
Carbon-supported WOx–Ru-based catalysts for the selective hydrogenolysis of glycerol to 1,2-propanediol†
Alessandro
Bellè
a,
Kohei
Kusada
 ab,
Hiroshi
Kitagawa
b,
Alvise
Perosa
a,
Lidia
Castoldi
c,
Daniele
Polidoro
a and
Maurizio
Selva
*a
ab,
Hiroshi
Kitagawa
b,
Alvise
Perosa
a,
Lidia
Castoldi
c,
Daniele
Polidoro
a and
Maurizio
Selva
*a
aDepartment of Molecular Sciences and Nanosystems, Scientific Campus, Ca’ Foscari University of Venice, Via Torino, 155 – Venezia Mestre, Italy. E-mail: selva@unive.it
bGraduate School of Science, Kyoto University Yoshida-honmachi, Sakyo-ku, Kyoto, 606-8501, Japan
cDepartment of Energy, Milan Polytechnic, Campus Bovisa – Via Lambruschini, 4a – 20156 Milano, Italy
First published on 9th November 2021
Abstract
New C-supported bimetallic Ru–WOx catalysts, prepared by co-impregnation of RuCl3 and Na2WO4, proved highly efficient for the liquid-phase hydrogenolysis of aqueous glycerol into 1,2-propanediol (1,2-PDO). The tuning of the catalyst composition and major reaction parameters, specifically operating at 150 °C, 5 bar H2, and Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W = 4:1 mol/mol, allowed conversion of glycerol and 1,2-PDO selectivity of 73≥99% and 88–98%, respectively, with a carbon loss of <5%. Ru–WOx/C offered a steady performance for up to 7 subsequent recycles during which leaching of Ru was negligible, while loss of W decreased from an initial 5 wt% (1st run) to 0.1 wt% after 5 runs. The catalyst characterization, in particular EDX analysis and high-resolution TEM images, confirmed a uniform dispersion of Ru and W on the C surface with the presence of small Ru nanoparticles (below 2 nm) and randomly aggregated dots which could be ascribed to WOx clusters of size below 100 nm. Based on both the Brønsted and the Lewis acidity of WOx species, a reaction mechanism was proposed through an initial dehydration of glycerol followed by a Ru-catalysed hydrogenation process.
:W = 4:1 mol/mol, allowed conversion of glycerol and 1,2-PDO selectivity of 73≥99% and 88–98%, respectively, with a carbon loss of <5%. Ru–WOx/C offered a steady performance for up to 7 subsequent recycles during which leaching of Ru was negligible, while loss of W decreased from an initial 5 wt% (1st run) to 0.1 wt% after 5 runs. The catalyst characterization, in particular EDX analysis and high-resolution TEM images, confirmed a uniform dispersion of Ru and W on the C surface with the presence of small Ru nanoparticles (below 2 nm) and randomly aggregated dots which could be ascribed to WOx clusters of size below 100 nm. Based on both the Brønsted and the Lewis acidity of WOx species, a reaction mechanism was proposed through an initial dehydration of glycerol followed by a Ru-catalysed hydrogenation process.
Introduction
The majority of glycerol available in the current market, also called native glycerol, comes from renewable sources.1 Thanks to its non-toxicity, physicochemical properties and flexible reactivity, native glycerol is one of the top bio-based platform chemicals with literally hundreds of applications and dozens of review articles published every year on its chemical and biological upgrading.2,3 Among the most attractive reactions, the catalytic hydrogenolysis of glycerol plays a preeminent role in the synthesis of high value-added commodity chemicals as 1,2-propanediols (both 1,2- and 1,3-propanediol isomers: 1,2- and 1,3-PDO) which are excellent solvents, intermediates, and monomers.4,5This subject, with particular reference to 1,2-PDO, has been exhaustively reviewed in recent papers where the most used catalysts based on noble and other transition metals have been described focusing on the critical issue of the process, i.e., the diol selectivity.6–8Scheme 1 highlights the three pathways (1–3) of dehydration–hydrogenation, dehydrogenation–dehydration–hydrogenation, and direct hydrogenolysis which are generally accepted for the conversion of glycerol to both 1,2- and 1,3-PDO, along with some major side reactions (in red) including further dehydration–hydrogenation and C–O/C–C bond cleavage processes which bring about the formation of a range of liquid derivatives (primary and secondary alcohols) and gaseous products (mostly CO and CH4).
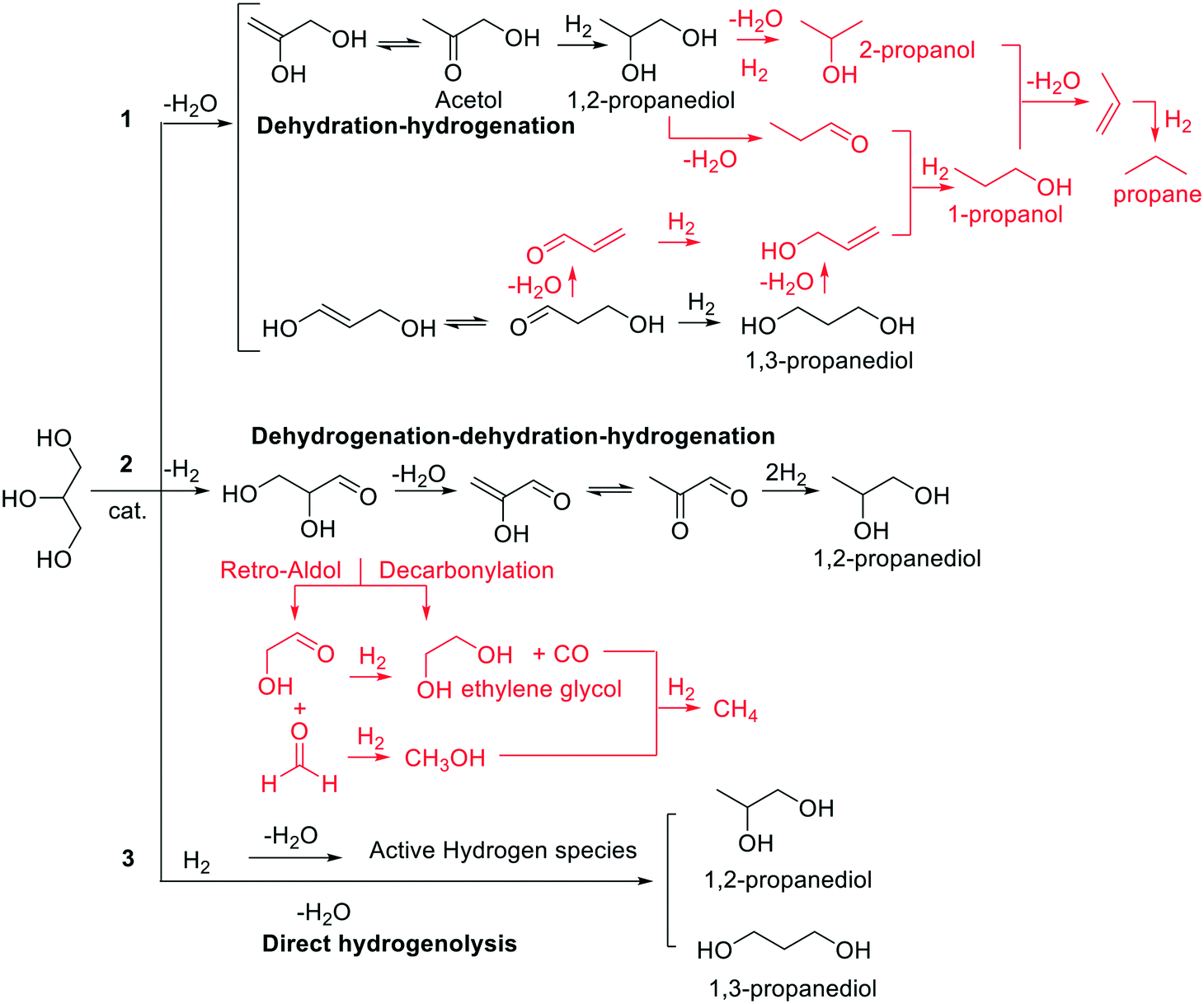 | ||
| Scheme 1 Major pathways (1–3) for the hydrogenolysis of glycerol to 1,2- and 1,3-PDO, including the formation of light liquids and gaseous side-products (red insets). | ||
Typical catalysts for the hydrogenolysis of glycerol are therefore heterogeneous bifunctional systems composed of a metal or multimetallic component acting as an oxidation–reduction functionality for the activation of hydrogen, and an acidic or basic support providing reactive sites for the removal of hydroxyl groups (dehydration processes). The benchmark system was copper chromite (Cu2Cr2O5) by which 1,2-propanediol was obtained in yields >70% under comparatively mild conditions (200 °C, 14 bar H2).9 Notwithstanding the good results, the presence of the highly toxic chromium posed environmental and safety concerns. A variety of alternatives have been proposed using Cu-, Ni- and Co-based catalysts or even their bimetallic combinations, supported on several solids including SiO2, MgO, ZnO, Al2O3, and others.6,7,10 These studies often reported a satisfactory 1,2-PDO selectivity (85–90%), but in a range of moderate (glycerol) conversions not exceeding 70%. If the hydrogenolysis proceeded further, the onset of side-reactions of Scheme 1 made the formation of the diol drop considerably, even far below 70%. Almost quantitative conversion (90–100%) and high selectivity (91–97%) were described only in a very limited number of papers: most relevant examples included the use of a CuAl2O4 spinel,11 Cu/B2O3/SiO2,12 Cu/ZnO,13 Cu/MgO,14 and Cu–Mg/SiO2 (ref. 15) as catalysts.
Noble metals have also been extensively investigated for the hydrogenolysis of glycerol. Due to the vastness of this topic, the discussion here is limited to the representative cases of Pt and Ru. The comparison of Pt-based bifunctional catalysts prepared by a variety of acid or basic supports demonstrated that one of the most high-performance systems was achieved by using Pt on Mg/Al basic hydrotalcites:7,16 at 220 °C and 30 bar H2, the conversion of glycerol was 92% and the 1,2-PDO selectivity was 93% after 20 h. The dehydrogenation–dehydration–hydrogenation route (path 2 in Scheme 1) was plausibly followed in this case. More recently, however, a conceptually different catalyst design allowed obtaining a Pt–In alloy which offered even better results (conversion and selectivity >99% and 91%, respectively, at 200 °C, 20 bar, and 24 h).17 The authors proposed that the Pt sites at the Pt–In alloy interface served as intrinsic catalytic centers for the activation and cleavage of C–H bonds and hydroxyl groups, while (other) discrete Pt sites suppressed the undesired C–C bond cleavage.
