
Mechanistic insight into construction of axially chiral biaryls via palladium/chiral norbornene cooperative catalysis: a DFT-based computational study†
Xuexiang
Ma
 ,
Aili
Feng
,
Chengbu
Liu
and
Dongju
Zhang
*
,
Aili
Feng
,
Chengbu
Liu
and
Dongju
Zhang
*
Key Lab of Colloid and Interface Chemistry, Ministry of Education, Institute of Theoretical Chemistry, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: zhangdj@sdu.edu.cn; Tel: +86 0531 88365833
First published on 12th November 2021
Abstract
Density functional theory calculations were performed on a prototype of three-component reactions involving aryl iodides, 2,6-substituted aryl bromides, and acrylates to understand the construction of axially chiral biaryls through the palladium/chiral norbornene cooperative catalysis (Nat. Catal.2020, 3, 727–733). The reaction is established to occur in sequence through oxidative addition of aryl iodide to Pd(0), norbornene insertion, C–H activation, oxidative addition of aryl bromide to Pd(II), C–C reductive elimination, norbornene extrusion, alkene insertion, and β-H elimination. The C–H activation is characterized as the rate-determining step, and the oxidative addition of aryl bromide on Pd(II) control the enantioselectivity of the reaction. The construction of more favorable (S)-C–C axial chirality is attributed to the smaller steric repulsion between the ortho-methyl group of aryl bromide and the norbornene moiety. The theoretical results rationalize the reaction sequence of aryl iodide and aryl bromide on both Pd(0) and Pd(II) and clarify the substantial role of the ortho-ester group in the aryl bromide.
1. Introduction
Axially chiral skeletons, as prevalent and privileged frameworks, are commonly employed in various fields such as medicinal chemistry,1 chiral materials2 and natural products.3 Moreover, taking advantage of their precise structure and rigidity, axially chiral compounds have also emerged as the attractive chiral ligands or catalyst motifs in asymmetric synthesis.4,5 During the past decades, much effort has been devoted to developing efficient approaches for directly constructing the axially chiral skeletons (especially biaryl motifs), including C–H activation,6 cross-coupling,7,8 central-to-axial chirality conversion,9,10 functionalization of prochiral biaryls (kinetic resolution, desymmetrization),11,12 and so forth.13–16 However, these types of reactions have several potential disadvantages, such as some of them usually require complex catalysts and/or specially functionalized substrates, which unfortunately restrict the practical applications of these strategies. Therefore, development of efficient synthetic strategies to improve these drawbacks are highly desired.The palladium/norbornene (Pd/NBE) cooperative catalysis pioneered by Catellani17 and later modified by Lautens,18 Yu,19,20 Dong,21,22 and others,23–25 is expected to be a promising approach to the construction of biaryl motifs. However, the asymmetric version of the Catellani reaction has barely been reported, until the first enantioselective construction of axially chiral biaryls was achieved by using a chiral phosphine ligand.26 Inspired by the recent progress, Zhou et al.27 successfully achieved axially chiral biaryls by using palladium and chiral norbornene (NBE*) catalyzed three-component cross-coupling reactions. As indicated in Scheme 1 by a representative example, this new strategy transforms aryl iodide 1, 2,6-substituted aryl bromide 2 and acrylate 3 (terminating reagent) into the axially chiral biaryl P in good yield (63%) with excellent enantioselectivity (97% ee). The optimal reaction conditions used Pd(OAc)2 as the catalyst, (1S,4R)-2-ethyl ester-substituted NBE* as the mediator, and K2CO3 as the base at 105 °C in acetonitrile (MeCN).
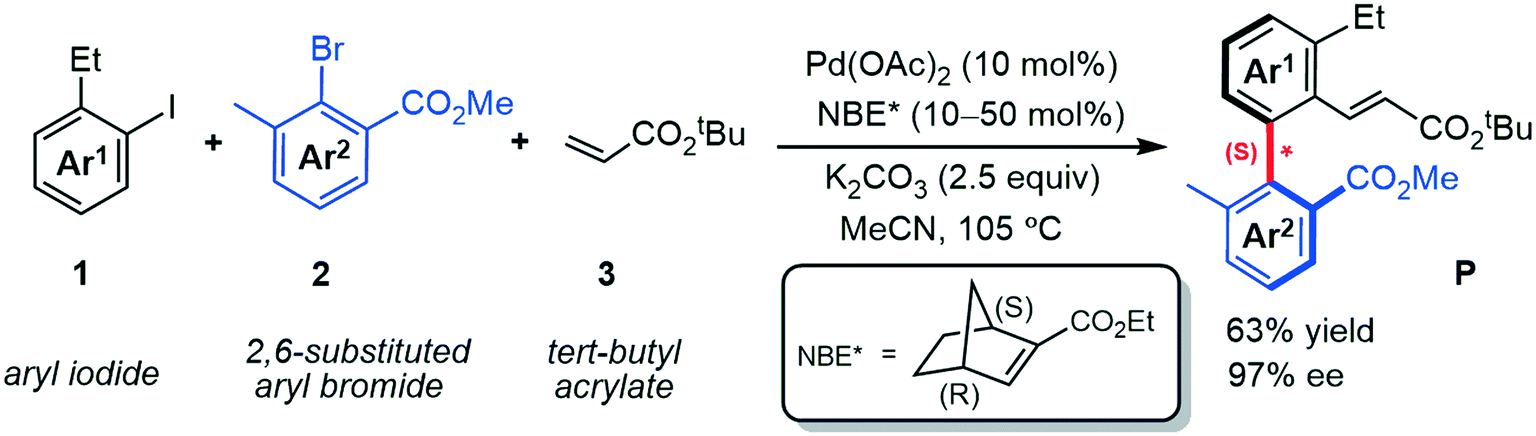 | ||
| Scheme 1 Pd/NBE* cooperative three-component cascade reaction for the construction of axially chiral biaryls. | ||
To understand the reactivity, Zhou and co-workers27 have tentatively proposed a plausible mechanism. As shown in Scheme 2, the reaction is assumed to start from the oxidative addition of aryl iodide 1 on Pd(0) species, generating the aryl Pd(II) intermediate A. Subsequently, the chiral norbornene (NBE*) inserts into Pd–C bond to give intermediate B, which then evolves into the five-membered aryl–norbornyl–palladacycle (ANP) intermediate Cvia C–H bond activation. Oxidative addition of 2,6-substituted aryl bromide 2 on Pd(II) center of C leads to Pd(IV) species D, from which C–C reductive elimination occurs to form axially chiral Pd(II) species E. Then ipso-carbopalladation intermediate F can be obtained via extrusion of NBE* from E (β-carbon elimination). Finally, F reacts with the terminating reagent acrylate 3 to furnish the final axially chiral biaryl P with regeneration of Pd(0) catalyst. The proposed mechanism provides a fundamental understanding of the construction of axial chirality. However, the mechanism details keep unexplored, and the observed excellent enantioselectivity still has not been rationalized well. In addition, it is noted that the proposed mechanism involves two oxidation addition steps: the first is for the substrate (aryl iodide) on Pd(0) and the second is for the arylation reagent (2,6-substituted aryl bromide) on Pd(II). Why is it such a reaction sequence? In other words, with simultaneous presence of two aryl halides, why the first oxidation addition gives preference to aryl iodide and the second to aryl bromide. Herein, we report a density functional theory (DFT) based computational study, from which we hope to clarify these essential questions.
