
In situ coating of a N, S co-doped porous carbon thin film on carbon nanotubes as an advanced metal-free bifunctional oxygen electrocatalyst for Zn–air batteries†
Xin
Wang
ab,
Guang-Lan
Li
 *ab,
Zhong-Fa
Lu
ab,
Shuo
Cao
ab,
Ce
Hao
ab,
Suli
Wang
c and
Gongquan
Sun
c
*ab,
Zhong-Fa
Lu
ab,
Shuo
Cao
ab,
Ce
Hao
ab,
Suli
Wang
c and
Gongquan
Sun
c
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116023, PR China. E-mail: guanglanli@dlut.edu.cn
bSchool of Chemical Engineering, Dalian University of Technology, Panjin 124221, PR China
cDivision of Fuel Cells and Battery, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Key Laboratory of Fuel Cells & Hybrid Power Sources, Chinese Academy of Sciences, Dalian 116023, China
First published on 19th November 2021
Abstract
Reversible oxygen catalysts with good catalytic performance and economic efficiency are urgently needed for the rapid development of metal–air batteries and reversible fuel cells. Herein, a metal-free N, S co-doped porous carbon thin film (NSCF) was coated in situ on carboxylic carbon nanotubes (CNT@NSCF) via a simple and scalable low temperature polymerization–high temperature pyrolysis strategy. By analyzing the surface structure of comparable carbon supports including ketjenblack, Vulcan XC-72 carbon, and activated carbon, it is found that the surface oxygen-containing functional groups of the support are the key factor to obtaining a uniform coating of NSCF with the formation of a large amount of efficient N, S active species. As a result, the optimal CNT@NSCF shows an excellent bifunctional oxygen catalytic activity with a narrow potential difference (ΔE = Ej = 10,OER − E1/2,ORR) value of 0.74 V, which is superior to that of Pt/C + RuO2 (0.75 V) and most metal-free carbon-based oxygen catalysts. Notably, the CNT@NSCF-based rechargeable Zn–air battery delivers a remarkable performance with a high open-circuit voltage and outstanding charge–discharge stability after 213 h, which fully demonstrates the promising practical application potential of the CNT@NSCF catalyst.
Introduction
To address the increasing energy demands and environmental pollution issues, various renewable energy systems have been widely studied for application. Rechargeable Zn–air batteries are one of the most promising systems for the next-generation energy conversion and storage devices, and this is mainly because of the advantages of them being pollution-free, having a high energy density, safe to operate, and having a low cost.1–6 However, the sluggish oxygen redox kinetics and high overpotential of the oxygen electrode still largely hamper their rapid development.7 Although Pt-based and RuO2/IrO2 catalysts deliver robust activities for oxygen reduction and evolution reactions (ORR/OER), respectively, they suffer drawbacks of high cost, insufficient reserves, poor stability, and single functional catalytic activity.8,9 Therefore, it is necessary and significant to develop ORR/OER catalysts with outperforming catalytic activity, ultra strong stability, and of low cost.In recent decades, carbon-based nanomaterials have been widely used as oxygen electrocatalysts because of their remarkable electrical conductivity and structural stability. In particular, carbon-based samples could be modulated with B, Br, Cl, N, P, S heteroatoms, which could fully encourage their catalytic potential as efficient multifunctional catalysts.10–13 Among the various metal-free materials, N-doped carbon materials (NC) have been widely exploited for use in ORR. Effective doping of N can break the electroneutrality of the adjacent C atoms and create positively charged C+ active centers, which are beneficial to the adsorption of O2 and thereby enhance their ORR activity.14–17 However, the further application of NC is limited by the poor OER performance.18,19 Introducing a suitable second non-metal heteroatom to NC may significantly improve the bifunctional oxygen catalytic activity through the synergistic effect of different dopants on charge density and spin density.20–24
Recently, S has often been introduced into NC to form a thiophene-S configuration, which will interact with pyridinic-N and graphitic-N to enhance the OER activity.25–27 Furthermore, the doped-S atom can also alter the charge and spin distribution of the adjacent C atoms, thereby promoting the ORR process.28–32 Therefore, a tremendous amount of research has been concentrated on the exploration of an efficient synthesis procedure with a suitable S precursor. For example, Guo et al. prepared a S, N dual-doped carbon nanosheet catalyst by a simple heat-treatment of L-cysteine.33 They demonstrated that the N and S active species synergistically improved the overall ORR/OER catalytic performance. Zhou et al. synthesized a N, S co-doped carbon catalyst by a fast deflagration method.34 They reported that the doped-N, S could change the charge density of the carbon atoms to form efficient catalytic active sites, thus enhancing the bifunctional oxygen performance. The N, S co-doped carbon materials have shown excellent catalytic potential in catalyzing the ORR/OER, but it is still a great challenge to get controllable synthesis of electrocatalysts with abundant high-efficiency active sites to achieve a satisfactory performance for practical applications.
In this work, a low temperature polymerization–high temperature pyrolysis strategy is described which coats in situ a N, S-co-doped porous carbon thin film (NSCF) on a carboxyl carbon nanotube (CNT@NSCF) as a bifunctional oxygen catalyst. The effect of the oxygen-containing functional groups (OFGs) on the CNT surface, and on the fabrication of the structure and morphology of NSCF was studied contrastively by analyzing the samples prepared as controls: ketjenblack (KB), Vulcan XC-72 carbon (VXC), and activated carbon (AC) supports. By systematically optimizing the synthetic parameters, the CNT@NSCF obtained exhibits a remarkable ORR/OER activity and ultra strong durability in both half-cells and a rechargeable Zn–air battery (ZAB). The outstanding ORR/OER performance of CNT@NSCF indicates its promising prospect as an electrocatalyst for energy conversion devices.
Materials and methods
Materials
