
Mechanistic insights into CoOx–Ag/CeO2 catalysts for the aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid†
Mengchen
Fu
a,
Weiyao
Yang
a,
Chenyu
Yang
a,
Yiwen
Zhang
a and
Chun
Shen
 *ab
*ab
aBeijing Key Laboratory of Bioprocess, College of Life Science and Technology, Beijing University of Chemical Technology, No. 15 of North Three-Ring East Road, Chaoyang District, Beijing 100029, P.R. China. E-mail: shenchun@mail.buct.edu.cn
bThe State Key Laboratory of Chemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, P.R. China
First published on 5th November 2021
Abstract
Although Ag catalysts have potential for 5-hydroxymethylfurfural (HMF) oxidation to 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), further oxidation into 2,5-furandicarboxylic acid (FDCA) remains a severe challenge. Herein, we propose a strategy to synthesize CoOx–Ag/CeO2 catalysts where Ag+–Ov–Ce3+ interfacial sites are generated. For the first time, a mono-noble metal Ag catalyst, namely, CoOx–Ag/CeO2-2, achieves a satisfactory FDCA yield of 92.8%. Even under base-free conditions, a yield of 71.2% could still be obtained. A combination study unveils the harmonious cooperation between Ag+ and Ov–Ce3+ sites during the rate-determining step: the Ag+ site facilitates cleavage of the C–H bond, and the Ov–Ce3+ site enhances the adsorption of reactants and accelerates the dissociation of H2O and the transformation of the adsorbed O2 into superoxide radicals. The present work sheds light on the mechanism study on the HMF oxidation over Ag-based catalysts and may contribute to the rational design of high-performance heterogeneous catalysts.
Introduction
Excessive depletion of fossil resources has ignited interest in production of renewable chemicals from biomass, which is recyclable and carbon-neutral.1–4 Being a notoriously versatile platform chemical, 5-hydroxymethylfurfural (HMF) could be transformed into diverse products, including fuel additives and chemical building blocks.5 2,5-Furandicarboxylic (FDCA), which is derived from HMF oxidation, has received significant attention for its wide applications in many fields, especially for the production of bio-based polymers.6Intensive efforts have been devoted to FDCA production from HMF using noble metal-based catalysts (Pt, Pd, Au, and Ru-based catalysts) on account of their ability for oxygen activation.7–18 Besides the noble metals mentioned above, it is well accepted that Ag catalysts with lower price allow the partial oxidation of HMF, yielding HMFCA or DFF in water.19,20 A methodology mediated by Ag2O was reported for the preparation of HMFCA from HMF using aqueous H2O2 as an oxidizing agent in aqueous Na2CO3 solution, and a yield of 98% was achieved.21 As reported by Grunwaldt, HMFCA could be produced with a yield over 98% with the addition of NaOH using Ag/ZrO2 and Ag/TiO2 as catalysts; however, further oxidation of HMFCA into FDCA was infeasible.22,23 The only reported catalysts which could realize HMF oxidation to FDCA are gold rich bimetallic Au–Ag/ZrO2 in the presence of NaOH, while the silver rich catalysts are inactive in FDCA formation.24 Up to now, HMFCA could be formed via oxidation of the aldehyde group in HMF, but further transformation into FDCA by oxidation of the hydroxymethyl group over Ag-based catalysts has not been realized yet.13 Moreover, aerobic oxidations of HMF over Ag catalysts are usually conducted in the presence of excess base.25 From the viewpoint of biomass valorization, it is of significant importance to develop novel Ag-based catalysts with boosted catalytic activity for FDCA production. Besides, a clear fundamental understanding about the catalytic mechanism (rate-determining step, active oxygen species, and active sites) is also extremely desirable.26,27
In our previous studies,28–31 efficient heterogeneous catalysts have been designed and applied in the conversion of biomass into bio-based chemicals and fuels, and the structure–property correlations have been established, motivating us to go a step further by focusing on an incisive understanding about the promotion in the catalytic performance and the corresponding catalytic mechanism of Ag-based catalysts for FDCA production from HMF.
In this work, a series of CoOx–Ag/CeO2-X catalysts (X represents the molar ratio of Co/Ag) have been prepared and evaluated for HMF oxidation. The proposed catalysts may feature three possible advantages. First, as far as we know, it is the first exploration of FDCA production over mono-metal Ag-based catalysts, which has not been reported yet. Second, given the easily reversible redox cycle of cerium ions (Ce3+/Ce4+) and the abundant oxygen vacancies (Ov),32 choosing CeO2 as the support may contribute to better catalytic performances. Third, the addition of the oxyphilic metal oxide CoOx may facilitate the adsorption of oxygen-containing reactants,33,34 thus enhancing the heterogeneous catalytic reaction. Satisfactory yields of FDCA were achieved over CoOx–Ag/CeO2-2, even under base-free conditions. A combination study including an adsorption experiment, kinetic study and selective poisoning experiment unveils that the Ag+–Ov–Ce3+ interface governs the catalytic performance of the rate-determining step of HMF oxidation, namely hydroxyl group oxidation (oxidation of HMFCA into FFCA): the Ag+ site facilitates cleavage of the C–H bond,35–38 and the Ov–Ce3+ site plays multiple roles, namely, enhancing the adsorption of HMFCA, promoting the O2 adsorption and activation,39,40 and accelerating the dissociation of H2O.41 Therefore, this work paves the way for rational design of Ag-based catalysts for FDCA production from HMF, which would be useful for other novel heterogeneous catalysts.