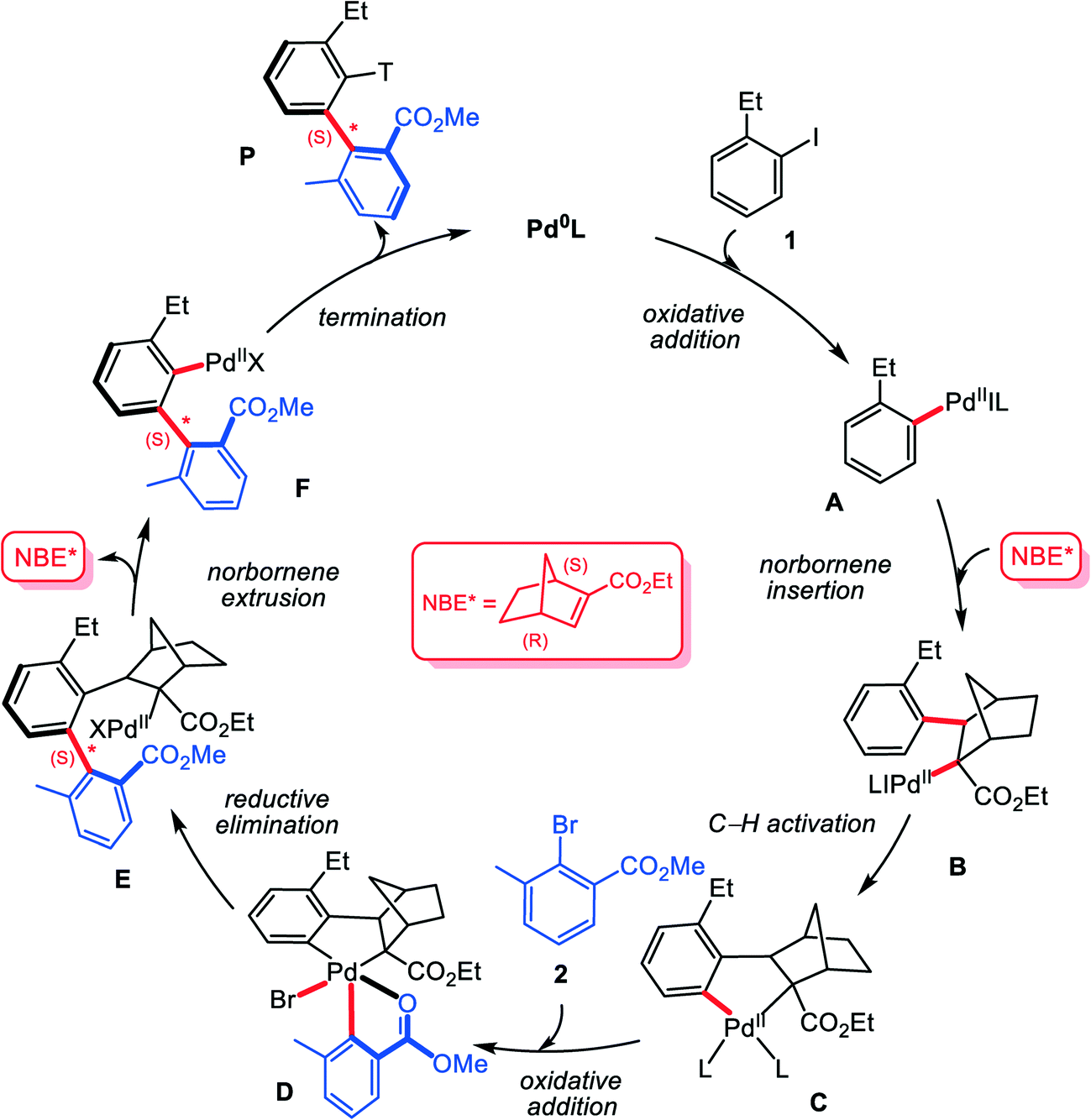 | ||
| Scheme 2 Plausible mechanism for the Pd/NBE* cooperative three-component cascade process to construct axial chirality biaryls. | ||
2. Computational details
All calculations were conducted with the Gaussian 16 software package.28 Geometry optimizations were carried out using the B3LYP29,30-D3(BJ)31 functional together with the SMD solvation model32 (acetonitrile was used as the solvent according to Scheme 1). Pd and I atoms were described with the LANL2DZ effective core potential (ECP) basis set33,34 augmented by two addition polarization functions, the d-type for I (ζd = 0.289) and the f-type for Pd (ζf = 1.472), and all other atoms were characterized by the 6-31G(d,p)35 basis set. Frequency calculations were performed at the same level of theory to identify all the stationary points as minima (zero imaginary frequency) or transition state (only one imaginary frequency) and to provide thermal corrections at 378.15 K (the experiment temperature). Intrinsic reaction coordinate (IRC)36 calculations were carried out to ensure that each transition state indeed connects its relevant minima on the potential energy surface. The relative Gibbs energies for all structures were refined by performing single-point energy calculations at the theory B3LYP-D3(BJ)/SDD-6-311++G(d,p) level in implicit acetonitrile solvent based on the optimized geometries above. The 3D diagrams of selected structures were prepared using CYLview.37 Natural bond orbital (NBO) analysis was performed by the GenNBO 5.0 program.383. Results and discussion
For the present Pd/NBE* cooperative three-component reaction (Scheme 1), the original catalyst used in the experiment is Pd(OAc)2. It is well known that Pd(OAc)2 under alkaline conditions can spontaneously afford palladium(0) complexes, which act as the catalytically active species.39,40 Starting from Pd(0) species, several different reaction sequences have been taken into account, as shown in Scheme 3, where the pathway with solid arrows corresponds to the mechanism in Scheme 2, and those with dotted arrows describe other potential possibilities. We carried out calculations for all these pathways to better understand the reactivity. Based on calculated results, our discussion is divided into three subsections, which describe formations of ANP intermediate, the C–C chiral axis, and the biaryl product, respectively. | ||
| Scheme 3 Possible reaction sequences of three-component cascade process. | ||
3.1 Formation of ANP
As indicated in Scheme 2, ANP denotes the five-numbered aryl–norbornyl–palladacycle formed via the reaction of aryl iodide with Pd(0) and NBE*. The calculated results for this process is summarized in Fig. 1, where the L2Pd(0)-olefin species 4, the most stable initial complex identified from the present calculations, is set as the energy reference point. Initially, aryl iodide 1 coordinates to the Pd(0) center with release of a ligand (MeCN) and olefin, resulting in intermediate 5, from which aryl iodide carries out oxidative addition on Pd(0) viaTS1 with a relative energy of 12.8 kcal mol−1, resulting in a three-coordinate aryl–palladium(II) halide 6. Subsequently, NBE* coordinates to the Pd(II) center in 6 to form intermediate 7 with a binding energy of 5.4 kcal mol−1. Then, NBE* inserts into the Pd(II)–C(aryl) bond viaTS2 with an energy barrier of 8.7 kcal mol−1 to afford intermediate 8. It should be noted that there are four possible pathways of NBE* insertion process depending on the coordination orientation of NBE* towards to Pd(II) center, and the most advantageous in energy is shown in Fig. 1. Other three energetically less favorable pathways are given in Fig. S1 in ESI.†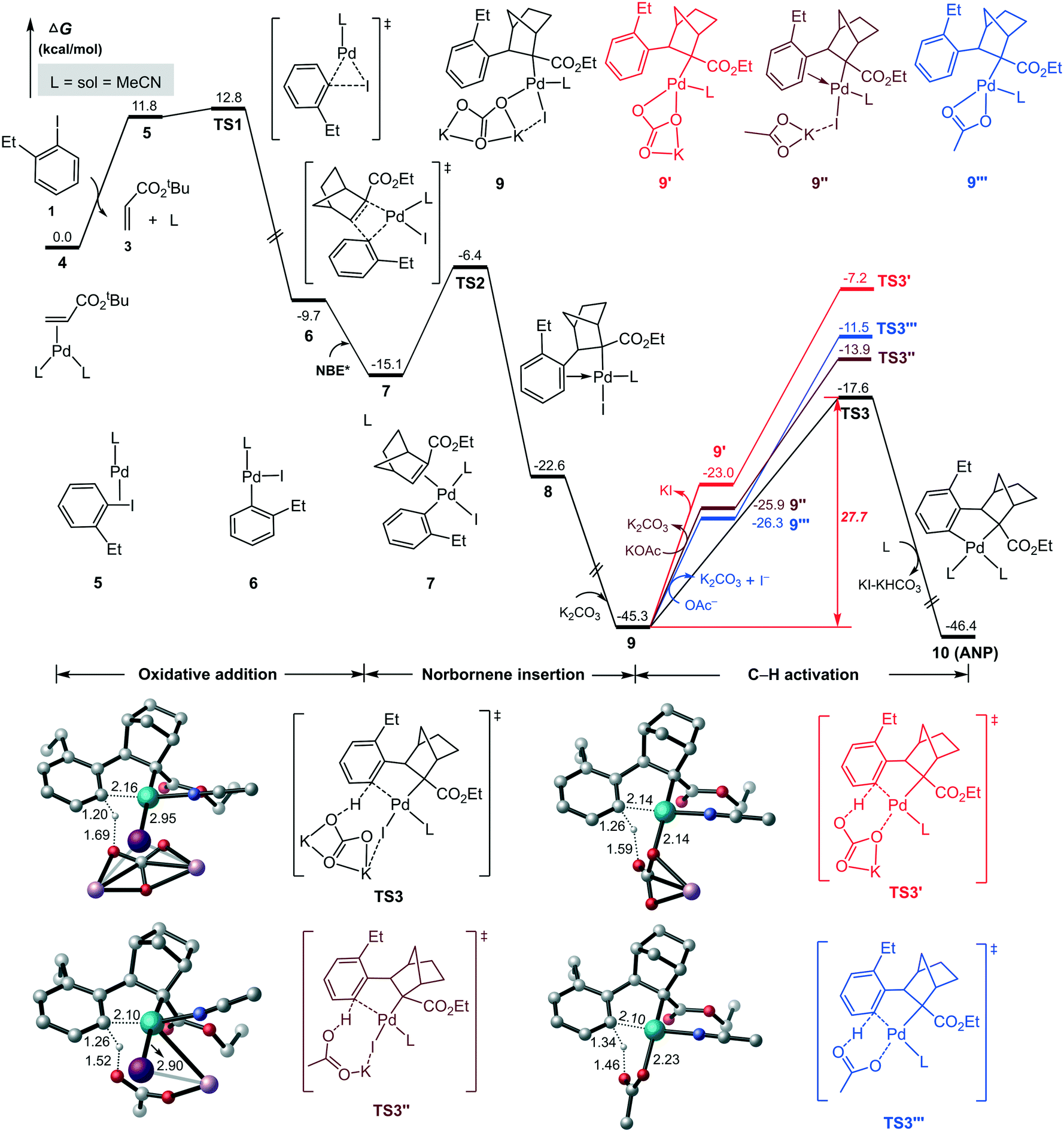 | ||
| Fig. 1 Calculated Gibbs energy profiles for the formation of the aryl–norbornyl–palladacycle (ANP) intermediate (10). Selected bond distances are given in Å and irrelevant hydrogen atoms are omitted for clarity. | ||
According to Scheme 2, the next step is the C–H bond activation to form the ANP intermediate. Based on previous studies by groups of Lan41 and Hong42 on the similar Pd/NBE cooperative Catellani reactions, this process is expected to occur with the assistance of a base. Here K2CO3 (2.5 equiv., Scheme 1) was used as proton abstraction reagent in the C–H bond activation. Upon coordination of K2CO3, intermediate 8 is brought to a tetracoordinated Pd(II) complex 9 with an energy release of 22.7 kcal mol−1. The subsequent C(sp2)–H activation proceeds according to a concerted metalation–deprotonation (CMD) mechanism43,44 to form the ANP (10) viaTS3 with a barrier of 27.7 kcal mol−1.