The CNT, KB, VXC, and AC were purchased from Xianfeng Nano Material Technology. Aniline (AN), ammonium persulfate ((NH4)2S2O8), sulfur powder, and sodium sulfide (Na2S·9H2O) were purchased from Sinopharm Chemical Reagent Co. Hydrochloric acid (HCl, 35%) was obtained from Beijing Chemical Reagents. The deionized water used in all the experiments was obtained using an ion-exchange and filtration system. Nafion® (5 wt%) solution was obtained from Sigma-Aldrich. The RuO2 and Pt/C (20 wt%, Johnson Matthey) was obtained from Aladdin Chemical Reagent Factory.Preparation of the S precursor
A portion (280 mg) of Na2S·9H2O and 64 mg of S powder were simultaneously dissolved in 24.0 mL of water. After ultrasonic mixing for 5 h at room temperature, a yellow transparent solution was obtained, which was then heated at 80 °C for 12 h in a Teflon autoclave. After natural cooling to ambient temperature, the Na2Sx solution was achieved.Preparation of catalysts
As shown in Scheme 1, 50 mg of CNT, 100 mg of AN, and 200 mg of Na2Sx solution were dispersed in 100 mL of 0.2 M HCl solution to obtain a uniform and transparent solution, which was stirred in an ice bath for 30 min. At the same time, 125 mg of (NH4)2S2O8 was also dissolved into 20 mL of 0.2 M HCl solution to obtain another transparent solution. The two solutions were then mixed and stirred continuously in an ice bath for another 30 min. Next, the mixture obtained was transferred into the cold closet (3–5 °C) of a refrigerator for 24 h, which was then filtered and dried at 80 °C for 12 h. The powder precursor obtained was named as CNT@NSCF0. The CNT@NSCF0 was finally carbonized at 900 °C for 2 h under temperature programming of 5 °C min−1 in a N2 flow to obtain the CNT@NSCF. For comparison, the catalysts were also heat-treated at 800 °C and 1000 °C under the same preparation conditions, and were named as CNT@NSCF−t (t = 800, 1000). The catalyst without polysulfide added was prepared, and was named as CNT@NCF. The catalyst without added support was synthesized and labeled as NSCF. The control KB@NSCF, VXC@NSCF, and AC@NSCF samples were synthesized by replacing the CNT with KB, VXC, and AC as support under the same preparation conditions, respectively.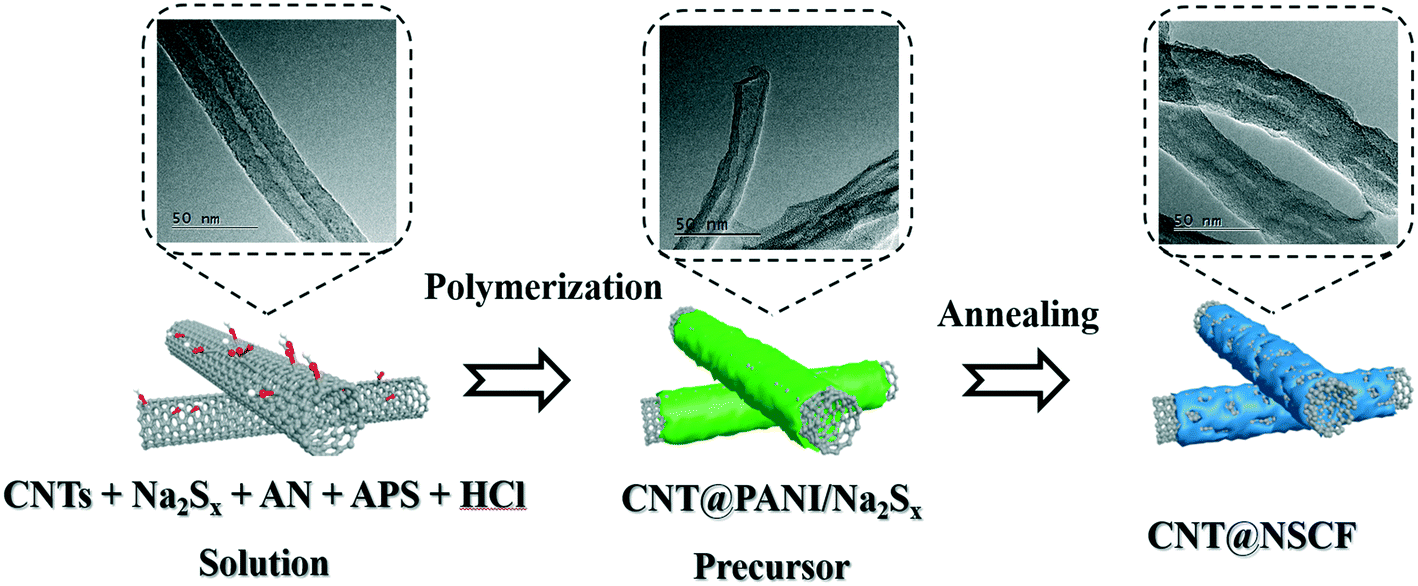 | ||
| Scheme 1 A schematic of the procedure for the synthesis of CNT@NSCF. | ||
Physicochemical characterization methods
The surface morphologies of the synthesized materials were obtained by using transmission electron microscopy (TEM, FEI Tecnai G2 F30), and field-emission scanning electron microscopy (FE-SEM, FEI Nova NanoSEM 450). Degree of graphitization of the samples was characterized using a confocal Raman microscopy. The crystallinity of the catalysts was obtained by X-ray powder diffraction (XRD, Shimadzu XRD-7000S) with Cu Kα radiation (λ = 1.5405 Å) with a scanning range of 10–60°. The detailed element types and chemical characterization of the prepared materials were determined by X-ray photoelectron spectroscopy (XPS, ThermoFisher ESCALAB 250Xi). The superficial functional groups of the carbon materials were characterized by Fourier transform infrared spectrophotometry (FTIR, Thermo Nicolet AVATAR 370). The surface area and pore structure of the obtained carbon materials were determined on a volumetric adsorption analyzer, and then calculated by the Brunauer–Emmett–Teller (BET) method and the Barrett–Joyner–Halenda (BJH) models, respectively.Electrochemistry measurements
All the electrochemical measurements were conducted on a three-electrode system on an electrochemical workstation (CH Instruments CHI 660E). An Ag/AgCl electrode was used as a reference electrode and a Pt wire as a counter electrode. The working electrode was made in the laboratory by the following procedure: 4 mg of catalyst was first dispersed into a Nafion (5 wt%, 30 μL)–ethanol (99.8 vol%, 2000 μL) mixed solution with ultrasonic dispersion. Next, 20 μL of catalyst ink was dropped onto the glassy carbon electrode with a catalyst loading of 0.56 mg cm−2. All the potentials were referenced to a standard reversible hydrogen electrode (RHE) using the formula: E(RHE) = E(Ag/AgCl) + 0.059 pH + 0.198 V.The ORR performance of the obtained materials was tested in 0.1 M KOH at room temperature. Cyclic voltammetry (CV) curves were measured in N2 or O2-saturated 0.1 M KOH solution at 50 mV s−1. Linear sweep voltammetry (LSV) was conducted at 10 mV s−1 at a speed of 400 to 2500 rpm. The electron-transfer number (n) was calculated using the Koutecký–Levich (K–L) equations:35
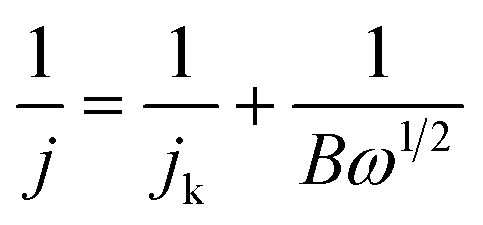
| B = 0.2nFD2/30v−1/6C0 |
| j k = nFkC0 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), the ORR rate constant (m s−1), saturation concentration of O2 (C0 = 1.21 mol m−3), diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm−2 s−1), and the dynamic viscosity of O2 (v = 0.01 cm2 s−1) in 0.1 M KOH, respectively.
485 C mol−1), the ORR rate constant (m s−1), saturation concentration of O2 (C0 = 1.21 mol m−3), diffusion coefficient of O2 (D0 = 1.9 × 10−5 cm−2 s−1), and the dynamic viscosity of O2 (v = 0.01 cm2 s−1) in 0.1 M KOH, respectively.
The ORR stability of the catalyst was tested using an accelerated aging test (AAT) and a chronoamperometry (i–t) measurement. The AAT was performed by a CV cycle of 20000 cycles at 100 mV s−1, and then the LSV curve was recorded at a sweep rate of 10 mV s−1. The i–t was determined at a fixed voltage of 0.57 V at 400 rpm. The OER performance was tested in 1 M KOH solution at 10 mV s−1 at 1600 rpm. The electrochemical impedance spectroscopy (EIS) was carried out at a potential of 1.62 V with an AC potential amplitude of 10 mV. The AAT was performed at 1.2–1.7 V for 2000 cycles and the OER polarization curves before and after the cycles were recorded.
The practical application performance of the CNT@NSCF catalyst was operated using self-made ZAB at room temperature. The catalyst ink was a mixture of CNT@NSCF catalyst and Nafion solution (5%), with the same ratio as mentioned previously. A carbon cloth coated with CNT@NSCF catalyst (active area: 1.23 cm2, mass loading: 2 mg cm−2) was used as an air cathode and a polished zinc plate was used as the anode (active area: 4.91 cm2). In the ZAB test, a mixed solution of 6 M KOH and 0.2 M Zn(Ac)2 was used as the electrolyte. For comparison, commercial Pt/C + RuO2 (m:m = 1:1) mixed catalysts were also used to make an air anode electrode (mass loading: 1 mg cm−2). All the ZABs were operated under ambient conditions. A battery testing system (Landt CT2001A) was used to evaluate the discharge–charge cycling stability and cycling reversibility. The discharge–charge polarization curves were recorded on an electrochemical work station (CH Instruments CHI 660E).