Results and discussion
Catalyst characterization
Fig. 1 shows the XRD patterns of CeO2, Ag/CeO2 and CoOx–Ag/CeO2-2, and Fig. S1† shows the XRD patterns of the rest of the catalysts. For all the three catalysts (Ag loading amount = 10 wt%), the intensive diffraction peaks at 2θ = 28.5, 33.1, 47.5, 56.3, 59.1 and 69.3° could be attributed to the (1 1 1), (2 0 0), (2 2 0), (3 1 1), (2 2 2), (4 0 0), (3 3 1) and (4 2 0) diffraction planes of the fluorite structure of CeO2 (JCPDS 34-0394).42,43 Two additional weak peaks at 2θ = 38.2° and 44.4° are observed on Ag/CeO2 and CoOx–Ag/CeO2-2, which can be ascribed to the (1 1 1) and (2 0 0) Bragg characteristic reflections of the face-centered cubic structure of Ag (JCPDS 65-2871).44,45 Notably, the body-centered cubic structure of Ag2O could also show the (2 0 0) reflection peak at 2θ = 38.2° (JCPDS 65-6811).46 Therefore, Ag2O nanoparticles may exist in the Ag/CeO2 and CoOx–Ag/CeO2-2 catalysts and are evenly loaded onto the surface of CeO2.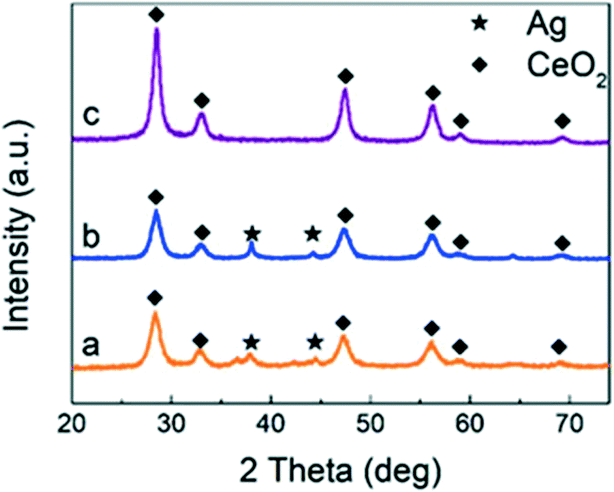 | ||
| Fig. 1 XRD patterns of the catalysts: (a) CoOx–Ag/CeO2-2, (b) Ag/CeO2, and (c) CeO2. | ||
TEM was conducted, and the results are shown in Fig. 2 to visually investigate the morphological characteristics of the Ag/CeO2 and CoOx–Ag/CeO2-2 catalysts. As shown in Fig. 2A and E, Ag nanoparticles can be clearly observed and evidenced by a lattice fringe of 0.24 nm, corresponding to the {111} lattice spacing of Ag.47 As for CeO2, {111} and {100} are observed to be the exposed planes with a lattice spacing of 0.31 and 0.27 nm, respectively.48 In Fig. 2E, metal Co shows a lattice fringe of 0.48 nm, which corresponds to the {111} facet of Co.49Fig. 2B–D and F–I show the elemental mapping images of the Ag/CeO2 and CoOx–Ag/CeO2-2 catalysts, respectively, indicating that Ag and Co species are uniformly dispersed on the surface of CeO2, in agreement with the XRD result. The addition of Co shows no great influence on the morphology of Ag and CeO2.
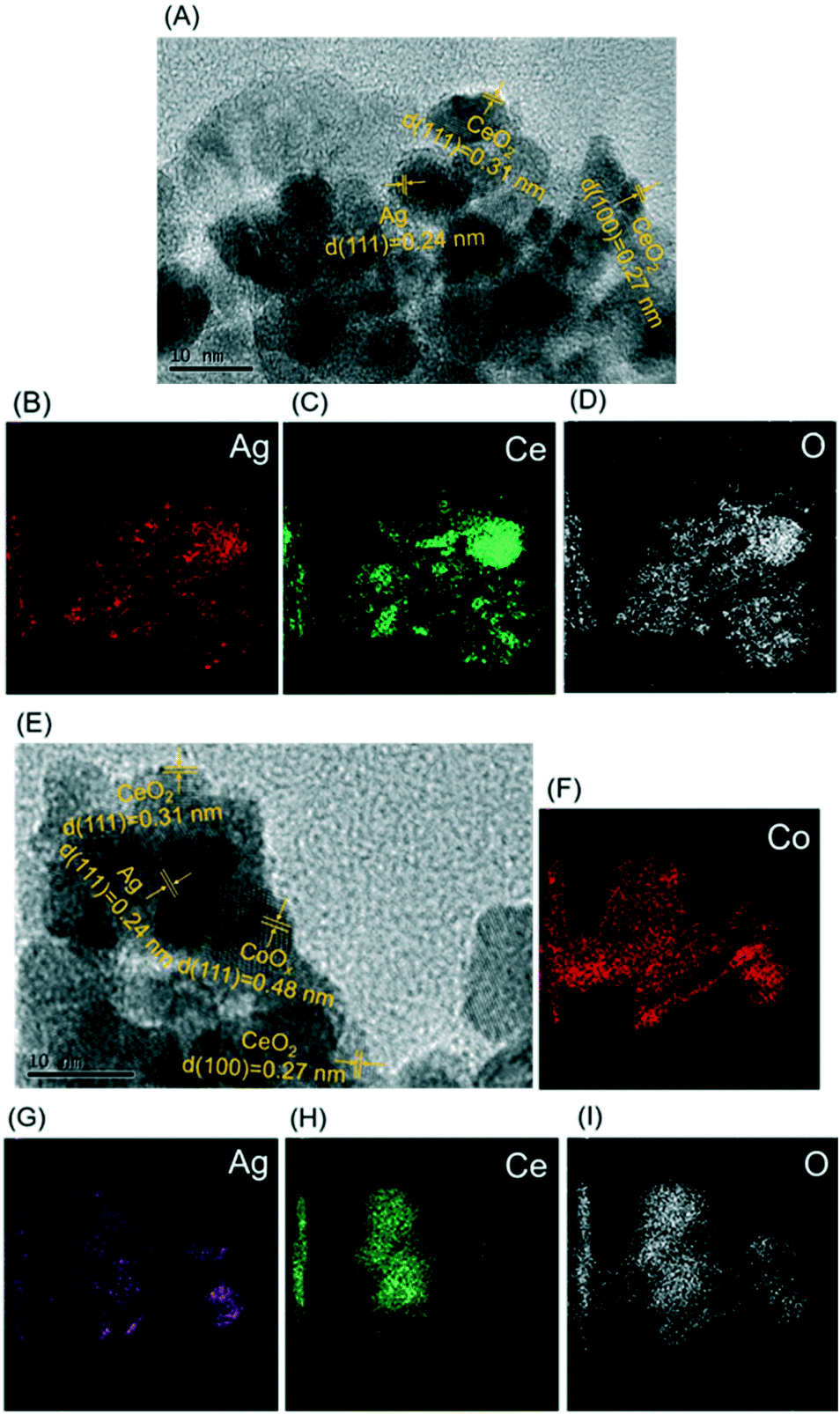 | ||
| Fig. 2 (A) TEM image of Ag/CeO2; (B)–(D) elemental mapping images of Ag/CeO2; (E) TEM image of CoOx–Ag/CeO2-2; (F)–(I) elemental mapping images of CoOx–Ag/CeO2-2. | ||
The contents of Ag and Co were determined by ICP-MS, and the result is listed in Table S1 in the ESI.†
To fully understand the electronic structure of the CoOx–Ag/CeO2-X and Ag/CeO2 catalysts, X-ray photoelectron spectroscopy (XPS) was performed. Deconvoluted by a Gaussian peak fitting method, the Ag 3d spectra (Fig. 3A) consist of two bands corresponding to the 3d3/2 and 3d5/2 transitions, which could be deconvoluted into four peaks. The two peaks at ∼367.3 and ∼373.3 eV are assigned to metallic Ag (Ag0), and the other two peaks at 368.1 and 374.1 eV are attributed to Ag+.44–46 The Ce 3d3/2–Ce 3d5/2 spectra in Fig. 3B could be resolved into six groups. The peaks at 885.6 eV and 904.0 eV are attributed to the Ce3+ cations, while the other peaks at ca. 882.7, 889.8, 898.6, 901.3 and 907.7 eV are assigned to the Ce4+ species.45,48 The Co 2p spectra in Fig. 3C is deconvolved into two peaks at around 780.1 and 782.6 eV, which could be ascribed to the Co2+ and Co3+ species, respectively.49 In Fig. 3D, the XPS spectra in the O 1s region are fitted into four peaks at around 532.1, 531.0, 530.1 and 529.4 eV, which could be assigned to the surface-adsorbed H2O molecules, surface hydroxyl groups, lattice oxygen and Ov, respectively.50 As shown in Table S2,† the ratios of Ag+, Ce3+, Co2+ and oxygen vacancy concentration of the doped catalysts increase first and then decrease gradually with that of CoOx–Ag/CeO2-2 being the highest. To confirm the relevance between oxidized Ag species and reactivity, the XPS spectra of Ag/CeO2-red are shown in Fig. S2.† After the addition of Co(NO3)3, the proportion of Ag+ and Ce3+ increases obviously, while Co2+ species are observed, indicating that electrons transfer from Ag to the support and Co species.
