
Engineering regioselectivity in the hydrosilylation of alkynes using heterobimetallic dual-functional hybrid catalysts†
Max
Roemer
 *a,
Sinead T.
Keaveney
b,
Vinicius R.
Gonçales
c,
Jiaxin
Lian
c,
James E.
Downes
d,
Shreedhar
Gautam
c,
J. Justin
Gooding
c and
Barbara A.
Messerle
*bc
*a,
Sinead T.
Keaveney
b,
Vinicius R.
Gonçales
c,
Jiaxin
Lian
c,
James E.
Downes
d,
Shreedhar
Gautam
c,
J. Justin
Gooding
c and
Barbara A.
Messerle
*bc
aSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: max.roemer@sydney.edu.au
bDepartment of Molecular Sciences, Macquarie University, NSW 2109, Australia. E-mail: barbara.messerle@mq.edu.au
cSchool of Chemistry and the Australian Centre for NanoMedicine, The University of New South Wales, Sydney, NSW 2052, Australia
dDepartment of Physics and Astronomy, Macquarie University, NSW 2109, Australia
First published on 8th November 2021
Abstract
The synthesis and characterization of carbon black supported rhodium and iridium heterobimetallic catalysts, termed hybrid catalysts, and their application in the hydrosilylation of alkynes is described. An aryl diazonium grafting procedure was applied to simultaneously immobilize Rh and Ir pyrazole–triazole complexes with tethers of varying lengths to carbon black, yielding the hybrid catalysts. The complexes differ in metal centre oxidation state and co-ligands, which are CO or Cp*Cl for the Rh complexes and Cp*Cl for the Ir complexes. The immobilization results in simultaneous surface binding and modification of the Rh complexes bearing CO-ligands. In this process, the CO ligands are removed and the overall structure of the catalytically active complex is altered. Analysis of the hybrid catalysts by XPS and SEM/EDX shows that the catalysts bear both surface bound Rh- and Ir-complexes. The Rh content is substantially higher than the Ir content. This is due to more efficient binding of the modified Rh complexes to the carbon black, as they feature two potential binding sites. Synchrotron based X-ray absorption spectroscopy (XAS) at the Rh K- and Ir L3 edges further confirms the presence of the surface bound metal complexes. There is no indication that the presence of a secondary metal affects the electronic structure of the adjacent metal in the systems under investigation, for either the long or short tether derivatives. The performance of the different catalysts was assessed for promoting the hydrosilylation of alkynes, an important industrially relevant reaction. All catalysts are highly efficient. The modified Rh sites are α-selective in the product formation on activation of terminal alkynes, while the RhCp*Cl and IrCp*Cl sites are β(Z)-selective. When operating at mild conditions with high metal loadings, the surface bound Rh catalyst is the active species, while the Ir sites are inactive. At a lower overall surface coverage or higher temperature, the Ir sites become active, which allows engineering of the regioselectivity by adjusting surface coverages and metal loadings.
Introduction
Hybrid catalysts combine several advantages of homogeneous and heterogeneous catalysts by heterogenization of organometallic catalysts.1–3 Organometallic complexes can be efficiently tailored as homogenous catalysts through metal choice and ligand design. However catalyst recovery and recycling is challenging. Immobilization of homogeneous catalysts on solid supports such as glassy carbon,4 carbon black (CB),5,6 reduced graphene oxide,5 carbon nanotubes7 or silica8 affords hybrid catalysts, which are also known as single atom catalysts. The catalysts are insoluble in common organic solvents and maintain, in the ideal case, the same reactivity as the homogeneous analogues, while their heterogenous nature results in the reaction occurring at the solid–liquid interface. The insolubility of the precious catalyst allows removal after reaction completion by simple centrifugation.Dual-functional catalysts – catalysts that consist of two different catalytic species – are potentially attractive multitools for the synthesis of complex organic compounds, as they can facilitate two or more processes simultaneously. A dual-functional catalyst can lower the activation barrier for a single catalyzed chemical reaction when compared to a mono-functional catalyst. Furthermore, the two catalysts present in a dual-functional catalyst can operate synergistically as the one catalytically active sites may enhance the activity of the other.9 Examples of dual-functional catalysts which have been constructed using different catalytically active moieties include those bearing two different transition metal complexes,10 a transition metal complex in combination with an organocatalyst11,12 or a transition metal complex in combination with a photocatalyst.13 Furthermore, dual catalysis has been achieved with combinations of homogeneous and heterogeneous catalysts.14,15 Despite the wealth of different dual catalysts reported, there are only a limited number of examples of dual-functional hybrid catalysts. Having two active transition metal complexes on the same carbon surface, which exhibit different reactivity, could allow selectivity and/or multi-step reaction sequences to be engineered using one hybrid material (Fig. 1).
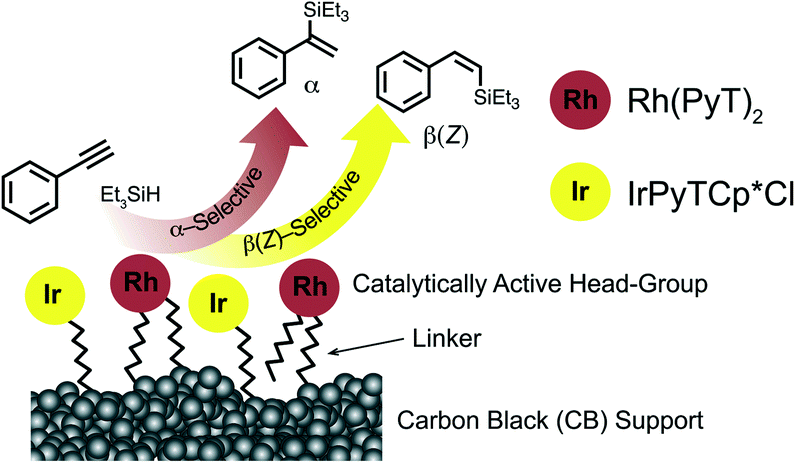 | ||
| Fig. 1 Representation of heterobimetallic (dual) hybrid catalysts. Engineered α- or β(Z)-regioselectivity in the hydrosilylation of a terminal alkyne is achieved by tailoring the composition of the hybrid catalysts in which the Rh sites are α-selective and the Ir sites are β(Z)-selective. | ||
We have recently attached Rh and Ir pyrazole–triazole (PyT) complexes to CB to give either Rh- or Ir-hybrid catalysts, which are highly efficient in either the α- or β(Z)-selective hydrosilylation of alkynes.16 Hydrosilylation of alkynes is an important industrially relevant and atom economical reaction giving access to silyl alkenes. In particular, generation of the α-isomer is challenging, as has been reported recently.17–20 Examples of α-selective homogeneous transition metal catalysts include platinum-,20 cobalt-,18,21 ruthenium,22–24 copper25 and iridium19 catalysts, with some α-selective hybrid Rh catalysts also reported.26 There is a broader range of β(Z)-selective homogeneous catalysts for hydrosilylation, which include iridium-,27 ruthenium-,28 rhodium-,29,30 palladium-31 and platinum32 catalysts. In addition, there are examples of carbon surface immobilized rhodium complexes which catalyse the hydrosilylation efficiently.33,34
Here we show that mixed heterobimetallic Rh and Ir dual hybrid catalysts with a range of tether lengths can be obtained by simultaneous immobilization of the appropriate Rh and Ir complexes on CB. We have examined these heterobimetallic hybrid catalysts in the hydrosilylation of a series of terminal and internal alkynes. For the case of terminal alkynes, the Rh and Ir sites exhibit different reactivity, producing either the α- or the β(Z) products, which can be fine-tuned by altering the composition of the layer, metal loading and reaction conditions.