Results and discussion
The structure and detailed morphology of the optimal catalysts were studied by SEM and TEM. As shown in Fig. S1 (ESI†), KB@NSCF, VXC@NSCF, and AC@NSCF exhibited irregular spherical particle accumulation morphology, which may be caused by the deposition of NSCF on the supports. Notably, no self-polymerization of the precursor was found (according to the large-area SEM images of the four catalysts), and this indicated that the polymerization of the precursor mainly occurred on the surface of the corresponding support. Unlike the KB@NSCF, VXC@NSCF, and AC@NSCF, the CNT@NSCF0, CNT@NCF, and CNT@NSCF showed a typical CNT crosslinked 3D network structure (Fig. 1a–c and S2, ESI†). Obviously, compared with pure CNT (Fig. S3, ESI†), the surfaces of CNT@NSCF0, CNT@NCF, and CNT@NSCF were all uniformly covered with a porous carbon film. Furthermore, compared with the diameter of CNT (31.1 nm), the average diameter of CNT@NSCF was about 37.4 nm, implying that the average thickness of NSCF film was about 3.15 nm. The CNT@NSCF was used for further detailed studies (Fig. 1d and e); two parts, including the inner CNT and the surface of the homogeneously coated NSCF, can be clearly seen in the HRTEM images. The lattice spacing distance of the inner CNT was 0.3376 nm, which corresponded to the (002) plane of graphitized carbon, demonstrating the intrinsic high crystalline structure of CNT@NSCF, and as for the surface of NSCF, no clear crystal structure was detected but many defects were observed, such as a positive topological disclination at the corner and the broken fringes on the shells,36,37 which might be mainly attributed to the removal of parts of N, S elements. The energy-dispersive spectroscopy (EDS) elemental mapping images (Fig. 1f) of CNT@NSCF definitively showed that the C, N, O, and S elements were distributed evenly in the full detection area, and this implied the successful doping of N, S into the encapsulated carbon thin film. | ||
| Fig. 1 (a) The SEM image of CNT@NSCF, (b–e) the TEM images, and (f) the EDS elemental mapping of C, N, and S of CNT@NSCF. | ||
The XRD patterns were obtained to determine the exhaustive graphitization of carbon materials. As shown in Fig. 2a, the two main peaks corresponding to the (002) and (101) planes of graphitic carbon for the CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF samples, appeared. Notably, their corresponding peak positions are all negatively shifted in different degrees compared to those of the standard card for graphite (PCPDF # 41-1487), which may be mainly caused by the large S dopants, who make the unit cell parameters larger and the increase the interplanar spacing.38,39 Furthermore, the C (002) peak of CNT@NSCF shows the narrowest half-wave width when compared to those of the other three samples, indicating that it has the highest crystallinity of all these catalysts, and this was mainly attributed to the CNT. In the Raman spectra of CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF, and CNT@NCF catalysts (Fig. 2b and S4, ESI†), all samples exhibited two dominant peaks at 1345 and 1590 cm−1, which were assigned to the D-band (defective sp3 hybridized carbon) and G-band (crystallized graphitic sp2 carbon), respectively.40 The intensity ratios (ID/IG peak area) of CNT@NSCF, CNT@NCF, KB@NSCF, AC@NSCF, and VXC@NSCF were 1.10, 1.04, 1.08, 0.98, and 0.96, respectively. The maximum ID/IG value of CNT@NSCF means that it possesses more defects on the NSCF surface than the other three catalysts, which is in agreement with the HRTEM results. The specific surface areas and pore volumes of the catalysts had a profound influence on the ORR/OER catalytic performance. As shown in Fig. 2c, all the CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF samples exhibited type-IV isotherms and H3 hysteresis loops, which verified the presence of the mesopores. Moreover, the obvious N2 absorption can be observed at p/p0 < 0.10, which indicating that there were also many micropores in the four samples.41 Their detailed pore size distribution curves (Fig. 2d and S5, ESI†) confirmed the presence of an abundant meso-microporous structure with a mainly bimodal range of 0.8–2 and 2–10 nm. Their corresponding BET specific surface areas (Table S1, ESI†) decreased in the sequence of KB@NSCF > CNT@NSCF > VXC@NSCF > AC@NSCF, which may be mainly from the specific surface area of the corresponding carbon support. In particular, the CNT@NSCF had a relatively larger BET surface area (748.4 m2 g−1) and a total pore volume (1.45 cm3 g−1), which may enable the exposure of more efficient active sites, accelerate the mass transfer of the intermediate products and reactants of ORR/OER, and thereby enhance the ORR/OER catalytic performance.40,42,43
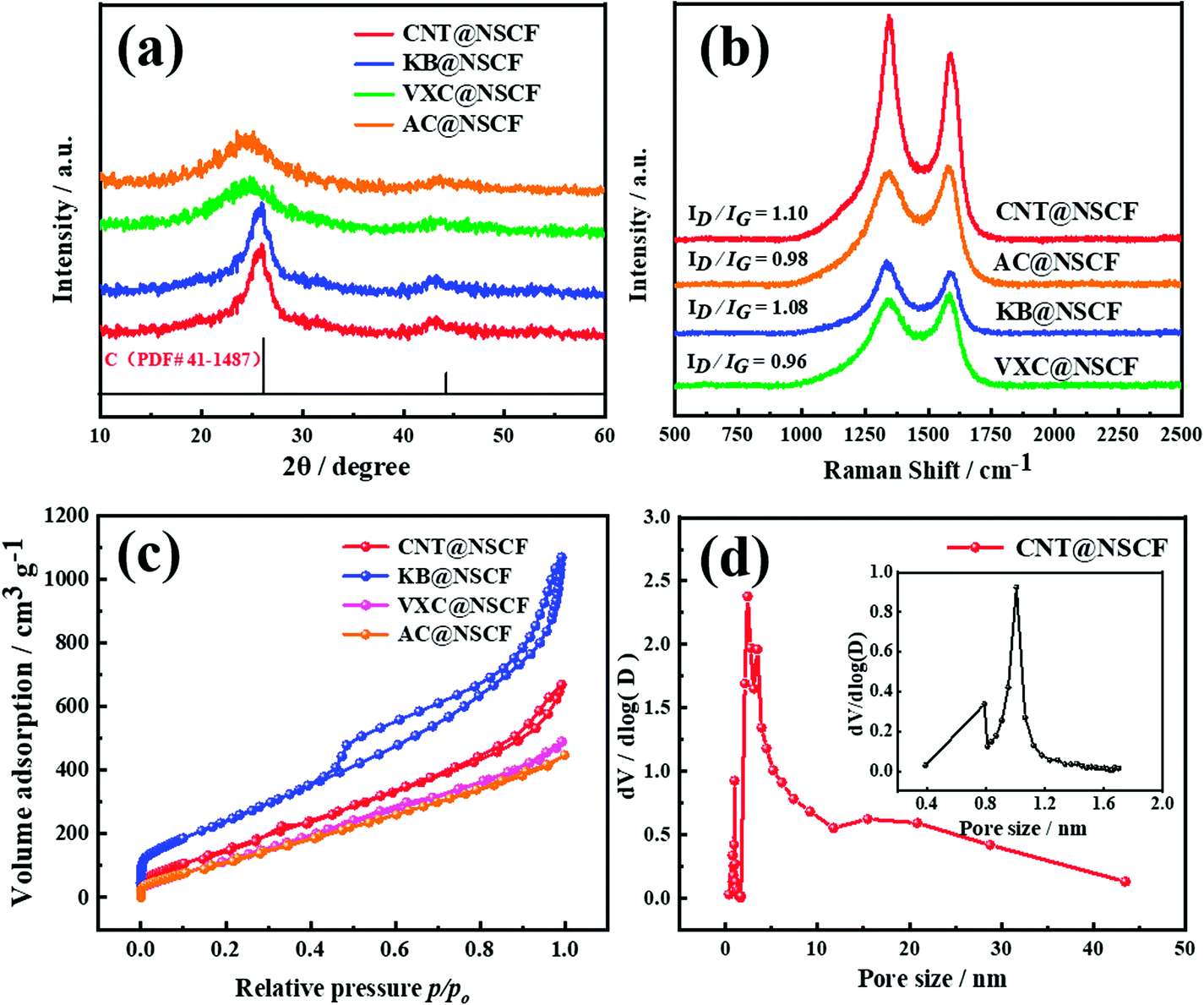 | ||
| Fig. 2 (a) The XRD patterns, (b) the Raman spectra, and (c) the N2 adsorption–desorption isotherms of CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF catalysts, (d) the pore size distribution curve of CNT@NSCF (the inset is an enlarged view of the micropore distribution curve). | ||
In order to discover the structure differences of NSCF among the CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF catalysts, the surface property of the four supports, which is one of the key factors that influence the character of NSCF, were carried out using XPS. As shown in Table S2 (ESI†), the relative surface oxygen content of CNT (8.25 at%) was much higher than those of KB (0.89 at%), VXC (1.72 at%), and AC (0.98 at%). Meanwhile, in their O 1s spectra (Fig. S6, ESI†) all peaks could be ascribed to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds,44,45 and the peaks in the C 1s spectra (Fig. S7, ESI†) could be attributed to the CC, C–O, and O–CO bonds.46 The FTIR spectra of the four supports are shown in Fig. S8 (ESI†). The absorption peaks at 1141, 1630, and 1718 cm−1 of the four supports could be ascribed to the C–O, CC, and CO stretching,34 respectively, which is in agreement with the XPS results. The previous results proved that the surfaces of the four supports were mainly distributed with OFGs, and the content of OFGs on CNT was the largest.