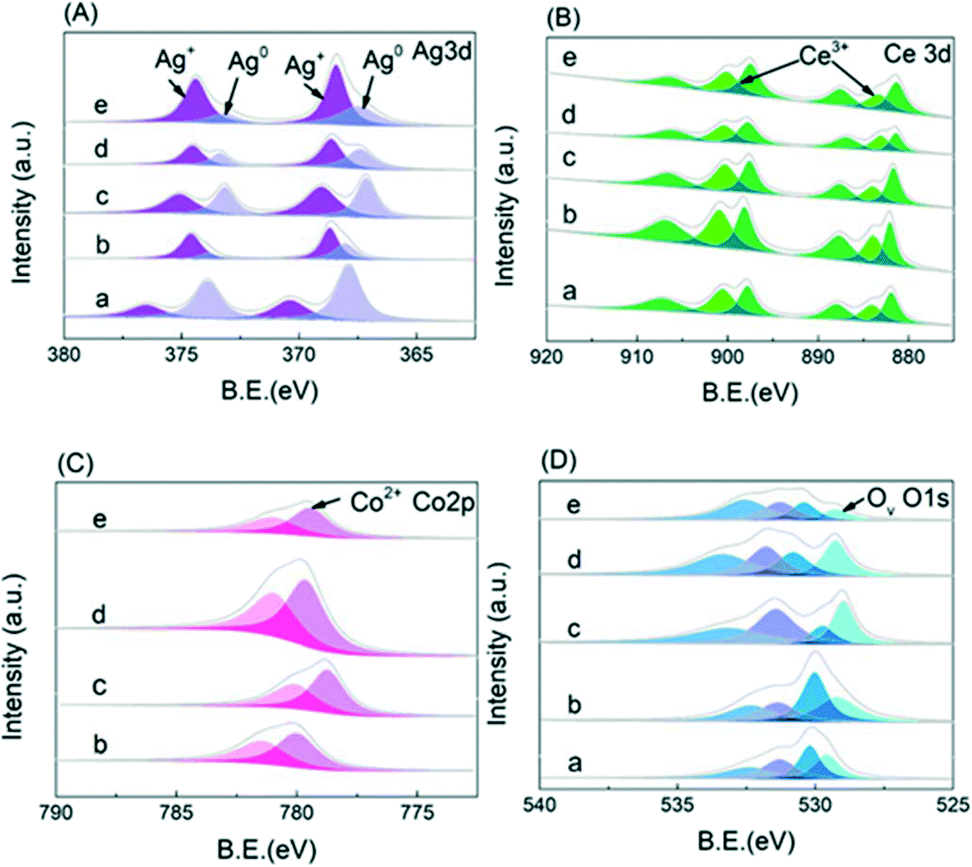 | ||
| Fig. 3 XPS spectra of (A) Ag 3d, (B) Ce 3d, (C) Co 2p, and (D) O 1s: (a) Ag/CeO2, (b) CoOx–Ag/CeO2-1, (c) CoOx–Ag/CeO2-2, (d) CoOx–Ag/CeO2-3, and (e) CoOx–Ag/CeO2-4. | ||
The Raman spectra are shown in Fig. S3;† the two peaks at 463 and 576 cm−1 are assigned to the symmetric stretching vibration mode (F2g) and the band of the defect-induced mode (D) of the cubic CeO2 fluoride phase,51 respectively, and the peak at 676 cm−1 could be attributed to the presence of Co3O4.52 The relative intensity ratio of ID/IF2G which could represent the relative Ov concentration is calculated, and the results are listed in Table S2,† in line with the XPS results. Obviously, the Ov concentration could be significantly improved via doping, in line with the report by Han.53
H2-TPR was conducted to characterize the redox behavior of the catalysts and the CeO2 support (Fig. S4†). The profile of the CeO2 support indicates hydrogen consumption in a range of 250 to 400 °C, which is related to the reduction of the surface oxygen of CeO2.41 The reduction peak of the catalysts from 200 to 350 °C is assigned to the reduction of strongly adsorbed oxygen on the large particles of silver.54 Compared with the pure CeO2, a significant decrease in the reduction temperature and increase in the H2 consumption are evident for the catalysts, which have been widely reported for other Ag-based catalysts.47 For CoOx–Ag/CeO2-2, the presence of a low-temperature peak at 150–200 °C is associated with the reduction of highly dispersed silver oxide on the ceria surface.50 With the gradual addition of CoOx, shifts to a lower value are observed for the peaks as compared to Ag/CeO2, suggesting great facilitation of the surface reduction. As listed in Table S3,† the H2 consumption amount increases first and then decreases. A plausible explanation is that an appropriate amount of CoOx would promote the generation of Ov and the formation of oxygen species adsorbed on the catalysts, while excess CoOx may cover the relative species, leading to a decrease in the amount of H2 consumption.
Combining the XPS, Raman and H2-TPR results, it is concluded that proper Co doping induces the generation of Ag+–Ov–Ce3+ interface sites.
