGCN can easily activate molecular oxygen (O2) via photogenerated electrons. This activated molecular oxygen with sequential energy transfer oxidized the FUR, which will be discussed in detail below. In agreement with previous literature reports, MAN and HFO were found to be the major products.37 A set of experiments were performed under different reaction conditions in order to confirm the photoactivity of SGCN, which is summarized in Table 1. The synthesized SGCN showed an excellent photocatalytic activity in which FUR was photo-oxidized with more than 95% conversion in 30 h (Table 1, entry 1). Product identification by GC-MS analysis revealed that, in addition to MAN and HFO, other minor products including SAN and β-FAA were also identified (Fig. S2–S6, ESI†). Meanwhile, some gaseous products, i.e., COx (CO2 and CO), could also be generated, which was confirmed by GC (Fig. S7, ESI†). Furthermore, no transformation was observed without light irradiation (Table 1, entry 2) and an O2 atmosphere (Table 1, entry 3). These results depicted that light and O2 are the requisite parameters for the reaction. The selectivity to the products was confirmed by GC-MS as such in the ACN reaction medium by means of further dilution as mentioned in Table 1. MAN and HFO could achieve the maximum selectivity of 42% and 33%, respectively (Table 1, entry 1).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Light | O2 | Conv.a% | Selectivitya% | ||||
| MAN | HFO | SAN | β-FAA | COxb | |||||
| Reaction conditions: 50 mg SGCN, 5 mL ACN, 0.11 mmol of substrate, reaction time: 30 h, room temperature (25 °C) and light source: solar simulator (AM 1.5G, 100 mW cm−2).a The conversion and selectivity were obtained from GC-MS.b COx selectivity was confirmed by GC.c Reaction without a catalyst.d Under a solar simulator (intensity: 65 mW cm−2) and reaction time 8 h.e Under natural sunlight (intensity: 65 mW cm−2) and reaction time 8 h.f Thermal reaction without light at 50 °C. | |||||||||
| 1 | SGCN | ✓ | ✓ | >95 | 42 | 33 | 8 | 11 | >5.5 |
| 2 | SGCN | ✗ | ✓ | — | — | — | — | — | — |
| 3 | SGCN | ✓ | ✗ | — | — | — | — | — | — |
| 4c | — | ✓ | ✓ | — | — | — | — | — | — |
| 5d | SGCN | ✓ | ✓ | 11 | 54 | — | — | 39 | <2 |
| 6e | SGCN | ✓ | ✓ | 5 | 66 | — | 32 | <1 | |
| 7f | SGCN | ✗ | ✓ | ∼2 | n.d | n.d. | |||
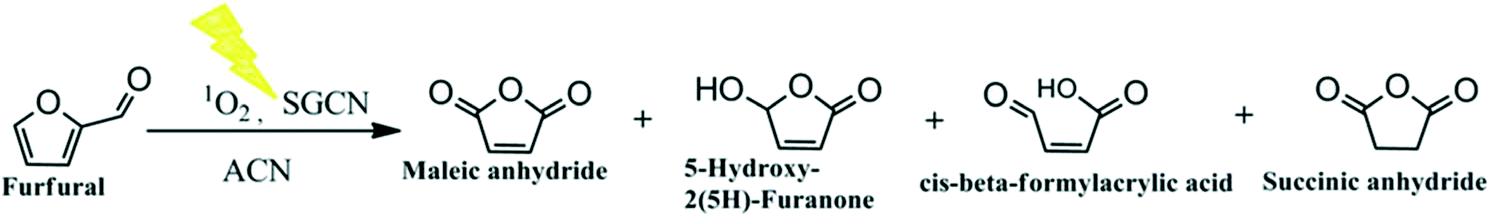
The real action spectrum was also investigated at different wavelengths to explore the wavelength-dependent FUR oxidation. The results showed that the activity of SGCN correlated well with its optical absorption spectrum (Fig. 4). It is interesting to highlight that utilization of natural sunlight for chemical or biomass valorization rather than simulated sunlight (artificial light sources) is the major step to achieve sustainability.38,39 Therefore, we decided to scrutinize the activity of SGCN for FUR photo-oxidation under natural sunlight which is described in the following paragraph.