O bonds,44,45 and the peaks in the C 1s spectra (Fig. S7, ESI†) could be attributed to the CC, C–O, and O–CO bonds.46 The FTIR spectra of the four supports are shown in Fig. S8 (ESI†). The absorption peaks at 1141, 1630, and 1718 cm−1 of the four supports could be ascribed to the C–O, CC, and CO stretching,34 respectively, which is in agreement with the XPS results. The previous results proved that the surfaces of the four supports were mainly distributed with OFGs, and the content of OFGs on CNT was the largest.
The surface OFGs on the support have a great influence on the structure and morphology of the catalyst. In particular, they not only can affect the dispersion of active species, but also impact on the interaction between the active species and the support.34,47–49 Therefore, assuming that the mass loss of the supports is not taken into account in comparison to the total catalyst mass loss during the preparation process, (i.e., the mass loss mainly from the decomposition of the N, S precursors), the NSCF loading of each catalyst was correlated with their relative content of OFGs on the corresponding support. As shown in Fig. 3a, the weight percentages of NSCF of each catalyst had a strong correlation with the relative content of OFGs in the support. For pristine NSCF, the yield after pyrolysis was about 25.97 wt% (not shown in Fig. 3a), which was obviously higher than that for the samples with carbon supports. This may be due to the fact that dispersion of the NSCF precursor is increased on the carbon supports, which helps to fully decompose the polymer precursor during pyrolysis. Furthermore, except for the AC@NSCF catalyst, the loading of NSCF in CNT@NSCF, KB@NSCF, and VXC@NSCF catalysts decreased with the increase of surface OFG content. The lowest weight percentage of NSCF on CNT@NSCF may be a result of the highest relative content of OFGs. More precursors could be uniformly anchored on the support by the abundant OFGs, which may be more fully decomposed during the pyrolysis process. It is worth emphasizing that the more evenly distributed NSCF had a larger specific surface area, and was also prone to produce more defects and accessible effective active centers, which will be conducive to an improvement of the activity of the samples.
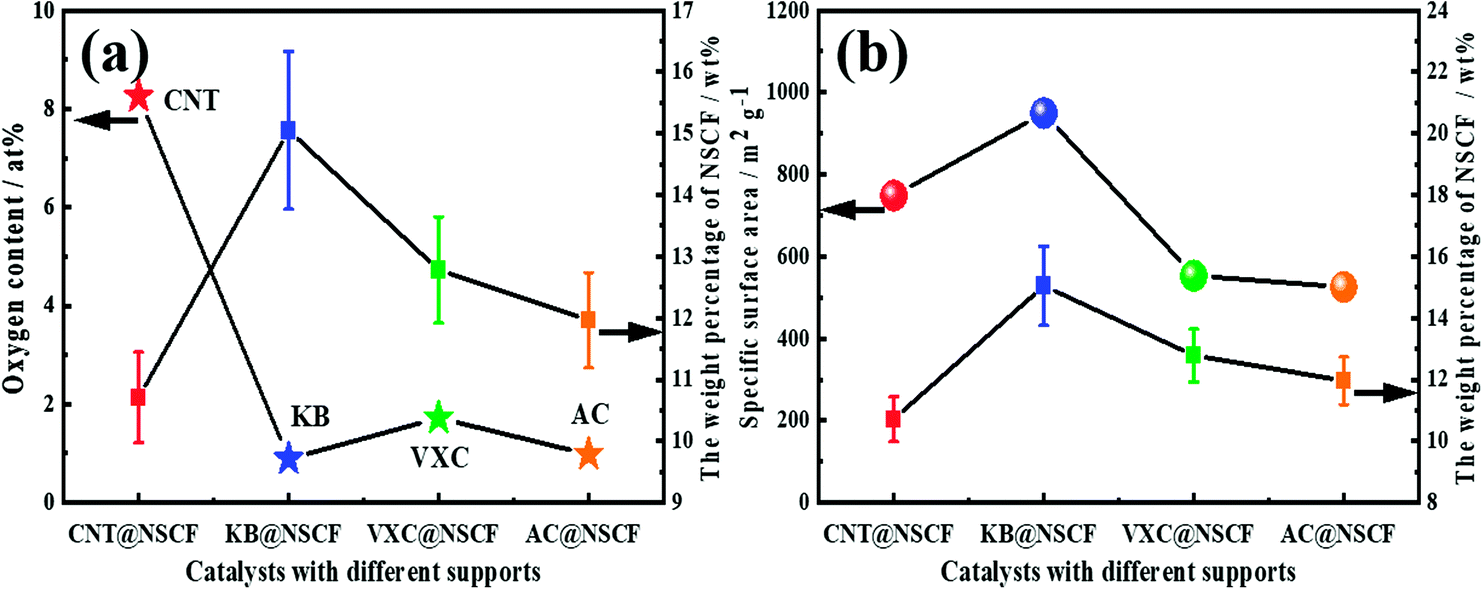 | ||
| Fig. 3 (a) The NSCF percentage weight change curve of different catalysts and the oxygen content curve of the corresponding supports. (b) The NSCF percentage weight change curve of different catalysts and the specific surface area curve of the corresponding catalysts. | ||
As for the AC@NSCF catalyst, the reason for the exception was related to it having the lowest specific surface area (Table S1, ESI†). Although the oxygen content of AC was close to that of KB, the small specific surface area may adsorb less precursor polymer on the pore surface, and therefore had the lowest relative content of NSCF. The weight percentages of NSCF for the four catalysts and their specific surface area of the corresponding supports were also related, which showed a good positive relationship trend except for the CNT@NSCF catalyst (Fig. 3b), i.e., the specific surface area of CNT@NSCF was larger than that of VXC@NSCF and AC@NSCF but it was smaller than that of KB@NSCF, but its NSCF loading is the smallest of the four catalysts. Generally, a larger specific surface area could provide more loading sites, and the corresponding NSCF loading should also be larger, which was confirmed by the KB@NSCF, VXC@NSCF, and AC@NSCF catalysts. The exception, CNT@NSCF was also due to the abundant OFGs on the CNT surface, which promoted the uniform distribution of the precursor polymer, which became fully decomposed during the pyrolysis process, unlike the other three catalysts. All these further prove that the NSCF coated on CNT may have the most uniformly distributed structure among the four samples.
It is well known that the doping type and surface chemical structure of heteroatoms will greatly affect the catalytic performance of metal-free electrocatalysts. Therefore, the surface chemical structure of the prepared catalysts was explored using XPS. Fig. S9 (ESI†) shows that the full survey spectra of CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF, and CNT@NCF samples all show N, S, C, and O peaks. Table S3 (ESI†) lists the relative atomic content of the N, S, C, and O species. Notably, the total content of N and S for CNT@NSCF (5.56 at%) was significantly higher than those of KB@NSCF (4.22 at%), VXC@NSCF (3.44 at%), and AC@NSCF (3.14 at%). This may mainly be due to the even distribution of NSCF on CNT@NSCF, which will be exposed to the efficient active species. In addition, the C, N, and O contents in CNT@NCF were close to those of CNT@NSCF. The high-resolution N 1s spectra of all the samples (Fig. 4a and S10 and S11, ESI†) can be fitted as pyridinic-N (398.2 eV), pyrrolic-N (400.5 eV), graphitic-N (401.3 eV), and oxidized-N (403.5 eV).50,51 Pyridinic-N and graphitic-N could offer a lone pair of electrons and then affect the charge density of the adjacent carbon matrix, thus effectively enhancing the catalytic activity of ORR and OER.10 The overall relative content of the pyridinic-N and the graphitic-N of the CNT@NSCF catalyst is the highest as shown in Fig. 4c, which may lead to an excellent bifunctional oxygen catalytic activity. The high-resolution S 2p spectra of the four samples (Fig. 4b and S12, ESI†) can be divided into S 2p3/2 (163.8 eV) and S 2p1/2 (165.1 eV) under the –C–S–C- configuration of thiophene-S, and oxidized-S (168.5 eV), respectively.33,52 Evidently, the thiophene-S content of the CNT@NSCF catalyst is also slightly higher than those of the other samples (Fig. 4d and Table S4, ESI†). Moreover, the configuration and the overall relative content of active pyridinic-N and graphitic-N of CNT@NCF were also similar to those of CNT@NSCF according to the N 1s XPS spectrum. The C 1s spectra (Fig. S13, ESI†) can be deconvoluted as CC (284.6 eV), C–N (285.2 eV), C–O (286.3 eV), and O–CO (289.2 eV) bonds.53,54 The formation of the CC bond assures the conductivity of the four catalysts. The FTIR spectra of the CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF, CNT@NCF, NSCF, and NSCF0 also confirmed the existence of CC, C–O, and CO stretching (Fig. S14 and S15, ESI†).49 Obviously, CNT@NSCF has the highest content of pyridinic-N, graphitic-N, and thiophene-S together with excellent conductivity when compared to the four other catalysts. These results indicate that the CNT@NSCF may also have the highest ORR/OER catalytic activity.