Catalytic performance evaluation
The catalytic performances for HMF oxidation were evaluated (Fig. 4). Although 100% HMF conversion could be achieved over the five catalysts, there are differences in FDCA yields: Ag/CeO2 exhibits the lowest FDCA yield of 88.3%. Increasing the CoOx ratio, the FDCA yield exhibits a volcano tendency, reaching the highest FDCA yield of 92.8% over CoOx–Ag/CeO2-2. These results undisputedly demonstrated the high catalytic activity of the as-prepared catalysts for FDCA production from HMF, which has not been reported ever (Table S4†). Nevertheless, as the temperature was decreased to 90 °C, the yield of FDCA decreased to 87.3% and 76.4% over CoOx–Ag/CeO2-2 and Ag/CeO2, respectively. This result is consistent with the findings of S. E. Davis,55 who claimed that the production of FDCA could occur at a much higher temperature.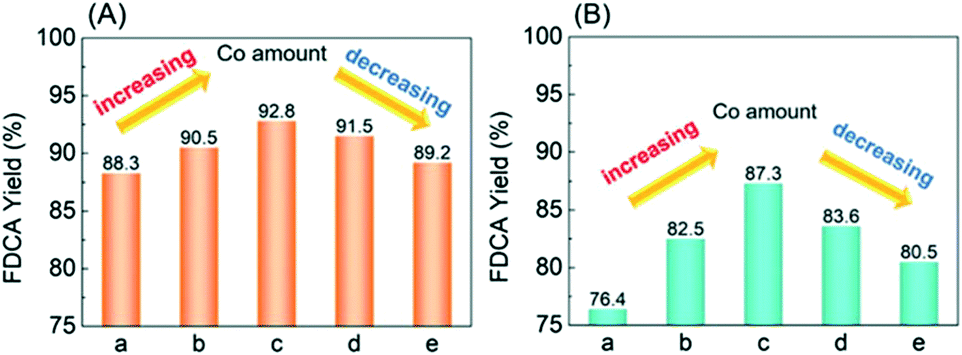 | ||
| Fig. 4 Catalytic performances for HMF oxidation at (A) 130 °C and (B) 90 °C: (a) Ag/CeO2, (b) CoOx–Ag/CeO2-1, (c) CoOx–Ag/CeO2-2, (d) CoOx–Ag/CeO2-3, and (e) CoOx–Ag/CeO2-4. Reaction conditions: HMF (0.5 mmol), catalyst (0.3 g), NaOH (2 mmol), H2O (20 mL), O2 (2 MPa), 720 min. | ||
To verify the catalytic activity of the as-prepared catalysts, the catalytic test results of CoOx/CeO2 (the molar ratio of Co/Ag was set to 2) and Ag/CeO2-red are listed in Table S5.† The data supported the correlation between Ag+ and the excellent yield of FDCA. Besides, oxidation of HMF was performed with different concentrations and types of bases, which shows that 4 eq of NaOH is the most suitable for our catalysts (Table S5†). The catalytic test results of different preparation methods prove the necessity of a hydrothermal method for the synthesis of Ag catalysts in this work (Table S6†).
The reusability of CoOx–Ag/CeO2-2 was studied by conducting five runs (Table S7†). After each experiment the catalyst was filtered, washed with deionized water and ethanol several times, and dried at 80 °C in air overnight to remove any adsorbed material from the catalyst surface. There are inevitable losses of particles during filtration; hence, the actual amount of catalyst used each time was 5% less than that from the previous experiment. Fresh catalyst makes up for the loss of the catalyst, and the HMF conversion and FDCA yield were found to be similar even after the 5th run. According to the ICP result, the content of Co was lower than 10 ppm in the liquid product even after five cycles. Combining the catalyst leaching results in Fig. S5† and TON comparisons in Table S9† shows that Co was prevented from leaching, which may be the reason for no reduction in selectivity and is consistent with the conclusion of the reusability experiment.
In order to reveal the underlying cause for the differences in catalytic performance and offer a deeper insight into the catalytic mechanism, the adsorption properties of the reactants, rate-determining step, active oxygen species, and active sites will be discussed in detail in the following section.
Mechanism investigation
Besides HMF, the activation capacity of oxygen is also a pivotal factor influencing the catalytic performance. Fig. S6B† shows the O2-TPD profile, which could be deconvoluted into three peaks via a Gaussian peak fitting method according to the desorption temperature: adsorbed oxygen species (<300 °C), lattice oxygen species (300–400 °C), and bulk lattice oxygen species (>400 °C).57,58 As listed in Table S10,† CoOx–Ag/CeO2-2 shows both the highest adsorption capacity of 194.8 μmol g−1 and the ratio of adsorbed oxygen species to all oxygen species of 34.3%. Among the three kinds of oxygen species, only the adsorbed oxygen species can directly participate and play a vital role in the reaction.59 The promoted O2 and HMF adsorption capacity after the addition of CoOx may result in gathering of more reactants around the active sites and thus the excellent catalytic activity for HMF oxidation of the Ag-based catalysts.