 | ||
| Fig. 4 Real action spectrum of FUR oxidation at different wavelengths. | ||
In order to explore the photo-oxidation of FUR, the photocatalytic activity of SGCN was even further extended under natural sunlight (intensity: 65 mW cm−2). The photo-oxidation ability of SGCN towards FUR under natural sunlight showed improved selectivity to MAN (66%) and β-FAA (32%), although the conversion was observed to be almost 5% in 8 h of sunlight illumination (Table 1, entry 6 and Fig. S8, ESI†). It is essential to mention that the absence of HFO and SAN formation contributes to the improved selectivity to MAN. Comparatively, another reaction was performed at 65 mW cm−2 under a solar simulator to realize the exact phenomenon towards the improved selectivity to MAN. In this study, we observed similar product distribution with a selectivity to MAN and β-FAA of 58 and 39%, respectively (Table 1, entry 5). Interestingly, the conversion of FUR was enhanced up to 11% (comparing Table 1, entries 5 and 6) which could be ascribed to the continuous collimated light of the solar simulator. Therefore, we concluded that the selectivity to MAN and the low product distribution could be enhanced at a lower intensity of light. However, the low intensity may hamper the overall conversion of FUR. Particularly, this result seems to provide an interesting and efficient protocol for upgrading FUR biomass towards selective production of fine chemicals under natural sunlight by the use of porous carbon nitrides. Lastly, to direct the role of thermal heat generated from the incident light during the all experiments was further corroborated by performing the FUR oxidation under similar reaction conditions at a temperature of 50 °C (Table 1, entry 7). A negligible conversion of FUR was observed under the thermal reaction conditions which further revealed no additional role of thermal heat in the photo-oxidation of FUR.
3.3. Time-dependent studies
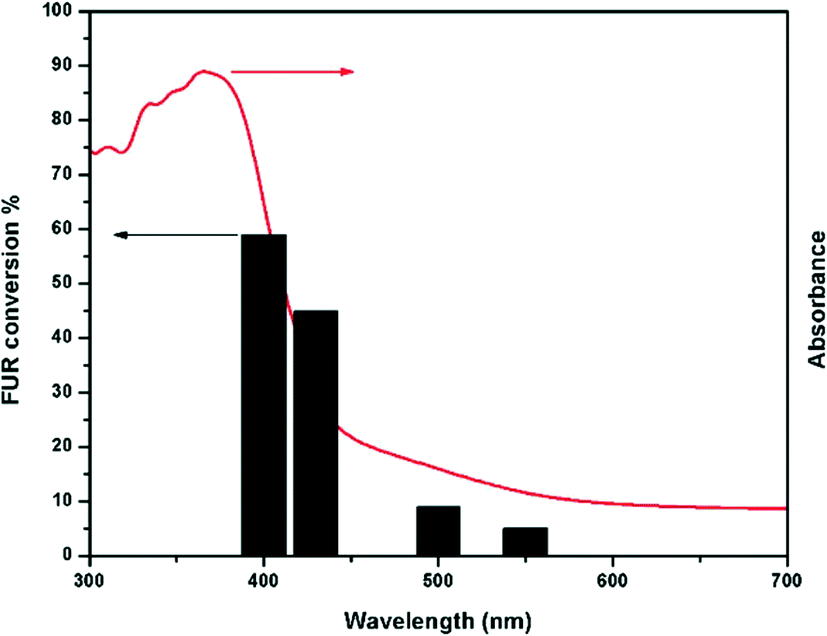
In order to determine the maximum conversion for FUR photo-oxidation under optimized conditions, time-dependent studies have been performed for 30 h at intervals of 12, 24, and 30 h. Experimental studies demonstrated that the FUR conversion increased with time and after 30 h, >95% conversion was achieved. Notably, there was not much change towards the selectivity to the products throughout the reaction as shown in Fig. 5. The GC-MS profile of the reaction mixtures at different time intervals is shown in Fig. S9, ESI.†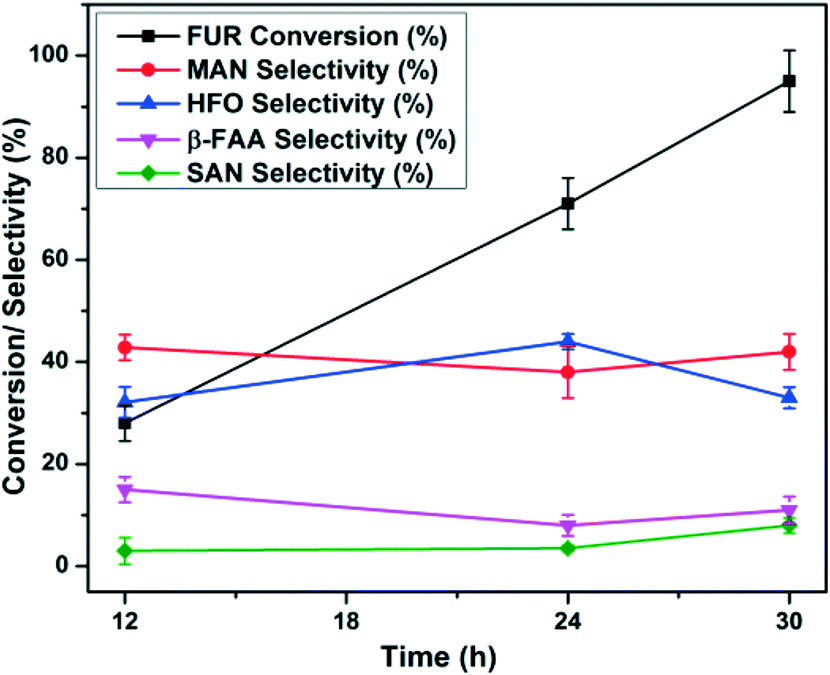 | ||
| Fig. 5 Photo-oxidation of FUR at different time intervals. | ||
3.4. Recycling and stability studies