Results and discussion
Syntheses of heterobimetallic hybrid catalysts
We have previously reported the syntheses of Rh- and Ir-pyrazole (PyT) complexes, bearing tethers of different lengths and different co-ligands, viz. carbonyl (CO), chloride and pentamethylcyclopentadienyl (Cp*).16 Briefly, the PyT ligands featuring a long-alkyl chain were prepared by tin-mediated Friedel–Crafts reaction35,36 of a halobenzene precursor, generation of the PyT-moiety through click-chemistry, followed by conversion to an amine. The required Rh and Ir complexes (Scheme 1a) were then synthesized by metal coordination to the ligands.16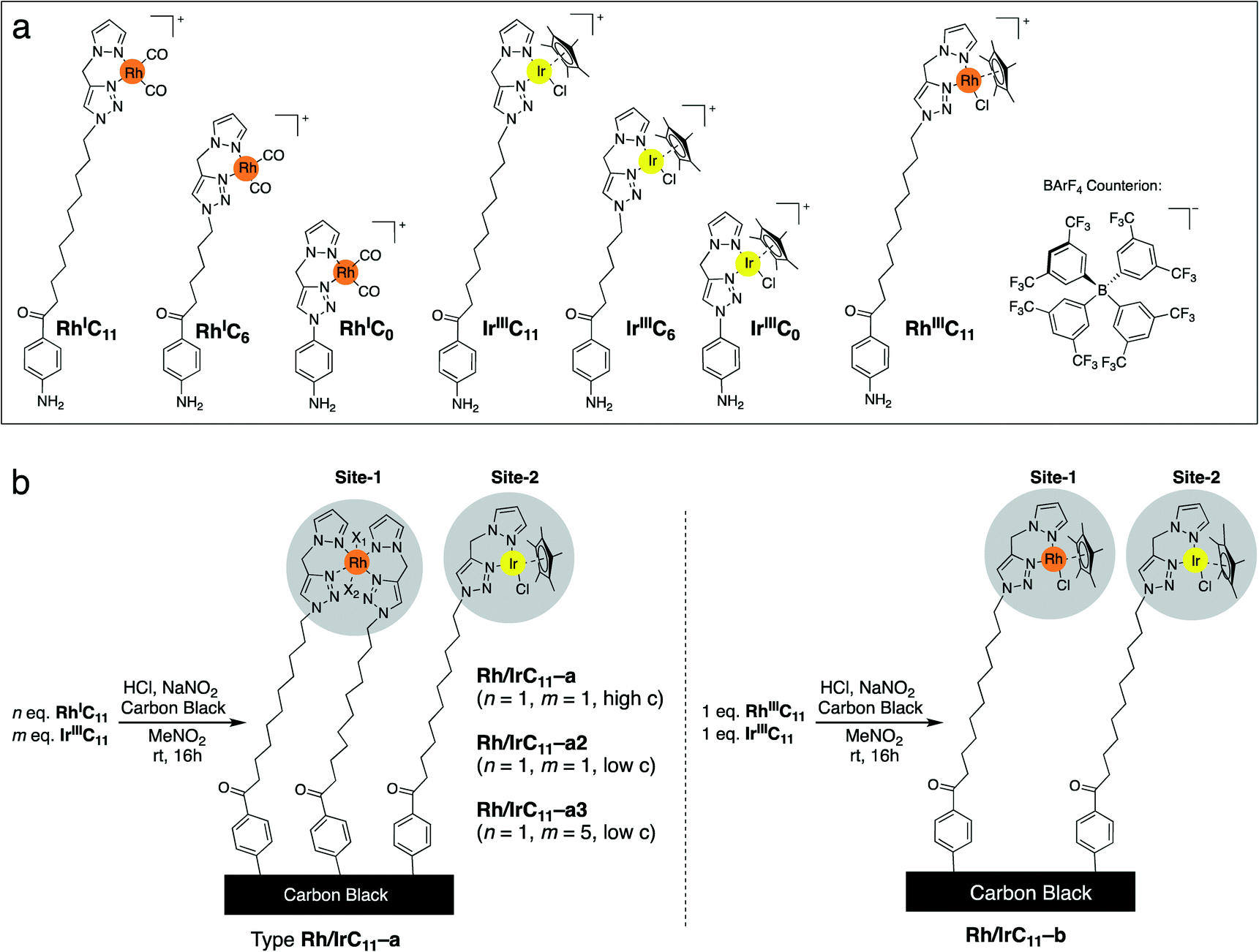 | ||
| Scheme 1 (a) The cationic Rh and Ir complexes with tethers of different alkyl chain lengths for surface immobilization. All complexes bear the BArF4 counterion. (b) Simultaneous surface immobilization of two different metal complexes with the same tether length on the carbon black support, example shown of the C11-carbon chain derivatives, to yield the heterobimetallic hybrid catalysts. The types of Rh/IrC11–a differ in overall concentration of the complexes during the grafting procedure, and the ratios of the Rh and Ir complexes. | ||
Here, we have immobilized each of the monometallic RhI- and RhIII-complexes simultaneously with IrIII-complexes of the same tether lengths on carbon black, which afforded heterobimetallic hybrid catalysts bearing Rh and Ir centres. The immobilization was conducted using the aryl diazonium strategy (Scheme 2a), generating a phenyl radical in situ from the anilines, which binds to the bulk of the CB surface.37
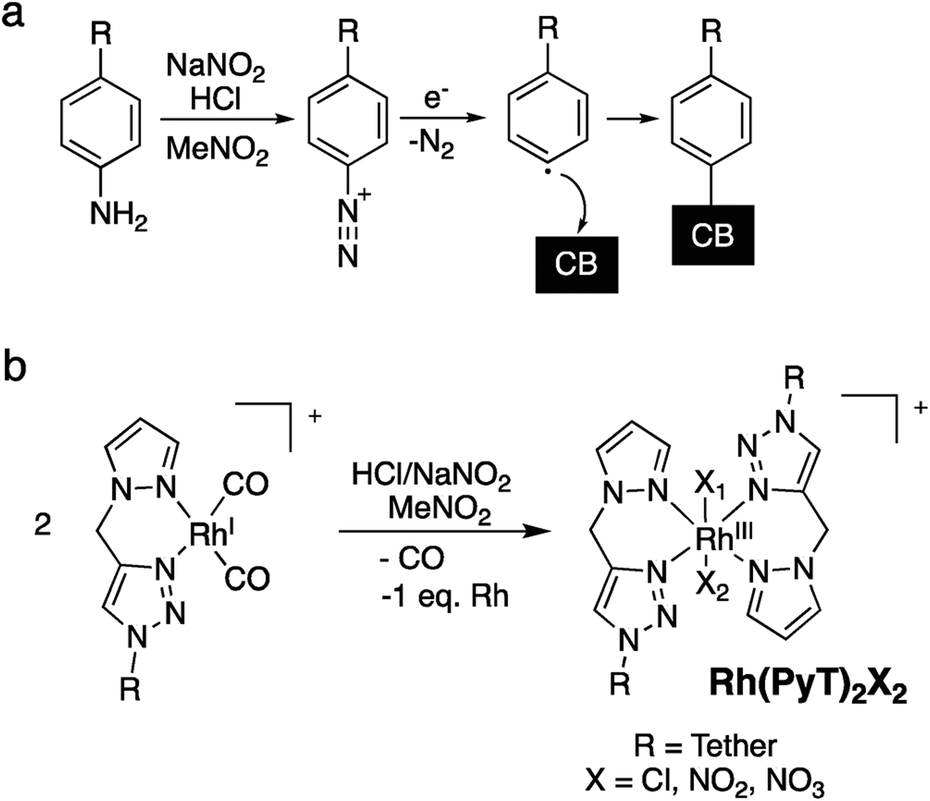 | ||
| Scheme 2 (a) Aryl diazonium surface immobilization via generation of a phenyl radical, which binds to the bulk of the carbon black solid support. (b) In situ modification of the RhI catalysts during the immobilization procedure. | ||
Structure of the catalysts
We have previously established that the herein applied RhI-complexes are chemically modified during the immobilization process, under loss of the CO co-ligands.16 The modification results in oxidation of the metal centres from RhI to RhIII and coordination of an extra PyT-ligand to the RhIII centre, with two further co-ligands present, X1 and X2 (Scheme 2b). The co-ligands X include Cl−, NO2−, and NO3−, originating from the highly reactive oxidative mixture generated from HCl and NaNO2. The newly formed surface bound RhIII(PyT)2X2 species are a mixture due to the number of different possible isomers and varying co-ligands X.16 As the modified complex bears two PyT ligands, it consequently features two tethers for possible surface binding.In the current work we initially applied the same total molar amount of a Rh and an Ir complex, in an equimolar ratio, as for the conditions we optimized previously for the synthesis of mono-metallic hybrid catalysts that feature a saturated surface layer (Scheme 1b).16 Using this methodology, we have synthesized different types of heterobimetallic hybrid catalysts (Table 1). It should be noted that in all cases the same tether length was used for the pairs of Rh and Ir species being immobilised. One set of heterobimetallic hybrid catalysts, Rh/IrCn–a, was prepared from equimolar amounts of RhICn and IrIIICn where n is the number of carbon atoms in the linker chain (11, 6 or 0).
| Hybrid catalyst | Rh precursor | Head group | Ir precursor | Head group |
|---|---|---|---|---|
| a The RhI complexes are modified during the immobilization, resulting in the Rh(PyT)2X2 head groups. b The Rh(PyT)Cp*Cl and Ir(PyT)Cp*Cl head groups remain unmodified during immobilization. | ||||
| Rh/IrC 11 –a | Rh I C 11 | Rh(PyT)2X2a | Ir III C 11 | Ir(PyT)Cp*Clb |
| Rh/IrC 6 –a | Rh I C 6 | Rh(PyT)2X2a | Ir III C 6 | Ir(PyT)Cp*Clb |
| Rh/IrC 0 –a | Rh I C 0 | Rh(PyT)2X2a | Ir III C 0 | Ir(PyT)Cp*Clb |
| Rh/IrC 11 –b | Rh III C 11 | Rh(PyT)Cp*Clb | Ir III C 11 | Ir(PyT)Cp*Clb |
The Rh/IrCn–a hybrid catalysts therefore features two different catalytically active sites – RhIII(PyT)2X2 and IrIII(PyT)Cp*Cl. Furthermore, we have prepared another set of heterobimetallic metal hybrid catalyst, Rh/IrC11–b, which is composed of a pair of Rh and Ir complexes, each with identical ligand environments, i.e. RhIII(PyT)Cp*Cl and IrIII(PyT)Cp*Cl (Scheme 1b). These hybrid catalysts were synthesized using equimolar amounts of RhIIIC11 and IrIIIC11.