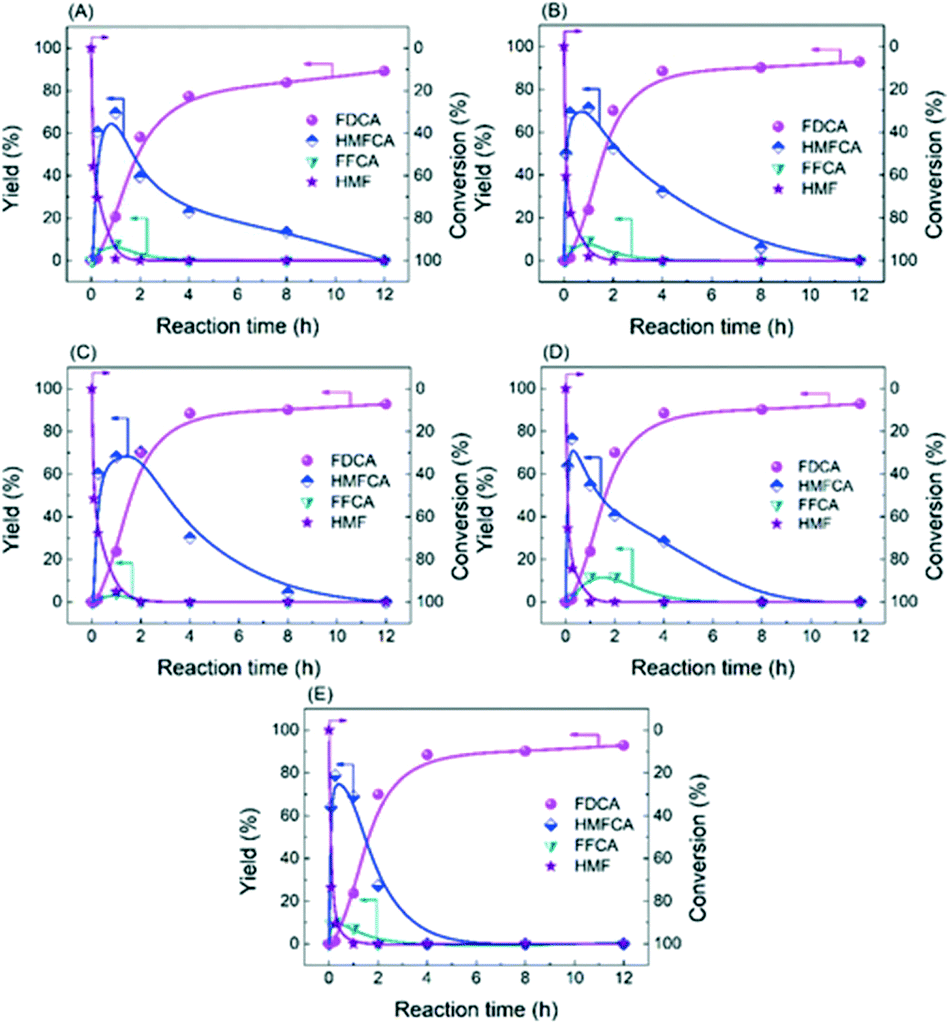 | ||
| Fig. 5 Time course of catalysts: (A) Ag/CeO2, (B) CoOx–Ag/CeO2-1, (C) CoOx–Ag/CeO2-2, (D) CoOx–Ag/CeO2-3, and (E) CoOx–Ag/CeO2-4. Reaction conditions: HMF (0.5 mmol), catalyst (0.3 g), NaOH (2 mmol), H2O (20 mL), O2 (2 MPa), 130 °C, 720 min. | ||
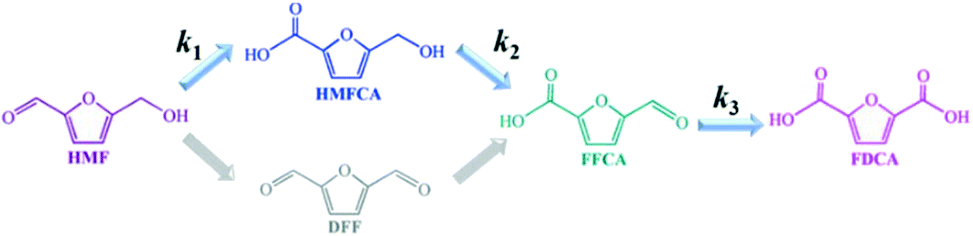 | ||
| Scheme 1 Reaction network for the HMF oxidation. | ||
Obviously, the result of the time course proves that the transformation of HMF follows the first one over the as-prepared Ag catalysts. The kinetic parameters of each step over Ag/CeO2 and CoOx–Ag/CeO2-X were calculated and are listed in Table S11,† including k1 (HMF → HMFCA), k2 (HMFCA → FFCA), and k3 (FFCA → FDCA). In comparison with the other two steps, the conversion of HMFCA to FFCA is quite slow, indicating that the oxidation of the hydroxyl group in HMFCA is the rate-determining step during the whole reaction network for FDCA production. The rate of the rate-determining step obviously increases by adding CoOx, with the rate constant k2 increasing from 0.14 min−1 over Ag/CeO2 to 0.47 min−1 over CoOx–Ag/CeO2-2, in good agreement with the result of the adsorption experiment.
Considering that the oxidation of the hydroxyl group in HMFCA is the rate-determining step during HMF oxidation, the catalytic mechanism will be discussed focusing on the transformation of HMFCA to FFCA.
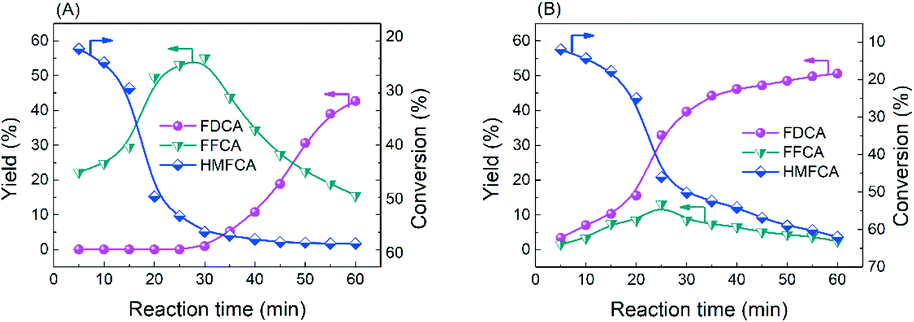 | ||
| Fig. 6 The reaction of HMFCA catalyzed by CoOx–Ag/CeO2-2 in (A) base-free and (B) basic environments. Reaction conditions: HMFCA (0.5 mmol), catalyst (0.3 g), NaOH (2 mmol), H2O (20 mL), O2 (2 MPa), 130 °C, 60 min. | ||
On the basis of the above results, it is envisioned that radicals instead of a base serve as the active oxygen species. There are four peaks with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 corresponding to the signals of DMPO–˙OH− adducts in the ESR result (Fig. 7A) and four peaks with an intensity ratio of 1:1:1:1 attributed to the signals of DMPO–˙O2− adducts in Fig. 7B, confirming the generation of ˙O2− (superoxide radicals) and ˙OH− (hydroxyl radicals).26 In order to find out which kind of radical governs the catalytic activity, a selective poisoning experiment was carried out with isopropanol as a trapping agent for ˙OH− and p-benzoquinone as the scavenger for ˙O2− (Fig. 8). The conversion of HMFCA to FDCA is inhibited by the addition of p-benzoquinone, while isopropanol has little influence on the reaction, demonstrating that ˙O2− is the determining active oxygen species.
:2:2:1 corresponding to the signals of DMPO–˙OH− adducts in the ESR result (Fig. 7A) and four peaks with an intensity ratio of 1:1:1:1 attributed to the signals of DMPO–˙O2− adducts in Fig. 7B, confirming the generation of ˙O2− (superoxide radicals) and ˙OH− (hydroxyl radicals).26 In order to find out which kind of radical governs the catalytic activity, a selective poisoning experiment was carried out with isopropanol as a trapping agent for ˙OH− and p-benzoquinone as the scavenger for ˙O2− (Fig. 8). The conversion of HMFCA to FDCA is inhibited by the addition of p-benzoquinone, while isopropanol has little influence on the reaction, demonstrating that ˙O2− is the determining active oxygen species.