With respect to Pt-based (and even Re- or Rh-based) catalysts, Ru-based systems usually display a higher activity for the conversion of glycerol, meaning that the hydrogenolysis may be run under comparatively milder reaction conditions,18 although Ru is also efficient in the C–C bond breaking. Studies have clearly highlighted how the reaction is affected not only by the metal/support combination but also by the metal precursors and the reduction conditions.19,20 Several strategies have been aimed to improve the formation of 1,2-PDO over Ru catalysts by increasing the density of acid groups or, alternatively, the basic features of the support. Examples include the combination of Ru/AC (activated carbon) and a solid organic resin such as Amberlyst 70 as an acid co-catalyst21 and by impregnating RuCl3 on basic oxides as CeO2 and Mg(OH)2:17,22 at 120–180 °C, 50–80 bar, and 10 h, the glycerol conversion ranged between 50% and 85%, but the 1,2-PDO selectivity did not exceed 70%. Better results, however, have been achieved with bimetallic systems where a second metal component (i.e. Cu, Re) was used to restrain the cleavage of C–C bonds.6,23 To date, the most efficient catalysts were obtained by co-dispersing Ru and Cu (in 3–10:1 ratio) on bentonite or zirconia as supports: at 180–230 °C and 80–100 bar, a quantitative conversion was reached with a yield/selectivity of 1,2-PDO up to 85%.24,25
In this lively context, as a part of our research interest on the Ru/C-catalysed processes for the valorisation of bio-based compounds,26 we were prompted to design multifunctional catalysts composed of a binary mixture of Ru and a solid acid such as WOx bearing strong Brønsted acid sites,27 dispersed on C, indicated as Ru–WOx/C. WOx has been widely described as a support for Pt in the hydrogenolysis of glycerol to 1,3-propanediol,28 but Ru–WOx systems have been much less investigated. To the best of our knowledge, only three pertinent examples (a–c) have been reported so far (Scheme 2).29–31
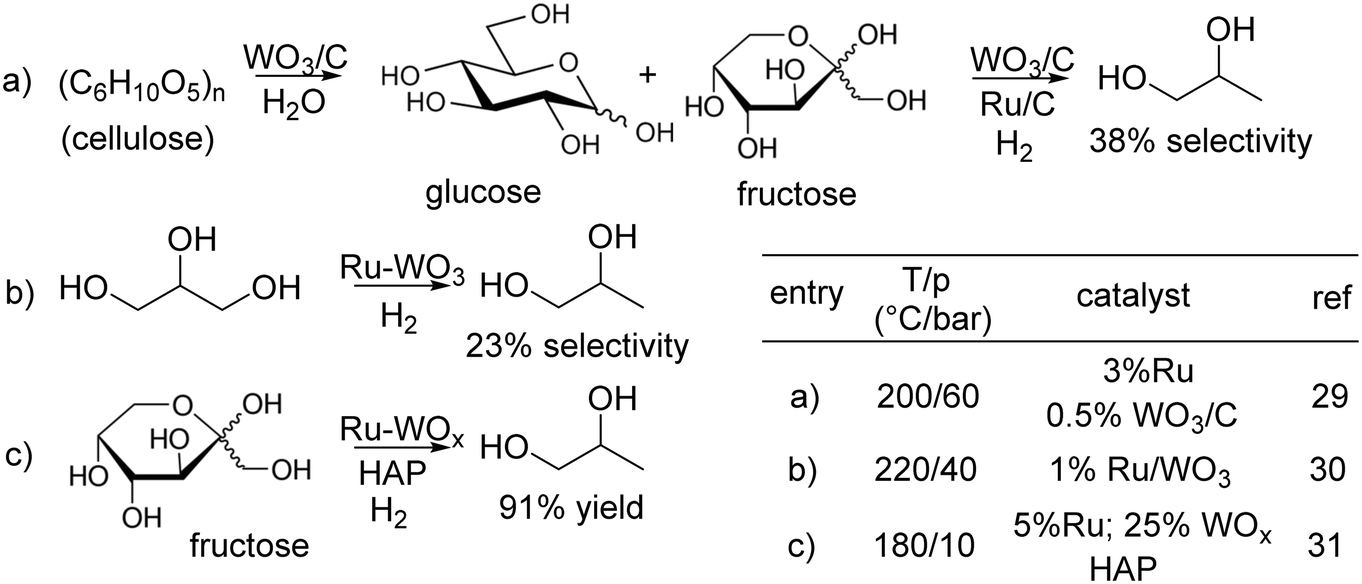 | ||
| Scheme 2 Ru–WOx systems in the production of 1,2-PDO. | ||
In the first two cases, the reaction of cellulose and glycerol was catalysed by a mechanical mixture of Ru/C and WO3/C or 1% Ru supported on WO3, respectively: the corresponding 1,2-PDO selectivity was 38% and 23% (paths a and b). The third very recent study claimed that the conversion of fructose to 1,2-PDO was achieved on a Ru–WOx system supported on hydroxyapatite (HAP; path c); albeit the product was obtained in a remarkably high yield (91% by GC), issues with enzymes necessary for synthesizing fructose (from isomerization of glucose) made the latter a less convenient substrate than glycerol to produce 1,2-PDO.32
In this paper, we wish to report that by co-impregnating simple commercial precursors such as RuCl3 and Na2WO4 on carbon, a series of Ru–WOx/C catalysts was prepared exhibiting excellent performance for the selective hydrogenolysis of glycerol. In particular, at 170 °C and 50 bar, a sample obtained with a Ru:W molar ratio of 4 (5% Ru) allowed a conversion >99% and a 1,2-PDO selectivity of 92%, and it proved recyclable for at least seven subsequent reactions without any loss of activity. Albeit the characterization of Ru–WOx/C systems was challenging, analyses indicated that the no metal alloy was obtained, but a strong Ru/W interaction was more than plausible due to the close co-presence of very small Ru-nanoparticles (below 2 nm) highly dispersed on C and aggregates randomly distributed on the same support, ascribed to WOx clusters below 100 nm.
Results and discussion
The catalysts
5% Ru/C is described in the literature as a reference catalyst for the reduction of glycerol,33 and it is among the preferred commercially available systems to carry out hydrogenation/hydrogenolysis of biobased compounds in water solutions.34 For these reasons, in this paper, four bimetallic Ru–WOx catalysts supported on C were prepared with a constant Ru loading of 5 wt% and a variable W content. The latter was changed sample by sample to achieve a Ru:W molar ratio in the range of 1–16. The metal oxide was indicated as WOx due to the coexistence of different oxidized phases of W, as described later in the characterization section. These solids were synthesized through the adjustment of co-impregnation–precipitation methods reported in the literature for the synthesis of Ru nanoparticles decorating WO335 and Pt–Ru–Sn–W/C.36 RuCl3·H2O and Na2WO4 were used as the metal precursors and a commercial powdered carbon (NORIT SX 1G) was the support. The properties of such a C support were described by us in a previous paper.24 In a typical synthesis, a suspension of carbon (NORIT SX 1G), RuCl3·H2O, Na2WO4, and water was stirred at rt, to which was added concentrated aq. HCl, and heated at 80 °C. After the removal of water, the solid was dried, reduced with H2 (25 mL min−1, 300 °C, 3 h), washed with Milli-Q water, and filtered. It was then dried again and stored. The samples achieved by this procedure had a nominal metal loading of 5 wt% for Ru and of 9, 2, 1 and 0.5 wt% for W. They were labelled according to the different W content as Ru–9WOx/C, Ru–2WOx/C, Ru–1WOx/C, and Ru–0.5WOx/C.
Two other bimetallic catalysts were prepared by a different approach. Following the impregnation method described above, Na2WO4 was first dispersed on carbon powder (NORIT SX 1G) to obtain a nominal W loading of 2 wt%. The sample was labelled as 2WOx/C and it was used as a support to introduce the second metal component, Ru, either through a subsequent impregnation or via a mechanical mixing. In the first case (impregnation), a suspension of RuCl3·H2O and 2WOx/C in water was stirred at rt, concentrated aq. HCl was added, and the mixture was heated at 80 °C. The solid was then dried, reduced and washed through the protocol used above for co-impregnated catalysts. This sample was labelled as [Ru–2WOx/C]ts (ts = two-step synthesis): the corresponding metal loadings were 5 and 2 wt% for Ru and W, respectively. In the second case (mechanical mixing), the catalyst was obtained by mixing 2WOx/C (150 mg) and commercial 5% Ru/C (150 mg). The sample was labelled as [Ru–2WOx/C]mm (mm = mechanical mixture) with metal loadings of 2.5 and 1 wt% for Ru and W, respectively.
Further details on the catalyst synthesis are described in the ESI.†
After the preparation, an aliquot (50 mg) of each solid was dissolved under strong acid/oxidizing conditions in the presence of aqua regia (5 mL) and H2O2 (1 mL) under MW irradiation, and the recovered aqueous solutions were subjected to ICP/MS analyses (other details are given in the Experimental section). Results are summarized in Table 1.
| Entry | Sample label | Synthetic method | Rua (wt%) | Wa (wt%) | Ru:W (molar ratio) |
||
|---|---|---|---|---|---|---|---|
| N | D | N | D | ||||
| a Metal (Ru or W) loading of the catalyst as wt%; N: nominal loading from the preparation; D: loading determined by ICP measures. b Nd: not determined. | |||||||
| 1 | Ru–0.5WOx/C | Co-impregnation | 5 | 4.8 | 0.5 | 0.4 | 16:1 |
| 2 | Ru–1WOx/C | Co-impregnation | 5 | 4.7 | 1 | 1 | 8:1 |
| 3 | Ru–2WOx/C | Co-impregnation | 5 | 4.8 | 2 | 2 | 4:1 |
| 4 | Ru–9WOx/C | Co-impregnation | 5 | 4.5 | 9 | 6 | 1.3:1 |
| 5 | 2WOx/C | Impregnation | — | — | 2 | 2 | — |
| 6 | [Ru–2WOx/C]ts | Two-step impregnation | 5 | 4.8 | 2 | 2.1 | 4:1 |
| 7 | [Ru–2WOx/C]mm | Mechanical mixing | 2.5 | Ndb | 1 | Ndb | 4:1 |
For three out of the four solids obtained by the co-impregnation procedure, specifically for Ru–2WOx/C, Ru–1WOx/C and Ru–0.5WOx/C, the metal loading (Ru or W; wt%) determined by ICP analyses (D values) matched the nominal metal content (N values) expected from the amounts of RuCl3·H2O and Na2WO4 used during the synthesis (entries 1–3). From the same (ICP) analyses, the residual Na was measured in each sample: the corresponding quantity (≤0.1%) equalled that of the commercial 5% Ru/C.
A deviation was instead observed for the sample Ru–9WOx/C, where the measured loadings of metals, especially for W, were lower than expected (entry 4). The Ru:W molar ratio was 1.3 rather than 1. Moreover, a remarkably high Na quantity (5 wt%) was detected. Attempts to repeat the preparation of this catalyst gave unsatisfactory and not reproducible results which led us to conclude that the synthetic protocol was not suitable to make materials with W loading exceeding 2 wt%. High relative amounts of metal precursors plausibly interfered with each other in the co-adsorption on the C support, making their dispersion not effective. Whichever the reason, the Ru–9WOx/C system was abandoned.
ICP analyses of other samples obtained by single or two-step impregnation such as 2WOx/C and [Ru–2WOx/C]ts gave a good correspondence between nominal and actual metal content for both Ru and W (entries 5 and 6).