Surface characterization
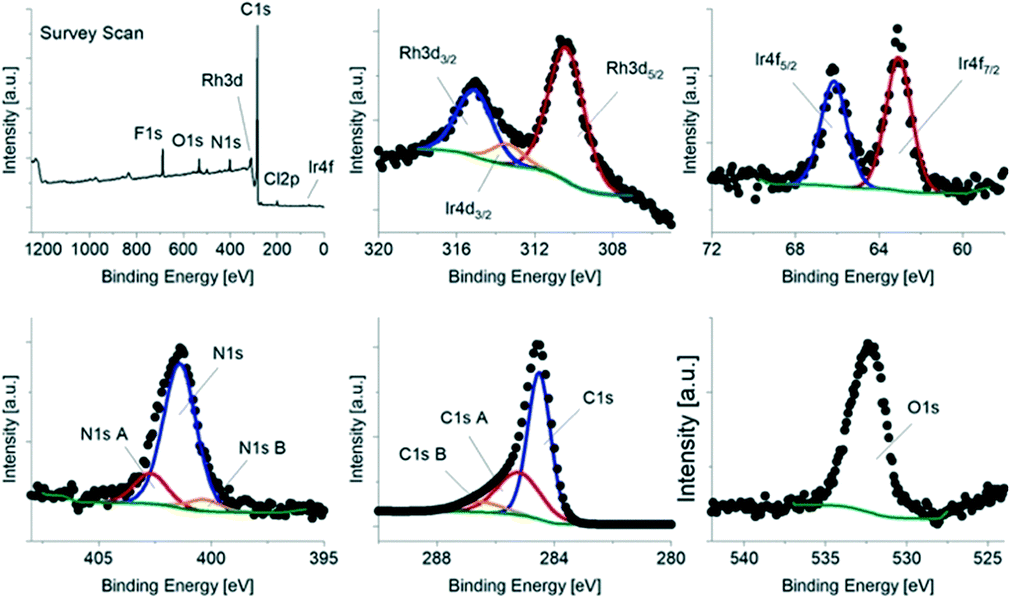 | ||
| Fig. 2 X-ray photoelectron spectra of the hybrid catalyst Rh/IrC11–a. Survey scan, narrow band scans for the Rh3d3/2/Rh3d5/2 signals, the Ir4f5/2/Ir4f7/2 signals, the N1s signal, the C1s signal and the O1s signal. | ||
The XPS data shows the presence of both Rh and Ir, as expected, as well as N, C and O. In order to calculate the relative surface composition of each sample, the narrow band scans were fitted for each element. The Ir 4d signal overlaps with the Rh 3d signals (Fig. 2), which was taken into account in the fitting. Interestingly, for Rh/IrC11–a, Rh/IrC6–a and Rh/IrC0–a, the determined concentrations of Rh (0.77 at%) are approximately one order of magnitude higher than those of Ir (0.07 at%) (Table 2), indicating that a higher amount of the Rh catalyst is immobilised on CB. This finding is consistent with our previous investigations of heterobimetallic mixed layers on CB involving the RhIC0 and IrIIIC0 precursors, where significantly higher Rh loadings were also observed on immobilization of mixed pairs of complexes.38 For Rh/IrC11–b, the ratio of Rh to Ir is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with an overall lower metal concentration of 0.36 at% and 0.12 at%, respectively.
:1, with an overall lower metal concentration of 0.36 at% and 0.12 at%, respectively.
| Hybrid Catalyst | Rh I C 11 a [mmol] | Ir III C 11 a [mmol] | at% Rh (XPS) | at% Ir (XPS) | at% Rh (EDX) | at% Ir (EDX) | Ratio Rh:Irb |
|---|---|---|---|---|---|---|---|
| a Reported amounts of precursors in mmol are relative to 1 mg CB. b The ratio was calculated from the EDX results and is rounded to whole numbers. | |||||||
| Rh/IrC 11 –a | 3.5 | 3.5 | 0.77 | 0.07 | 0.41 | 0.06 | 7:1 |
| Rh/IrC 11 –a2 | 0.35 | 0.35 | 0.39 | 0.02 | 0.09 | 0.04 | 2:1 |
| Rh/IrC 11 –a3 | 0.35 | 1.75 | 0.38 | 0.10 | 0.07 | 0.12 | 1:2 |
| Rh/IrC 6 –a | 3.5 | 3.5 | 0.76 | 0.09 | 0.40 | 0.07 | 6:1 |
| Rh/IrC 0 –a | 3.5 | 3.5 | 0.83 | 0.06 | 0.42 | 0.06 | 7:1 |
| Rh/IrC 11 –b | 3.5 | 3.5 | 0.36 | 0.12 | 0.20 | 0.23 | 1:1 |
The observed N to metal ratio (Σ (Rh + Ir)) is approximately 5:1 for Rh/IrCn–a (n = 11, 6, 0) and Rh/IrC11–b, which agrees with the expected ratio from the molecular structures of the precursor complexes (Tables S2–S4, ESI†).
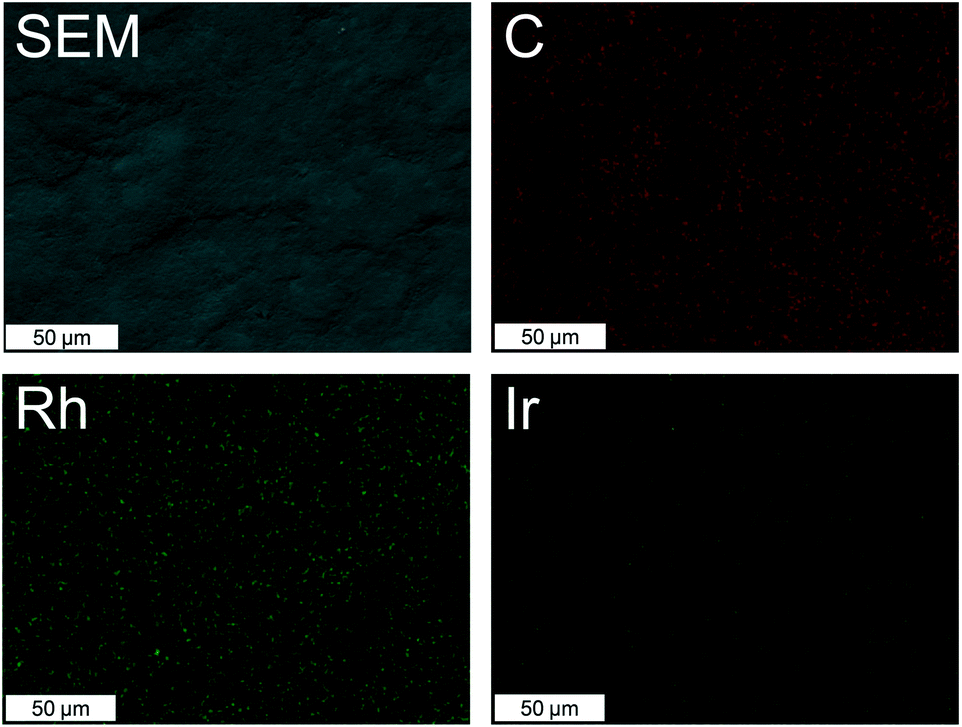 | ||
| Fig. 3 SEM image of Rh/IrC11–a and EDX maps for carbon, rhodium and iridium. | ||
For the Rh/IrCn–a hybrid catalysts the Rh:Ir ratio was 7:1, in analogy to the data obtained from XPS, confirming that significantly more Rh complexes bind to the surface than Ir. In contrast, the metal ratios for Rh/IrC11–b are 1:1, indicating that when the metal has the same ligands the extent of surface binding is comparable (Table 2).