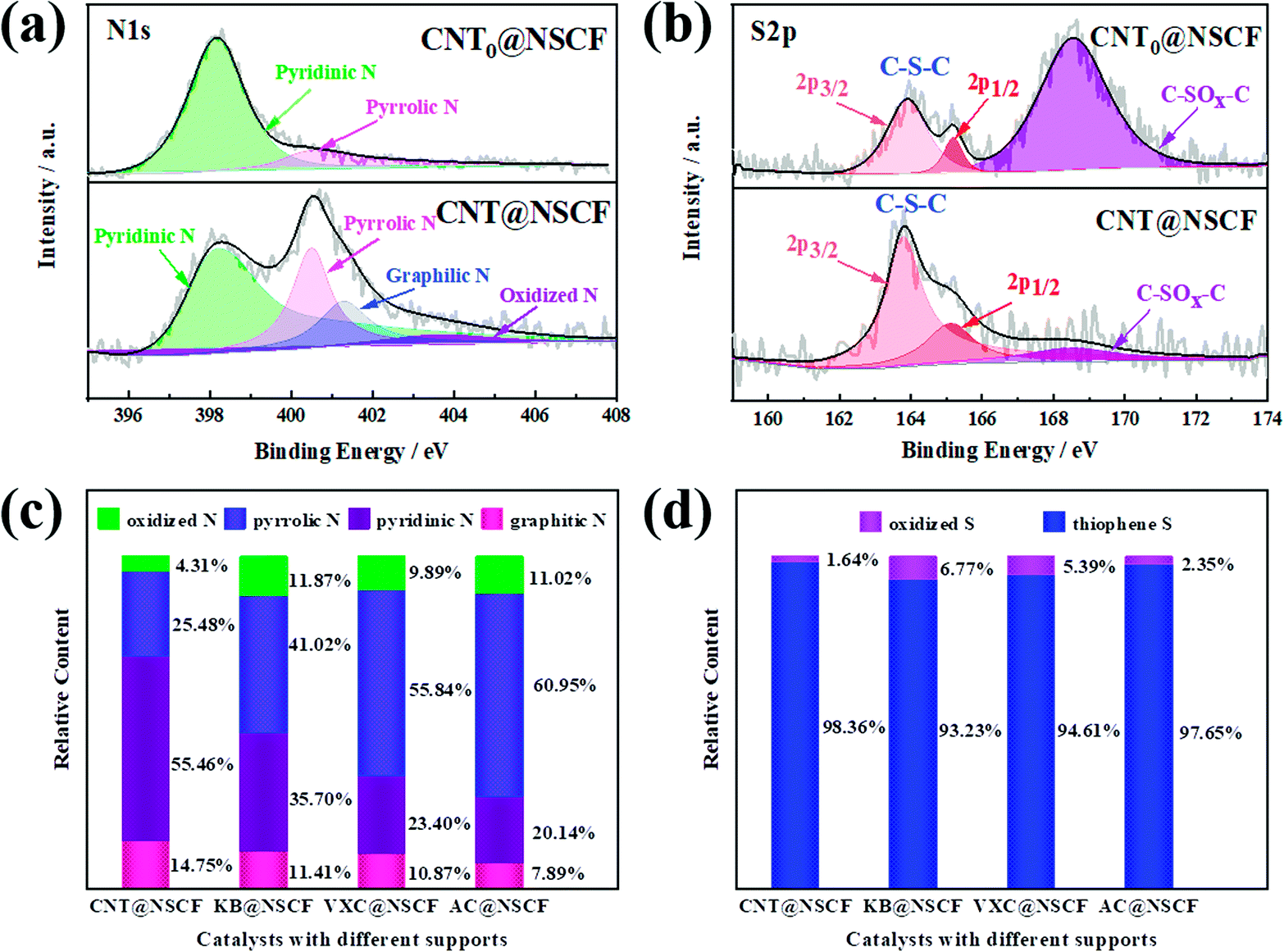 | ||
| Fig. 4 (a) The N 1s spectra of CNT@NSCF0 and CNT@NSCF, (b) the S 2p spectra of CNT@NSCF0 and CNT@NSCF, (c) the columnar statistics of various nitrogen active species of different carbon-supported catalysts, and (d) the columnar statistics of various active sulfur species of different carbon-supported catalysts. | ||
The ORR performances of CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF, and Pt/C were tested in 0.1 M KOH at room temperature. As expected, the LSV curves (Fig. 5a) showed that CNT@NSCF exhibited a relatively high onset potential (Eonset) of 0.94 V and a half-wave potential (E1/2) of 0.82 V, which were close to those of Pt/C (Eonset = 0.97 V, and E1/2 = 0.84 V), and which surpassed those of the KB@NSCF, VXC@NSCF, AC@NSCF, and NSCF catalysts (Fig. S16 and Table S5, ESI†). At the same time, CNT@NSCF (pyrolyzed at 900 °C) showed the best ORR activity compared to NSCF@CN−800 and NSCF@CN−1000 (Fig. S17, ESI†). This may be due to the fact that the active sites and proper hierarchical porous structure can be formed at 900 °C, whereas the degree of graphitization is poor at lower temperatures, and the active sites and porous structure would be destroyed at higher temperatures. In addition, CNT@NSCF displayed a better ORR catalytic activity than CNT@NCF (Fig. S18, ESI†), which may be due to the synergy between the N, S dopants especially when considering their surface structure characterization (Fig. S11 and Tables S3 and S4, ESI†). To eliminate the influence of the metal ions, SCN− ions were introduced to determine if there were any iron-based catalytic sites (Fig. S19, ESI†). Obviously, the current density did not change significantly after adding KSCN, which supported the excellent inherent activity of this N, S co-doped metal-free catalyst.55 The corresponding Tafel slopes of CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF, and Pt/C were 82.6, 86.7, 95.3, 98.1, and 79.9 mV dec−1 (Fig. 5b), respectively. The lower Tafel slope of CNT@NSCF indicated the rapid mass/charge transfer occurring on the catalyst surface.