Other three potential pathways for the C–H bond activation are also shown in Fig. 1 for comparison. The red describes the pathway with the removal of KI prior the C–H activation to give species 9′, which then carries out the C–H bond activation viaTS3′. However, TS3′ is indicated to be energetically less favorable than TS3 by 10.4 kcal mol−1. This can be attributed to the large energy requirement for cleaving the strong Pd–I bond to release of KI.
The blue and brown pathways correspond to the C–H bond activation processes by using acetate anion as the proton abstraction reagent, as done in several recent works of Pd/NBE cooperative Catellani reactions.41,42 Identified transition states in these two pathways are denoted as TS3′′ and TS3′′′, which are counterparts of TS3 and TS3′, respectively. Our calculations indicate that these two pathways are also energetically less favorable than the black one. This fact, together with the low concentration of the acetate anion from a catalytic amount of Pd(OAc)2, suggests that the acetate anion is not appropriate for assisting the proton abstraction process. Thus, we believe that the C–H metalation occurs with assistance of K2CO3 which was used in the experiment in a stoichiometric amount (Scheme 1). The situation is similar to previous theoretical reports by Bi45 and Liu.46
After clarifying the formation mechanism of ANP intermediate, we now consider the question mentioned above: why the aryl iodide (as substrate) rather than the aryl bromide (as arylation reagent) carries out the initial oxidation addition on the Pd(0) center. To clarify the question, we carried out calculations for the oxidative addition of aryl bromide and the results are compared with that of the aryl iodide in Fig. 2a. It is found that the oxidation addition of aryl bromide viaTS1′ needs to overcome a barrier of 16.1 kcal mol−1, which is larger by 3.3 kcal mol−1 than that for the aryl iodide. Thus the Pd(0) species selectively reacts with aryl iodide instead of with aryl bromide, as confirmed by previous experimental and theoretical studies.47,48 This can be attributed to the larger bond energy of C–Br bond than C–I bond (78.4 vs. 67.3 kcal mol−1 from the present calculations) and the strong electron-withdrawing capacity of the –CO2Me group in the aryl bromide, which remarkably reduces the negative charge of the carbon atom of the C–Br bond (−0.08 vs. −0.23 e in the C–I bond) and thus reduces its nucleophilic attack ability on the positively charged palladation center.
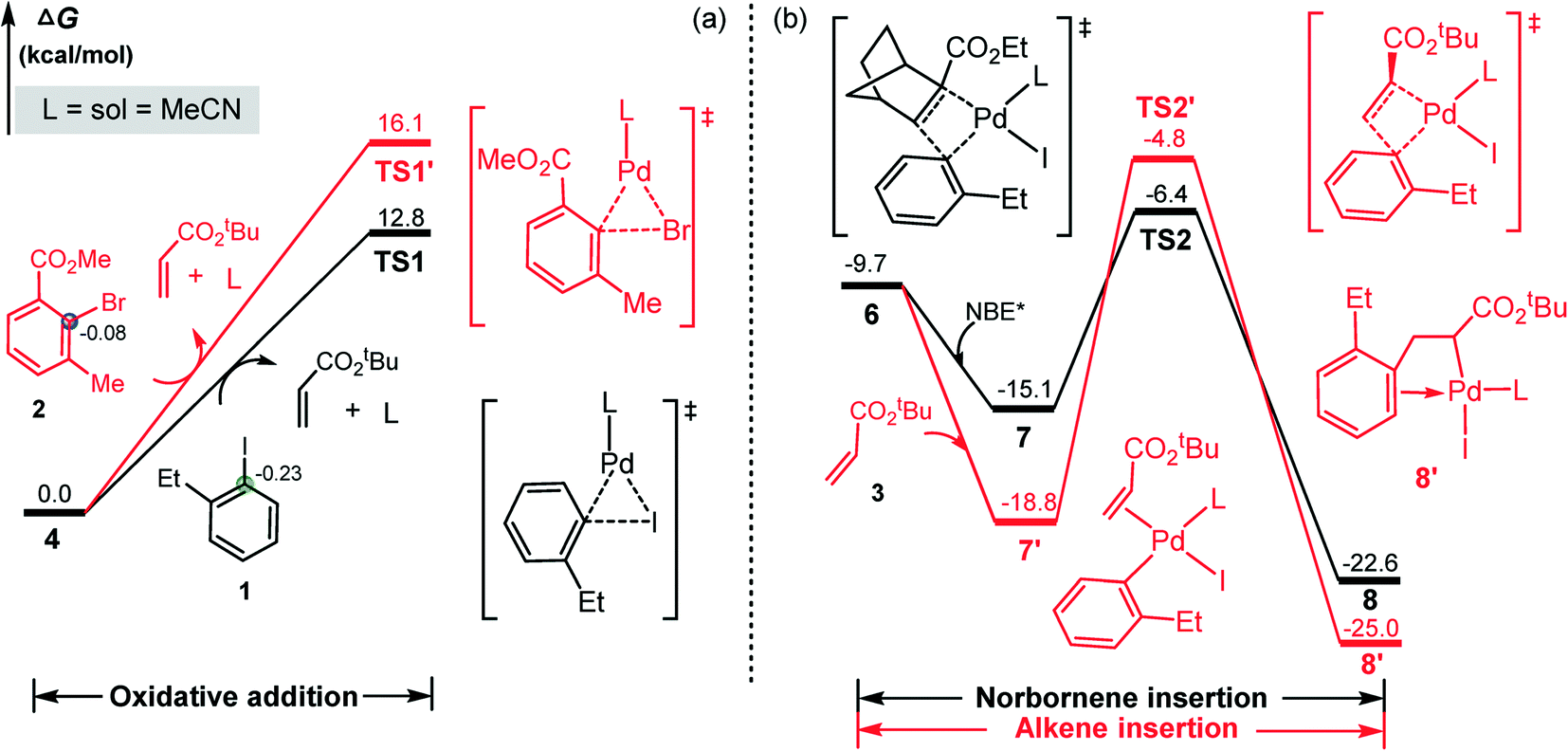 | ||
| Fig. 2 (a) Aryl iodide vs. aryl bromide oxidative addition, and (b) NBE*vs. alkene insertion into the Pd(II)–C(aryl) bond. Calculated NBO charges are given in e. | ||
It should be noted that instead of NBE*, the alkene (3) can also insert into the Pd–C bond of aryl–palladium iodine intermediate 6, resulting in the coupling product of 1 with 3. This pathway would result in the direct coupling product of 1 with 3. The calculated results are shown in Fig. 2b, and the reaction occurs viaTS2′. This pathway is located to be 1.6 kcal mol−1 less favorable than the NBE* insertion pathway viaTS2. Inserting NBE* or 3 into the Pd–C bond involves the charge transfer from the highest occupied molecular orbital (HOMO) of NBE* or 3 to the lower unoccupied molecular orbital (LUMO) of 6. As shown in Fig. 3, the calculated HOMO–LUMO gap is 4.24 eV for the NBE* insertion and 4.74 eV for the alkene insertion, implying that the NBE* insertion is more favorable than the alkene insertion. This is in agreement with the experimental obsevation27 where the direct Heck coupling product between 1 and 3 is in very low yield.