The SGCN catalyst has been applied for the FUR photo-oxidation and further we have elucidated the stability of this SGCN, as shown in Fig. 6. Four subsequent cycles have been performed for FUR photo-oxidation in the presence of SGCN. After the 4th cycle, the catalyst was washed with water followed by ethanol and subsequently dried and characterized (Fig. S10 and S13†). The general trend showed a slight decline in activity which may be ascribed to the saturation of the adsorption sites over the catalyst surface. This decline in activity can be correlated to the reduced SSA of the catalyst, and after the 4th cycle it was measured to be 140 m2 g−1 (Fig. S10, ESI†). No significant change in the crystallinity of the structure was observed even after the 4th cycle (Fig. S11, ESI†). During investigation of the surface oxidation of SGCN under deep oxidation conditions, it was observed that no obvious oxidation took place even after the 4th cycle which was corroborated by the XPS analysis. However, little increase in the atomic% of O was observed after the 4th cycle, which might be due to the adsorbed moisture over the surface of SGCN during sample handling (Fig. S12 and Table S1, ESI†). | ||
| Fig. 6 Recycling studies of SGCN for FUR photo-oxidation: a) FUR conversion during different cycles and b) product selectivity at different cycles. | ||
3.5. Scavenger, control, and ESR studies
Photo-oxidation of FUR may possibly be initiated by holes (h+) and reactive oxygen species (ROS), e.g., superoxide radicals (O2˙−) and singlet oxygen (1O2), which are generally produced via multiple redox processes. To examine the exact pathway for FUR photo-oxidation, some control or scavenging studies were performed, which are included in Table 2. In addition, the electron spin resonance (ESR) technique was carried out to further verify the existence of ROS species especially for 1O2.| Entry | Substrate | Scavenger | O2 | Conversiona (%) | Oxidized/identified productsa |
|---|---|---|---|---|---|
| Reaction conditions: 50 mg SGCN, 5 mL ACN, 0.11 mmol of substrate. Reaction time = 12 h, room temperature (25 °C). Light source: solar simulator (AM 1.5G).a Conversion and oxidized products were confirmed by GC-MS or GC.b Reaction conditions: 50 mg SGCN, 5 ml ACN, 4.5 mol of substrate. Reaction time = 30 h, room temperature (25 °C) and light source: solar simulator (AM 1.5G).c Reaction conditions: 50 mg SGCN, (4 ml ACN: 1 ml substrate). Reaction time = 30 h, room temperature (25 °C), and light source: solar simulator (AM 1.5G). | |||||
| 1 | FUR | — | + | 28 | MAN, HFO, β-FAA, SAN, and COx |
| 2 | FUR | BQ (O2˙−) | + | 13 | MAN, HFO, and traceable β-FAA and COx |
| 3 | FUR | NaN3 (1O2) | + | — | — |
| 4 | FUR | KI (h+) | + | — | — |
| 5b | Furoic acid | — | + | 100 | MAN, SAN and β-FAA and COx |
| 6c | Furan | — | + | >70 | MAN, HFO, SAN, β-FAA, 2(5H)-furanone and COx |
The prerequisite nature of molecular O2 for FUR conversion was proven as shown in Table 1, entries 1 and 3. O2˙− and 1O2 were detected in the reaction medium by treating with their respective quenchers. In this regard, p-benzoquinone (BQ) was used as the O2˙− quencher, and sodium azide (NaN3) as the 1O2 quencher.40 If the FUR oxidation is stimulated via the generation of any of these ROS, then the photo-oxidation of FUR would be impeded in the presence of their respective quenchers. To assess the role of O2˙−, BQ was added into the reaction mixture, and the oxidation of FUR was observed to be only 13% (Table 2, entry 2) which confirmed the active role of O2˙− in the reaction.