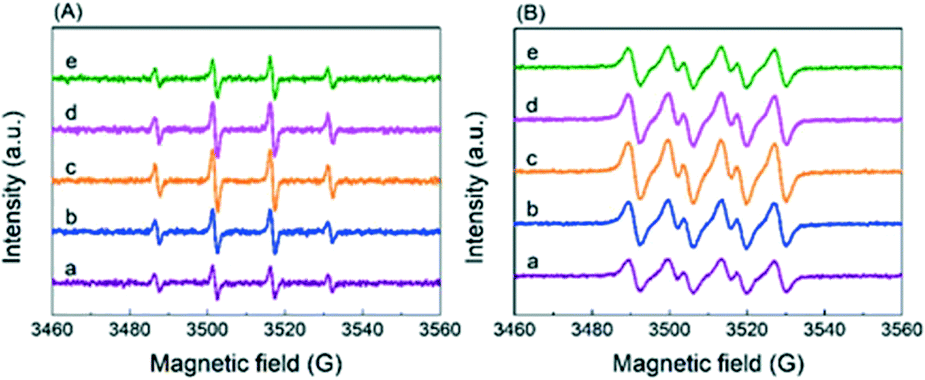 | ||
| Fig. 7 ESR spectra (130 °C, in an aqueous solution with an O2 inlet) of the catalysts using (A) hydroxyl radicals and (B) superoxide radicals: (a) Ag/CeO2, (b) CoOx–Ag/CeO2-1, (c) CoOx–Ag/CeO2-2, (d) CoOx–Ag/CeO2-3, and (e) CoOx–Ag/CeO2-4. | ||
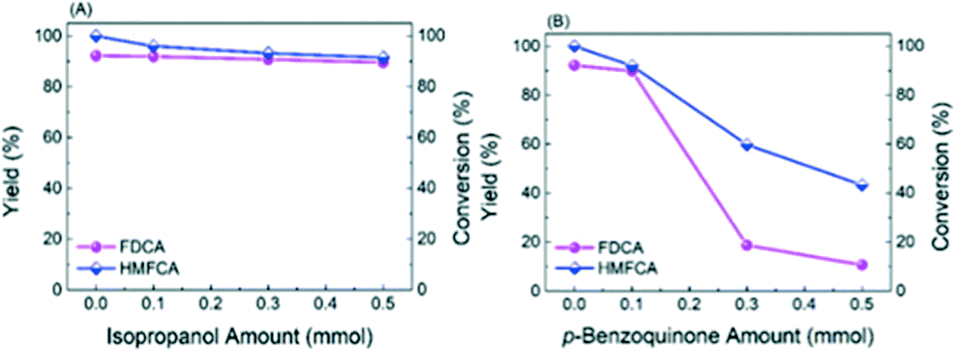 | ||
| Fig. 8 The influence of adding different free radical scavengers on the yield of FDCA and the conversion of HMFCA: (A) isopropanol and (B) p-benzoquinone. Reaction conditions: HMF (0.5 mmol), catalyst (0.3 g), NaOH (2 mmol), H2O (20 mL), O2 (2 MPa), 130 °C, 720 min. | ||
Considering that the superoxide radicals stem from oxygen adsorbed on catalysts (eqn (1) and (2)), O2-TPO was thus conducted to evaluate the ultimate oxygen storage capacity (OSC). As shown in Fig. S8,† the peak at 30 to ca. 100 °C could be attributed to surface adsorption oxygen refilling the oxygen vacancies.42,61 As listed in Table S12,† the OSCs are calculated from the O2 consumption amount, with the order CoOx–Ag/CeO2-2 > CoOx–Ag/CeO2-3 > CoOx–Ag/CeO2-1 > CoOx–Ag/CeO2-4 > Ag/CeO2. This result is in line with the XPS and Raman results, demonstrating that the high-activity catalysts possess more oxygen vacancies and consequently exhibit higher reactivity of oxygen species.
| O2 + e− = ˙O2− | (1) |
| ˙O2− + H2O + e− = OOH− + OH− | (2) |
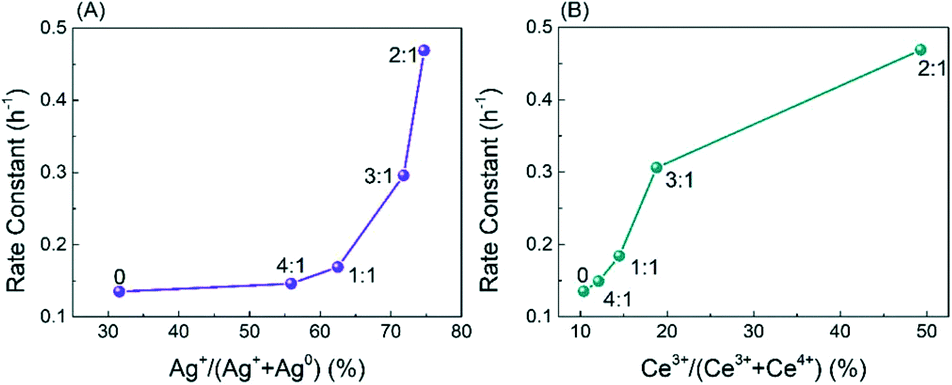 | ||
| Fig. 9 k 2 as a function of (A) Ag+/(Ag0 + Ag+) and (B) Ce3+/(Ce3+ + Ce4+). | ||
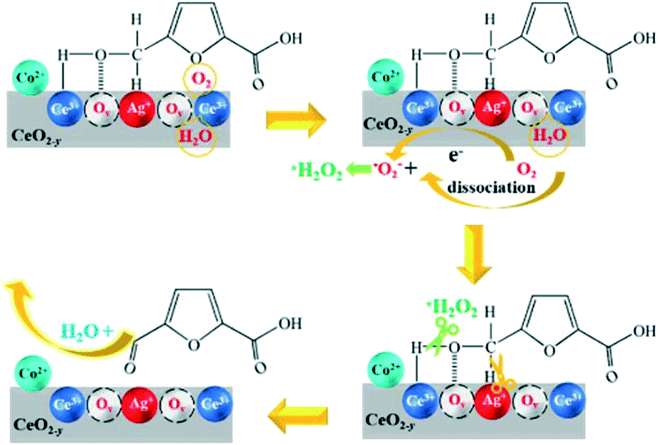 | ||
| Scheme 2 Proposed reaction mechanism for the oxidation of HMFCA. | ||
Conclusions
In summary, Ag/CeO2 and CoOx–Ag/CeO2-X catalysts were prepared and evaluated for FDCA production from HMF. CoOx–Ag/CeO2-2 exhibits the best catalytic performance toward HMF oxidation with a FDCA yield of 92.8%. A combination study identifies that the Ag+–Ov–Ce3+ interface is formed, which provides sites for reactant adsorption and active oxygen species generation. Defined by the kinetic study, the hydroxyl oxidation in HMFCA is the rate determining step, which is dominated by superoxide free radicals stemming from O2 instead of a base. On this basis, HMF oxidation under base-free conditions has been carried out, achieving a FDCA yield of 71.2%. The structure–activity correlation reveals that the Ag+–Ov–Ce3+ interface acts as the active sites for O–H bond scission and C–H bond scission in HMFCA. The Ag+ site contributes to the C–H bond cleavage, and the Ov–Ce3+ site plays the roles of gathering HMFCA and activating and dissociating O2 and H2O. It is the first report in which high FDCA yields are obtained over Ag catalysts, and a fundamental understanding about the reaction mechanism is achieved, which would be useful for rational design of other heterogeneous catalysts.Author contributions
Mengchen Fu: conceptualization, methodology, and writing – original draft. Chun Shen: supervision, and writing – review and editing. Weiyao Yang: software and data curation. Chenyu Yang: visualization. Yiwen Zhang: investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of the State Key Laboratory of Chemical Engineering (No. SKL-ChE-20A02) and the support of the International Clean Energy Talent Program by China Scholarship Council.References
- B. Op De Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morré, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240, 10.1039/c4ee01523a\.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453, 10.1039/c8gc02680g.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546, DOI:10.1016/j.chempr.2019.05.022.
