
Tantalum based single, double, and triple atom catalysts supported on g-C2N monolayer for effective nitrogen reduction reaction: a comparative DFT investigation†
Anjumun
Rasool
a,
Insha
Anis
a,
Mudit
Dixit
 b,
Ashakiran
Maibam
cd,
Afshana
Hassan
a,
Sailaja
Krishnamurty
*cd and
Manzoor Ahmad
Dar
*a
b,
Ashakiran
Maibam
cd,
Afshana
Hassan
a,
Sailaja
Krishnamurty
*cd and
Manzoor Ahmad
Dar
*a
aDepartment of Chemistry, Islamic University of Science and Technology, Awantipora, Jammu and Kashmir-1920221, India. E-mail: manzoor.dar@islamicuniversity.edu.in
bDepartment of Chemistry, Lovely Professional University, Phagwara, Punjab, India
cPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune 411 008, India. E-mail: sailaja.raaj@gmail.com
dAcademy of Scientific and Innovative Research, CSIR-Human Resource Development Centre (CSIR-HRDC) Campus, Postal Staff College area, Gaziabad, 201 002, Uttar Pradesh, India
First published on 12th November 2021
Abstract
Design of efficient and low cost electrocatalysts for the reduction of N2 molecule to NH3 in a green manner remains a great challenge in the 21st century. Herein, we have used density functional theory based first principle simulations to systematically investigate the nitrogen reduction reaction (NRR) ability of single, double, and triple Ta-atom catalysts anchored to C2N monolayer. Our results demonstrate that the single and triple Ta-atom catalysts anchored to C2N monolayer act as superior catalysts for the NRR via alternating and distal pathways as compared to the Ru(0001) stepped surface. In particular, the triple Ta-atom catalyst anchored to C2N shows enhanced NRR performance with a limiting potential of −0.72 V which is comparable to the experimentally reported Ru based single atom catalyst. Further, all the three catalysts were found to be highly selective for NRR with an enhanced ability to suppress the competitive hydrogen evolution reaction. Electronic structure analysis revealed that the enhanced ability of Ta3@C2N catalyst to effectively capture and reduce N2 molecule could be attributed to the built up of localized d states near the fermi level, thereby aiding in strong electron transfer into the antibonding orbitals of N2. Thus, our findings propose a highly active catalyst for the NRR with an emphasis on the importance of triple atom-based catalysts for electrocatalytic applications.
1. Introduction
Conversion of atmospheric nitrogen (N2), one of the most abundant and chemically inert molecules in nature, into ammonia (NH3) at a large scale is of paramount importance for producing fertilizers to speed up agricultural growth for sustenance of life on earth.1–3 Further, a high gravimetric hydrogen density coupled with ease of liquification makes ammonia a safe carbon free energy storage intermediate with no emission of harmful greenhouse gases like carbon dioxide into the atmosphere.4,5 At present the industrial conversion of N2 to NH3 follows the conventional Haber–Bosch process which is highly energy-intensive and results in release of significant amount of harmful greenhouse gases into the atmosphere.6–8 To achieve more sustainable and environmentally friendly way of ammonia production on an industrial scale, design of efficient electrocatalysts is the core component to make the nitrogen reduction reaction (NRR) kinetically and thermodynamically feasible.Over the years, transition metal-based systems at different length scales have been at the facade of research for electrocatalyst design for efficient reduction of N2. For example, two-dimensional transition metal dichalcogenides,9 nitrides and carbides,10 and boron nanosheets11–13 have demonstrated improved electrocatalytic activity for activation and reduction of N2 molecule to NH3. In addition, size selected monometallic and bimetallic transition metal clusters14,15 supported on variable supports have been thoroughly explored as catalysts for NRR. The catalytic performance of nanomaterials for NRR has been shown to be highly dependent on factors such as size, shape, crystal phase and degree of amorphization.16 Although a wide variety of nanomaterials have been explored and developed for the electrocatalytic NRR, the efficiency of NRR is still not comparable for the industrial synthesis of NH3.