It has been reported that 1O2 molecules could be generated over the surface of carbon nitrides by two processes during photocatalytic reactions.41,42 One way is that conjugated carbon nitride acts as a photosensitizer which involves energy transfer between excited triplet excitons and ground-state oxygen molecules (O2) to give 1O2. Another way is photoreduction of O2 from the LUMO of carbon nitrides via photogenerated electrons transmitted to O2 to produce O2˙− which subsequently oxidized further via h+ into 1O2 molecules as follows:43,44
| 3O2 + e− → O2˙− | (1) |
| O2˙− + h+ → 1O2 | (2) |
In our study, the generation of 1O2 in the reaction was determined by adding NaN3 to the reaction mixture (Table 2, entry 3). Interestingly, no conversion of FUR took place when NaN3 was added into the reaction mixture, which proves that the major ROS species involved in the reaction mechanism was 1O2. Furthermore, the generation of 1O2 over the SGCN surface was also confirmed by selective photo-oxidation of 1,5-dihydroxynaphthalene (1,5-DHN) instead of FUR. In addition, the UV-vis spectra of the reaction mixture were recorded for 60 min (Fig. S14, ESI†), which confirmed the formation of juglone (absorption peak at 423 nm).41 This is the characteristic reaction of 1O2, thus proving the generation of 1O2 over the SGCN surface.
Of note, to reinforce the control experiments for the generation of 1O2, ESR analysis has been performed in the presence of 2,2,6,6-tetramethylpiperidine (TEMP) as a trapping agent (shown in Fig. 7a), which is in good agreement with the results based on the scavenger or control experiments. Moreover, the O2˙− generation was analyzed in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as shown in Fig. 7b.
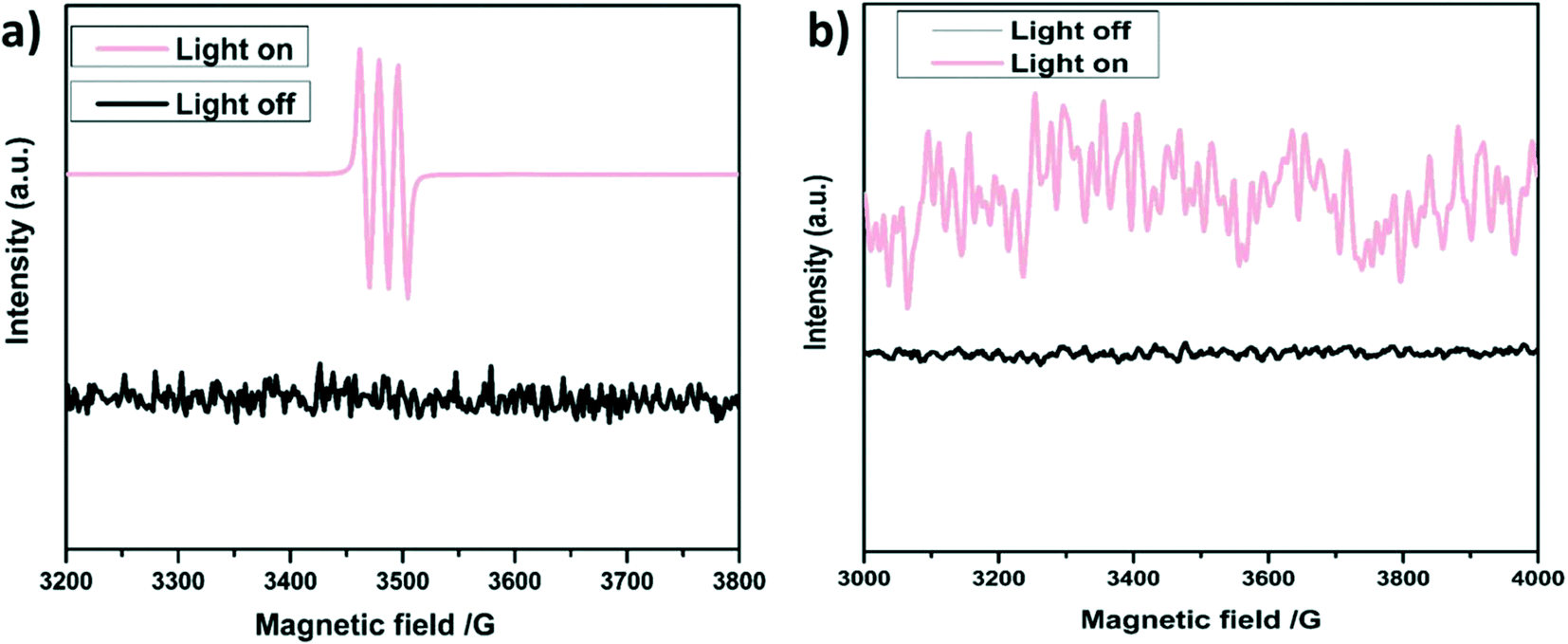 | ||
| Fig. 7 ESR spectra of a) SGCN–TEMP and b) SGCN–DMPO for 1O2 and O2˙− generation, respectively. | ||
In order to define the role of h+ in the oxidation process, potassium iodide (KI) was added to the reaction medium (Table 2, entry 4) and it was found that there is a negligible conversion of FUR. Indirectly, it proved that the major role of h+ is towards the generation of 1O2 (eqn (2)),44 which further subsequently proceeded for the oxidation of FUR.