The catalytic activity of Ru–WOx/C systems
The performance of Ru–WOx/C systems was investigated for the hydrogenolysis of aqueous glycerol. Due to the complexity of the reaction and the vast body of literature available, a preliminary screening was conducted using a commercial 5% Ru/C sourced by Aldrich, under the conditions more often reported for the process, in particular T and p in the range of 100–200 °C and 40–80 bar H2, respectively (see the ESI,† Fig. S1 and S2).37–39 In light of these results and other findings on the effect of the H2 pressure,40 experiments were initially performed at 150 °C and 5 bar H2 for 6 h in a stainless-steel autoclave charged with an aqueous solution of glycerol (5 mL, 5 wt%) and the chosen catalyst (150 mg).The reaction provided both liquid and gaseous products (cf.Scheme 1). The attention was focused on liquid derivatives including 1,2-PDO, EG (ethylene glycol), and lighter alcohols (1- and 2-propanol, ethanol and methanol). The formation of these compounds and the conversion of glycerol were determined by GC, using triglyme as an external standard, and their structure was confirmed by GC/MS through comparison with authentic commercial compounds. Other products, named as “others”, included gases (mostly CO, CO2, and CH4) whose total amount was the complement to 100 of the overall reaction selectivity. The carbon loss in the liquid phase (%Closs) was evaluated from the carbon balance (%Cbalance) as the difference of initial moles of glycerol and the total molar amount of all liquid products.
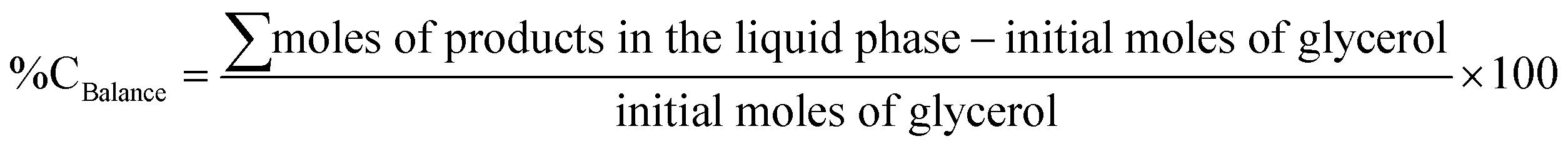
| %Closs in the liquid phase = 100 − %CBalance |
| Entry | Catalyst | Conv. (%) | Liquid products (selectivity, %) | Closs (%) | ||||
|---|---|---|---|---|---|---|---|---|
| 1,2-PDO | EG | 1-PrOH | 2-PrOH | MeOH + EtOH | ||||
| All reactions were carried out using an aqueous solution of glycerol (5 mL, 5 wt%) and the chosen catalyst (150 mg). | ||||||||
| 1 | Ru/C | 60 | 13 | 80 | — | — | 7 | 51 |
| 2 | Ru–0.5WOx/C | 40 | 98 | 0.5 | 1 | 0.5 | 1 | 5 |
| 3 | Ru–1WOx/C | 61 | 98 | 0.5 | 0.5 | 1 | 1 | 2 |
| 4 | Ru–2WOx/C | 73 | 97 | 0.5 | 0.5 | 1 | 1 | 3 |
| 5 | [Ru–2WOx/C]ts | 35 | 96 | 1 | 0.5 | 0.5 | 1 | 5 |
| 6 | [Ru–2WOx/C]mm | 15 | 97 | 1 | 1 | 0.5 | 0.5 | 5 |
| 7 | 2WOx/C | 0 | ||||||
The commercial Ru/C catalyst allowed a 60% conversion, but it clearly favoured the multiple hydrogenolysis of glycerol: EG (80%) was the major liquid product formed along with a sizeable amount of gaseous derivatives. A high carbon loss of 51% was determined (entry 1). In contrast, a striking improvement in the reaction selectivity and carbon balance was manifested when co-impregnated WOx-modified Ru/C systems were used. All such catalysts provided the almost exclusive formation of 1,2-PDO (97–98%) and displayed negligible C–C bond cleavage, producing only traces (≤3%) of liquid by-products (entries 2–4). Another salient aspect was the effect of the amount of WOx: glycerol conversion showed a progressive increase from 40% to 61% and 73% when lowering the Ru:W molar ratio from 16:1 to 8:1 and 4:1.
On balance, the best-performing system was Ru–2WOx/C (Ru:W = 4 mol/mol) by which the highest conversion (73%) was reached with high 97% selectivity towards 1,2-PDO (entry 4).
Other experiments in Table 2 further corroborated this observation: 2WOx/C was totally ineffective for the reaction (entry 7), while both [Ru–2WOx/C]ts and [Ru–2WOx/C]mm allowed a high 1,2-PDO selectivity (96%) with low carbon loss (≤5%), although the corresponding glycerol conversion was only 35% and 15%, respectively (entries 5 and 6). This led to the conclusion that W oxide was not involved in the catalysis, but its contribution was crucial to limit C–C bond cleavage by Ru. Moreover, interactions between the metal catalyst components were optimized when the metal precursors were simultaneously impregnated on the C support. Multiple effects improving both the glycerol conversion and the product distribution could be anticipated from the literature, including the formation of (i) WOx clusters on the carbon surface, providing Brønsted acid sites;41 (ii) small metal particles originating from an acid chloride precursor of Ru.19
Based on these results, the study was continued using the best system identified in Table 2: Ru–2WOx/C (Ru:W 4:1 molar ratio). The influence of temperature and pressure as major reaction parameters was investigated in more detail.
Effects of T and p
The hydrogenolysis of glycerol catalysed by Ru–2WOx/C was explored at different T and p in the range of 120–170 °C and 5–35 bar H2, respectively. Other conditions were kept unchanged with respect to Table 2 (aqueous glycerol solution: 5 mL, 5 wt%; t = 6 h; catalyst: 150 mg). Results are reported in Fig. 1.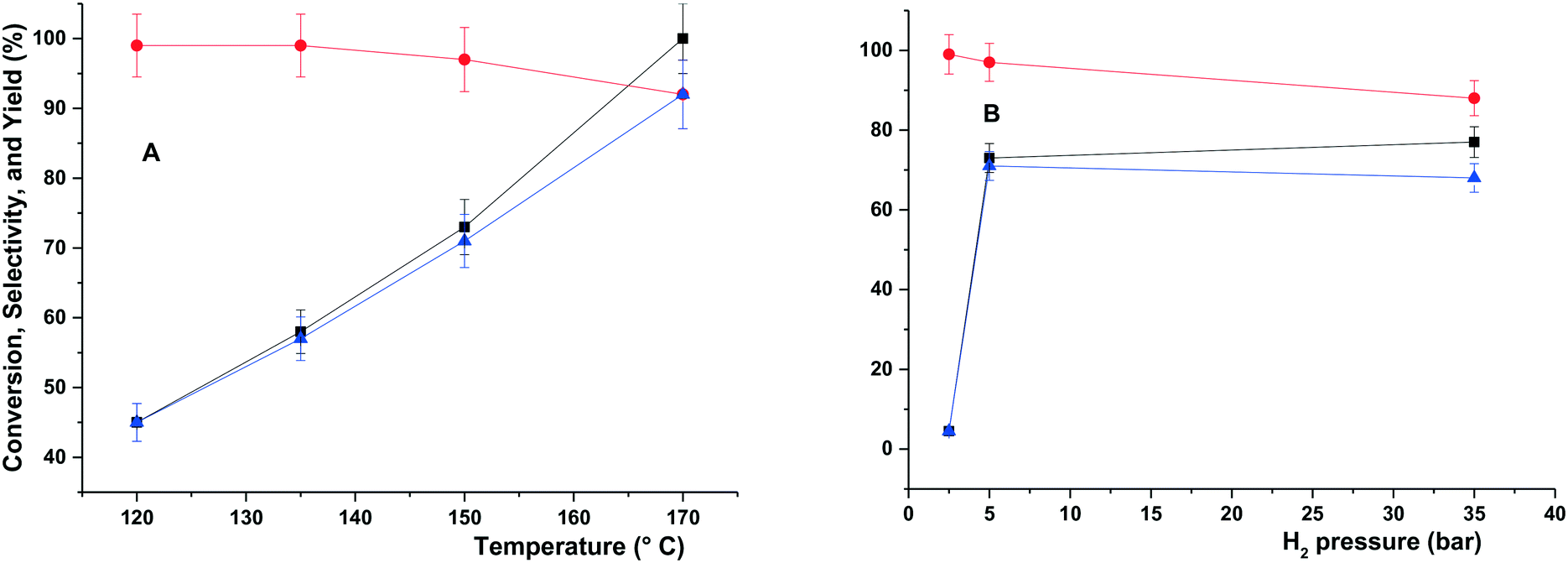 | ||
Fig. 1 Influence of the temperature (A, p = 5 bar) and pressure (B, T = 150 °C) on the conversion (–■–) of glycerol, and the selectivity ( ) and yield ( ) and yield ( ) towards 1,2-PDO. Other conditions: aqueous glycerol = 5 mL, 5 wt%, Ru–2WOx/C = 150 mg; 6 h. ) towards 1,2-PDO. Other conditions: aqueous glycerol = 5 mL, 5 wt%, Ru–2WOx/C = 150 mg; 6 h. | ||
At a constant pressure of 5 bar (Fig. 1A), increasing the temperature allowed a quantitative conversion of glycerol (black profile) and improved the yield of 1,2-PDO from 45% to >92% (blue profile), while the corresponding selectivity (red profile) remained substantially steady and above 97% at 120–150 °C, with a slight drop to 92% at 170 °C due to the formation of ethylene glycol (EG: 2%) and light alcohols (MeOH, EtOH and propanols: 6% in total) as by-products. In all cases, the carbon loss was negligible (≤5%).
At a constant temperature of 150 °C (Fig. 1B), a sharp increase of both the glycerol conversion and the yield of 1,2-PDO from 3% to 73% and 3% to 71%, respectively, was noticed when the pressure was raised from 2 to 5 bar. This was consistent with the availability of gaseous H2 in the reactant solution: at 50 °C for example, the H2 solubility in water has been reported to increase almost linearly with pressure from 1 to 5 bar.42 Minimal, if any, effects were observed on the 1,2-PDO selectivity (97–99%). A further rise of the pressure up to 35 bar had limited consequences on the conversion, but it reduced the selectivity and the yield to 88% and 68%, respectively, in favour of EG (1%) and light alcohols (11% in total). The further increase of the H2 solubility in water, which triples in the interval 5–35 bar,43 plausibly accounted for an improved activity of the catalyst towards C–C bond cleavage reactions. Similar effects of the H2 pressure were described for the hydrogenolysis of glycerol in the presence of both Cu- and Ru-based systems.44,45 Moreover, for the same reaction catalyzed by Ru/CsPW [Ru-doped acidic heteropoly salt, Cs2.5H0.5[PW12O40] (CsPW)], over-reduction of W(VI) was reported at pressure >10 bar, resulting in a decrease of the catalyst acidity and a poorer performance.39
Additional experiments demonstrated that even at 150 °C and 5 bar (conditions of Fig. 1, left), by prolonging the hydrogenolysis for up to 12 hours, a quantitative conversion of glycerol was achieved with a 1,2-PDO selectivity of 87% (other liquid by-products were EG and light alcohols) and a carbon loss below 5%. This result demonstrated that the WOx-modified Ru catalyst allowed an outcome comparable to the best existing methods for the synthesis of 1,2-PDO from glycerol (based on Ru/Cu-based systems23,24), with the advantage of requiring far milder reaction conditions (150 °C and 5 bar vs. 180–230 °C and 80–100 bar).