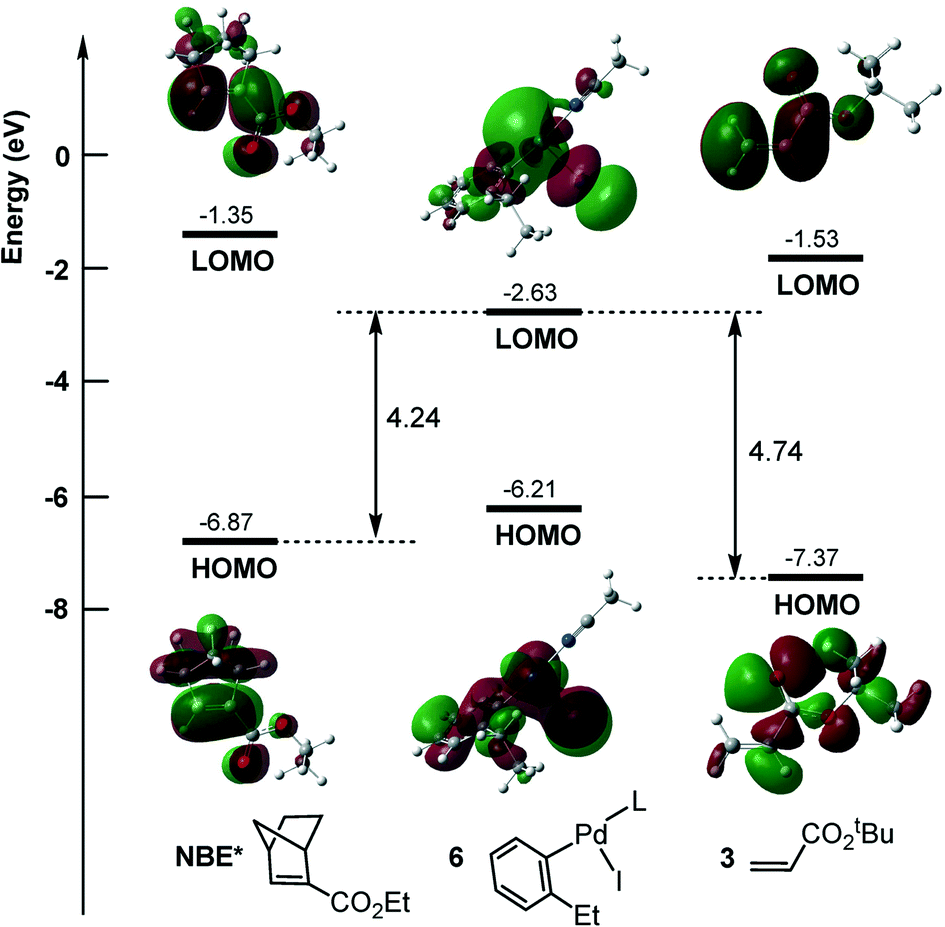 | ||
| Fig. 3 Calculated HOMO and LUMO energy levels for NBE*, 3 and 6. | ||
3.2 Formation of C–C chiral axis
According to Scheme 2, the axially chiral intermediate can be obtained via the reaction of ANP with the aryl bromide. Fig. 4 shows the calculated mechanism details for construction of both (R)- and (S)-C–C axial chiralities, involving oxidative addition of the C(aryl)–Br bond on the Pd(II) center followed by the C–C reductive elimination on the Pd(IV) center. Our attention first focuses on the construction of (S)-C–C axial chirality (the right profile in Fig. 4). The coordination of 2,6-substituted aryl bromide 2 to ANP results in the quasi-planar tetracoordinated complex 11, where the O and Br atoms in 2 were located at the trans orientations of Cphenyl and Cnorborneyl, respectively. Subsequently, the oxidative addition of C–Br bond on the Pd(II) occurs viaTS4, resulting in the Pd(IV) intermediate 12, a bis-palladacyclic species. Finally, intermediate 12 undergoes the reductive elimination viaTS5 to form the stable (S)-C–C axially chiral intermediate 13. The barriers of oxidative addition and reductive elimination involved in this stage are 13.5 and 20.2 kcal mol−1, respectively.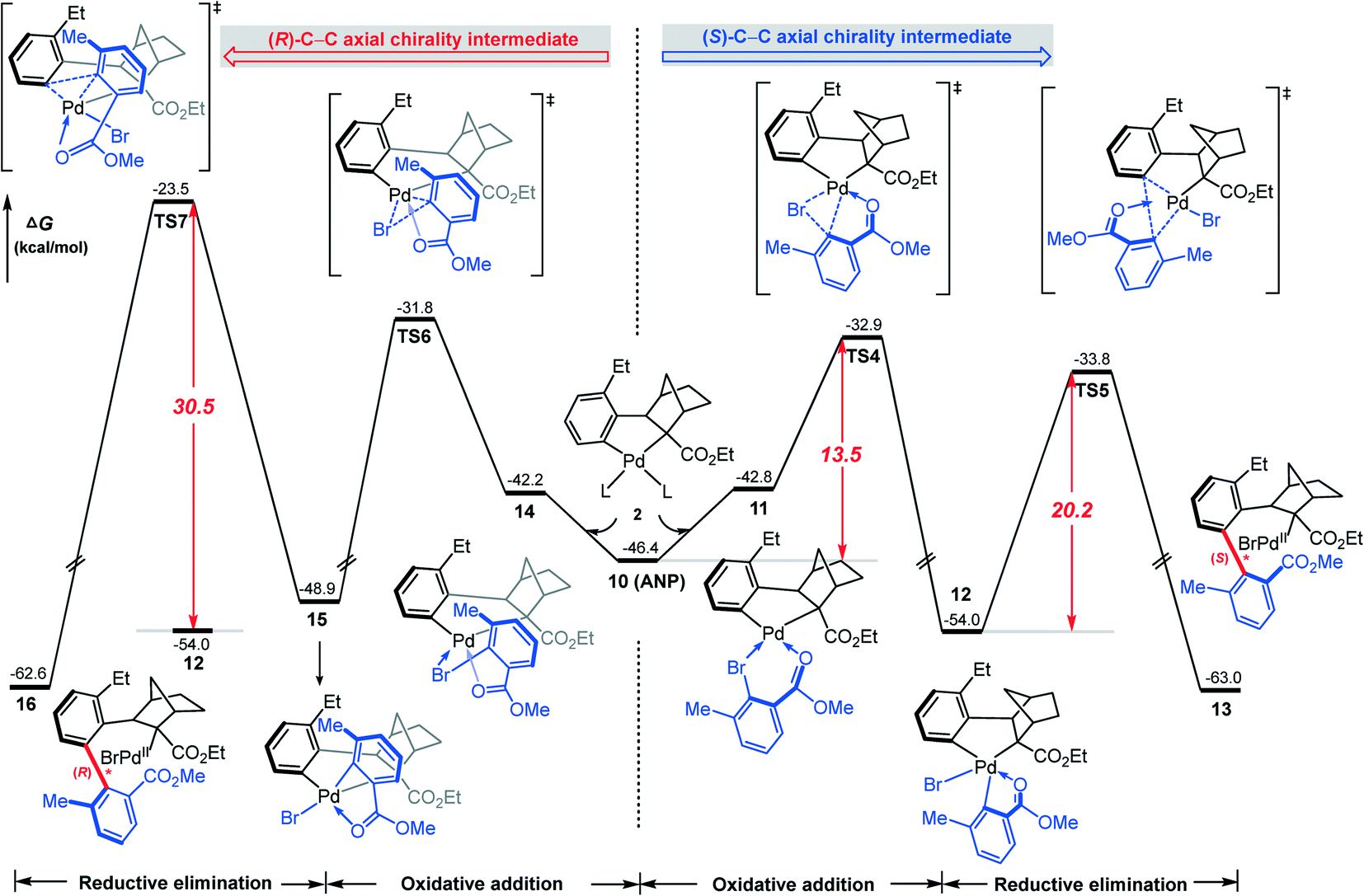 | ||
| Fig. 4 Calculated Gibbs energy profiles for the formation of (S)-C–C chiral axis (the right profile) in comparison with that of (R)-C–C chiral axis (the left profile). | ||
In contrast, the calculated result for the construction of (R)-C–C axial chirality is shown in the left profile of Fig. 4. The barriers of the C–Br bond oxidative oxidation viaTS6 and the C–C reductive elimination viaTS7 are 22.2 and 30.5 kcal mol−1 (from 12 to TS6/TS7). Thus, the construction of (R)-C-C axial chirality involves much higher barrier than that of the (S)-C–C axial chirality, 30.5 vs. 20.2 kcal mol−1, exclusively favoring the latter. The optimized geometries show that the enantioselectivity mainly originates from the different steric repulsions in transition states involved in these two branches. As shown in Fig. 5, in TS7, there is a distinct steric repulsion between the ortho-methyl group of aryl bromide and the bridged methylene of the norbornene (H–H distances: 1.90 and 2.13 Å), while in TS4 and TS5, no remarkable steric hindrance is observed (the shortest H–H distance is 2.16 Å in TS5). Therefore, the (S)-C–C axial chirality pathway is more favored than the (R)-C–C axial chirality pathway. We also investigated the other two oxidation addition pathways (TS4′ and TS6′), which involve the same reaction scheme but with opposite Br, O coordination position compared to TS4 and TS6. It is found that TS4′ and TS6′ are energetically less favorable by ∼8 kcal mol−1 than TS4 and TS6, respectively (Fig. S2†).