The moderate oxidation ability of the photogenerated h+ of SGCN can rule out the formation of the non-selective ˙OH radicals, which is more likely due to the less positive valence band potential of SGCN. In addition, non-aqueous ACN as a solvent also inhibits the production of ˙OH via h+ oxidation. However, simulated light contains 4% UV light (like natural sunlight) which may break down H2O2, if generated, into ˙OH radicals and can also help in the oxidation of FUR. Therefore, we have performed a control experiment under visible light (>420 nm) by using N,N-diethyl-1,4-phenylenediamine (DPD) as a probe indicator45 (Fig. S15, ESI†). This confirmed that there was no generation of H2O2 during the reaction, hence eliminating the possibility of ˙OH radical generation during the reaction.
Based on the above discussed results, we confirmed that 1O2 and h+ displayed a dominant role in photo-oxidation of FUR. The oxidized products of FUR were MAN, HFO, SAN and β-FAA along with gaseous COx.
3.6. Mechanistic studies
From the above detailed experimental results and observations, we may conclude that the 1O2 generation probability via the energy transfer mechanism (oxidizing O2˙− by h+) plays a major role since in the presence of the h+ scavenger (KI), no FUR conversion took place. However, the probability of 1O2 generation through triplet energy transfer from photoexcited SGCN* to ground-state oxygen 3O2 was minor. Therefore, in the presence of the O2˙− scavenger (BQ), still 13% (Table 2, entry 2) conversion took place, which might suggest that the reaction proceeded differently via triplet energy transfer from photoexcited SGCN*.In order to prove the plausible mechanistic pathway of FUR photo-oxidation, it is imperative to gain more insight into the reactive intermediate species generated during the reaction. Furoic acid and 2(5H)-furanone could be the possible intermediates for MAN and HFO formation in the FUR photo-oxidation. To address these intermediates in the mechanistic pathway of FUR photo-oxidation, additional experiments were performed under control conditions (Table 2, entries 5 and 6). It was found that when furoic acid was taken as a substrate (Table 2, entry 5), MAN, SAN and β-FAA were identified in the GC-MS analysis and HFO was not detected (Fig. S16, ESI†). In addition, gaseous COx was also generated and detected by GC.