Finally, the effect of glycerol concentration was explored using 5–20 wt% aqueous solutions at 150 °C and 5 bar. This study showed a five-fold drop of the conversion, from quantitative to ca. 20%, with the higher concentration solution (20 wt%), while 1,2-PDO selectivity increased from 88% to >95%. Overall, the extent of undesired C–C cleavage reactions and dehydration/hydrogenation of 1,2-PDO (paths 1 and 2, Scheme 1) could be tuned below 5% also by the concentration effect, providing that the conversion did not exceed 50% (details of this investigation are described in the ESI,† Fig. S3).
Recycle of the catalyst
The catalyst in a liquid-phase reaction may affect up to one third of the total cost of the process, meaning that its recovery and reuse are fundamental to the economic and environmental sustainability of any catalytic protocol.46 With the aim of exploring this aspect, recycle experiments were carried out using the reference catalyst, Ru–2WOx/C, under conditions chosen to control the reaction outcome at a final conversion not exceeding 75%. Hydrogenolysis tests were therefore performed for 6 hours at 150 °C and 5 bar H2, in the presence of Ru–2WOx/C (150 mg) and an aqueous solution of glycerol (5 mL, 5 wt%) (conditions of Fig. 1, left). Once a (first) reaction cycle was complete, the catalyst was filtered, washed with water (15 mL), dried overnight under vacuum (70 °C at 5 mbar), and recycled for a second hydrogenolysis run. The overall sequence was repeated for 7 subsequent experiments. Of note, the mass loss of dried catalyst was on average 0.01% from one recycle to another (with respect to the initial sample, the weight of the residual catalyst after the 7th test was 140 mg). Results are reported in Fig. 2. The catalytic performance of Ru–2WOx/C was unaltered over 7 subsequent recycles: both the conversion (black profile) and the 1,2-PDO selectivity (green bars) were substantially steady at about 70% and >95%, respectively, with changes within the experimental error (≤5%). However, ICP analyses of aq. solutions from the 1st, 2nd and 5th recycles proved that the chemical composition of the catalyst changed: leaching of Ru was always negligible (<0.01 wt%), but a significant loss of W (ca. 5 wt%) occurred after the first reaction and then it quickly decreased to 0.3 wt% and tended to stabilize at 0.1 wt% after the second and fifth runs, respectively (Table 3).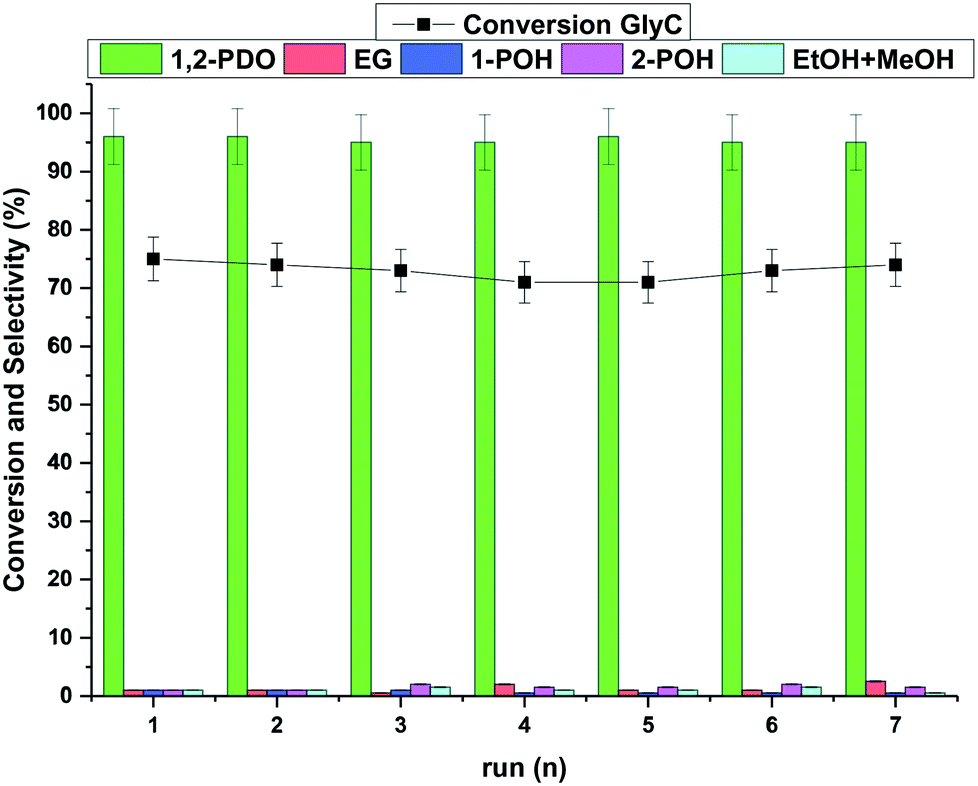 | ||
| Fig. 2 Recycle tests of Ru–2WOx/C (150 mg). Reaction conditions were those of Fig. 1, left: aq. glycerol (5 mL, 5 wt%), 150 °C, 5 bar H2, 6 h. | ||
These results were confirmed by additional recycle experiments (carried out under the conditions of Fig. 2) where the recovered catalyst was subjected to ICP analyses as described in Table 1. Although leaching of WOx species was detected, this gradually diminished in the repeated uses and had no effects on the active sites of the catalyst.
A similar behavior was described in previous studies: (i) in the hydrogenolysis of 1,4-anhydroerythritol catalyzed by Pt–WO3/SiO2, where albeit a W leaching of 3.6, 1.8, and 0.2 wt% was observed after the first reaction and two subsequent recycles, no loss of Pt and a stable catalytic activity were reported;47 (ii) in the conversion of cyclopentene to glutaraldehyde catalysed by mesoporous WO3/SBA-15. In this latter case, leaching of WO3 determined a slight decrease of catalyst performance which was offset through thermal regeneration of WO3/SBA-15 before its reuse.48 The authors concluded that the presence of polymeric tungsten species or crystalline WO3, which weakly interacted with the support (during catalyst preparation and use), were responsible for the observed leaching effects. This was consistent with the behaviour of Ru-2WOx/C (Fig. 2 and Table 3); such an aspect, however, was not further investigated by us.
A final set of recycle experiments was designed by exposing Ru–2WOx/C to harsher, more stressful conditions, specifically by carrying out five subsequent hydrogenolysis reactions at 150 °C, but at a higher pressure (35 bar) and for a longer time (12 h) than those of Fig. 2. The catalyst proved stable over time: a quantitative conversion of glycerol was observed in all repeated tests, while the 1,2-PDO selectivity slightly decreased from 88% (run 1) to 82% (run 5): this drop was in line with pressure effects described in Fig. 1 (details of this study are in the ESI,† Fig. S4).
Catalyst characterization: morphological and structural properties
The best-performing catalyst for the hydrogenolysis of glycerol, Ru–2WOx/C, was considered for this study. A fresh and a used sample (before and after the reaction) of Ru–2WOx/C were characterised by XPS, TEM, and XRD. The “used” specimen was the catalyst recovered after two hydrogenolysis tests carried under the conditions of Fig. 1 (150 °C, 5 bar, 6 h). For comparison, XPS analysis was performed also for a sample containing only W, 2WOx/C (entry 5, Table 1).XPS analyses
Spectral line binding energies were referred to the C at 284.4 eV. The three investigated samples (fresh and used Ru–2WOx/C and 2WOx/C) did not show surface charging during analysis, though due to the large amount of C with respect to Ru (the C/Ru ratio is around 20), only the Ru 3p doublet band was recorded, while the Ru 3d band was overlapped by the much more intense C 1s band. The standard energy differences between different tungsten oxides and ruthenium species were considered in the curve filling process of the Ru 3p and W 4f energy region of the investigated systems. The following section summarizes the salient aspects inferred from XPS analysis, while spectra are reported in the ESI† (Fig. S5–S7).In the fresh Ru–2WOx/C sample, the W 4f7/2 signal at 35.1 eV was characteristic of WO3 and matched the literature data.49 The presence of some W(V) could not be excluded, but the very low amount of W with respect to oxygen in the analysed region prevented any significant fit on the O 1s band involving contributions from different W oxides. The Ru 3p3/2 signal at 462.6 eV was in agreement with the presence of Ru oxide, RuO2.50,51 However, the small difference in BE between oxidized and metallic species in the Ru 3p3/2 band could not rule out the presence of Ru in oxidation states lower than +4.
Notwithstanding the W-leaching (Table 3) observed for the used Ru–2WOx/C sample, the corresponding XPS spectra did not show significant changes of W 4f7/2 and Ru 3p3/2 signals compared to those of the fresh catalyst, except for a slightly smaller FWHM (full width at half-maximum) of the Ru 3p3/2 band. This suggested a preference for a defined chemical/oxidation state of Ru.
In the 2WOx/C sample, the W 4f7/2 signal was consistent with the structure of WO3, and the reduced FWHM suggested a better-defined chemical state of tungsten. This made the presence of W(V) less probable compared the Ru-doped samples, albeit it could not be completely excluded also in this case.52
TEM analysis
High-resolution scanning transmission electron microscopy (HRSTEM), high-angle annular dark-field (HAADF)-STEM and EDX analyses were carried out on fresh and used Ru–2WOx/C.The EDX mapping analysis of fresh Ru–2WOx/C indicated a uniform dispersion of both metals (Ru and W) on the surface of the C support (Fig. 3A), while TEM images showed the presence of very small Ru nanoparticles with an average size of 1.5 ± 0.2 nm and 1.6 ± 0.4 nm for both the fresh and the used catalyst, respectively (Fig. 3B).