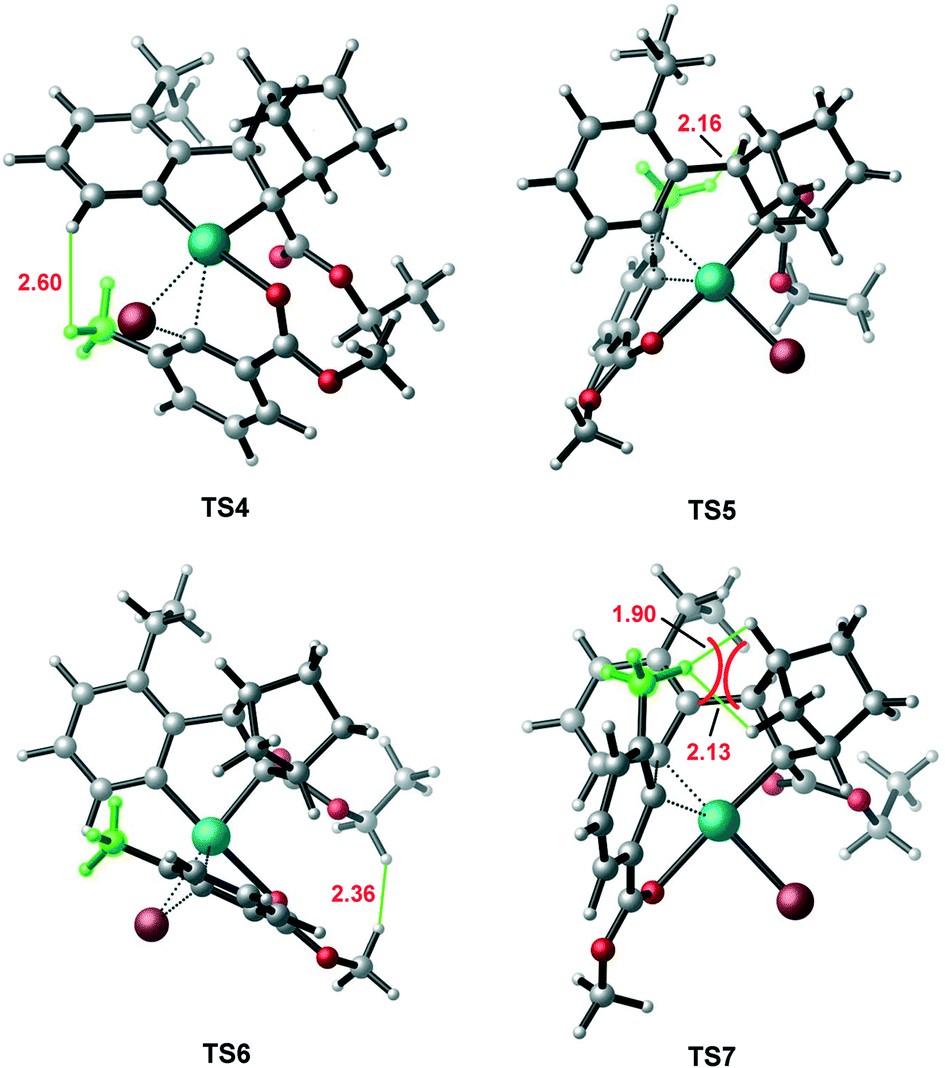 | ||
| Fig. 5 Optimized transition state geometries involved in the construction of C–C chiral axis. The selected bond distances are given in Å. | ||
Furthermore, the other three potential pathways from ANP according to Scheme 3 were also calculated: the C–C reductive elimination (Fig. 6a), the oxidative addition of aryl iodide to the Pd(II) center (Fig. 6b), and the terminating reagent insertion into one of two Pd–C bonds in ANP (Fig. S3 and S4†). The C–C reductive elimination viaTS8 is found to involve a barrier as high as 41.9 kcal mol−1, explaining the fact that no the C–C reductive elimination by-product was observed in the experiment. This may be attributed to the large strain of the 3,4-bridged ring and the steric repulsion between the palladium center and –CO2Et group of the norbornene moiety in TS8. The oxidative addition of aryl iodide to the Pd(II) center viaTS9 requires to overcome a barrier of 21.2 kcal mol−1, which is higher by 7.7 kcal mol−1 in comparison with the oxidative addition of aryl bromide viaTS4, implying that this process is also inaccessible. This situation is different from the oxidative addition of aryl iodide and aryl bromide on the Pd(0) center discussed above, where the aryl iodide is more prone to oxidative addition than aryl bromide due to the weaker C–I bond. Furthermore, the possibility for the terminating reagent insertion into Pd–C bond must also be ruled out due to high barriers involved (Fig. S3 and S4†). These results indicate that from ANP, the energetically most favorable reaction is the oxidation addition of aryl bromide on Pd(II).
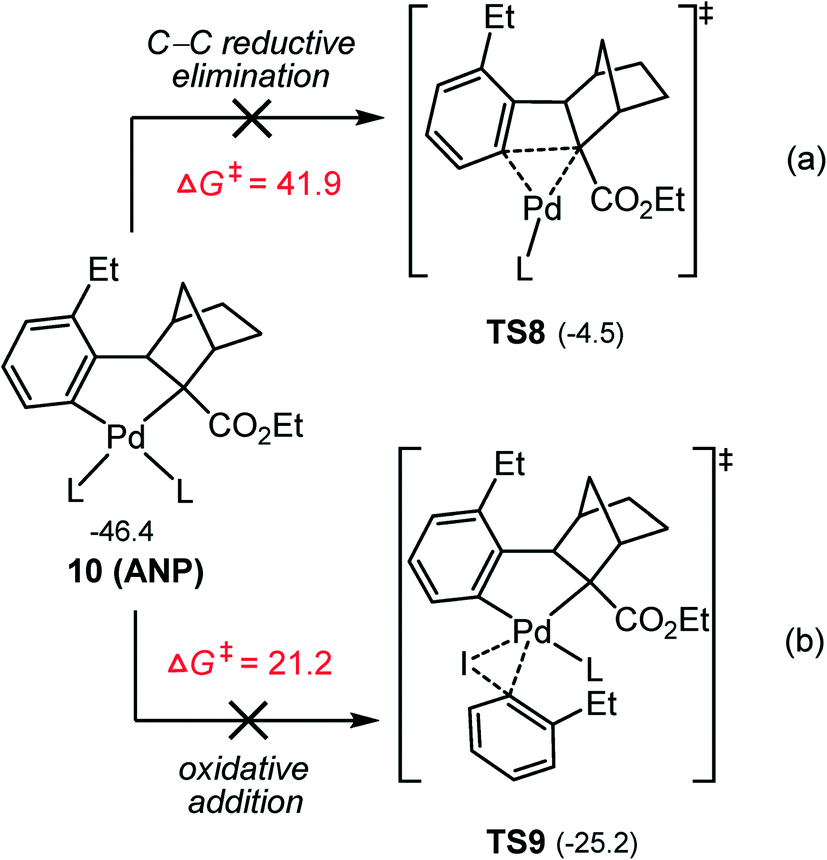 | ||
| Fig. 6 Calculated Gibbs energies (in kcal mol−1) of the direct reductive elimination (a) and oxidative addition (b) pathways from ANP. | ||
The different reactivity of these two aryl halides towards the Pd(II) center can be understood by comparing the optimized geometries of TS4 and TS9. In TS4, the ortho-ester group of reacting aryl bromide coordinates to Pd(II) center to form a fused five–five-membered palladacycle structure, while such a fused bis-palladacycle structure is absent in TS9. Therefore, we assume that the bis-palladacycle structure plays a substantial role for stabilizing the oxidative addition transition state of aryl bromide (TS4). To test this hypothesis, we replaced the ortho-ester group in TS4 with a hydrogen atom and added an acetonitrile (solvent) molecule which enables the Pd(II) to keep its quasi-planar tetracoordinated geometry. Upon these substitutions, the optimized oxidative addition transition state is indicated as TS4′′, a mono-palladacycle structure and is compared with the bis-palladacyclic TS4 in Fig. 7. It is found that the barrier of the oxidative addition increases to 24.6 kcal mol−1 (from ANP to TS4′′), confirming the intrinsic role of the bis-palladacycle for stabilizing the oxidative addition transition state. On the other hand, the calculated barriers crossing TS4′′ and TS9, 24.6 and 21.2 kcal mol−1 respectively, support the generally accepted view that the oxidative