We note that there are deviations between the EDX and XPS results, which is unsurprising and likely due to differences in the covered depth of the two different techniques. EDX provides information about the bulk of the hybrid catalyst as it covers elemental composition of the material to a micron-range depth, while XPS gives information about the composition of the outer surface and several atomic layers beneath. Based on this data, we conclude that the head group plays a key role. The total metal concentration used during the immobilization procedure matched that used previously to achieve saturation of a Rh complex on CB.16 Therefore, the heterobimetallic hybrid catalysts prepared here are also close to surface saturation, and thus the Rh and Ir complexes likely compete for surface binding.
While the possible origin of the differential Rh/Ir metal loadings have been postulated previously for similar systems,38 our new insight into the modification of the Rh(CO)2 head group during immobilization16 is essential to understanding this divergent binding behaviour. As mentioned above, the RhI complexes are converted to RhIII(PyT)2X2 during immobilization, thus they feature two surface binding groups. As such, it is likely that the RhIII(PyT)2X2 complexes in Rh/IrC11–a, Rh/IrC6–a, Rh/IrC0–a bind more efficiently to CB compared to the Ir complexes that bear only a single PyT-ligand (and thus only one surface binding group).
It would be expected that competition between the RhIII(PyT)2X2 and IrIII(PyT)Cp*Cl species binding to CB would become less important when a lower concentration of the homogeneous precursors are used, as the CB surface would not be saturated. Using more a dilute concentration of the metal complexes produced the hybrid catalyst Rh/IrC11–a2, which featured a changed ratio in favour of Ir compared to Rh/IrC11–a. The Rh:Ir ratio was 2:1 on the CB surface, as determined by EDX (Table 2). Lastly, using a five-fold excess of Ir complex, at a low overall concentration, gave Rh/IrC11–a3 which featured a reversed Rh:Ir ratio of 1:2. This highlights that to achieve a higher relative amount of Ir, an excess of the Ir precursor complex is required. Overall, these results further confirm that the different metal complexes compete for vacant sites during the binding, and that the RhIII(PyT)2X2 species generated in situ from RhIC11 binds better to the carbon support than IrIIIC11 (which features the IrIII(PyT)Cp*Cl head group).
We have illustrated several plausible binding situations for the different types of hybrid catalysts on the surface, depending on the complex concentrations during the immobilization, in Fig. 4. Rh/IrC11–a prepared using higher concentrations of RhIC11 and IrIIIC11 is shown in panel a: as the Rh head-group bears two PyT ligands, there are two possible surface binding groups, which leads to a high loading of Rh on the surface, compared to Ir. Panels b and c illustrate the binding situation when the concentration is lowered for RhIC11 and IrIIIC11, as the surface is not saturated a more even ratio of Rh and Ir can be achieved. Panel d shows Rh/IrC11–b prepared from RhIIIC11 and IrIIIC11; as these complexes feature the same ligands (MIII(PyT)Cp*Cl), both complexes compete evenly for surface binding resulting in near equivalent Rh and Ir loadings on the surface.
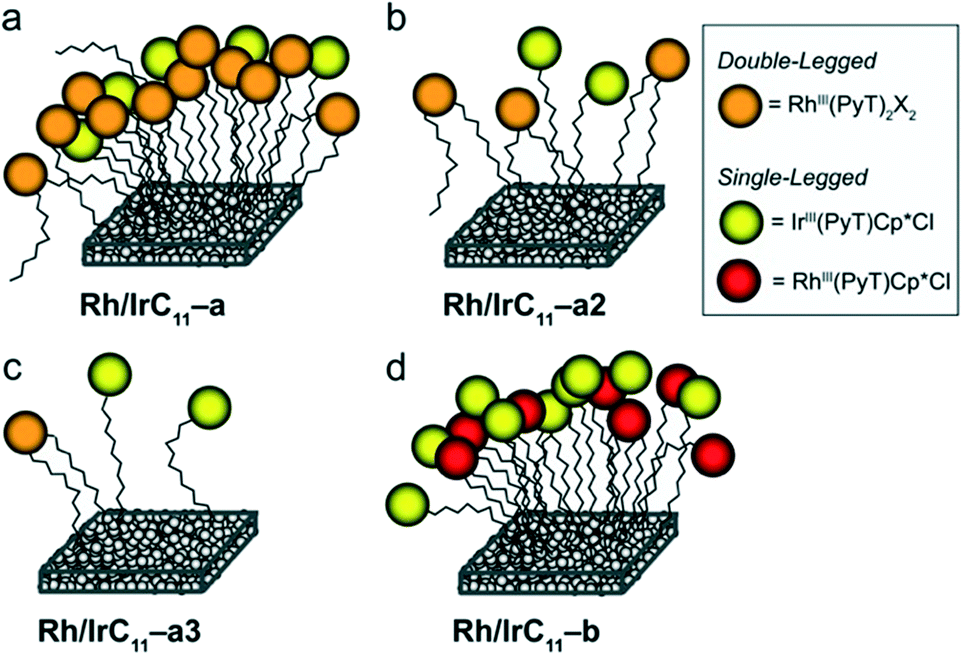 | ||
| Fig. 4 Plausible representations of different levels of surface coverages and Rh/Ir ratios of the heterobimetallic hybrid catalysts synthesized from RhIC11 and IrIIIC11 (a–c) and from RhIIIC11 and IrIIIC11 (d). For the cases of a–c, the in situ generated Rh(PyT)2 head-group is double-legged and may bind bidentate to the surface. The surface binding phenyl groups are omitted for clarity in the simplified representations. | ||
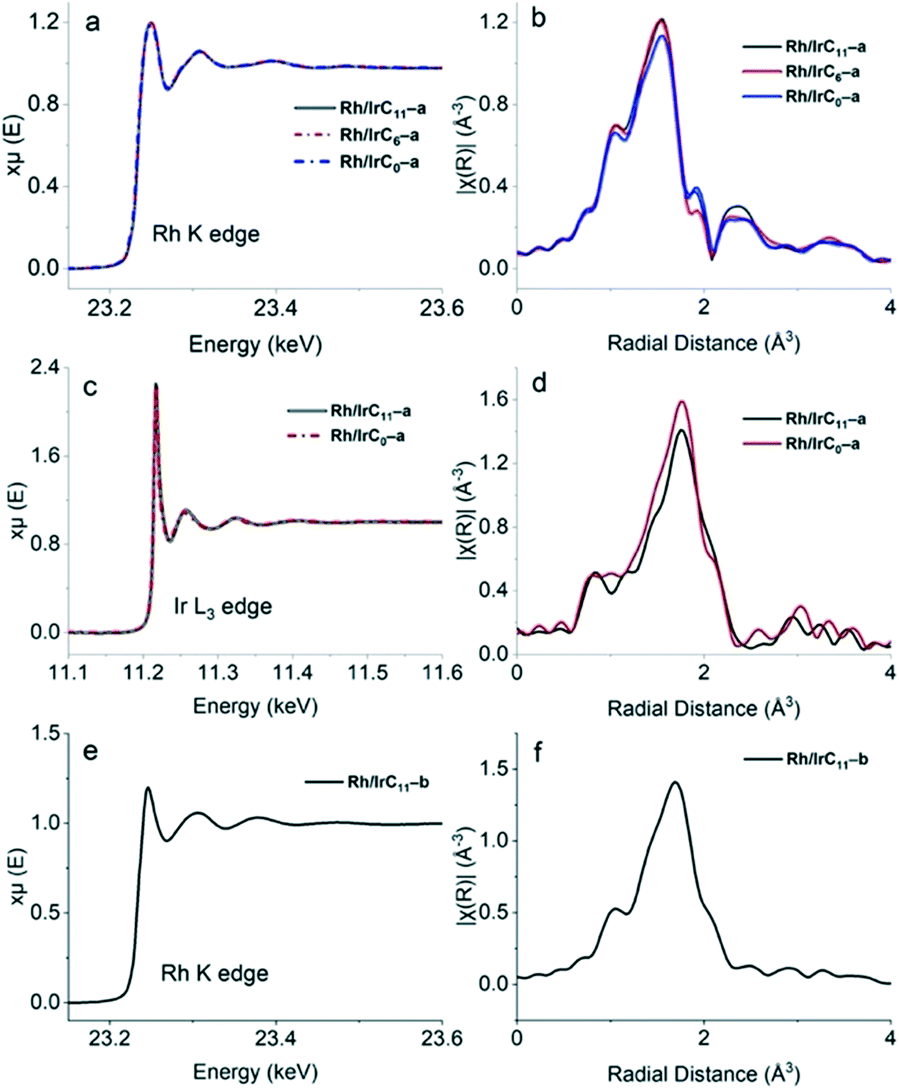 | ||
| Fig. 5 EXAFS spectra of the heterobimetallic hybrid catalysts. (a) Rh K edge spectra of the series Rh/IrCn–a (n = 11, 6, 0) and the corresponding k2 weighted radial distributions (b). (c) Ir L3 edge spectra of the series Rh/IrCn–a (n = 11, 0) and the corresponding k2 weighted radial distributions (d). (e) Rh K edge spectrum of Rh/IrC11–b and the corresponding k2 weighted radial distribution (f). | ||
The obtained XAS data further confirms the presence of both metals on the surface. Furthermore, the Rh and Ir EXAFS spectra of the hybrid catalysts featuring different tether lengths are consistent in pre-edge, edge- and post-edge regions, confirming that there are no changes in the metal environment when changing the tether length. The previously established head-group modification for the RhI complexes during immobilization, to RhIII(PyT)2X2,16 is well reflected in the current EXAFS data (Fig. 5), with significant differences in the spectra for the species before immobilization (RhICn) and after immobilization (Rh/IrCn–a).