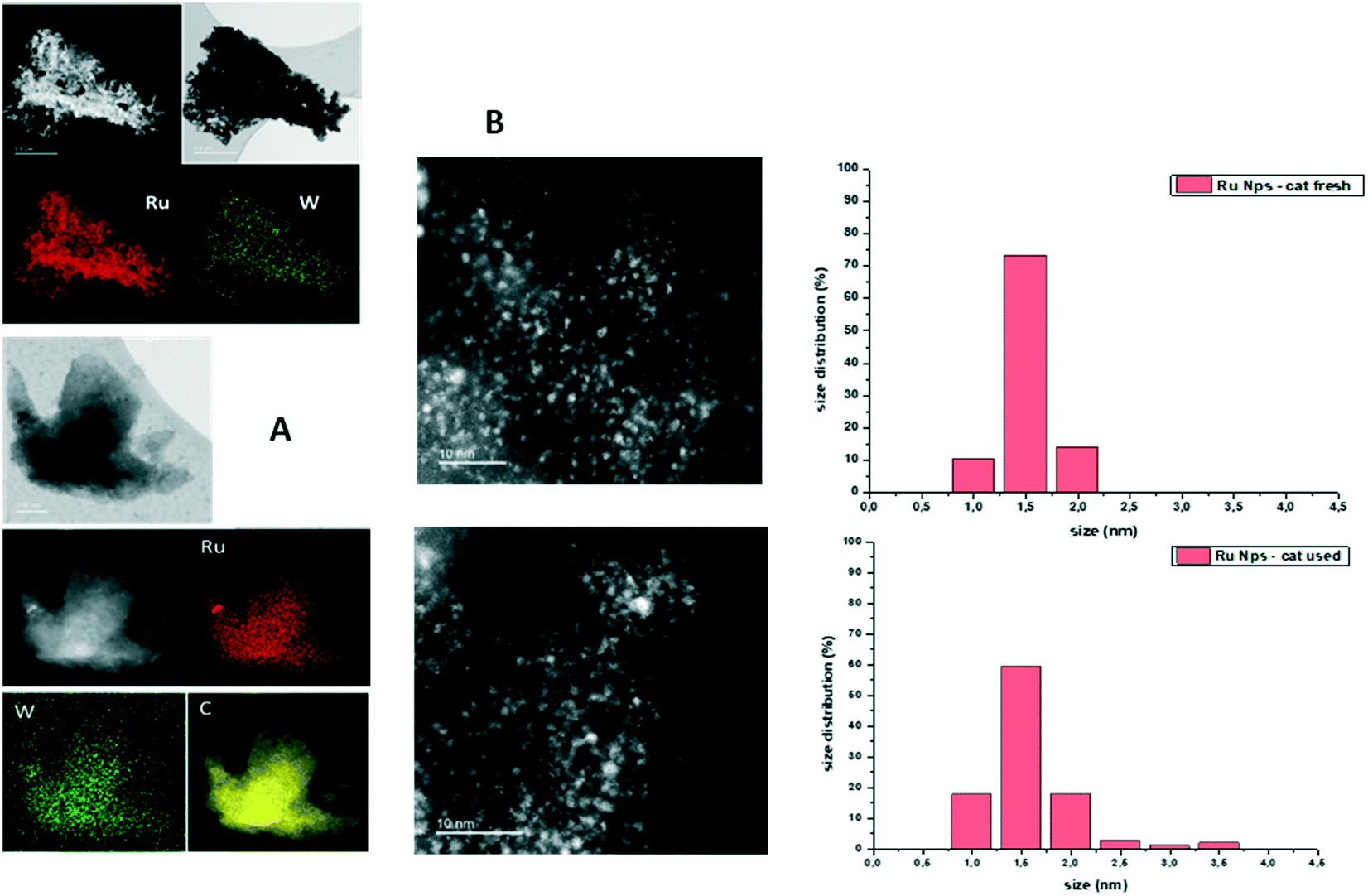 | ||
| Fig. 3 (A) EDX mapping analysis of Ru–2WOx/C. (B) STEM images (left) and size distribution of Ru nanoparticles (right) of Ru–2WOx/C. The top and bottom of (A) and (B) refer to the fresh and the used catalyst. The size of Ru was averaged on more than 100 nanoparticles. | ||
This evidence not only highlighted how the catalyst preparation was effective in achieving a homogeneous distribution of the metal components on the catalyst but it corroborated the results of recycle tests: the stable performance of Ru–2WOx/C during its reuses (Fig. 2) was consistent with the size preservation of metal (Ru) nanoparticles from the fresh to the used sample. A further indication of the nature and composition of nanoparticles came from the cross-sectional analyses reported in Fig. 4.
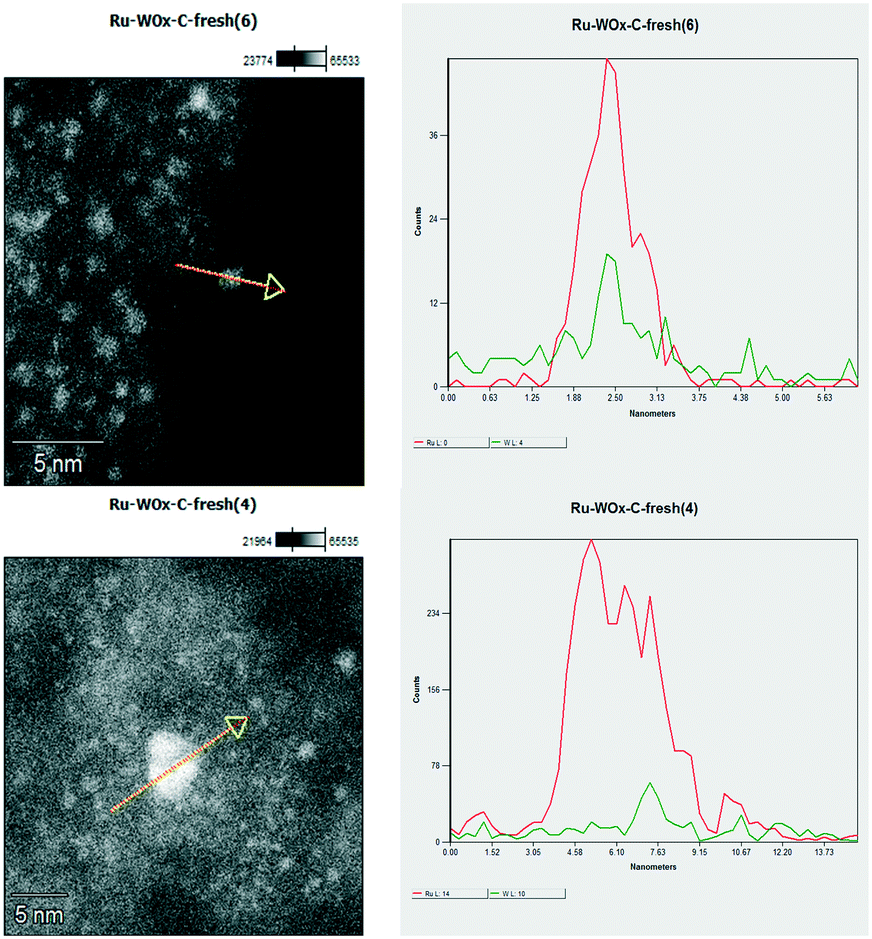 | ||
| Fig. 4 Cross-sectional analysis of nanoparticles in the Ru–2WOx/C catalyst. Top: a single dot; bottom: an agglomerate present on the catalyst surface. | ||
High-resolution images showed the presence of crystalline ordered dots (top) along with single or randomly aggregated dots (bottom) which might be ascribed to WOx clusters smaller than 100 nm. The close proximity of the two metals indicated that a strong Ru–W interaction was possible, although the formation of any bimetallic alloy could not be detected. Moreover, the amount of W in the chosen particles/agglomerates (top and bottom) was lower than expected for Ru–2WOx/C (Ru:W = 4 mol/mol; compare profiles of Fig. 5, left). Recent studies on the activity of a multimetallic Pt–WOx/ZrO2 system (Pt–WOx supported on ZrO2) for the hydrogenolysis of glycerol demonstrated that the reaction was structurally sensitive to the domain size of surface WOx clusters.53 WOx/ZrO2 was categorised as a superacid solid where medium-sized clusters (medium polymerized WOx domains) imparted a strong Brønsted acidity to boost the selective formation of 1,3-PDO, while smaller clusters (isolated WOx and/or with a low degree of polymerization) behaved as Lewis acid sites. Another investigation described similar acid features for a Pt–WOx/Al2O3 catalyst which were attributed to different WOx species composed of monotungstate, polytungstate and crystalline clusters with variable proportions depending on the W loading.54 In analogy to these results, whichever the structure of WOx, the acidity of the Ru–2WOx/C catalyst should plausibly favour the dehydration of glycerol during hydrogenolysis (path 1 in Scheme 1), thereby improving the selectivity towards 1,2-PDO.
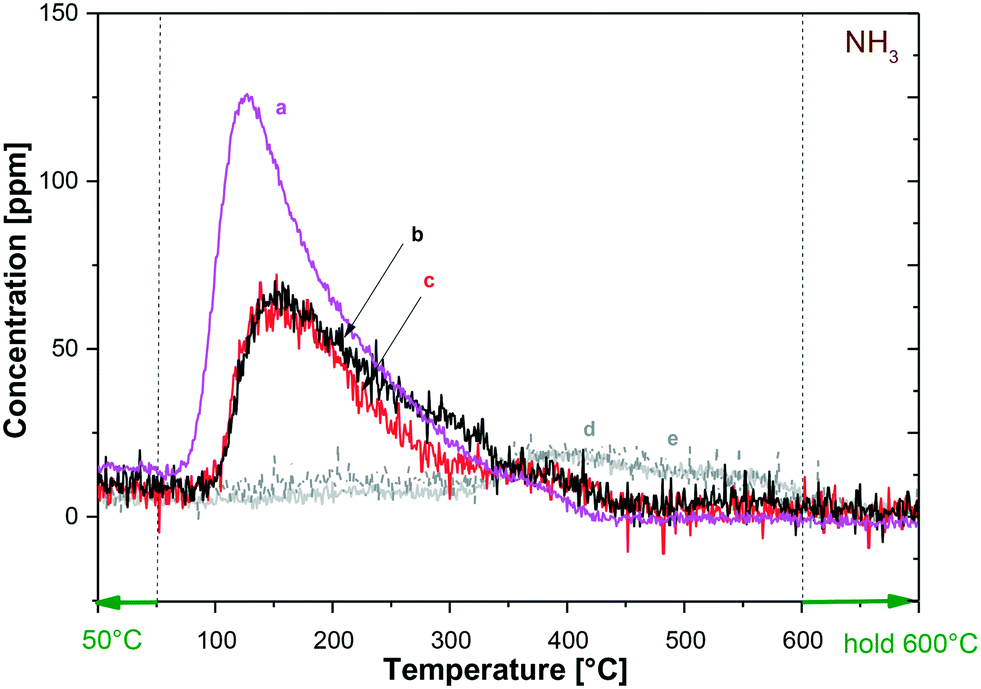 | ||
| Fig. 5 Temperature-programmed desorption (TPD) up to 600 °C after NH3 (1000 ppm in He) adsorption at 50 °C for different catalysts: (a) Ru2WOx/C; (b) Ru1WOx/C; (c) Ru0.5WOx/C; (d) fresh carbon support; (e) acid-treated carbon support. | ||
XRD analyses
XRD spectra of fresh and used Ru–2WOx/C did not offer information on either WOx or Ru species on the catalyst, since diffraction profiles of both samples matched that of the carbon support (details are in the ESI,† Fig. S8).55 This result was consistent with TEM analyses that showed a homogeneous dispersion of small nanoparticles undetectable by XRD.Catalyst characterization: surface acidity and Ru dispersion
The acidic nature of the catalysts and the dispersion of the metal active for hydrogenolysis (Ru) were investigated by NH3 temperature-programmed desorption (NH3-TPD) and CO chemisorption. This study was aimed at comparing the properties of the bimetallic catalysts used in this work, particularly to explore any effect induced by the change of the W content. Tests were carried out as described in the Experimental section, starting from samples (60 mg) of Ru–2WOx/C, Ru–1WOx/C and Ru–0.5WOx/C, where the Ru amount was constant (5 wt%), while the W loading was progressively decreased from 2 to 0.5 wt% (compare Table 1). Moreover, to shed light on the role of the surface acidity, additional NH3-TPD experiments were carried out on the carbon support as such (NORIT SX 1G: Cfresh) and after treating it with aq. HCl according to the same procedure used for the catalyst preparation (NORIT SX 1G: Cacid-treated; details are in the Experimental section).Fig. 5 shows the results of NH3-TPD measures. The amount of acid sites was calculated from the desorbed NH3 as reported in Table 4.