This clearly supports that the FUR photo-oxidation to MAN and HFO proceeded via furoic acid as an intermediate followed by further decarboxylation (C–C cleavage). Further, when furan was taken as a substrate (Table 2, entry 6), MAN, HFO, SAN, β-FAA and 2(5H)-furanone were observed while monitoring the liquid reaction mixture via GC-MS analysis (Fig. S17 and S18, ESI†). Generation of 2(5H)-furanone was corroborated with further oxidation into HFO.46 Therefore, in this experiment, we presumed that 2(5H)-furanone was also generated along with the formation of MAN and HFO. With the above results, we arrive at the conclusion towards the plausible mechanistic pathway for FUR photo-oxidation over the SGCN surface, which is schematically illustrated in Scheme 2. Here, FUR was initially photo-oxidized via1O2 into furoic acid followed by decarboxylation into furan. Further oxidation led to the generation of 2(5H)-furanone which subsequently oxidized into HFO. Finally, MAN was feasibly generated through further oxidation of HFO. In addition, HFO showed partial tautomerism with the formation of SAN and β-FAA via in situ rearrangement during the reaction.
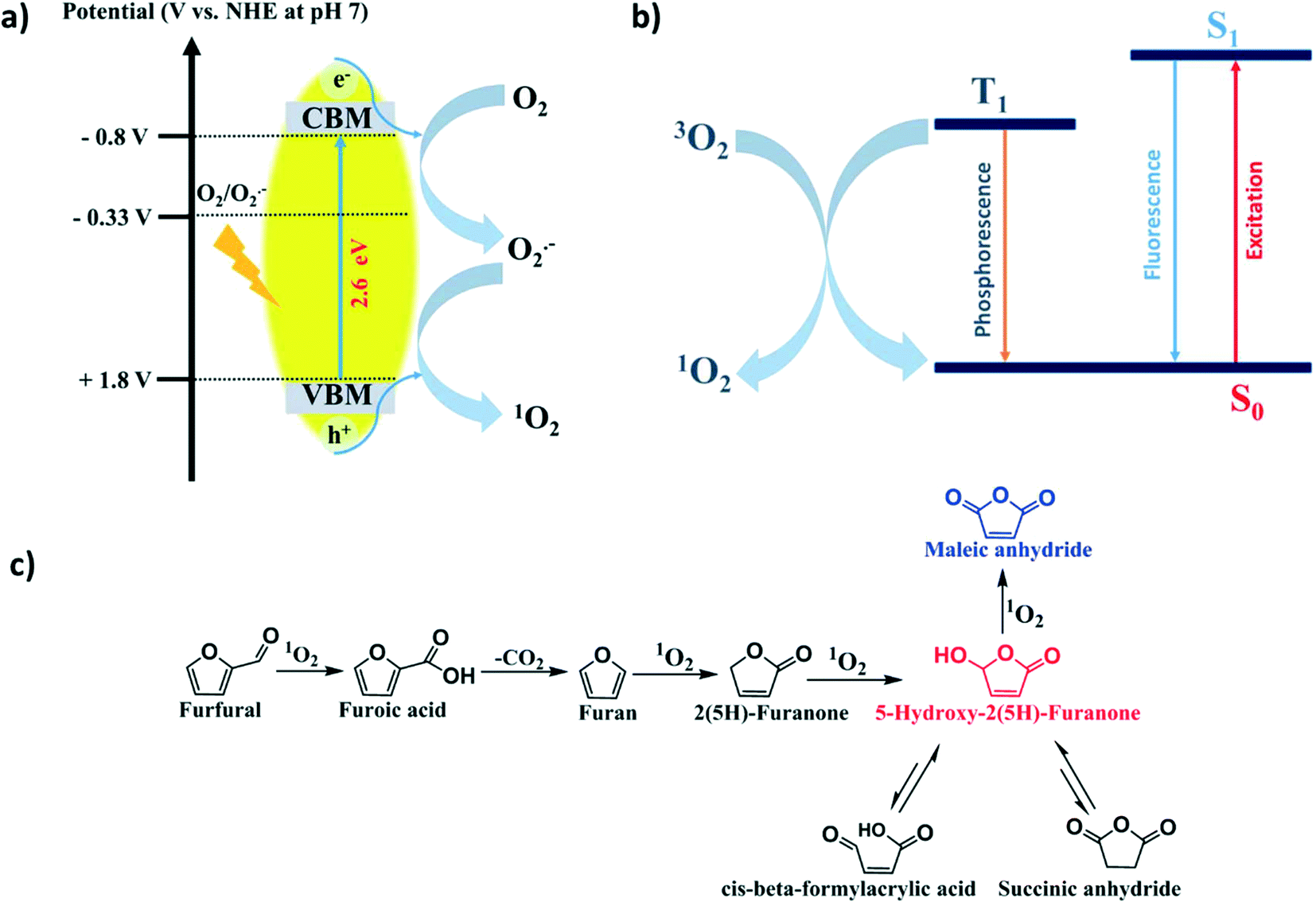 | ||
| Scheme 2 a) Generation of singlet oxygen on the surface of SGCN via h+, b) singlet oxygen generation via triplet state energy transfer and c) plausible mechanistic pathway of FUR photo-oxidation. | ||
4. Conclusion
In summary, photo-oxidation of biomass-derived FUR to high value-added MAN and HFO was achieved for the first time by employing mesoporous carbon nitride (SGCN) as an efficient polymeric organic semiconductor photocatalyst without any co-catalyst. This work showed excellent photo-oxidation ability and good selectivity to MAN (42%) and HFO (33%) which could be obtained with a FUR conversion of >95% in ACN under simulated solar light. The liquid reaction mixture shows the generation of MAN, HFO, SAN and β-FAA along with gaseous oxidized products COx. Moreover, the detailed photocatalytic mechanistic study of FUR oxidation indicates the reaction pathway through the generation of ROS and h+ and further subsequent oxidation, C–C