addition of aryl bromide on a metal center is more difficult than aryl iodine. It is noted that in TS4, there is another fused 5/6-ring, which presents a quasi-planar motif and thus remarkably increase the rigidity and stability of the structure. The result is consistent with the previous study by our group on the palladium-catalyzed carboxylate-assisted C(sp3)–H arylation, wherein the unique 5/6-ring transition state was found to account for the facile C–C reductive elimination.49
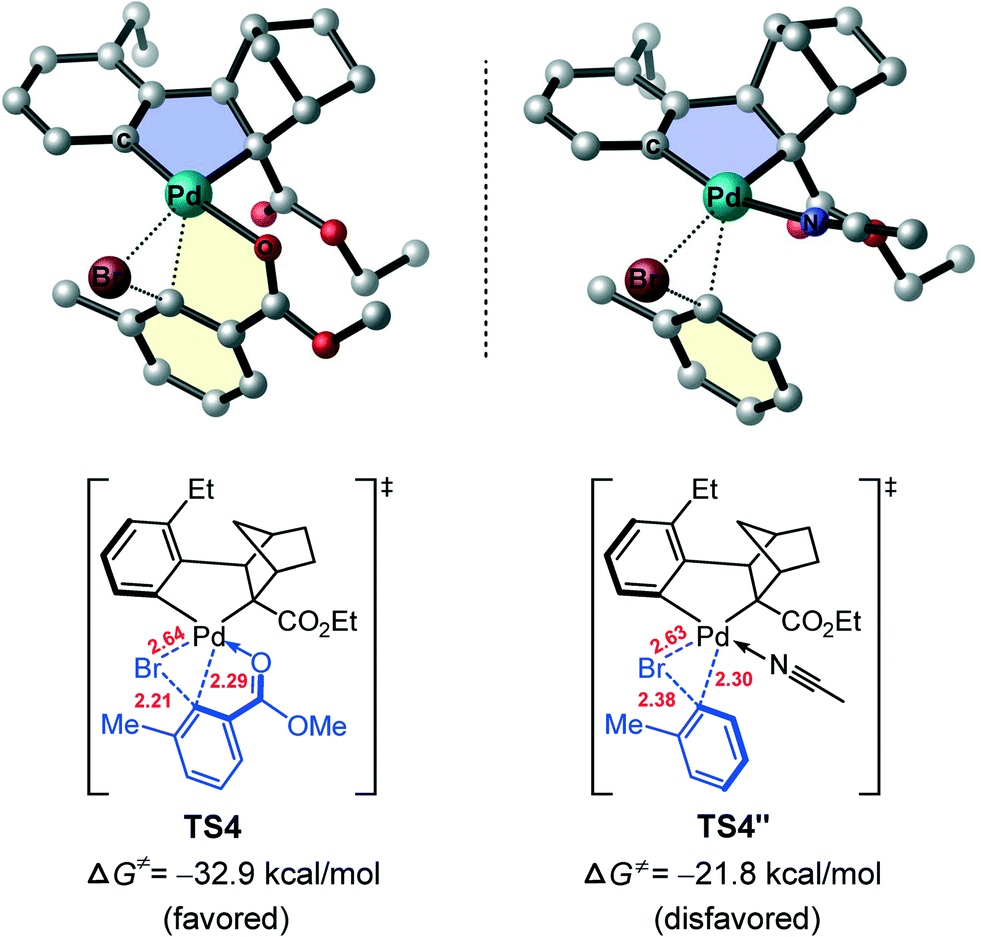 | ||
| Fig. 7 Optimized geometries of TS4 and TS4′′ with selected bond distances (red values, in Å). All hydrogen atoms have been omitted for clarity. | ||
From these discussion, it is clear that the ortho-ester group in the aryl bromide is of crucial importance, which makes the oxidative addition of the aryl bromide on the Pd(II) more facile than that of the aryl iodine. So the Pd(II) species can selectively react with the aryl bromide instead of the aryl iodide. This substrate characteristics of aryl bromine have been also shown in several reports.12,41,42
3.3 Intermolecular termination
Our attention now turns to the subsequent reaction of the (S)-C–C axially chiral intermediate 13 with the terminating reagent (tert-butyl acrylate, 3) to furnish the final axially chiral biaryl. Fig. 8 shows the calculated mechanism details that involve NBE* extrusion, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C insertion into the Pd–C(sp2) bond, and β-H elimination. The NBE* extrusion from 13 takes place via transition state TS10 with a barrier of 13.4 kcal mol−1, leading to intermediate 17. Note that the key geometrical parameters in TS10 is similar to those in TS2 where NBE* is inserting into the Pd–C(sp2) bond.
C insertion into the Pd–C(sp2) bond, and β-H elimination. The NBE* extrusion from 13 takes place via transition state TS10 with a barrier of 13.4 kcal mol−1, leading to intermediate 17. Note that the key geometrical parameters in TS10 is similar to those in TS2 where NBE* is inserting into the Pd–C(sp2) bond.
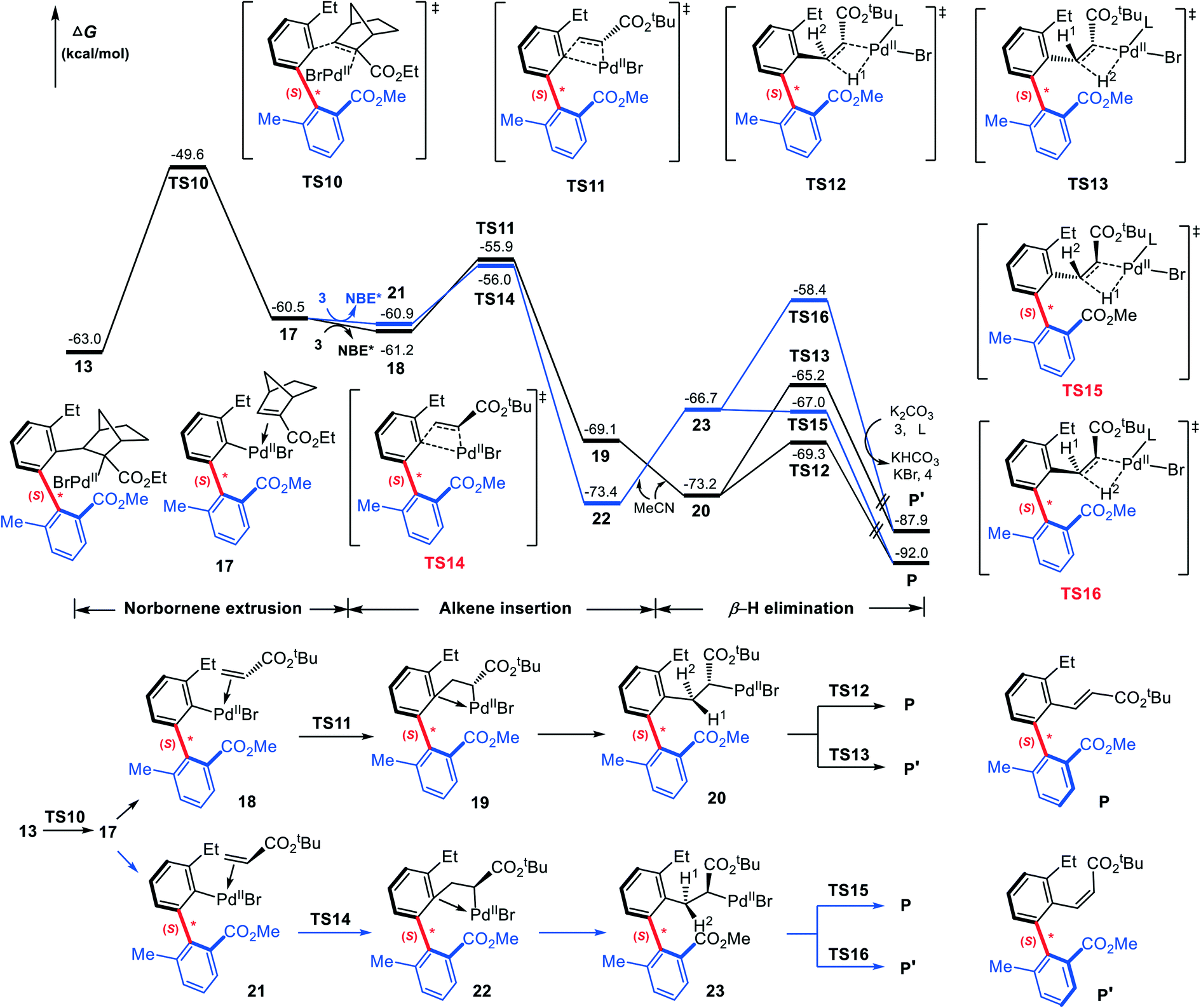 | ||
| Fig. 8 Calculated Gibbs energy profiles for the intermolecular termination. | ||
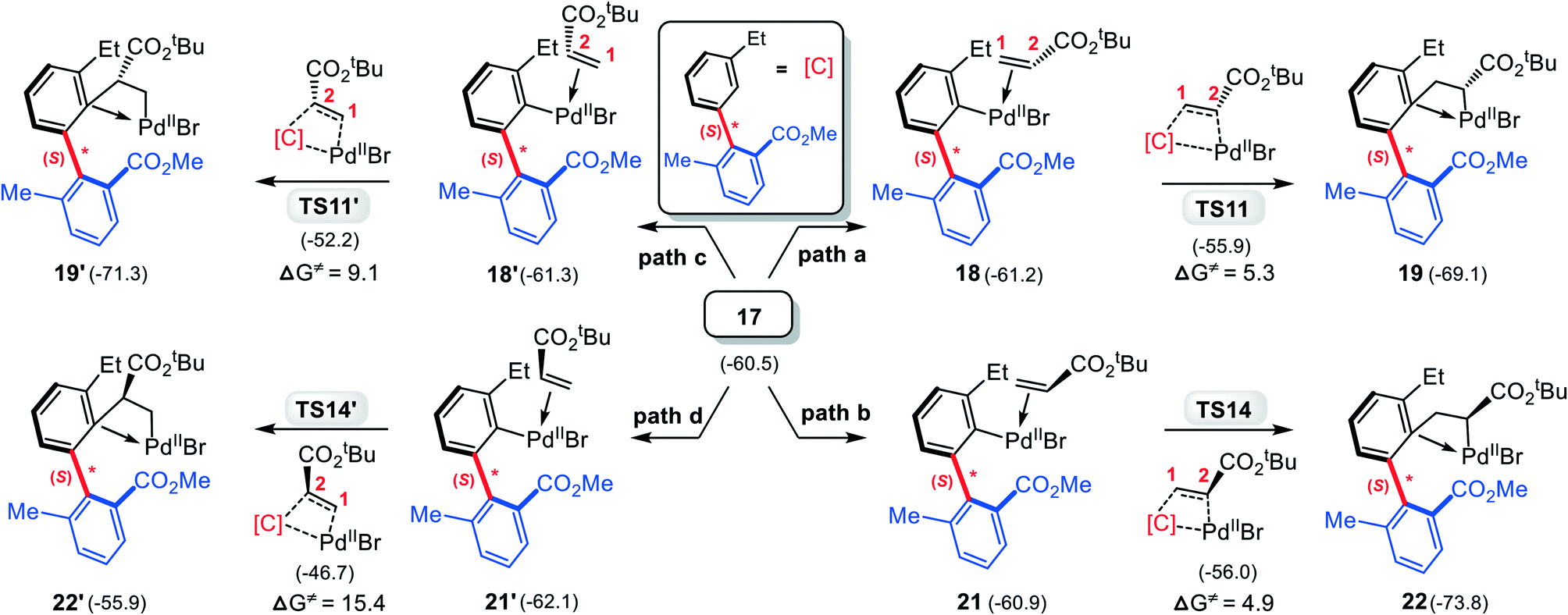
Subsequently, 3 replaces the NBE* in 17, giving the olefin-Pd complex 18, from which the CC inserts into the Pd–C(sp2) bond. Both the 1,2- and 2,1-insertion modes have been taken into account, as shown in Scheme 4, where each mode contains two different pathways depending on –CO2tBu group in 3 pointing to or away from the chiral axis. It is found that two 2,1-insertion modes involve almost isoenergetic transition states (−55.9 kcal mol−1 for TS11 and −56.0 kcal mol−1 for TS14), and are energetically more favorable than two corresponding 1,2-insertion modes (−52.2 kcal mol−1 for TS11′ and −46.7 kcal mol−1 for TS14′). This can be attributed to both the larger π electron density of C2 than C1 due to the electron-withdrawing property of –CO2tBu group, favoring the nucleophilic attack of C2 to Pd(II), and the smaller steric hindrance between the phenyl moiety and the –CO2tBu group in the 2,1-insertion mode than that in the 1,2-insertion.