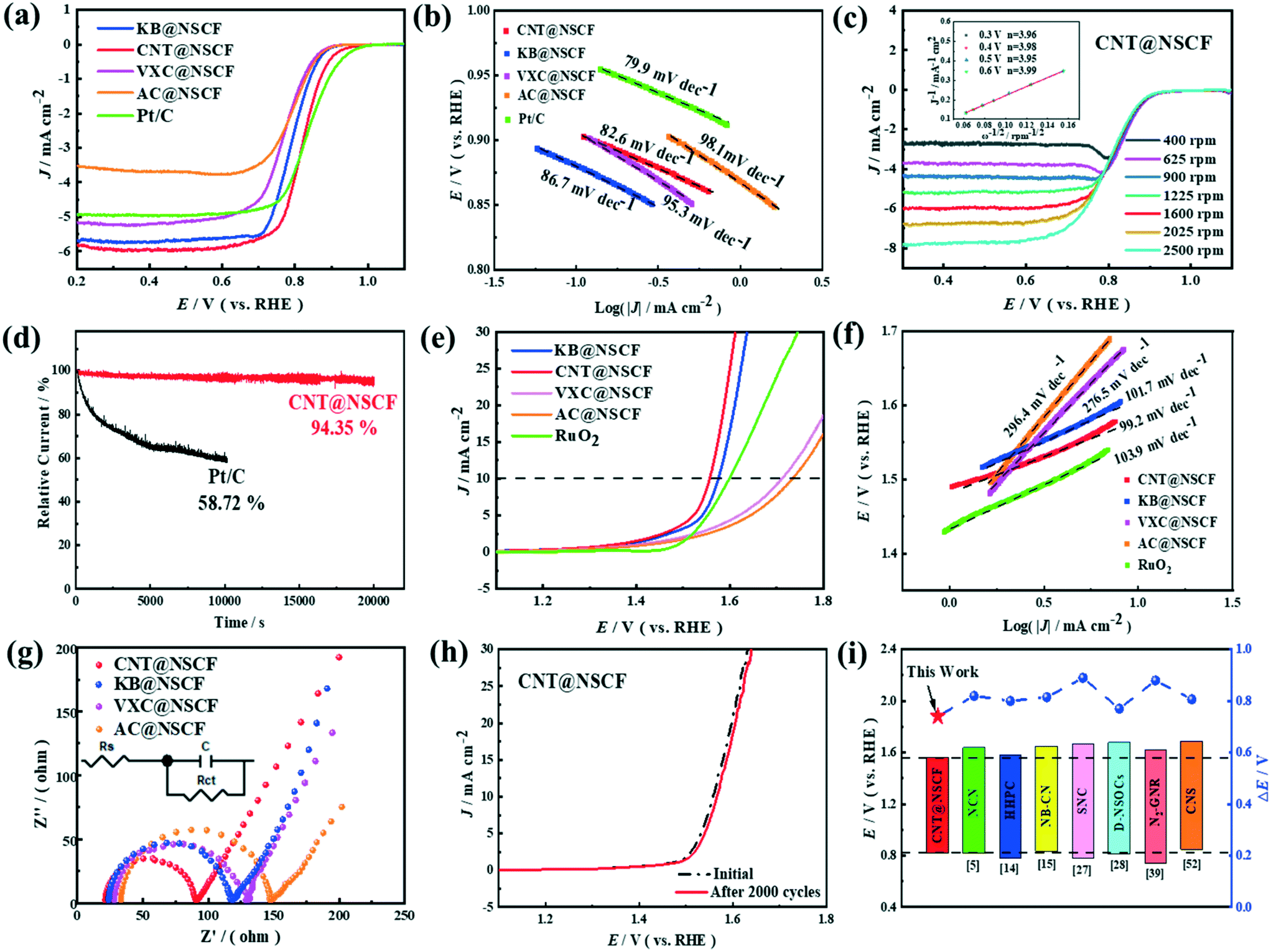 | ||
| Fig. 5 (a) The ORR LSV curves of different samples recorded at 1600 rpm and 10 mV s−1, (b) the corresponding Tafel plots from the ORR LSV curves for the CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF and Pt/C catalysts, (c) the ORR polarization curves of CNT@NSCF at different rotation speeds. The inset is the K–L plot curve at different potentials, (d) the chronoamperometric responses of CNT@NSCF and Pt/C electrocatalysts in an O2-saturated 0.1 M KOH solution at a rotational speed of 400 rpm, (e) the OER LSV curves of CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF catalysts recorded at 1600 rpm and 10 mV s−1, (f) the corresponding Tafel plots from the OER LSV curves for the CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF and RuO2 catalysts, (g) the EIS Nyquist plots for the CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF catalysts, (h) the OER LSV curves of the CNT@NSCF catalyst in O2-saturated 0.1 M KOH before and after 2000 cycles, and (i) a comparison of the oxygen electrode activities of the recently reported metal-free ORR/OER bifunctional catalysts. | ||
To further elucidate the ORR dynamics, the LSV curves of the four samples were tested at different rates of rotation as shown in Fig. 5c and S20 (ESI†). Obviously, the limiting current density of the four samples gradually increased with the increase of the rotational speed, which indicated that the ORR of the four samples was a first-order reaction.56 Furthermore, the K–L plots of CNT@NSCF, KB@NSCF, VXC@NSCF, and AC@NSCF showed good linearity and consistent slopes. The number of transferred electrons (n) of the CNT@NSCF was closer to four than the other samples, showing that the CNT@NSCF mainly followed the 4e− ORR process.57 Long-term durability and poisoning resistance are also key indicators for the evaluation of the catalysts' ORR performance. As shown in Fig. 5d, the CNT@NSCF attenuated slowly and still maintained a high relative current of 94.35% after 20000 s of testing. In contrast, the Pt/C decayed to 58.72% of the primary current after 10000 s. Furthermore, Fig. S21 and S22 (ESI†) also show that the CV curve of the accelerated aging test and the polarization curve recorded for CNT@NSCF after 8000 cycles with a negligible change of E1/2 and Eonset, confirmed the excellent cycling stability of the CNT@NSCF. To test the methanol resistance of CNT@NSCF, 3 M methanol was poured into the electrolyte at 500 s during the chronoamperometric test, and the ORR current density response was recorded (Fig. S23, ESI†). Compared to the large current density change of Pt/C, the almost unchanged current density of CNT@NSCF confirmed its excellent methanol tolerance.
The CNT@NSCF also showed excellent OER performance in an alkaline solution. From the OER polarization curves in Fig. 5e and S24 (ESI†), it can be seen that the CNT@NSCF achieved 1.56 V at 10 mA cm−2, which was less than that of the other samples including KB@NSCF (1.58 V), VXC@NSCF (1.71 V), AC@NSCF (1.73 V), NSCF (1.78 V), and commercial RuO2 (1.59 V). In addition, the CNT@NSCF (pyrolyzed at 900 °C) also exhibited the best OER activity compared to CNT@NSC−800 and CNT@NSC−1000 (Fig. S25†), which confirmed that a suitable pyrolysis temperature was also of great significance to increasing the reaction rate of the OER. Correspondingly, the Tafel slopes of CNT@NSCF, KB@NSCF, VXC@NSCF, AC@NSCF, and RuO2 were 99.2, 101.7, 276.5, 296.4, and 103.9 mV dec−1, respectively, which inferred that the CNT@NSCF had rapid OER kinetics (Fig. 5f). Moreover, the CNT@NSCF displayed the smallest Nyquist plot out of the four catalysts (Fig. 5g), and this indicated its fast charge transfer ability.58 Furthermore, there are significant differences in the diameter of the Nyquist plot between the catalyst and the corresponding support (Fig. 5g and S26, ESI†).
Considering the essential difference between the support and the catalyst is the fact that the NSCFs is coated on to the catalyst, and it can be deduced that the NSCF plays a major role in the EIS testing of all the catalysts. The difference of the charge transfer resistance (Rct) values between the catalyst and corresponding support can show the conductivity difference of the coated NSCFs to a certain extent. Therefore, according to the ΔRct values in Table S6 (ESI†), it can be concluded that the conductivity of the NSCFs coated on the different supports decrease with the sequence of NSCFCNT (ΔRct: 10.76 Ω) > NSCFAC (ΔRct: 15.95 Ω) > NSCFKB (ΔRct: 26.42 Ω) > NSCFVXC (ΔRct: 31.49 Ω). More importantly, the CNT@NSCF also retains a high stability with a smaller voltage drop after a 2000 cycle accelerated aging test (Fig. 5h and S27†), which showed the potential for further practical applications.
To assess the overall ORR/OER activity, the potential gap: (ΔE = Ej = 10,OER − E1/2,ORR) of CNT@NSCF and RuO2 + Pt/C was calculated. Remarkably, the CNT@NSCF showed the lowest ΔE of 0.74 V (Fig. S28 and Table S7, ESI†) than that of RuO2 + Pt/C (0.75 V), KB@NSC (0.79 V), VXC@NSC (0.94 V), AC@NSC (0.94 V), and NSCF (1.05 V). The changing trend of ΔE in 0.1 M KOH was in agreement with the activity changes in 1 M KOH. Notably, the ΔE value of CNT@NSCF was also much lower than most advanced metal-free heteroatom-doped carbon bifunctional electrocatalysts recently reported (Fig. 5i), indicating its superior bifunctional oxygen catalytic activity.10,20,21,33,34,45,59 All of the previous results indicated that the CNT@NSCF exhibited excellent ORR/OER bifunctional catalytic activity as well as good stability and durability. The main reason for these performances of CNT@NSCF may be because of: (i) the relatively large BET surface area and abundant hierarchical pore structure of CNT@NSCF made the electrolyte and catalyst fully contact, which was conducive to exposing more efficient active sites and reduced the mass transfer resistance while shortening the O2 and electrolyte transmission paths, and (ii) the uniformly dispersed NSCF on CNT provided the highest amount of effective ORR/OER active species, such as pyridine-N, graphite-N and thiophene-S, which synthetically catalyzed the ORR/OER.