cleavage (decarboxylation), and rearrangements/isomerization.These vast investigations provided novel insights to understand the photo-oxidation mechanisms of the biomass-derived FUR. Eventually, we believe that this could impart an efficient methodology to comprehend and investigate new pathways towards furan-based biomass platforms into value-added chemicals selectively through carbon nitride and other conjugated polymers as photocatalysts under light irradiation or under natural sunlight.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
D. K. Chauhan thanked INST Mohali for financial support. Dr. K. Kailasam thanked the Department of Science and Technology, India (DST) for the DST Nano Mission NATDP funded Technology Project, File No. SR/NM/NT-06/2016, and DST-CERI project, File No. TMD/CERI/BEE/2016/082, for the financial support. Dr. A. Patra acknowledges the DST-SERB funded project, File no. EMR/2017/000233, for the financial support.References
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- G. C. de Assis, I. M. A. Silva, T. G. dos Santos, T. V. dos Santos, M. R. Meneghetti and S. M. P. Meneghetti, Catal. Sci. Technol., 2021, 11, 2354–2360 RSC.
- Z. Yuan, B. Liu, P. Zhou, Z. Zhang and Q. Chi, Catal. Sci. Technol., 2018, 8, 4430–4439 RSC.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. López Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- X. Chen, L. Zhang, B. Zhang, X. Guo and X. Mu, Sci. Rep., 2016, 6, 28558 CrossRef PubMed.
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC.
- H. Zhang, S. Wang, H. Zhang, J. H. Clark and F. Cao, Green Chem., 2021, 23, 1370–1381 RSC.
- W. Zhu, F. Tao, S. Chen, M. Li, Y. Yang and G. Lv, ACS Sustainable Chem. Eng., 2019, 7, 296–305 CrossRef CAS.
- H. Wu, J. Song, H. Liu, Z. Xie, C. Xie, Y. Hu, X. Huang, M. Hua and B. Han, Chem. Sci., 2019, 10, 4692–4698 RSC.
- X. Li, J. Ko and Y. Zhang, ChemSusChem, 2018, 11, 612–618 CrossRef CAS.
- E. R. Nielsen, Ind. Eng. Chem., 1949, 41, 365–368 CrossRef CAS.
- D. Y. Murzin, E. Bertrand, P. Tolvanen, S. Devyatkov, J. Rahkila, K. Eränen, J. Wärnå and T. Salmi, Ind. Eng. Chem. Res., 2020, 59, 13516–13527 CrossRef CAS.