| Entry | Sample | Metal loading (wt%) | Desorption | NH3 desorbed (μmol gcat−1) | ||
|---|---|---|---|---|---|---|
| Ru | W | T (°C) | ||||
| The surface acidity was calculated from desorbed NH3 in TPD profiles.a Cfresh: the fresh carbon support (NORIT SX 1G) used in this study.b Cacid-treated: the carbon support treated under the same acid conditions used for the preparation of Ru–WOx/C systems.c Not determined. | ||||||
| 1 | Cfresha | — | — | 390 | 540 | 26.8 |
| 2 | Cacid-treatedb | 390 | Ndc | 29.8 | ||
| 3 | Ru–2WOx/C | 4.8 | 2.0 | 127 | 375 | 73.3 |
| 4 | Ru–1WOx/C | 4.7 | 1.0 | 155 | 360 | 58.8 |
| 5 | Ru–0.5WOx/C | 4.8 | 0.5 | 155 | 370 | 46.9 |
As expected, the fresh and the acid-treated carbon samples displayed some surface acidity which was due to the typical functionalities present on carbon, as carboxylic acids, anhydrides (hydrolysed in aqueous solutions), phenols, and quinones.26 The corresponding TPD profiles of Cfresh and Cacid-treated, however, were almost superimposed (curve d and e, respectively: entries 1 and 2), thereby suggesting that even though the acidity of the support could contribute to the reaction progress, it (acidity) was not substantially affected by the acid treatment used in the synthesis of Ru–WOx/C samples.56
Moving on to the bimetallic samples, the literature correlates the desorption temperature of NH3 in TPD profiles to the surface acidity by identifying weak (lower than 300 °C), medium (300–500 °C), and strong (>500 °C) sites.57 According to this classification, Fig. 5 and Table 4 indicate that the three examined catalysts were all characterized by the presence of acid sites of weak to medium strength (T desorption in the range of 127–375 °C: entries 2 and 3), but the total acidity, albeit far higher than that of the support, was considerably different between the samples. Indeed, experiments showed that the lower the W loading, the lower the concentration of acid sites: the amount of desorbed NH3 progressively decreased from 73.3 to 46.9 μmol gcat−1 as the W content was decreased from 2 to 0.5 wt% (entries 3–5: Ru–2WOx/C, Ru–1WOx/C, and Ru–0.5WOx/, respectively). An additional TPD run – not shown in Fig. 5 – on the Ru–2WOx/C catalyst recovered after its use (same sample of Fig. 3, bottom) confirmed a slight drop of surface acidity compared to the fresh system. This behaviour was consistent to the W loss observed during the recycle of the catalyst (Table 3).
CO chemisorption from the gas phase was used to determine the accessible Ru surface area.58Table 5 shows the results by reporting the Ru dispersion (DRu, %).
| Entry | Sample | Total CO ads (μmol gcat−1) | D Ru (%) |
|---|---|---|---|
| 1 | Ru–2WOx/C | 35 | 7 |
| 2 | Ru–1WOx/C | 70 | 14 |
| 3 | Ru–0.5WOx/C | 77 | 15 |
The decreasing of the W loading from 2 to 1 wt% brought about an evident increase of the Ru dispersion from 7% to 14% (entries 1 and 2). The latter, however, showed only a slight increase to 15% when the W content was further halved to 0.5 wt% (entry 3). This behaviour corroborated the results of TEM experiments: Ru and W not only interacted with each other because of their close proximity on the catalyst surface, but the entity/strength of this interaction was correlated to the W content. Other details of CO chemisorption measures are described in the ESI† (Table S1).
The reaction mechanism
In addition to the above-described Pt–WOx/ZrO2 and Pt–WOx/Al2O3 catalysts,54,55 a significant body of literature on bi- and multi-metallic systems including for example, mesoporous Ti–W oxides,59 Pt on WO3/silica-alumina,60 and Pt–HSiW/SiO2,61 led to conclude that WOx-doping increased the catalyst acidity. In aqueous solutions, especially Brønsted acid sites generated on the WOx surface promoted the dehydration of glycerol, while other WOx species (even slightly reduced W(V) sites formed in situ by H2 or glycerol) stabilized adsorption of glycerol and intermediate species during hydrogenolysis. Moreover, the surface acidity of carbon could play a role. This hypothesis was corroborated by several studies, among which two pertinent investigations reported an improved performance of a set of acid-activated Pd/C catalysts in the hydrogenolysis of hydroxymatairesinol (a known lignan) to matairesinol: it was observed that either on carbon or carbon nanofibers, the more acid the support (from 0.0078 to 0.0246 mmol g−1 of acidity), the more active and selective the catalyst, with product yields that increased up to a factor of 4.62,63These considerations applied also to the WOx-modified Ru catalysts on C studied here, for which TEM analyses (Fig. 3 and 4), NH3-TPD (Fig. 5 and Table 4) and CO chemisorption (Table 3) suggested the occurrence of a synergic effect of the two metal components in the form of Ru nanoparticles and (isolated) WOx acidic clusters. Moreover, the little, but not negligible, acidity of the carbon support (Table 4) could contribute to the activation of glycerol over the catalytic surface. The mechanism shown in Fig. 6 was therefore hypothesised.
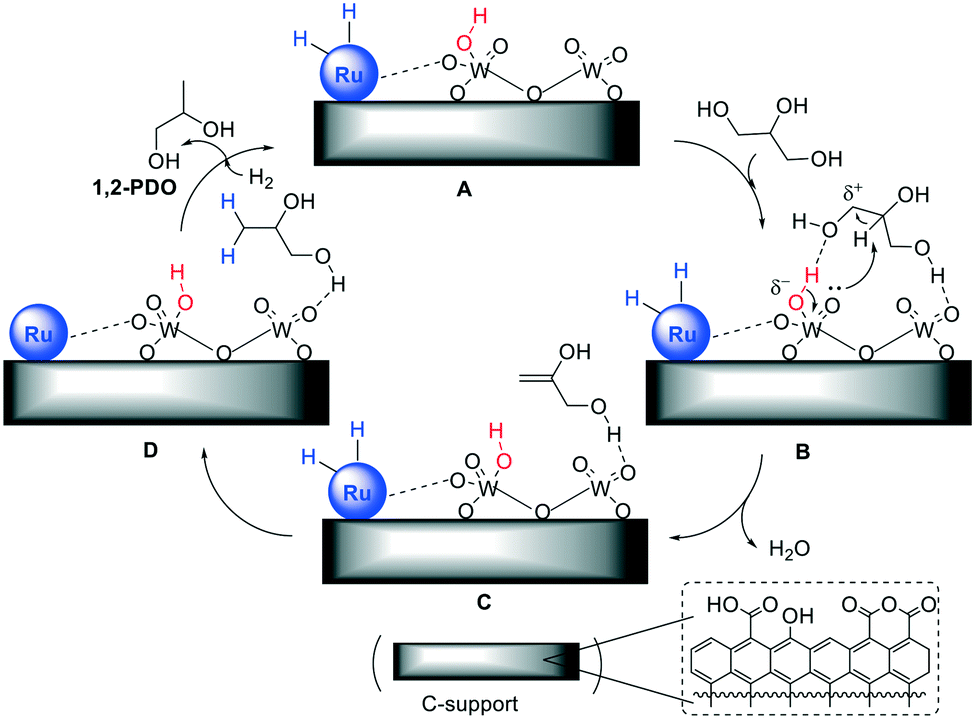 | ||
| Fig. 6 Pictorial view of the hydrogenolysis of glycerol over WOx-modified Ru catalysts on C. The dashed box (bottom): major oxygenated acid species of the support surface. | ||
In the first step (A → B), glycerol adsorbs on acid WOx species, acting either as a H-bond acceptor on Brønsted acid sites (red) or a H-bond donor on Lewis acid sites. The steric hindrance plausibly favours H-bonding of primary hydroxyls with respect to the secondary one. Although not explicitly shown to avoid overburdening Fig. 6, similar interactions with acid functions on the C support (see dashed box) are expected to further stabilize glycerol on the catalyst surface. This mode of adsorption has been described, for example, in the removal of glycerol from biodiesel wash waters using activated carbon materials.64
Thereafter, the moderate (Brønsted) acid strength of surface species is enough to assist dehydration which takes place concurrently to deprotonation and restoration of the catalyst acidity (B → C).
Prop-2-ene-1,2-diol [HOCH2CH(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)OH] forms on the catalyst surface (compare path 1, Scheme 1): this species reacts with H atoms deriving from H2 dissociation on the Ru nanoparticles, producing 1,2-PDO (C → D) which finally desorbs in the aqueous solution (D → A). The surface density of acid groups obviously affects the outcome of all steps (rate/selectivity) and it may offer an explanation for the change of the catalyst performance described in Table 2. In particular, the improved efficiency of the catalysts with the increase of the W loading from 0.5 to 2 wt% was correlated to the higher concentration of surface WOx sites, as demonstrated by NH3-TPD measurements. On the other hand, the total inactivity exhibited by WOx/C (entry 7, Table 2) indicates that the hydrogenolysis reaction becomes successful only on condition that Ru, WOx and the carbon support act synergistically. It should also be noted that at 150–200 °C, some acid-activated carbons have been reported to catalyse the dehydration of primary alcohols (ROH: Rn-Pr, n-Bu):65 these systems, however, display a surface acidity far higher (up to 1800 μmol g−1) than that of Ru–WOx/C samples.
CH2)OH] forms on the catalyst surface (compare path 1, Scheme 1): this species reacts with H atoms deriving from H2 dissociation on the Ru nanoparticles, producing 1,2-PDO (C → D) which finally desorbs in the aqueous solution (D → A). The surface density of acid groups obviously affects the outcome of all steps (rate/selectivity) and it may offer an explanation for the change of the catalyst performance described in Table 2. In particular, the improved efficiency of the catalysts with the increase of the W loading from 0.5 to 2 wt% was correlated to the higher concentration of surface WOx sites, as demonstrated by NH3-TPD measurements. On the other hand, the total inactivity exhibited by WOx/C (entry 7, Table 2) indicates that the hydrogenolysis reaction becomes successful only on condition that Ru, WOx and the carbon support act synergistically. It should also be noted that at 150–200 °C, some acid-activated carbons have been reported to catalyse the dehydration of primary alcohols (ROH: Rn-Pr, n-Bu):65 these systems, however, display a surface acidity far higher (up to 1800 μmol g−1) than that of Ru–WOx/C samples.
The mechanism of Fig. 6 cannot rule out that also secondary hydroxyl groups of glycerol are H-bonded to acid sites (both WOx and oxygenated species on C) as reported for W-doped Pt catalysts.53 However, the mild acidity of Ru–WOx/C systems hinders the dehydration of sec-OH since in our case the product expected from this reaction (1,3-PDO) has never been detected.
An additional aspect is the presence of Na traces in the catalytic samples. Although it is well known that the doping by alkali metals affects the hydrogenation performance of Ru-based catalysts,66 it seems unlikely that a similar effect favours the hydrogenolysis of glycerol towards 1,2-PDO in the case of the bimetallic systems investigated here. Indeed, the latter (Ru–WOx/C) have a Na content (<0.1 wt%) equal to that of the commercial 5% Ru/C which in no way is a selective catalyst.