- P. Sudarsanam, R. Zhong, S. Van Den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402, 10.1039/c8cs00410b.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544, DOI:10.1021/acscatal.5b01491.
- D. Yan, G. Wang, K. Gao, X. Lu, J. Xin and S. Zhang, Ind. Eng. Chem. Res., 2018, 57, 1851–1858, DOI:10.1021/acs.iecr.7b04947.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144, DOI:10.1002/cssc.200900137.
- H. Chen, J. Shen, K. Chen, Y. Qin, X. Lu, P. Ouyang and J. Fu, Appl. Catal., A, 2018, 555, 98–107, DOI:10.1016/j.apcata.2018.01.023.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185, DOI:10.1021/cs5003096.
- Z. Zhang, J. Zhen, B. Liu, K. Lv and K. Deng, Green Chem., 2015, 17, 1308–1317, 10.1039/c4gc01833h.
- P. V. Rathod and V. H. Jadhav, ACS Sustainable Chem. Eng., 2018, 6, 5766–5771, DOI:10.1021/acssuschemeng.7b03124.
- D. Lei, K. Yu, M. R. Li, Y. Wang, Q. Wang, T. Liu, P. Liu, L. L. Lou, G. Wang and S. Liu, ACS Catal., 2017, 7, 421–432, DOI:10.1021/acscatal.6b02839.
- C. Zhou, W. Deng, X. Wan, Q. Zhang, Y. Yang and Y. Wang, ChemCatChem, 2015, 7, 2853–2863, DOI:10.1002/cctc.201500352.
- X. Han, C. Li, Y. Guo, X. Liu, Y. Zhang and Y. Wang, Appl. Catal., A, 2016, 526, 1–8, DOI:10.1016/j.apcata.2016.07.011.
- G. Yi, S. P. Teong and Y. Zhang, Green Chem., 2016, 18, 979–983, 10.1039/c5gc01584g.
- S. Hameed, L. Lin, A. Wang and W. Luo, Catalysts, 2020, 10, 120–145, DOI:10.3390/catal10010120.
- S. Albonetti, A. Lolli, V. Morandi, A. Migliori, C. Lucarelli and F. Cavani, Appl. Catal., B, 2015, 163, 520–530, DOI:10.1016/j.apcatb.2014.08.026.
- J. F. Nie, J. H. Xie and H. C. Liu, J. Catal., 2013, 301, 83–91, DOI:10.1016/j.jcat.2013.01.007.
- D. Zhao, T. Su, Y. Wang, R. S. Varma and C. Len, Mol. Catal., 2020, 495, 111133, DOI:10.1016/j.mcat.2020.111133.
- G. D. Yadav and R. V. Sharma, Appl. Catal., B, 2014, 147, 293–301, DOI:10.1016/j.apcatb.2013.09.004.
- D. Zhao, D. Rodriguez-Padron, R. Luque and C. Len, ACS Sustainable Chem. Eng., 2020, 8, 8486–8495, DOI:10.1021/acssuschemeng.9b07170.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J. D. Grunwaldt, Green Chem., 2018, 20, 3530–3541, 10.1039/c8gc01340c.
- O. R. Schade, A. Gaur, A. Zimina, E. Saraçi and J. D. Grunwaldt, Catal. Sci. Technol., 2020, 10, 5036–5047, 10.1039/d0cy00878h.
- O. R. Schade, F. Stein, S. Reichenberger, A. Gaur, E. Saraçi, S. Barcikowski and J. D. Grunwaldt, Adv. Synth. Catal., 2020, 362, 5681–5696, DOI:10.1002/adsc.202001003.
- J. An, G. Sun and H. Xia, ACS Sustainable Chem. Eng., 2019, 7, 6696–6706, DOI:10.1021/acssuschemeng.8b05916.
- Y. Liu, H. Y. Ma, D. Lei, L. L. Lou, S. Liu, W. Zhou, G. C. Wang and K. Yu, ACS Catal., 2019, 9, 8306–8315, DOI:10.1021/acscatal.9b02115.
- J. F. Nie and H. C. Liu, J. Catal., 2014, 316, 57–66, DOI:10.1016/j.jcat.2014.05.003.
- J. Wang, W. Yang, C. Wu, Y. Gong, J. Zhang and C. Shen, ACS Sustainable Chem. Eng., 2020, 8, 16960–16967, DOI:10.1021/acssuschemeng.0c07070.
- Q. Zhu, C. Shen, J. Wang and T. Tan, ACS Sustainable Chem. Eng., 2017, 5, 8181–8191, DOI:10.1021/acssuschemeng.7b01837.
- Y. Gong, C. Shen, J. Wang, C. Wu and T. Tan, ACS Sustainable Chem. Eng., 2019, 7, 10323–10331, DOI:10.1021/acssuschemeng.9b00190.
- C. Wu, C. Shen, Y. Gong and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 18943–18954, DOI:10.1021/acssuschemeng.9b04311.
- L. Lei, Y. Wang, Z. Zhang, J. An and F. Wang, ACS Catal., 2020, 10, 8788–8814, DOI:10.1021/acscatal.0c01900.
- G. D. Yadav and R. K. Mewada, Chem. Eng. Res. Des., 2012, 90, 86–97, DOI:10.1016/j.cherd.2011.08.007.
- S. Song, Y. Wu, S. Ge, L. Wang, Y. Wang, Y. Guo, W. Zhan and Y. Guo, ACS Catal., 2019, 9, 6177–6187, DOI:10.1021/acscatal.9b01679.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2012, 51, 136–139, DOI:10.1002/anie.201106244.