Encouraged by the excellent half-cell ORR/OER performance of CNT@NSCF, a ZAB utilizing a carbon cloth loaded with CNT@NSCF as an air cathode and a polished zinc sheet as an anode (detailed structure shown in Fig. S29, ESI†) were assembled. As shown in Fig. 6a, the ZAB using the CNT@NSCF catalyst gave a high open circuit voltage of 1.46 V. Its charge–discharge overpotential was 0.78 V at 10 mA cm−2 (Fig. 6b), which was smaller than that of the Pt/C + RuO2 (m:m = 1:1) counterpart (0.79 V), thus confirming its dominant ORR/OER performance in the actual application process. Furthermore, the ZAB using the CNT@NSCF catalyst achieved a peak power density of 96.7 mW cm−2, which was about 1.15 times that of Pt/C + RuO2 (84.0 mW cm−2, Fig. 6c). Moreover, the CNT@NSCF-based ZAB also delivered a small voltage drop of 0.141 V from 10 to 50 mA cm−2, with very little difference from that of the Pt/C + RuO2 catalysts (0.123 V, Fig. 6d). When cycled at 10 mA cm−2 (10 min per cycle), the CNT@NSCF-based ZAB exhibited a discharge–charge gap of only 0.78 V and an extraordinary cycling stability (Fig. 6e). Even after continuous running for 1278 cycles (∼213 h), the polarization voltage only slightly increased to 0.85 V, which outperformed the most recently reported metal-free oxygen electrocatalysts. Conversely, the discharge–charge gap of a Pt/C + RuO2-based ZAB showed a significant increase after only 300 cycles (∼50 h). More importantly, the LED panel light (2.5 V) could be lit by two CNT@NSCF-based ZABs (Fig. 6f). These results fully demonstrate that CNT@NSCF is a remarkable bifunctional oxygen catalyst for metal–air batteries.
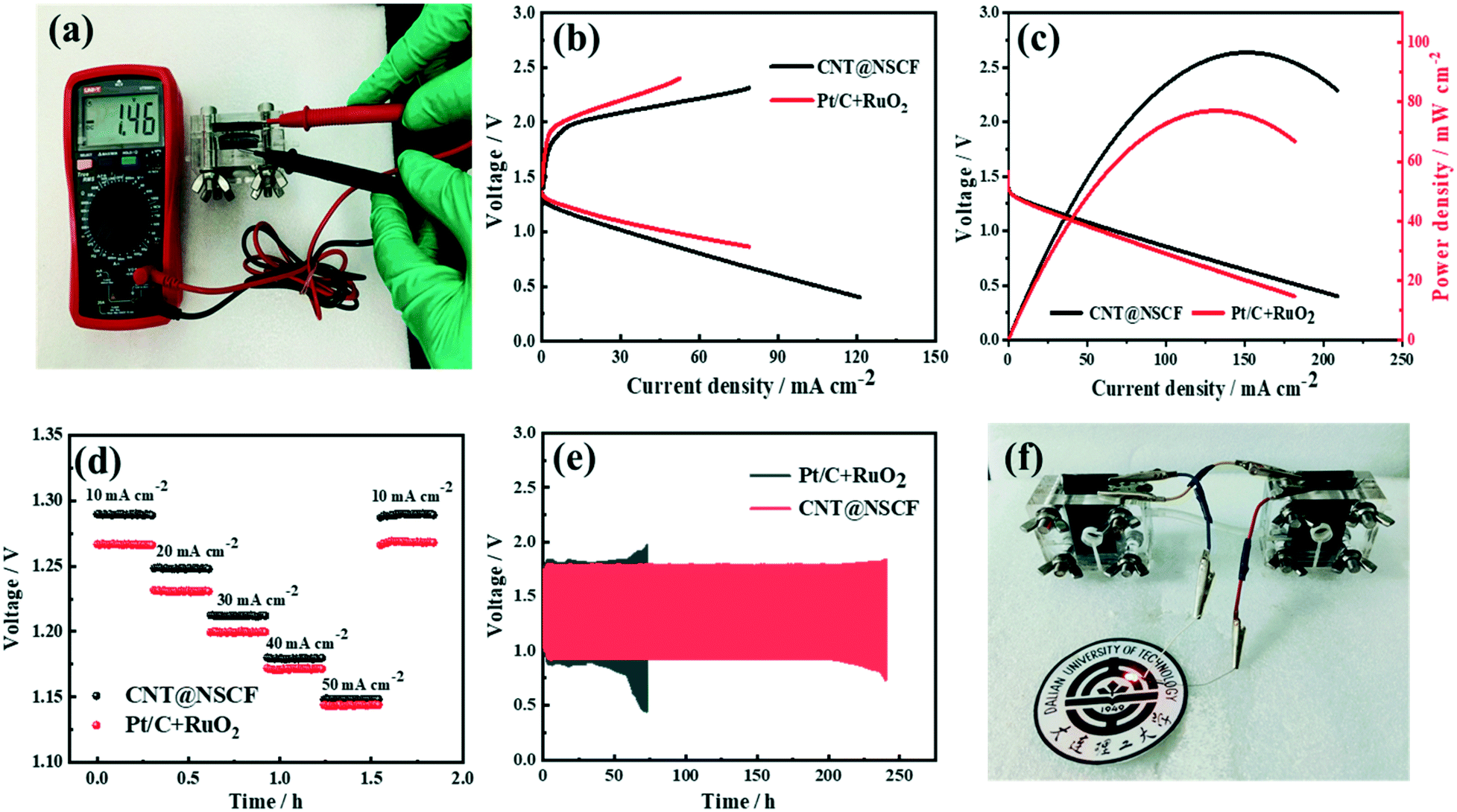 | ||
| Fig. 6 (a) The open-circuit potential of the ZAB equipped with the CNT@NSCF catalyst, (b) the charge and discharge polarization curves, (c) the discharge polarization curves and corresponding power densities, (d) the galvanostatic discharge voltage-time curves of batteries assembled with CNT@NSCF or Pt/C + RuO2 at different current densities, (e) the long-term cycling stability at 10 mA cm−2 of rechargeable ZABs assembled with CNT@NSCF and Pt/C + RuO2, and (f) a photograph of a red LED light (2.5 V) powered by two integrated ZABs in series assembled with a CNT@NSCF catalyst. | ||
Conclusion
In conclusion, an advanced ORR/OER bifunctional metal-free electrocatalyst has been developed by coating NSCF on to carboxylic CNT (CNT@NSCF) by a low temperature polymerization–high temperature pyrolysis strategy. The surface OFGs on CNT clearly promote the uniform dispersion of NSCF with the formation of abundant high-efficiency active species (pyridinic-N, graphitic-N and thiophene-S) and defects. Both the unique NSCF structure and the large amount of pyridinic-N, graphitic-N, and thiophene-S of CNT@NSCF are responsible for its remarkable ORR/OER catalytic activity. The CNT@NSCF catalyst shows an ORR half-wave potential of 0.82 V, which is approaching that of Pt/C (0.84 V), and an OER overpotential of 330 mV (10 mA cm−2), giving an extremely small ΔE of 0.74 V. Furthermore, the CNT@NSCF-based ZAB displays a dominant power density of 96.7 mW cm−2 and a long-term stability of over 1278 cycles (∼213 h). This study shows a new way to design robust metal-free bifunctional carbon materials for use in renewable energy devices.Author contributions
Xin Wang: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft. Guang-Lan Li: project administration, writing – review and editing. Zhong-Fa Lu: data curation, formal analysis. Shuo Cao: data curation, formal analysis. Ce Hao: formal analysis. Suli Wang: conceptualization, data curation, formal analysis. Gongquan Sun: formal analysis.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Key Laboratory of Fuel Cell & Hybrid Power Sources, CAS (KLFC-2019-02), the National Natural Science Foundation of China (No. 21805026), and the Fundamental Research Funds for the Central Universities (DUT20JC10).References
- W. Si, Z. Yang, X. Hu, Q. Lv, X. Li and F. Zhao, et al. , J. Mater. Chem. A, 2021, 9, 14507–14514 RSC.
- Y. Tian, X. Liu, L. Xu, D. Yuan, Y. Dou and J. Qiu, et al. , Adv. Funct. Mater., 2021, 31, 2101239 CrossRef CAS.
- P. He, Q. Chen, M. Yan, X. Xu, L. Zhou and L. Mai, et al. , EnergyChem, 2019, 1, 1 CrossRef.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai and J. Gao, et al. , Adv. Funct. Mater., 2018, 28, 1800499–1800506 CrossRef.
- X. Guo, S. Zheng, Y. Luo and H. Pang, Chem. Eng. J., 2020, 401, 126005 CrossRef CAS.
- B. Li, H. Xue, H. Pang and Q. Xu, Sci. China: Chem., 2020, 63, 475–482 CrossRef CAS.
- F. Yang, X. Gao, J. Xie, X. Liu, J. Jiang and X. Lu, Small Struct., 2021, 2100144 CrossRef.
- M. Ha, D. Y. Kim, M. Umer, V. Gladkikh, C. W. Myung and K. S. Kim, Energy Environ. Sci., 2021, 14, 3455–3468 RSC.