 | ||
| Scheme 4 Four different modes of tert-butyl acrylate insertion into Pd–C(sp2) bond with calculated relative Gibbs energies (in kcal mol−1). | ||
In Fig. 8, only two favorable 2,1-insertion modes viaTS11 and TS14 are given, and the calculated barriers are 5.3 and 4.9 kcal mol−1, respectively. The resultant intermediates are 19 and 22. Finally, the catalytic cycle is closed through the β-H elimination process. From 19, β-H1 elimination viaTS12 (−69.3 kcal mol−1) affording the E-configuration product P is calculated to be more favorable by 4.1 kcal mol−1 than β-H2 elimination viaTS13 (−65.2 kcal mol−1) affording the Z-configuration product P′. The fact can be attributed to the greater steric hindrance between the phenyl moiety and the ester group –CO2tBu in TS13. Similarly, from 22, transition states resulting P and P′ are denoted as TS15 and TS16, which are less stable by 2.3 and 6.8 kcal mol−1 than TS12 and TS13, respectively. Thus 22 is expected to be less possible for providing the desired axially chiral product. From Fig. 8, it is clear that the β-H elimination viaTS12, offering the (S)-type, E-configuration axially chiral biaryl product, is the most favorable pathway.
To show a complete mechanistic picture of the Pd/NBE* cooperative three-component cascade reaction, we combine the main results in Fig. 1, 4, and 8 into Fig. 9. The construction of axial chirality is achieved via eight steps: Ar–I oxidative addition to Pd(0), norbornene insertion, C–H activation, Ar–Br oxidative addition to Pd(II), C–C reductive elimination, norbornene extrusion, alkene insertion, and β-H elimination. The C–H activation is identified as the rate-determining step of the reaction with a barrier of 27.7 kcal mol−1, which matches the experimental conditions (105 °C). The oxidative addition of Ar–Br on Pd(II) followed by the C–C reductive elimination favors the construction of the (S)-C–C axial chirality. This situation is different from the recent report by Lan et al.41 for a similar three-component Catellani reaction, where the rate-determining step of the reaction was identified as the oxidative addition of aryl bromide.
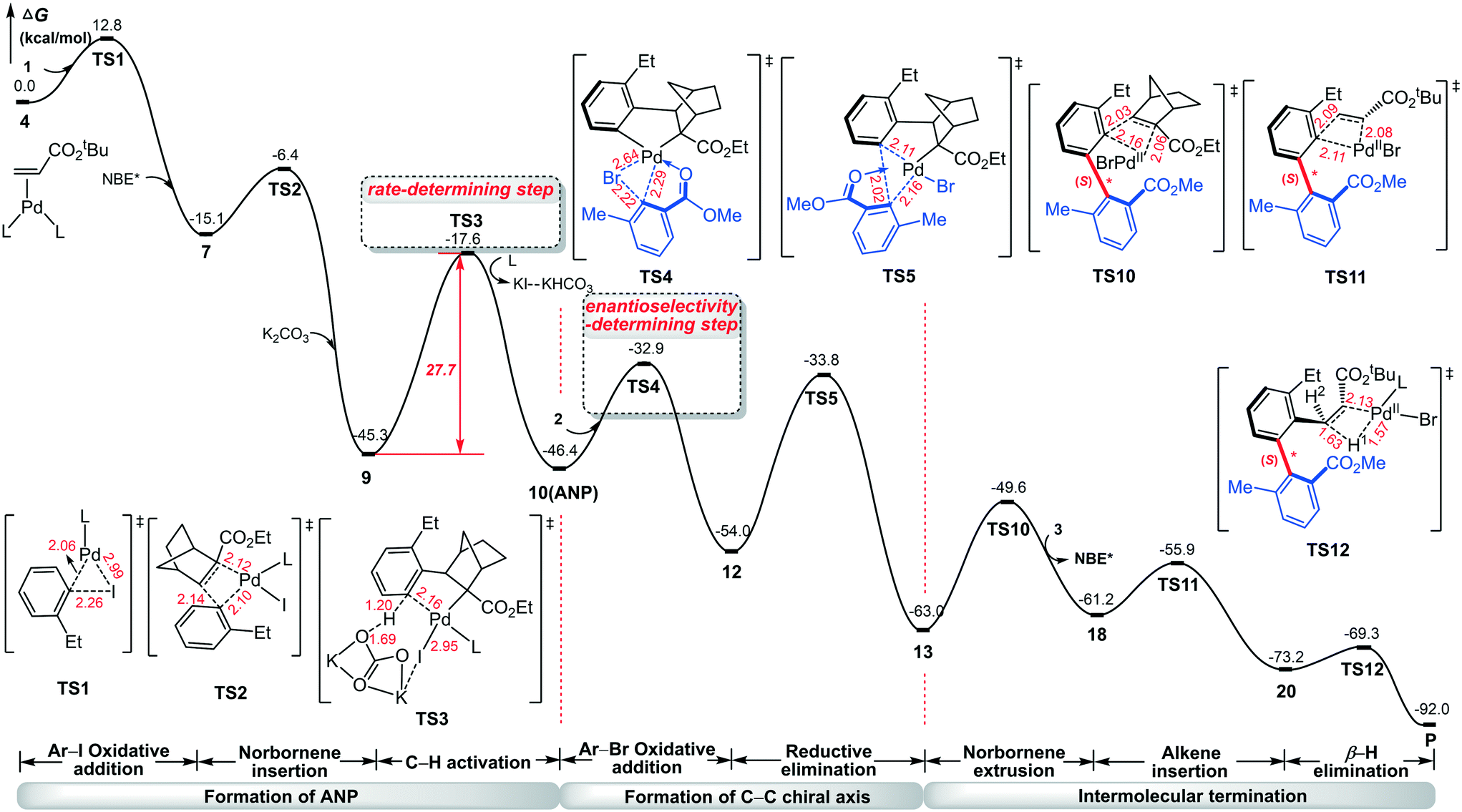 | ||
| Fig. 9 Complete mechanistic picture of the Pd/NBE* cooperative three-component cascade reaction for construction of the (S)-C–C axially chiral biaryl. | ||
Finally, we try to rationalize the experimentally observed enantiomeric excess (ee) value of the product, 97%. From our calculations (Fig. 4), the barriers for constructing the (S)- and (R)-C–C chiral axes are 20.2 and 30.5 kcal mol−1, respectively. The large difference (10.3 kcal mol−1) implies the reaction exclusively favors the (S)-C–C axially chiral biaryl. In this case, how is a small amount of R-type product produced? We conjecture that the R-type product may originate from the S–R isomerization. The calculated barrier of the C–C chiral axis rotation is 34.1 kcal mol−1 (Fig. 10). The present reaction was carried out under 105 °C, which provides a potential possibility of the S–R isomerization.