- T. Mitsudome, A. Noujima, Y. Mikami, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2010, 49, 5545–5548, DOI:10.1002/anie.201001055.
- M. V. Grabchenko, G. V. Mamontov, V. I. Zaikovskii and O. V. Vodyankina, Kinet. Catal., 2017, 58, 642–648, DOI:10.1134/S0023158417050056.
- S. Klacar and H. Grönbeck, Catal. Sci. Technol., 2013, 3, 183–190, 10.1039/c2cy20343j.
- T. Gao, J. Chen, W. Fang, Q. Cao, W. Su and F. Dumeignil, J. Catal., 2018, 368, 53–68, DOI:10.1016/j.jcat.2018.09.034.
- Z. Miao, Y. Zhang, X. Pan, T. Wu, B. Zhang, J. Li, T. Yi, Z. Zhang and X. Yang, Catal. Sci. Technol., 2015, 5, 1314–1322, 10.1039/c4cy01060d.
- C. Zhang, S. Li, M. Li, S. Wang, X. Ma and J. Gong, AIChE J., 2011, 58, 516–525, DOI:10.1002/aic.12599.
- R. Peng, X. Sun, S. Li, L. Chen, M. Fu, J. Wu and D. Ye, Chem. Eng. J., 2016, 306, 1234–1246, DOI:10.1016/j.cej.2016.08.056.
- L. Ma, D. Wang, J. Li, B. Bai, L. Fu and Y. Li, Appl. Catal., B, 2014, 148–149, 36–43, DOI:10.1016/j.apcatb.2013.10.039.
- M. V. Grabchenko, G. V. Mamontov, V. I. Zaikovskii, V. La Parola, L. F. Liotta and O. V. Vodyankina, Appl. Catal., B, 2020, 260, 118148, DOI:10.1016/j.apcatb.2019.118148.
- L. Yu, R. Peng, L. Chen, M. Fu, J. Wu and D. Ye, Chem. Eng. J., 2018, 334, 2480–2487, DOI:10.1016/j.cej.2017.11.121.
- W. Liu, J. Sun, X. Zhang and M. Wei, Ind. Eng. Chem. Res., 2018, 57, 15606–15612, DOI:10.1021/acs.iecr.8b03987.
- X. Zhang, Y. Yang, L. Song, Y. Wang, C. He, Z. Wang and L. Cui, Mol. Catal., 2018, 447, 80–89, DOI:10.1016/j.mcat.2018.01.007.
- S. Liu, X. Wu, W. Liu, W. Chen, R. Ran, M. Li and D. Weng, J. Catal., 2016, 337, 188–198, DOI:10.1016/j.jcat.2016.01.019.
- L. Ding, W. Yang, L. Chen, H. Cheng and Z. Qi, Catal. Today, 2020, 347, 39–47, DOI:10.1016/j.cattod.2018.04.069.
- N. Liu, M. Xu, Y. Yang, S. Zhang, J. Zhang, W. Wang, L. Zheng, S. Hong and M. Wei, ACS Catal., 2019, 9, 2707–2717, DOI:10.1021/acscatal.8b04913.
- S. Chang, M. Li, Q. Hua, L. Zhang, Y. Ma, B. Ye and W. Huang, J. Catal., 2012, 293, 195–204, DOI:10.1016/j.jcat.2012.06.025.
- H. Wang, L. Wang, J. Zhang, C. Wang, Z. Liu, X. Gao, X. Meng, S. J. Yoo, J. G. Kim, W. Zhang and F. S. Xiao, ChemSusChem, 2018, 11, 3965–3974, DOI:10.1002/cssc.201801709.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849, 10.1039/c9gc03698a.
- G. V. Mamontov, V. V. Dutov, V. I. Sobolev and O. V. Vodyankina, Kinet. Catal., 2013, 54, 487–491, DOI:10.1134/S0023158413040137.
- S. E. Davis, A. D. Benavidez, R. W. Gosselinkc, J. H. Bitter, K. P. de Jongc, A. K. Datye and R. J. Davis, J. Mol. Catal. A: Chem., 2014, 388–389, 123–132, DOI:10.1016/j.molcata.2013.09.013.
- K. Ji, C. Shen, J. Yin, X. Feng, H. Lei, Y. Chen, N. Cai and T. Tan, Ind. Eng. Chem. Res., 2019, 58, 10844–10854, DOI:10.1021/acs.iecr.9b01522.
- S. Mo, Q. Zhang, J. Li, Y. Sun, Q. Ren, S. Zou, Q. Zhang, J. Lu, M. Fu, D. Mo, J. Wu, H. Huang and D. Ye, Appl. Catal., B, 2020, 264, 118464, DOI:10.1016/j.apcatb.2019.118464.
- B. Chen, B. Wu, L. Yu, M. Crocker and C. Shi, ACS Catal., 2020, 10, 6176–6187, DOI:10.1021/acscatal.0c00459.
- J. Xiong, K. Wu, J. Yang, P. Liu, L. Song, J. Zhang, M. Fu, L. Chen, H. Huang, J. Wu and D. Ye, Appl. Surf. Sci., 2021, 539, 148211, DOI:10.1016/j.apsusc.2020.148211.
- X. Han, C. Li, X. Liu, Q. Xia and Y. Wang, Green Chem., 2017, 19, 996–1004, 10.1039/c6gc03304k.
- C. G. Rotaru, G. Postole, M. Florea, F. Matei-Rutkovska, V. I. Pârvulescu and P. Gelin, Appl. Catal., A, 2015, 494, 29–40, DOI:10.1016/j.apcata.2015.01.024.
- K. Yu, L. L. Lou, S. Liu and W. Zhou, Adv. Sci., 2020, 7, 1–8, DOI:10.1002/advs.201901970.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098, DOI:10.1021/acscatal.0c03034.
- Q. Wang, L. Chen, S. Guan, X. Zhang, B. Wang, X. Cao, Z. Yu, Y. He, D. G. Evans, J. Feng and D. Li, ACS Catal., 2018, 8, 3104–3115, DOI:10.1021/acscatal.7b03655.
- S. P. Bates, M. J. Gillan and G. Kresse, J. Phys. Chem. B, 2017, 2, 2017–2026, DOI:10.1021/jp9804998.
- E. Farfan-Arribas and R. J. Madix, J. Phys. Chem. B, 2002, 106, 10680–10692, DOI:10.1021/jp020729p.
- Z. Zhang, O. Bondarchuk, J. M. White, B. D. Kay and Z. Dohnalek, J. Am. Chem. Soc., 2006, 128, 4198–4199, DOI:10.1021/ja058466a.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185, DOI:10.1021/cs5003096.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01599k |
| This journal is © The Royal Society of Chemistry 2022 |