A brief comment is finally addressed to the metal dispersion (Table 5). Although the investigation of the decrease of the Ru dispersion with the increase of the WOx content is beyond the scope of the present paper, the best performance of the least dispersed catalyst might suggest that the overall reaction is structure-sensitive.
Experimental
General
Glycerol, 5% Ru/C, RuCl3·H2O, Na2WO4, aq. HCl (37 wt%), HNO3 (70 wt%), H2O2 (30 wt%), 1,2-propanediol (1,2-PDO), 1-PrOH, 2-PrOH, ethylene glycol (EG), EtOH, MeOH, triglyme, and carbon NORIT SX 1G were commercially available compounds sourced from Aldrich. If not otherwise specified, they were employed without further purification. Water was Milli-Q grade. H2 was from SIAD, Italy.Catalyst preparation
WOx-modified Ru catalysts were prepared by a co-impregnation method using RuCl3·H2O and Na2WO4 as precursors. Finely powdered carbon (1 g, NORIT SX 1G) was suspended and stirred in Milli-Q water (50 mL). In a separate flask, RuCl3·H2O (0.1 g) and Na2WO4 (0.128–0.09 mg; Ru:W = 1–16 mol:mol) were dissolved in Milli-Q water (50 mL). The aqueous solution of metal precursors was added to the carbon suspension and the resulting mixture was stirred overnight at rt. It was then heated at 80 °C, aq. HCl (1 mL; 15 wt%) was added, and the mixture was kept at 80 °C for 3 h. Thereafter, water was slowly evaporated at 95 °C. The black powder was collected, dried at 180 °C overnight, reduced at 300 °C in a H2 atmosphere (25 mL min−1) for 3 h, and cooled to rt under N2 (25 mL min−1) for 30 minutes. The sample was then washed with Milli-Q water (50 mL) and dried under vacuum (5 mbar) at 70 °C for 12 hours before any use.
Catalyst characterization
Once prepared, Ru–WOx/C systems were characterized by ICP, XPS, TEM, XRD, and NH3-TPD.ICP-MS analyses were performed using a Perkin Elmer Optima 5300DV instrument. Analyses of the fresh and used (post-reaction) catalysts (Tables 1 and 3) were performed after digestion in the presence of a highly oxidant solution under MW irradiation. Details of the digestion procedure and the analytical protocol for ICP measures are reported in the ESI.†
High-resolution scanning transmission electron microscopy (HRSTEM), high-angle annular dark-field (HAADF)-STEM and EDX analyses were recorded on a Hitachi HD-2700 STEM instrument and a JEM-ARM 200F STEM instrument both operated at 200 kV. The samples for TEM were prepared by directly dispersing the fine powders of the products onto a micro-grid carbon polymer supported on a copper grid.
X-ray powder diffraction (XRD) patterns of the catalysts were recorded using a Philips X'Pert powder diffractometer (Bragg–Brentano parafocusing geometry). Nickel-filtered CuKα1 radiation (λ = 0.15406 nm) and a voltage of 40 kV were employed.
X-ray photoelectron spectroscopy (XPS) was performed on a Perkin Elmer Φ 5600ci spectrometer using nonmonochromatic Al Kα radiation (1486.6 eV) in the 10−7 Pa pressure range (other details are in the ESI†).
NH3 temperature-programmed desorption (TPD) was performed using a powdered sample of the catalyst (60 mg) which was sieved at 70–100 μm (140–200 mesh) and then loaded in a quartz microreactor with an internal diameter of 8 mm. The outlet of the reactor was directly connected to a UV analyzer specific for NH3 analysis (Limas 11HW, ABB). The sample was pretreated under a He flow at a flow rate of 60 Ncm3 min−1 in the microreactor at 300 °C for 1 h. Then, the catalyst was cooled to 50 °C and at this temperature, NH3 (1000 ppm) was added stepwise to the gas mixture for 30 minutes (reaching the steady state). The NH3 supply was closed and it was purged in He until the baseline was stable. Finally, the catalyst was heated under temperature programming (TPD) up to 600 °C (heating rate 10 °C min−1).
CO chemisorption experiments were carried out on a powdered sample of the catalyst (60 mg). A first CO adsorption isotherm (@40 °C) was achieved to measure the total amount of adsorbed carbon monoxide (chemisorbed and physisorbed). The catalyst was then outgassed, and a second CO adsorption isotherm was measured to evaluate the amount of physisorbed CO. The total amount of chemisorbed CO was obtained by subtracting the second isotherm from the first one. It should be noted that before the adsorption isotherm measurements, the sample surface was exposed to a H2 atmosphere (4% v/v) for 0.5 h at 500 °C and 1 h at the same temperature in He so as to reduce the surface metal oxide possibly formed during the catalyst storage under air. From chemisorption measurements, the proportion of accessible metal located at the surface of the Ru particles, i.e. the dispersion DRu, was calculated from the following expression:
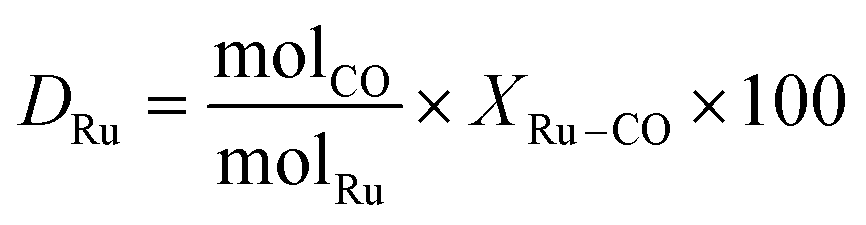
Acid-treated carbon (Cacid-treated: Table 4)
Finely powdered carbon (300 mg, NORIT SX 1G) was suspended and stirred in Milli-Q water (15 mL). The aqueous suspension and the resulting mixture was stirred overnight at rt. It was then heated at 80 °C, aq. HCl (0.3 mL; 15 wt%) was added, and the mixture was kept at 80 °C for 3 h. Thereafter, water was slowly evaporated at 95 °C. The black powder was collected, dried at 180 °C overnight, reduced at 300 °C in H2 a atmosphere (25 mL min−1) for 3 h, and cooled to rt under N2 (25 mL min−1) for 30 minutes. The sample was then washed with Milli-Q water (50 mL) and dried under vacuum (5 mbar) at 70 °C for 12 hours before any use.Reaction procedure
In a typical hydrogenolysis experiment, a 25 mL tubular reactor of borosilicate glass (Pyrex) was charged with 5 mL of a 5% w/w glycerol/water solution and the catalyst of choice (150 mg, selected among those reported in Table 2). The vessel was placed in a jacketed steel autoclave equipped with a manometer and two needle valves by which, at rt, H2 was admitted at the desired pressure. The autoclave was then heated by oil circulation at the desired temperature (120–170 °C), while the mixture was kept under magnetic stirring at a rate of 1300 rpm. After 6–12 hours, the autoclave was cooled to rt and purged. An aliquot (0.5 mL) of the water solution was collected, mixed with an aqueous solution of triethylene glycol dimethyl ether as external standard [triglyme, MeO(CH2CH2O)3Me; 0.01 M, 0.5 mL], and analysed by GC and GC/MS to determine the conversion of glycerol and the selectivity towards liquid products (Table 2) and to confirm their structure.GC-MS (EI, 70 eV) and GC/FID analyses were performed using an HP5-MS capillary column (L = 30 m, Ø = 0.32 mm, film = 0.25 mm) and an Elite-624 capillary column (L = 30 m, Ø = 0.32 mm, film = 1.8 mm), respectively.
GC calibration curves for the reactant (glycerol) and the major liquid products (EG, 1,2-PDO, 2-PrOH, and EtOH) are reported in the ESI† (Fig. S9–S13). Gaseous by-products were collected after the reaction catalysed by Ru/C was complete (Fig. S1†): an aliquot (ca. 2 L) of the gaseous mixture vented from the autoclave was conveyed to a rubber reservoir and analysed by GC/MS. This confirmed the formation of CO, CH4, and propane.
The same reaction procedure was repeated by changing the glycerol concentration from 5 wt% to 7.5, 10, 15 and 20 wt% (150 °C, 5 bar, 12 h; Fig. S3†).
Conclusions
The potential of new bimetallic Ru–WOx/C catalysts has been explored for the selective synthesis of 1,2-PDO by the hydrogenolysis of glycerol in aqueous solutions. Among the salient aspects of this study, it has been demonstrated that (i) glycerol conversion substantially improves up to 100% by reducing the Ru:W molar ratio up to 4:1 mol mol−1. Further reduction of this ratio is impracticable as it leads to catalyst reproducibility issues at W loadings >2 wt%; (ii) albeit WOx (supported on C) is totally ineffective for the reaction, W-doping of Ru-based systems is crucial to boost the reaction selectivity up to 97–98%. In contrast, Ru/C alone favors multiple C–C bond cleavage side-reactions, yielding ethylene glycol as the major product in the liquid phase along with a large amount of gaseous derivatives (carbon loss of 51%); (iii) Ru–WOx/C catalysts can be recycled without any loss of activity and selectivity. Leaching of W occurs in the aqueous solution, but it diminishes and stabilizes after a few repeated runs, and most of all, it is uninfluential in the catalyst performance.
Overall, the here described protocol offers conversion and selectivity equally efficient as those of the best existing methods for the synthesis of 1,2-PDO from glycerol (based on Ru/Cu-based systems), with the advantage of requiring far milder reaction conditions (150 °C and 5 bar vs. 180–230 °C and 80–100 bar). The result is consistent with the complementary information offered by STEM and TPD characterization techniques on Ru and WOx components of the bimetallic system. This leads one to hypothesize a synergic effect between the two metals in the form of Ru nanoparticles uniformly dispersed on the carbon support and in close proximity to different WOx species whose structure, although not conclusively defined, may include monotungstate, polytungstate and crystalline clusters. The proposed mechanism takes into account the acid character (both of Brønsted and Lewis type) of the WOx sites which allows for the adsorption via H-bonding and dehydration of glycerol to prop-2-ene-1,2-diol (PED), while Ru is responsible for the hydrogenation of PED to 1,2-PDO via hydrogen spillover onto the surface. The total sequence is positively affected by the concentration of acid sites which increases with the W-loading. However, in analogy to the behavior reported for other W-based catalysts, a high metal loading (>2 wt%) brings about the formation of tungsten species loosely adsorbed on the support that may cause leaching and adverse effects on the catalyst preparation.
Author contributions
Alessandro Bellè: conceptualization, investigation, methodology, writing – original draft preparation; Kohei Kusada: investigation, methodology; Hiroshi Kitagawa: supervision, validation; Alvise Perosa: writing – reviewing, funding acquisition, validation; Maurizio Selva: conceptualization, supervision, writing – reviewing and editing, funding acquisition; Daniele Polidoro: investigation; Lidia Castoldi: investigation, methodology, writing – draft preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr. K. Kusada gratefully acknowledges Ca′ Foscari University for granting a visiting scholarship for his stay in Venezia (June–August 2019; public grant no. 777/2018, profile 11J). Dr. T. Tabanelli at the University of Bologna, Prof. E. Cattaruzza at Ca′ Foscari University and are gratefully acknowledged for their help in the synthesis and characterization of some catalysts.References
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Glycerol, Ullmann's Encyclopedia of Industrial Chemistry, 2012, vol. 17, pp. 67–82 Search PubMed.