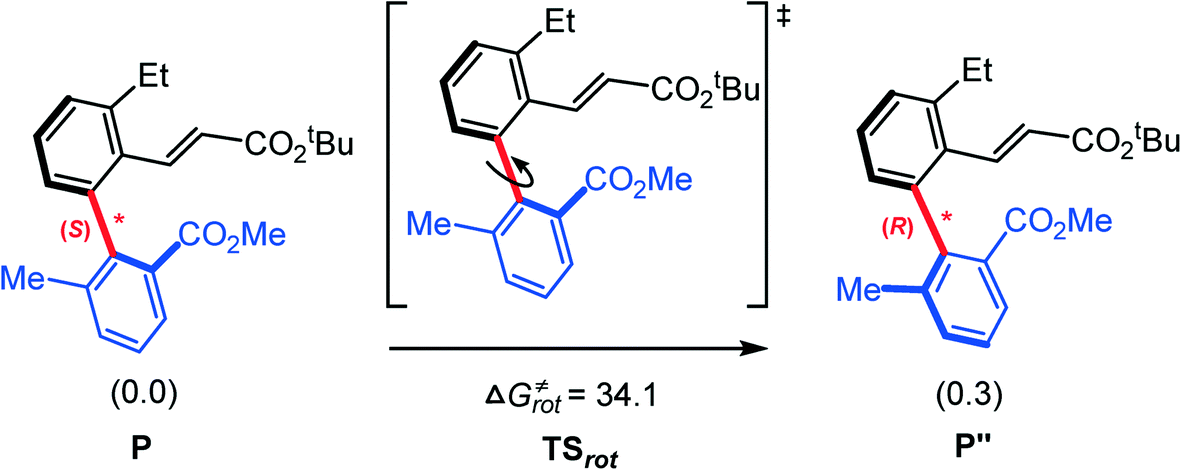 | ||
| Fig. 10 Calculated Gibbs energies (in kcal mol−1) for the S–R isomerization by rotating the chiral axis. | ||
4. Conclusions
In summary, by performing DFT calculations on the prototype system of the palladium/chiral norbornene cooperative three-component cascade reactions, we have shown the mechanistic details for the construction of axially chiral biaryls. The catalytic cycle is divided into three stages: (i) formation of the aryl–norbornyl–palladacycle (ANP) intermediate via the oxidation addition of the aromatic iodine on the Pd(0) center, norbornene insertion, and the potassium carbonate assisted C–H activation, (ii) construction of C–C chiral axis, involving the oxidation addition of aryl bromide on the Pd(II) and the C–C reduction elimination on Pd(IV), and (iii) intermolecular termination via norbornene extrusion, alkene insertion, and β-H elimination. The present calculations identify the C–H activation as the rate-determining step of the reaction. The oxidative addition of aryl bromide on Pd(II) controls the enantioselectivity and the barrier involved for the construction of the (S)-C–C axial chirality is calculated to be 20.2 kcal mol−1. The preferred formation of the atroposelective (S)-C–C axially chiral biaryl is attributed to the smaller steric repulsion between the ortho-methyl group of aryl bromide and the norbornene moiety than that in the (R)-C–C counterpart. The ortho-ester group on the aryl bromide plays a substantial role, which makes the oxidation addition of the aryl bromide on Pd(II) more facile than that of the aryl iodine. The regioselective insertion of the terminating reagent in the final intermolecular termination stage originates from both the larger π electron density of C2 in tert-butyl acrylate and the smaller steric hindrance between the phenyl moiety and the –CO2tBu group. The theoretical results rationalize the reaction sequence of the aryl iodide and the aryl bromide: the Pd(0) catalyst selectively reacts with the aryl iodide to provide the Pd(II) species, which then preferentially reacts with the aryl bromide to furnish the key (S)-axial chirality intermediate. The norbornene as a transient mediator also plays a substantial role, which assists the ortho-C–H bond activation of aryl iodide and inhibits the insertion of tert-butyl acrylate into Pd(II)–aryl intermediates to avoid direct coupling between aryl iodide and tert-butyl acrylate. The theoretical results rationalized the experimental observations and provide a deep understanding of the palladium/chiral norbornene cooperative catalysis in the synthesis of axially chiral biaryl compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21833004 and 21773139).References
- J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS.
- Q. Li, L. Green, N. Venkataraman, I. Shiyanovskaya, A. Khan, A. Urbas and J. W. Doane, J. Am. Chem. Soc., 2007, 129, 12908–12909 CrossRef CAS PubMed.
- J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC.
- M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC.
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC.
- D. Shen, Y. Xu and S.-L. Shi, J. Am. Chem. Soc., 2019, 141, 14938–14945 CrossRef CAS PubMed.
- L.-W. Qi, S. Li, S.-H. Xiang, J. Wang and B. Tan, Nat. Catal., 2019, 2, 314–323 CrossRef CAS.
- H. Yang, J. Chen and L. Zhou, Chem. – Asian J., 2020, 15, 2939–2951 CrossRef CAS.
- V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140–2143 CrossRef CAS PubMed.
- J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251–1255 CrossRef CAS.
- Y. Hua, Z.-S. Liu, P.-P. Xie, B. Ding, H.-G. Cheng, X. Hong and Q. Zhou, Angew. Chem., 2021, 60, 12824–12828 CrossRef CAS.
- L. Zhang, J. Zhang, J. Ma, D.-J. Cheng and B. Tan, J. Am. Chem. Soc., 2017, 139, 1714–1717 CrossRef CAS PubMed.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC.
- Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS.
- M. Lautens and S. Piguel, Angew. Chem., Int. Ed., 2000, 39, 1045–1046 CrossRef CAS PubMed.
- L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 14870–14877 CrossRef CAS.
- P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed.
- Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed.
- Z. Dong, G. Lu, J. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2018, 140, 8551–8562 CrossRef CAS.
- B.-S. Zhang, Y. Li, Z. Zhang, Y. An, Y.-H. Wen, X.-Y. Gou, S.-Q. Quan, X.-G. Wang and Y.-M. Liang, J. Am. Chem. Soc., 2019, 141, 9731–9738 CrossRef CAS PubMed.
- X. Li, J. Pan, S. Song and N. Jiao, Chem. Sci., 2016, 7, 5384–5389 RSC.
- R. Ferraccioli, Synthesis, 2013, 45, 581–591 CrossRef CAS.
- L. Ding, X. Sui and Z. Gu, ACS Catal., 2018, 8, 5630–5635 CrossRef CAS.
- Z.-S. Liu, Y. Hua, Q. Gao, Y. Ma, H. Tang, Y. Shang, H.-G. Cheng and Q. Zhou, Nat. Catal., 2020, 3, 727–733 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Jr. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- S. Huzinaga, Comput. Phys. Rep., 1985, 2, 281–339 CrossRef.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001 Search PubMed.
- I. Franzoni, H. Yoon, J.-A. García-López, A. I. Poblador-Bahamonde and M. Lautens, Chem. Sci., 2018, 9, 1496–1509 RSC.
- C. Amatore, A. Jutand and A. Thuilliez, Organometallics, 2001, 20, 3241–3249 CrossRef CAS.
- Q. Feng, X. Ma, W. Bao, S.-J. Li, Y. Lan and Q. Song, CCS Chem., 2021, 3, 377–387 CrossRef.
- Z.-S. Liu, P.-P. Xie, Y. Hua, C. Wu, Y. Ma, J. Chen, H.-G. Cheng, X. Hong and Q. Zhou, Chem, 2021, 7, 1917–1932 CAS.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- Y. Liang, Y.-Y. Jiang, Y. Liu and S. Bi, Org. Biomol. Chem., 2017, 15, 6147–6156 RSC.
- X. Qi, J. Wang, Z. Dong, G. Dong and P. Liu, Chem, 2020, 6, 2810–2825 CAS.
- F. Barrios-Landeros and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 6944–6945 CrossRef CAS PubMed.
- A. Ariafard and Z. Lin, Organometallics, 2006, 25, 4030–4033 CrossRef CAS.
- X. Ma, Z. Han, C. Liu and D. Zhang, Inorg. Chem., 2020, 59, 18295–18304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01863a |
| This journal is © The Royal Society of Chemistry 2022 |