- J. Balamurugan, T. T. Nguyen, N. H. Kim, D. H. Kim and J. H. Lee, Nano Energy, 2021, 85, 105987 CrossRef CAS.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu and L. Wang, et al. , Energy Environ. Sci., 2019, 12, 322–333 RSC.
- J. Guo, Y. Yu, J. Ma, T. Zhang and S. Xing, J. Alloys Compd., 2020, 821, 153484 CrossRef CAS.
- J. Kim, J. Park, J. Lee, W. G. Lim, C. Jo and J. Lee, Funct. Mater., 2021, 31, 2010882 CrossRef CAS.
- W. Chen, L. Xu, Y. Tian, H. Li and K. Wang, Carbon, 2018, 137, 458–466 CrossRef CAS.
- M. Vikkisk, I. Kruusenberg, U. Joost, E. Shulga, I. Kink and K. Tammeveski, Appl. Catal., B, 2014, 147, 369–376 CrossRef CAS.
- H. Han, Y. Noh, Y. Kim, W. S. Jung, S. Park and W. B. Kim, Nanoscale, 2019, 11, 2423–2433 RSC.
- E. Y. Choi, D. E. Kim, S. Y. Lee and C. K. Kim, Carbon, 2020, 166, 245–255 CrossRef CAS.
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao and X. Wang, et al. , Sci. Adv., 2016, 2, e1501122 CrossRef.
- S. K. Singh, K. Takeyasu and J. Nakamura, Adv. Mater., 2019, 31, 1804297 CrossRef PubMed.
- S. Kumar, S. Gonen, A. Friedman, L. Elbaz and G. D. Nessim, Carbon, 2017, 120, 419–426 CrossRef CAS.
- X. Xiao, X. Li, Z. Wang, G. Yan, H. Guo and Q. Hu, et al. , Appl. Catal., B, 2020, 265, 118603 CrossRef CAS.
- Z. Lu, J. Wang, S. Huang, Y. Hou, Y. Li and Y. Zhao, et al. , Nano Energy, 2017, 42, 334–340 CrossRef CAS.
- X. Lin, P. Peng, J. Guo and Z. Xiang, Chem. Eng. J., 2019, 358, 427–434 CrossRef CAS.
- H.-W. Zhang, Y.-Y. Li, W.-Q. Huang, B.-X. Zhou, S.-F. Ma and Y.-X. Lu, et al. , Carbon, 2019, 148, 231–240 CrossRef CAS.
- H. Fan, Y. Wang, F. Gao, L. Yang, M. Liu and X. Du, et al. , J. Energy Chem., 2019, 34, 64–71 CrossRef.
- X. Zhang, Y. Wang, K. Wang, Y. Huang, D. Lyu and F. Yu, et al. , Chem. Eng. J., 2021, 416, 129096 CrossRef CAS.
- Z. Li, Y. Yao, Y. Niu, W. Zhang, B. Chen and X. Zeng, et al. , Chem. Eng. J., 2021, 418, 129321 CrossRef CAS.
- J. Gu, S. Magagula, J. Zhao and Z. Chen, Small Methods, 2019, 3, 1800550 CrossRef.
- T. Asefa, Acc. Chem. Res., 2016, 49, 1873–1883 CrossRef CAS.
- A. L. Cazetta, L. Spessato, K. C. Bedin, I. P. F. A. Souza, R. A. Araújo and A. F. Martins, et al. , Appl. Surf. Sci., 2019, 467-468, 75–83 CrossRef CAS.
- Y. Li, J. Yang, J. Huang, Y. Zhou, K. Xu and N. Zhao, et al. , Carbon, 2017, 122, 237–246 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu and L. Dai, et al. , Adv. Mater., 2019, 31, 1804799 CrossRef.
- X. Zhu, R. Amal and X. Lu, Small, 2019, 15, 1804524 CrossRef PubMed.
- Y. B. Guo, S. Yao, L. X. Gao, A. Chen, M. G. Jiao and H. J. Cui, et al. , J. Mater. Chem. A, 2020, 8, 4386–4395 RSC.
- S. Zhou, J. Zang, H. Gao, X. Tian, P. Tian and S. Song, et al. , Carbon, 2021, 181, 234–245 CrossRef CAS.
- J. Geng, H.-J. Zhang, S. Yao, C. Cai, Z.-F. Ma and J. Wang, et al. , RSC Adv., 2019, 9, 24770–24776 RSC.
- M. H. Naveen, K. Shim, M. S. A. Hossain, J. H. Kim and Y.-B. Shim, Adv. Energy Mater., 2017, 7, 1602002 CrossRef.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu and O. Zhuo, et al. , ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- S. Zhong, L. Zhou, L. Wu, L. Tang, Q. He and J. Ahmed, J. Power Sources, 2014, 272, 344–350 CrossRef CAS.
- D. Lyu, S. Yao, Y. Bahari, S. W. Hasan, C. Pan and X. Zhang, et al. , Appl. Mater. Today, 2020, 20, 100737 CrossRef.
- W. Xia, J. Tang, J. Li, S. Zhang, K. C. Wu and J. He, et al. , Angew. Chem., Int. Ed., 2019, 58, 13354–13359 CrossRef CAS PubMed.
- Y. Pan, Y. Zhao, S. Mu, Y. Wang, C. Jiang and Q. Liu, et al. , J. Mater. Chem. A, 2017, 5, 9544–9552 RSC.
- Z. Zou, M. Cai, X. Zhao, J. Huang, J. Du and C. Xu, J. Mater. Chem. A, 2019, 7, 14011–14018 RSC.
- H. W. Liang, S. Bruller, R. Dong, J. Zhang, X. Feng and K. Mullen, Nat. Commun., 2015, 6, 7992 CrossRef CAS.
- T. Liu, Y.-F. Guo, Y.-M. Yan, F. Wang, C. Deng and D. Rooney, et al. , Carbon, 2016, 106, 84–92 CrossRef CAS.
- J. Lim, J.-W. Jung, N.-Y. Kim, G. Y. Lee, H. J. Lee and Y. Lee, et al. , Energy Stor. Mater., 2020, 32, 517–524 CrossRef.
- D. Li, B. Ren, Q. Jin, H. Cui and C. Wang, J. Mater. Chem. A, 2018, 6, 2176–2183 RSC.
- J. Sun, C. Hu, Z. Liu, H. Liu and J. Qu, Carbon, 2019, 145, 140–148 CrossRef CAS.
- H. Zhang, X. Quan, S. Chen, X. Fan, G. Wei and H. Yu, Environ. Sci. Technol., 2018, 52, 4827–4834 CrossRef CAS PubMed.
- L. Li, H. Yang, J. Miao, L. Zhang, H.-Y. Wang and Z. Zeng, et al. , ACS Energy Lett., 2017, 2, 294–300 CrossRef CAS.
- Y. Qiu, J. Huo, F. Jia, B. H. Shanks and W. Li, J. Mater. Chem. A, 2016, 4, 83–95 RSC.
- J. Yang, F. Xiang, H. Guo, L. Wang and X. Niu, Carbon, 2020, 156, 514–522 CrossRef CAS.
- J.-M. You, M. S. Ahmed, H. S. Han, Choe Je, Z. Üstündağ and S. Jeon, J. Power Sources, 2015, 275, 73–79 CrossRef CAS.
- N. Xu, Y. Zhang, T. Zhang, Y. Liu and J. Qiao, Nano Energy, 2019, 57, 176–185 CrossRef CAS.
- N. Chandrasekaran and S. Muthusamy, Langmuir, 2017, 33, 2–10 CrossRef CAS.
- C. Tang, H. F. Wang, X. Chen, B. Q. Li, T. Z. Hou and B. Zhang, et al. , Adv. Mater., 2016, 28, 6845–6851 CrossRef CAS.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng and M. Zhou, et al. , Adv. Funct. Mater., 2018, 28, 1805641–1805650 CrossRef.
- I. S. Amiinu, J. Zhang, Z. Kou, X. Liu, O. K. Asare and H. Zhou, et al. , ACS Appl. Mater. Interfaces, 2016, 8, 29408–29418 CrossRef CAS.
- J. Chen, F. Zheng, S.-J. Zhang, A. Fisher, Y. Zhou and Z. Wang, et al. , ACS Catal., 2018, 8, 11342–11351 CrossRef CAS.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang and M. Zhu, et al. , Energy Environ. Sci., 2017, 10, 742–749 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01818c |
| This journal is © The Royal Society of Chemistry 2022 |