- J. Kaur, A. K. Sarma, M. K. Jha and P. Gera, Biotechnol. Rep., 2020, 27, e00487 CrossRef.
- M. K. Aroua and P. Cognet, Front. Chem., 2020, 8, 69 CrossRef PubMed.
- G. N. Oliveira, C. Barbosa, F. C. Araújo, P. H. G. Souza, A. V. H. Soares, F. C. Peixoto, J. W. M. Carneiro and F. B. Passos, in Jatropha, Challenges for a New Energy Crop, ed. S. Mulpuri, N. Carels and B. Bahadur, Springer, Singapore, 2019, pp. 383–414 Search PubMed.
- A. D. da Silva Ruy, R. M. de Brito Alves, T. L. R. Hewer, D. de Aguiar Pontes, L. Sena Gomes Teixeira and L. A. Magalhães Pontes, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.06.035 , in the press.
- H. Zhao, L. Zheng, X. Li, P. Chen and Z. Hou, Catal. Today, 2019, 355, 84–95 CrossRef.
- Y. Wang, J. Zhou and X. Guo, RSC Adv., 2015, 5, 74611–74628 RSC.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Appl. Catal., B, 2016, 193, 75–92 CrossRef CAS.
- M. A. Dasari, P.-P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231 CrossRef CAS.
- A. Perosa and P. Tundo, Ind. Eng. Chem. Res., 2005, 44, 8535–8537 CrossRef CAS.
- B. K. Kwak, D. S. Park, Y. S. Yun and J. Yi, Catal. Commun., 2012, 24, 90–95 CrossRef CAS.
- S. H. Zhu, X. Q. Gao, Y. L. Zhu, Y. F. Zhu, H. Y. Zheng and Y. W. Li, J. Catal., 2013, 303, 70–79 CrossRef CAS.
- Y. Du, C. Wang, H. Jiang, C. Chen and R. Chen, Ind. Eng. Chem., 2016, 35, 62–267 Search PubMed.
- N. N. Pandhare, S. M. Pudi, P. Biswas and S. Sinha, Org. Process Res. Dev., 2016, 20, 1059–1067 CrossRef CAS.
- P. Kumar, A. K. Shah, J.-H. Lee, Y. H. Park and U. Stangar, Ind. Eng. Chem. Res., 2020, 59, 6506–6516 CrossRef CAS.
- Z. L. Yuan, P. Wu, J. Gao, X. Y. Lu, Z. Y. Hou and X. M. Zheng, Catal. Lett., 2009, 130, 261–265 CrossRef CAS.
- X. Zhang, G. Cui and M. Wei, Ind. Eng. Chem. Res., 2020, 59, 12999–13006 CrossRef CAS.
- J. Liu, L. Ruan, J. Liao, A. Pei, K. Yang, L. Zhu and B. Hui Chen, New J. Chem., 2020, 44, 16054–16061 RSC.
- R. B. Mane, S. T. Patil, H. Gurav, S. S. Rayalu and C. V. Rode, ChemistrySelect, 2017, 2, 1734–1745 CrossRef CAS.
- R. Mane, S. Patil, M. Shirai, S. Rayalu and C. Rode, Appl. Catal., B, 2017, 204, 134–146 CrossRef CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 329, 30–35 CrossRef CAS.
- J. Feng, W. Xiong, B. Xu, W. D. Jiang, J. B. Wang and H. Chen, Catal. Commun., 2014, 46, 98–102 CrossRef CAS.
- A. V. H. Soares, J. B. Salazar, D. D. Falcone, F. A. Vasconcellos, R. J. Davis and F. B. Passos, J. Mol. Catal. A: Chem., 2016, 415, 27–36 CrossRef CAS.
- T. Jiang, Y. X. Zhou, S. G. Liang, H. Z. Liu and B. X. Han, Green Chem., 2009, 11, 1000–1006 RSC.
- H. Liu, S. Liang, T. Jiang, B. Han and Y. Zhou, Clean: Soil, Air, Water, 2012, 40, 318–324 CAS.
- A. Bellè, T. Tabanelli, G. Fiorani, A. Perosa, F. Cavani and M. Selva, ChemSusChem, 2019, 12, 3343–3354 CrossRef.
- W. Zhou, J. Luo, Y. Wang, J. Liu, Y. Zhao, S. Wang and X. Ma, Appl. Catal., B, 2019, 242, 410–421 CrossRef CAS.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, ACS Catal., 2019, 9, 10913–10930 CrossRef CAS.
- Y. Liu, C. Luo and H. Liu, Angew. Chem., 2012, 124, 3303–3307 CrossRef.
- L. Wei, R. Bibi, W. Tian, L. Chen, Y. Zheng, N. Li and J. Zhou, New J. Chem., 2018, 42, 3633–3641 RSC.
- C. Li, G. Xu, X. Zhang and Y. Fu, Chin. J. Chem., 2020, 38, 453–457 CrossRef CAS.
- M. Yabushita, N. Shibayama, K. Nakajima and A. Fukuoka, ACS Catal., 2019, 9, 2101–2109 CrossRef CAS.
- M. Pagliaro and M. Rossi, in The Future of Glycerol, New Uses of a Versatile Raw Material, ed. J. H Clark and G. A. Kraus, RSC Green Chemistry Book Series, 2010 Search PubMed.
- S. Fulignati, C. Antonetti, D. Licursi, M. Pieraccioni, E. Wilbersb, H. J. Heeresb and A. M. Raspolli Galletti, Appl. Catal., A, 2019, 578, 122–133 CrossRef CAS.
- C. Rajkumar, B. Thirumalraj, S. M. Chen, P. Veerakumar and S.-B. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 31794–31805 CrossRef CAS PubMed.
- A. S. Aricò, Z. Poltarzewski, H. Kim, A. Morana, N. Giordano and V. Antonucci, J. Power Sources, 1995, 55, 159–166 CrossRef.
- W. Zang, G. Li, L. Wang and X. Zhang, Catal. Sci. Technol., 2015, 5, 2532–2553 RSC.
- A. M. Robinson, J. E. Hensley and J. Will Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- M. Balaraju, V. Rekha, P. S. Sai Prasad, B. L. A. Prabhavathi Devi, R. B. N. Prasad and N. Lingaiah, Appl. Catal., A, 2009, 354, 82–87 CrossRef CAS.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Catal. Lett., 2008, 120, 307–311 CrossRef CAS.
- D. G. Barton, S. L. Soled, G. D. Meitzner, G. A. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57–72 CrossRef CAS.
- V. I. Baranenko and V. S. Kirov, Sov. At. Energy, 1989, 66, 30–34 CrossRef.
- H. A. Pray, C. E. Schweickert and B. H. Minnich, Ind. Eng. Chem., 1952, 44, 1146–1151 CrossRef CAS.
- L. Ma, D. He and Z. Li, Catal. Commun., 2008, 9, 2489–2495 CrossRef CAS.
- L. Guo, J. Zhou, J. Mao, X. Guo and S. Zhang, Appl. Catal., A, 2009, 367, 93–98 CrossRef CAS.
- F. K. Kazi, A. D. Patel, J. C. Serrano-Ruiz, J. A. Dumesic and R. P. Anex, Chem. Eng. J., 2011, 169, 329–338 CrossRef CAS.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 2375–2380 RSC.
- X.-L. Yang, R. Gao, W.-L. Dai and K. Fan, J. Phys. Chem. C, 2008, 112, 3819–3826 CrossRef CAS.
- N. Tammanoon, T. Iwamoto, T. Ueda, T. Hyodo, A. Wisitsoraat, C. Liewhiran and Y. Shimizu, ACS Appl. Mater. Interfaces, 2020, 12, 41728–41739 CrossRef CAS PubMed.
- K. Qadir, S. H. Joo, B. S. Mun, D. R. Butcher, J. Russell Renzas, F. Aksoy, Z. Liu, G. A. Somorjai and J. Y. Park, Nano Lett., 2012, 12, 5761–5768 CrossRef CAS.
- W. Wang, Y. Li and H. Wang, React. Kinet., Mech. Catal., 2013, 108, 433–441 CrossRef CAS.
- Z. Cui, L. Feng, C. Liu and W. Xing, J. Power Sources, 2011, 196, 2621–2626 CrossRef CAS.
- W. Zhou, J. Luo, Y. Wang, J. Liu, Y. Zhao, S. Wang and X. Ma, Appl. Catal., B, 2019, 242, 410–421 CrossRef CAS.
- S. Zhu, X. Gao, Y. Zhu and Y. Li, J. Mol. Catal. A: Chem., 2015, 398, 391–398 CrossRef CAS.
- X.-Y. Liu, M. Huang, H.-L. Ma, Z.-Q. Zhang, J.-M. Gao, Y.-L. Zhu, X.-J. Han and X.-Y. Guo, Molecules, 2010, 15, 7188–7196 CrossRef CAS.
- The authors wish to thank a referee for having prompted further investigations on the acidity of the carbon support.
- H. Atia, U. Armbruster and A. Martin, J. Catal., 2008, 258, 71–82 CrossRef CAS.
- F. Micoud, F. Maillard, A. Bonnefont, N. Job and M. Chatenet, Phys. Chem. Chem. Phys., 2010, 12, 1182–1193 RSC.
- Y. Zhang, X.-C. Zhao, Y. Wang, L. Zhou, J. Zhang, J. Wang, A. Wanga and T. Zhang, J. Mater. Chem. A, 2013, 1, 3724–3732 RSC.
- S. Feng, B. Zhao, L. Liu and J. Dong, Ind. Eng. Chem. Res., 2017, 56, 11065–11074 CrossRef CAS.
- S. Zhu, Y. Zhu, S. Hao, L. Chen, B. Zhang and Y. Li, Catal. Lett., 2012, 142, 267–274 CrossRef CAS.
- H. Markus, P. Maki-Arvela, N. Kumar, N. V. Kulkova, P. Eklund, R. Sjoholm, B. Holmbom, T. Salmi and D. Yu. Murzin, Catal. Lett., 2005, 103, 125–131 CrossRef CAS.
- H. Markus, A. J. Plomp, P. Maki-Arvela, J. H. Bitter and D. Yu. Murzin, Catal. Lett., 2007, 113, 141–146S CrossRef CAS.
- S. Liua, S. Reddy Musukua, S. Adhikarib and S. Fernando, Environ. Technol., 2009, 30, 505–510 CrossRef.
- (a) J. Bedia, J. M. Rosas, J. Marquez, J. Rodrıguez-Mirasol and T. Cordero, Carbon, 2009, 47, 286–294 CrossRef CAS; (b) G. Szymajski and G. Rychlicki, Carbon, 1991, 29, 489–498 CrossRef.
- S. Cao, J. R. Monnier and J. R. Regalbuto, J. Catal., 2017, 347, 72–78 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S13 and Schemes S1 and S2. See DOI: 10.1039/d1cy00979f |
| This journal is © The Royal Society of Chemistry 2022 |