In analogy to the monometallic hybrid catalysts,16 the energy of the Rh K edge for the hybrid catalysts Rh/IrCn–a (n = 11, 6, 0) is 23.250 keV, indicating an edge shift to slightly higher energy compared to the RhI complexes before immobilization (23.242 keV). The FEFF calculations further confirm the presence of the RhIII(PyT)2X2 head group in hybrid catalysts Rh/IrCn–a, noting that the Rh fits all contained misfits with higher R values (0.04–0.05), which is unsurprising due to the modified head-groups. In contrast, the EXAFS spectra at the Ir L3 edge (11.218 keV) for Rh/IrCn–a matches the spectra of the Ir precursors, with high quality fits (R = 0.1), confirming that the IrIII(PyT)Cp*Cl head group stays intact during immobilization.
For the Rh/IrC11–b hybrid catalyst, the obtained XAS spectrum matches that of the homogeneous complex before immobilization, with a good fit observed (R = 0.2). For example, in both the homogeneous precursors and the hybrid catalyst the Rh K edge is at 23.245 keV.
This further suggests that the electronic structure of both metals on the surface remains unaltered when immobilized simultaneously together with another metal species, either Rh or Ir, and that the electronic structure of the metals on the surface is also independent of the tether length.
Overall, the XAS data confirms that during immobilization the RhICn precursor is modified to produce the new head group RhIII(PyT)2X2. In contrast, the RhIIICn and IrIIICn precursors remain intact during immobilization, with the MIII(PyT)Cp*Cl head group still intact after immobilization. This is consistent with that reported previously for monometallic hybrid catalysts.16 For all the heterobimetallic hybrid catalysts, Rh/IrCn–a and Rh/IrC11–b, the obtained data is comparable to the analogous monometallic hybrid catalysts,16 suggesting that the presence of another metal does not affect the electronic structure or coordination environment of the metal centre.
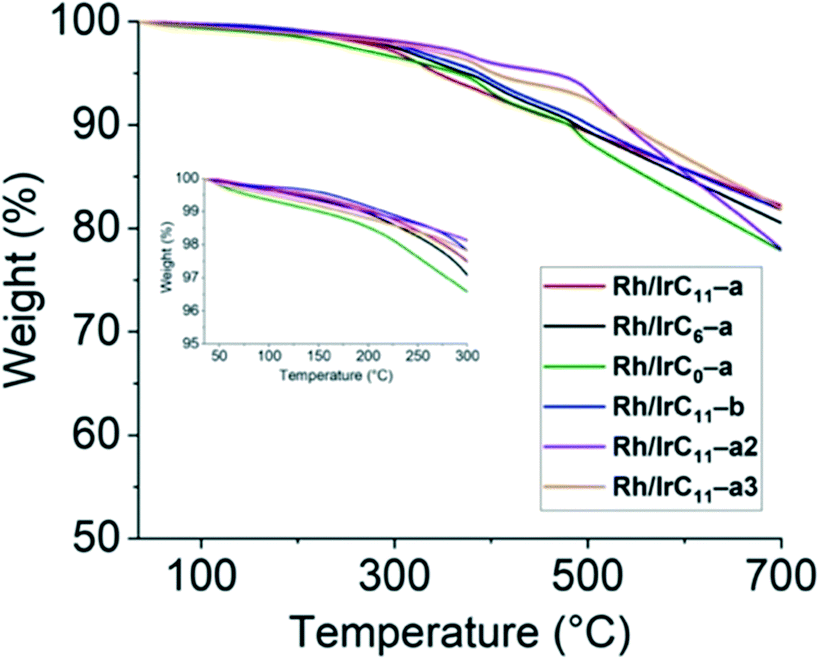 | ||
| Fig. 6 TGA curves of the different hybrid catalysts over a temperature range of 35 to 700 °C. The inset shows the temperature range of 35 to 300 °C. | ||
The curves are near featureless up to approximately 300 °C, with the short tether derivative Rh/IrC0–a showing a slightly larger weight loss compared to all other derivatives. The same was already observed for the monometallic Rh analogue.16 From 300 °C onwards, a gradual decrease in the weight of the samples is observed. There is no significant difference to the TGA curves of the monometallic analogues.16 The measurements confirm the thermostability of the hybrid catalysts at the operating range of the hydrosilylation reactions (75–120 °C), which is well below the observed decomposition onset at ca. 300 °C.
Application in catalysis – hydrosilylation of alkynes
We have tested the performance of the heterobimetallic hybrid catalysts in the hydrosilylation of two terminal alkynes, phenylacetylene and 1,4-diethynylbenzene, and two internal alkynes, diphenylacetylene and 1,4-diphenylbutadiyne.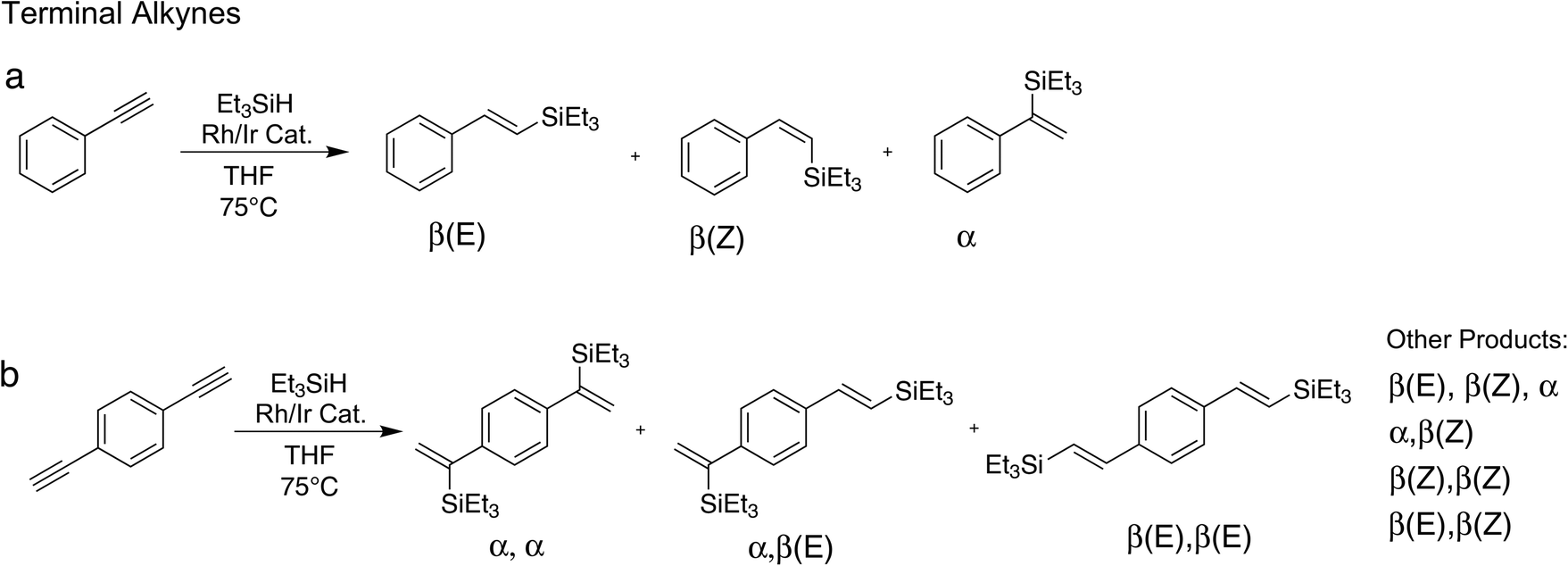 | ||
| Scheme 3 Different model reactions for the hydrosilylation of terminal alkynes using the heterobimetallic hybrid catalysts. The reactions generate different isomers of silylated alkenes. Substrates: (a) phenylacetylene; (b) 1,4-diethynylbenzene. | ||
| Hybrid Catalyst | Time [min] | Solvent | T [°C] | β(E)/β(Z)/α | Conversion [%] |
|---|---|---|---|---|---|
| a Monometallic Rh hybrid catalyst as control reaction. | |||||
| Rh/IrC 11 –a | 60 | THF | 75 | 10/3/87 | 99 |
| Rh/IrC 6 –a | 60 | THF | 75 | 12/7/82 | 90 |
| Rh/IrC 0 –a | 60 | THF | 75 | 9/3/87 | 99 |
| Rh/IrC 11 –a | 10 | Toluene-d8 | 120 | 15/23/62 | 20 |
| Rh/IrC 11 –a | 60 | Toluene-d8 | 120 | 20/14/66 | 100 |
| Rh/IrC 11 –a2 | 60 | THF | 75 | 6/39/55 | 54 |
| Rh/IrC 11 –a3 | 60 | THF | 75 | 3/93/4 | 51 |
| RhC 11 CB a | 10 | Toluene-d8 | 120 | 13/3/84 | 82 |
| Rh/IrC 11 –b | 60 | THF | 75 | 1/95/4 | 80 |
Previous work examining the reactivity of monometallic hybrid catalysts found that the RhIII(PyT)2X2 head group is α-selective, whereas the IrIII(PyT)Cp*Cl and RhIII(PyT)Cp*Cl head groups are β(Z)-selective. As such, the products observed for the Rh/IrCn–a (n = 11, 6, 0) hybrid catalysts suggest that the RhIII(PyT)2X2 head group is dominant, whereas for Rh/IrC11–b both head groups are β(Z)-selective. As established above, the relative Rh:Ir ratio in Rh/IrCn–a (n = 11, 6, 0) is 7:1. While the Rh content is significantly higher compared to Ir in the systems under investigation here, it is surprising that the outcome of the reaction matches that of the monometallic Rh(III) analogues16 with no increase in formation of the β(Z)-isomer (Fig. 7a) which would be expected due to the presence of IrIII(PyT)Cp*Cl.