- N. Araji, D. D. Madjinza, G. Chatel, A. Moores, F. Jérôme and K. De Oliveira Vigier, Green Chem., 2017, 19, 98–101 RSC.
- S. R. Kubota and K.-S. Choi, ACS Sustainable Chem. Eng., 2018, 6, 9596–9600 CrossRef CAS.
- U. Thubsuang, S. Chotirut, K. Nuithitikul, A. Payaka, N. Manmuanpom, T. Chaisuwan and S. Wongkasemjit, J. Colloid Interface Sci., 2020, 565, 96–109 CrossRef CAS PubMed.
- B. Liu, S. Xu, M. Zhang, X. Li, D. Decarolis, Y. Liu, Y. Wang, E. K. Gibson, C. R. A. Catlow and K. Yan, Green Chem., 2021, 23, 4034–4043 RSC.
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., A, 2013, 458, 55–62 CrossRef CAS.
- A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316 CrossRef.
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493–5501 CrossRef CAS.
- H. Zhao, S. Wang, F. He, J. Zhang, L. Chen, P. Dong, Z. Tai, Y. Wang, H. Gao and C. Zhao, Carbon, 2019, 150, 340–348 CrossRef CAS.
- N. K. R. Eswar, S. A. Singh and J. Heo, J. Mater. Chem. A, 2019, 7, 17703–17734 RSC.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- D. K. Chauhan, S. Jain, V. R. Battula and K. Kailasam, Carbon, 2019, 152, 40–58 CrossRef CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- A. Savateev and M. Antonietti, ChemCatChem, 2019, 11, 6166–6176 CrossRef CAS.
- C. Yang, B. Wang, L. Zhang, L. Yin and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 6627–6631 CrossRef CAS PubMed.
- Y. Li, S. Ouyang, H. Xu, X. Wang, Y. Bi, Y. Zhang and J. Ye, J. Am. Chem. Soc., 2016, 138, 13289–13297 CrossRef CAS.
- A. Gassama, C. Ernenwein and N. Hoffmann, ChemSusChem, 2009, 2, 1130–1137 CrossRef CAS.
- Y. Morita, H. Tokuyama and T. Fukuyama, Org. Lett., 2005, 7, 4337–4340 CrossRef CAS PubMed.
- K. Kailasam, J. D. Epping, A. Thomas, S. Losse and H. Junge, Energy Environ. Sci., 2011, 4, 4668–4674 RSC.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 RSC.
- W. Chen, T.-Y. Liu, T. Huang, X.-H. Liu and X.-J. Yang, Nanoscale, 2016, 8, 3711–3719 RSC.
- S. Gu, J. Xie and C. M. Li, RSC Adv., 2014, 4, 59436–59439 RSC.
- F. Chang, C. Li, J. Luo, Y. Xie, B. Deng and X. Hu, Appl. Surf. Sci., 2015, 358, 270–277 CrossRef CAS.
- S. Samanta, V. R. Battula, N. Sardana and K. Kailasam, Appl. Surf. Sci., 2021, 563, 150409 CrossRef CAS.
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4351–4358 RSC.
- V. R. Battula, H. Singh, S. Kumar, I. Bala, S. K. Pal and K. Kailasam, ACS Catal., 2018, 8, 6751–6759 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- W. Wu, C. Han, Q. Zhang, Q. Zhang, Z. Li, D. J. Gosztola, G. P. Wiederrecht and M. Wu, J. Catal., 2018, 361, 222–229 CrossRef CAS.
- I. Camussi, B. Mannucci, A. Speltini, A. Profumo, C. Milanese, L. Malavasi and P. Quadrelli, ACS Sustainable Chem. Eng., 2019, 7, 8176–8182 CrossRef CAS.
- T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2007, 111, 5259–5275 CrossRef CAS.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS.
- C. Su, R. Tandiana, B. Tian, A. Sengupta, W. Tang, J. Su and K. P. Loh, ACS Catal., 2016, 6, 3594–3599 CrossRef CAS.
- L. Gong, N. Agrawal, A. Roman, A. Holewinski and M. J. Janik, J. Catal., 2019, 373, 322–335 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01681d |
| This journal is © The Royal Society of Chemistry 2022 |