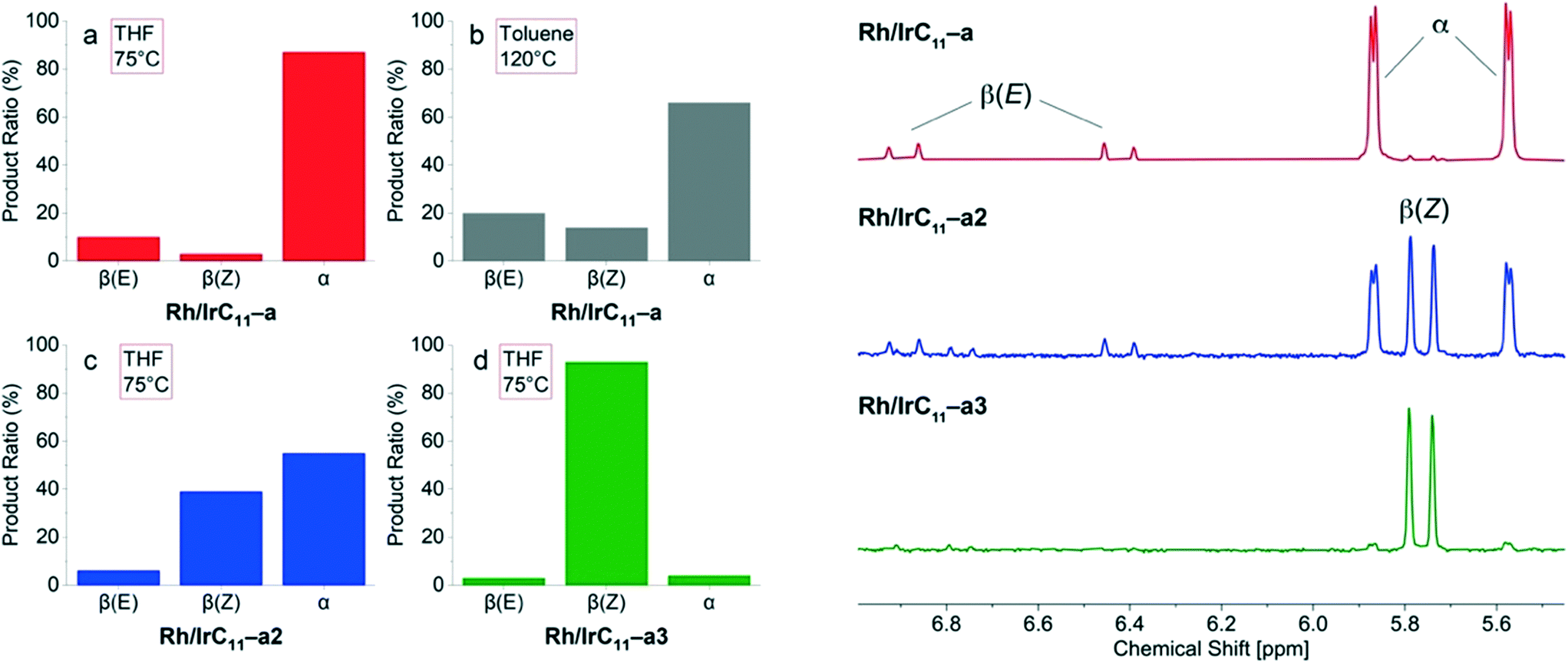 | ||
| Fig. 7 Product distributions for the hydrosilylation of phenylacetylene in THF and toluene-d8 for the heterobimetallic hybrid catalysts of type Rh/IrC11–a, which differ in Rh/Ir ratios (left; a–d). Selected ranges of the 1H NMR spectra of the corresponding product mixture from the reactions in THF, using the heterobimetallic hybrid catalysts (a) Rh/IrC11–a, (c) Rh/IrC11–a2 and (d) Rh/IrC11–a3 (right). | ||
We next examined how increasing the reaction temperature affects selectivity using this catalyst, using the higher boiling solvent toluene-d8. Interestingly, at a temperature of 120 °C, the distribution of isomers formed when using Rh/IrC11–a changes, with more of the β(Z)-isomer formed (14%), Fig. 7b. A control experiment, using the previously reported monometallic RhC11CB hybrid catalyst,16 showed that low quantities (3%) of the β(Z)-isomer forms in toluene-d8 at 120 °C. As such, the increased formation of the β(Z)-isomer when using the hybrid catalyst Rh/IrC11–a likely arises from greater activation of the IrIII(PyT)Cp*Cl head group at higher temperatures. In the next step we tested Rh/IrC11–a2 (Fig. 7c) and Rh/IrC11–a3 (Fig. 7d) as catalysts for the reaction, each of which have lower overall metal loadings and different Rh:Ir ratios, 2:1 for Rh/IrC11–a2 and 1:2 for Rh/IrC11–a3 respectively.
Interestingly, for the reaction catalysed by Rh/IrC11–a2, the product ratio changed in favour of the β(Z)-isomer (39%), however large quantities of the α-isomer (55%) still formed (Fig. 7c). In the case of Rh/IrC11–a3, the catalyst is highly β(Z)-selective (93%, Fig. 7d). The determined relative ratios of the products for the different catalysts and selected ranges of the corresponding 1H NMR spectra of the reactions conducted in THF are illustrated in Fig. 7, and S17 (ESI†).
These results show that is possible to engineer the regioselectivity of the hydrosilylation reaction by adjusting the composition of the heterobimetallic hybrid catalysts. In addition, it allows the selective activation of the catalyst head groups through temperature control, i.e. in the densely packed layer of Rh/IrC11–a the Rh sites are always active while the Ir sites are inactivated at lower temperature, but become active at higher temperature.
On using the heterobimetallic Rh/IrC11–b as catalyst, which is comprised of both metals in the same ligand environment (MIII(PyT)Cp*Cl), the observed outcome is that of high β(Z)-selectivity (95%), as was expected as both immobilized catalysts are β(Z)-selective.
It should be noted that the recyclability of Rh (ref. 5 and 16) and Ir (ref. 39) based hybrid catalysts, prepared using the same grafting procedure and solid support utilised here, has been demonstrated previously.
| Catalyst | Et3SiH [eq.] | Solvent | T [°C] | Time [h] | α | β(Z) | β(E) | α,α | α,β(E) | β(Z), β(Z) | Conversion [%] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Conditions: 2 mol% catalyst. a Conversions and compositions of the crude reaction mixtures were determined by 1H NMR spectroscopy through integration of the proton resonances of products and starting material, unless otherwise stated. b Conversions and compositions of the crude reaction mixtures were determined by GC-MS (EI) and percentages are based on the total ion current (TIC). | |||||||||||
| Rh/IrC 11 –a | 3.6 | THF | 75 | 15 | — | — | — | 84 | 16 | — | 100 |
| Rh/IrC 11 –a | 3.6 | THF | 75 | 24 | — | — | — | 80 | 20 | — | 100 |
| Rh/IrC 11 –a | 3.6 | THF | 75 | 15 | — | — | — | 59b | 22b | — | 100b |
| Rh/IrC 11 –b | 3.6 | THF | 75 | 15 | — | 38 | — | — | 62 | 92 | |
The close proximity of the signals to each other, and the presence of other signals, hampered unambiguous differentiation between the formed products thus we employed GC-MS for further analysis of the mixture (Fig. S19 and S20†). The GC-MS results, while qualitative, support the NMR data, showing a composition of α,α-isomer (59%), α,β(E)-isomer (22%) and small amounts of other components (19% in total). Among those, a small amount of triple hydrosilylated product was identified. Noteworthy, double hydrosilylation of a single alkyne has only recently been achieved through a copper catalysed reaction.40 The unexpected triple hydrosilylation of 1,4-diethynylbenzene highlights the high catalytic activity of the herein studied hybrid catalyst. α,α-Selectivity has been reported for the same substrate and cobalt based catalysts in solution.18,41,42
In contrast, the Rh/IrC11–b produces with very good selectivity the β(Z)-products. After a reaction time of 15 h, the reaction reached 92% completion with a ratio β(Z) to β(Z)β(Z)-isomers of 62:38 and only negligible amounts of other isomers.
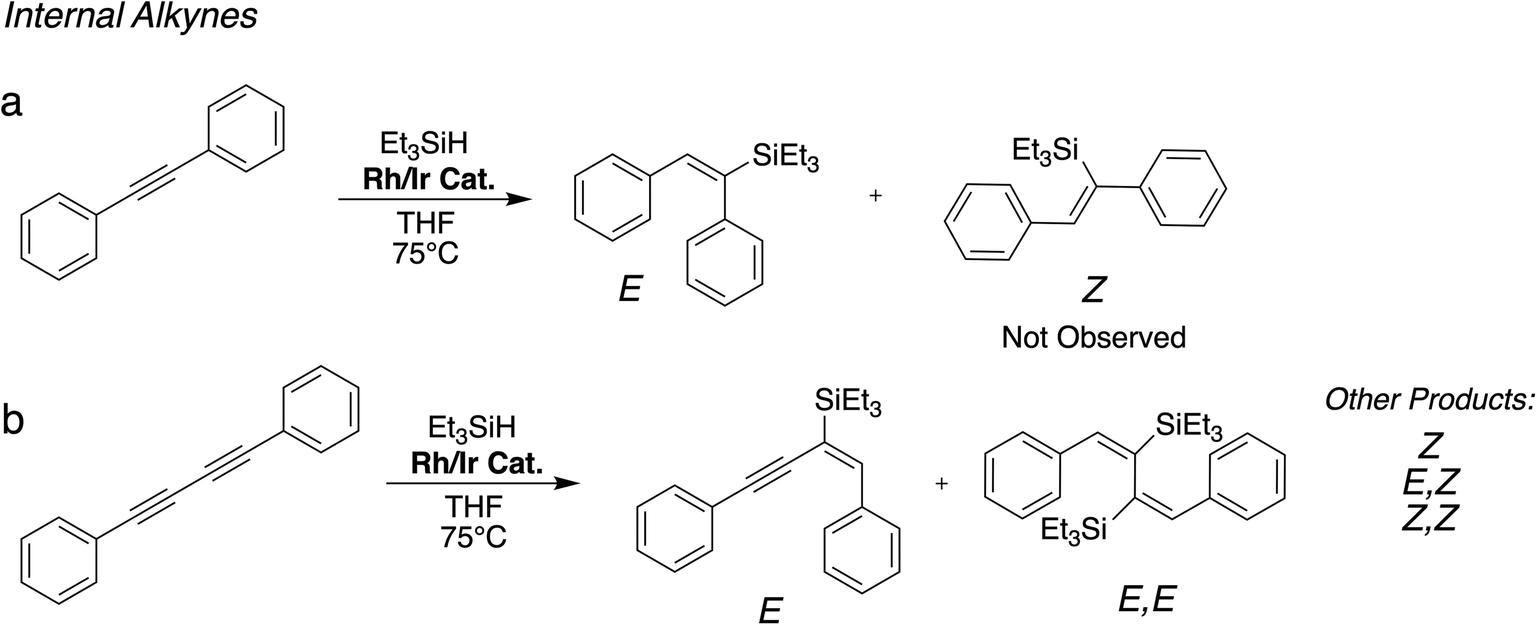 | ||
| Scheme 4 Model reactions for the hydrosilylation of internal alkynes using the heterobimetallic hybrid catalysts. Substrates: (a) diphenylacetylene; (b) 1,4-diphenylbutadiyne. | ||
:9 ratio.
| Catalyst | Et3SiH [eq.] | Solvent | T [°C] | Time [h] | E | E,E | Conversion [%] |
|---|---|---|---|---|---|---|---|
| Conditions: 2 mol% catalyst. a Compositions of the crude reaction mixtures were determined by 1H NMR spectroscopy through integration of the vinyl proton resonances of E and E,E products, unless otherwise stated. b Conversions of the crude reaction mixtures were determined by GC-MS (EI) and percentages are based on the total ion current (TIC). | |||||||
| Rh/IrC 11 –a | 3.6 | THF | 75 | 15 | 70 | 30 | 95b |
| Rh/IrC 11 –a | 3.6 | THF | 75 | 24 | 76 | 24 | ∼99a |
| Rh/IrC 11 –a | 3.6 | Toluene-d8 | 120 | 1 | 48 | 52 | 100b |
| Rh/IrC 11 –a | 3.6 | Toluene-d8 | 120 | 24 | — | 100 | 100b |
Reaction over 15 h using 3.8 eq. of Et3SiH resulted in near complete consumption of 1,4-diphenylbutadiyne and the major product formed was in both cases the E isomer, with minor amounts of E,E isomer also present (see ESI† for additional details and other minor products observed by GCMS). A similar product mixture was also obtained after heating for 24 hours.
Significantly improved conversion to the E,E product was obtained when using an elevated temperature of 120 °C in toluene-d8, with a E to E,E ratio of 48:52 obtained after 1 hour and no starting material remained, as determined by GC-MS analysis (Fig. S24 and S25, ESI†). Heating for 24 hours resulted in complete conversion of the E isomer to the E,E isomer, which became the major product, along with a mixture of other by-products (Fig. S26 and S27, ESI†). Regarding both the internal alkynes, the outcome of the reaction is directed by the steric bulk of the catalyst, resulting in formation of the less congested E-isomers as major products, which is agreement with previous work involving the studied substrates.5,43,44 Hydrosilylation of 1,4-diphenylbutadiyne has been previously achieved with supported Pt catalysts43 and Pt catalysts in solution.44 Here we show that this reaction is as well possible the Rh/Ir hybrid catalysts under investigation, with good conversion and selectivity.
Conclusions
In conclusion, we have prepared a number of heterobimetallic Rh and Ir hybrid catalysts by immobilization of Rh and Ir precursors on carbon black. The active metal catalysts are bound by alkyl tethers of varying chain lengths to the carbon black support. Densely packed mixed layers were obtained when immobilizing equimolar amounts of Rh and Ir complexes. The layers are enriched in Rh, which originates from more efficient binding of the Rh complexes due to the two surface bindings sites present in the RhIII(PyT)2X2 head group. In addition, we have prepared less densely packed layers by lowering the amount of the homogeneous complexes present during the surface immobilization procedure. We have characterised the catalysts using a suite of spectroscopic techniques, including XPS, EDX and XAS. The synchrotron-based XAS data showed no evidence of cooperative effects for the surface bound Rh and Ir complexes. This holds true for both long and short tether examples, thus indicating that the change in metal–metal distance does not alter the electronic structure of the systems under investigation.We applied the hybrid catalysts in the hydrosilylation of terminal and internal alkynes. For terminal alkynes, the catalytically active sites, Rh and Ir, exhibit different reactivity through ligand control, being either α- or β(Z)-selective. The outcome of the reaction can be controlled by adjusting the ratios of the metal complexes on the surface. The densely packed layers enriched in Rh are α-selective, whereas by tuning the hybrid catalyst composition to be Ir enriched allowed selective β(Z)-isomer formation. The internal alkynes are activated by the RhIII(PyT)2X2 head-group to selectively produce the E-isomers. This difference in selectivity of the catalysts tested here highlights that controlling the hybrid catalyst composition can be used to engineer regioselectivity.
Author contributions
M. R. synthesized the hybrid catalysts, characterized the hybrid catalysts by XAS, EDX, TGA and conducted the catalysis experiments. S. T. K. and J. E. D. characterized the surfaces by XAS. V. R. G, J. L., S. G, and J. J. G. characterized the surfaces by XPS. M. R. and B. A. M. conceived the experiments. All authors discussed the data, and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from The University of Sydney and Macquarie University is gratefully acknowledged. We gratefully acknowledge beamtime at the Australian Synchrotron (Projects 14297 and 15780) and thank Dr Peter Kappen and Dr Jessica Hamilton for scientific and technical assistance with X-ray absorption spectroscopy (XAS) measurements at the XAS beamline. J. J. G. acknowledges the financial support from the Australian Research Council Discovery Projects (DP210102698). We acknowledge the scientific and technical assistance of Sydney Analytical at The University of Sydney, the NMR and chemical analysis facilities at Macquarie University and the solid state and elemental analysis unit at the Mark Wainwright Analytical Centre, UNSW. We thank Dr Nicholas Proschogo for assisting with mass spectrometric measurements. We thank Dr Timothy Murphy for assisting with SEM/ EDX measurements.References
- A. Nodzewska, A. Wadolowska and M. Watkinson, Coord. Chem. Rev., 2019, 382, 181–216 CrossRef CAS.
- R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS.
- A. A. Tregubov, K. Q. Vuong, E. Luais, J. J. Gooding and B. A. Messerle, J. Am. Chem. Soc., 2013, 135, 16429–16437 CrossRef CAS.
- C. M. Wong, D. B. Walker, A. H. Soeriyadi, J. J. Gooding and B. A. Messerle, Chem. Sci., 2016, 7, 1996–2004 RSC.
- A. A. Tregubov, D. B. Walker, K. Q. Vuong, J. J. Gooding and B. A. Messerle, Dalton Trans., 2015, 44, 7917–7926 RSC.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Nat. Commun., 2017, 8, 14675 CrossRef.
- X. S. Zhao, X. Y. Bao, W. Guo and F. Y. Lee, Mater. Today, 2006, 9, 32–39 CrossRef CAS.
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- M. Sawamura, M. Sudoh and Y. Ito, J. Am. Chem. Soc., 1996, 118, 3309–3310 CrossRef CAS.
- I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2006, 45, 1952–1956 CrossRef CAS PubMed.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef CAS.
- D. Wang, R. Malmberg, I. Pernik, S. K. K. Prasad, M. Roemer, K. Venkatesan, T. W. Schmidt, S. T. Keaveney and B. A. Messerle, Chem. Sci., 2020, 11, 6256–6267 RSC.
- L. Chen, G. Chen, C.-F. Leung, S.-M. Yiu, C.-C. Ko, E. Anxolabéhère-Mallart, M. Robert and T.-C. Lau, ACS Catal., 2015, 5, 356–364 CrossRef CAS.
- M. Melchionna and P. Fornasiero, Chem, 2021, 7, 834–835 CAS.
- M. Roemer, V. R. Gonçales, S. T. Keaveney, I. Pernik, J. Lian, J. Downes, J. J. Gooding and B. Messerle, Catal. Sci. Technol., 2021, 11, 1888–1898 RSC.
- Z. Cheng, J. Guo, Y. Sun, Y. Zheng, Z. Zhou and Z. Lu, Angew. Chem., Int. Ed., 2021, 60, 22454–22460 CrossRef CAS.
- D. Wang, Y. Lai, P. Wang, X. Leng, J. Xiao and L. Deng, J. Am. Chem. Soc., 2021, 143, 12847–12856 CrossRef CAS.
- X. Xie, X. Zhang, W. Gao, C. Meng, X. Wang and S. Ding, Commun. Chem., 2019, 2, 101 CrossRef.
- M. A. Rivero-Crespo, A. Leyva-Perez and A. Corma, Chem. – Eur. J., 2017, 23, 1702–1708 CrossRef CAS.
- J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835–10838 CrossRef CAS PubMed.
- Y. Kawanami, Y. Sonoda, T. Mori and K. Yamamoto, Org. Lett., 2002, 4, 2825–2827 CrossRef CAS.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2001, 123, 12726–12727 CrossRef CAS PubMed.
- B. M. Trost and Z. T. Ball, J. Am. Chem. Soc., 2005, 127, 17644–17655 CrossRef CAS PubMed.
- P. Wang, X.-L. Yeo and T.-P. Loh, J. Am. Chem. Soc., 2011, 133, 1254–1256 CrossRef.
- J. Tian, D. Yang, J. Wen, A. S. Filatov, Y. Liu, A. Lei and X. M. Lin, Nanoscale, 2018, 10, 1047–1055 RSC.
- R. S. Tanke and R. H. Crabtree, J. Am. Chem. Soc., 1990, 112, 7984–7989 CrossRef CAS.
- Y. Na and S. Chang, Org. Lett., 2000, 2, 1887–1889 CrossRef CAS.
- J. W. Faller and D. G. D'Alliessi, Organometallics, 2002, 21, 1743–1746 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2008, 27, 224–234 CrossRef.
- A. Mori, E. Takahisa, Y. Yamamura, T. Kato, A. P. Mudalige, H. Kajiro, K. Hirabayashi, Y. Nishihara and T. Hiyama, Organometallics, 2004, 23, 1755–1765 CrossRef CAS.
- L. N. Lewis, K. G. Sy, G. L. Bryant and P. E. Donahue, Organometallics, 1991, 10, 3750–3759 CrossRef CAS.
- B. Sánchez-Page, M. V. Jiménez, J. J. Pérez-Torrente, V. Passarelli, J. Blasco, G. Subias, M. Granda and P. Álvarez, ACS Appl. Nano Mater., 2020, 3, 1640–1655 CrossRef.
- B. Sánchez-Page, J. Munarriz, M. V. Jiménez, J. J. Pérez-Torrente, J. Blasco, G. Subias, V. Passarelli and P. Álvarez, ACS Catal., 2020, 10, 13334–13351 CrossRef.
- M. Roemer, S. T. Keaveney and N. Proschogo, J. Org. Chem., 2021, 86, 9007–9022 CrossRef CAS PubMed.
- M. Roemer, B. Donnadieu and C. A. Nijhuis, Eur. J. Inorg. Chem., 2016, 1314–1318 CrossRef CAS.
- G. Liu, M. Chockalingham, S. M. Khor, A. L. Gui and J. J. Gooding, Electroanalysis, 2010, 22, 918–926 CrossRef CAS.
- S. C. Binding, I. Pernik, V. R. Gonçales, C. M. Wong, R. F. Webster, S. Cheong, R. D. Tilley, A. E. Garcia-Bennett, J. J. Gooding and B. A. Messerle, Organometallics, 2019, 38, 780–787 CrossRef CAS.
- C. M. Wong, R. T. McBurney, S. C. Binding, M. B. Peterson, V. R. Gonçales, J. J. Gooding and B. A. Messerle, Green Chem., 2017, 19, 3142–3151 RSC.
- H. Yamagishi, J. Shimokawa and H. Yorimitsu, Synlett, 2019, 30, 1551–1554 CrossRef CAS.
- S. Zhang, J. J. Ibrahim and Y. Yang, Org. Lett., 2018, 20, 6265–6269 CrossRef CAS.
- Z. Zong, Q. Yu, N. Sun, B. Hu, Z. Shen, X. Hu and L. Jin, Org. Lett., 2019, 21, 5767–5772 CrossRef CAS.
- F. Alonso, R. Buitrago, Y. Moglie, A. Sepúlveda-Escribano and M. Yus, Organometallics, 2012, 31, 2336–2342 CrossRef CAS.
- J. Walkowiak, K. Salamon, A. Franczyk, K. Stefanowska, J. Szyling and I. Kownacki, J. Org. Chem., 2019, 84, 2358–2365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available Experimental details, spectral and catalysis data. See DOI: 10.1039/d1cy01804c |
| This journal is © The Royal Society of Chemistry 2022 |