Recently size reduction of matter to a few atoms has been foreseen to be a valuable strategy to boost the catalytic ability of materials.17–21 Particularly single atom catalysts consisting of single metal atoms dispersed on variable supports has proven to be effective for catalysing diverse reactions. The drastic improvement in the catalytic ability of SACs has been ascribed to factors such as maximum atom utilization, unique quantum size effects and unsaturated catalytic sites.22–24 Due to these reasons increasing number of studies have been carried out to understand the NRR ability of transition metal-based SACs dispersed on different types of supporting materials. For example, Liu et al.25 using extensive density functional theory-based simulations on 20 different transition metal SACs dispersed on three different nitrogen-doped carbon substrates proposed a two-step strategy for improving the NRR activity. By further tuning the adsorption strength of the key intermediates as a function of the coordination environment of the already predicted promising SACs, the authors concluded that Ru@g-C3N4 and Rh@g-C3N4 act as best NRR catalysts. Sun and co-workers26 revealed that the coordination environment around the single molybdenum catalyst plays an important role in the NRR by tuning the d-band center around the Fermi level. Further, many theoretical works27–43 have been carried out to understand the underlying intrinsic factors controlling the activity of a wide variety of SACs anchored to different supports, leading to successful descriptors for designing promising SAC candidates for efficient NRR.
Beyond SACs, double and triple atom catalysts comprising two and three metal atoms anchored to suitable supports have emerged as potential catalysts.44 In the case of NRR, some of the recent theoretical studies45–49 concluded that mono and hetero-atomic double atom catalysts (DACs) act as better catalysts with enhanced activity and selectivity as compared to their corresponding SACs. Similarly, triple atom catalysts (TACs)50,51 of different metal atoms have also been investigated in some of the very recent research works for NRR. In one such interesting study,52 Fe3 and Fe4 catalysts supported on nitrogen-doped graphene demonstrated better catalytic activity with low limiting potential for nitrogen reduction compared to single and double Fe atom catalysts. Similarly, spin-polarized density functional theory calculations by Li et al.53 on Mox (x = 1–4) supported on graphdiyne (GDY) revealed that Mo3 acts as a superior catalyst for NRR. In contrast to the above studies, single and double atom catalysts of Cu were found to be proficient for catalysing NRR by Ma and co-workers54 during the computational evaluation of transition metal™ single, double and triple atom catalysts (TM = Mn, Fe, Co, Ni) supported on graphdiyne monolayers. The above studies inspire us to perform a comparative investigation on single, double, and triple atom catalysts for NRR. Thus, in this work, we have carried out spin-polarized density functional theory calculations to understand NRR activity of tantalum based single, double, and triple atom catalysts. Further, the justification for choosing tantalum in the current study lies in the fact that experimental studies have shown that small-sized gas-phase tantalum clusters strongly bind and activate nitrogen molecule. For example, Fryzuk and coworkers,55 recognized based on spectroscopic and quantum chemical analysis the ability of di-nuclear Ta complexes to bind N2 molecule in side-on and end-on geometries. Geng et al.56 using Fourier transform ion cyclotron resonance mass spectroscopy identified a planar four-membered Ta2N2+ structure to be the key intermediate for mediating N2 to NH3 conversion similar to a Haber–Bosch-like process. Geng et al.57 further revealed that N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N undergoes complete cleavage in a thermal reaction with Ta2N+ through a degenerate ligand exchange mechanism under standard conditions. More recently Ma and co-workers58 photoelectron spectroscopy results corroborated by quantum chemical calculations by Ma and co-workers showed that N2 is strongly activated by Ta3N3H and Ta3N2 cluster anions with the hydrogen atom playing a crucial role in the reactivity.
N undergoes complete cleavage in a thermal reaction with Ta2N+ through a degenerate ligand exchange mechanism under standard conditions. More recently Ma and co-workers58 photoelectron spectroscopy results corroborated by quantum chemical calculations by Ma and co-workers showed that N2 is strongly activated by Ta3N3H and Ta3N2 cluster anions with the hydrogen atom playing a crucial role in the reactivity.
To evaluate the NRR activity of Ta-based single, double, and triple atom catalysts, we employed the g-C2N monolayer as the substrate. Apart from exhibiting a very high thermal and kinetic stability,59 g-C2N possesses uniformly distributed nanopores with pyrazinic sp2 nitrogen atoms. Further, the benzene rings in the g-C2N monolayer are connected by nitrogen atoms generating a partial negative charge on the nitrogen atoms,60 thereby providing suitable sites for adsorption, and enhancing the propensity of stabilizing single, double, and triple atom catalysts.
2. Computational details
In this work, plane-wave density functional theory as implemented in Vienna ab initio simulation package (VASP)61–63 was used to perform spin-polarized first-principles calculations with the electron–ion interaction being described by the projector augmented wave (PAW) method.64 The exchange-correlation energy is treated by the Perdew–Burke–Ernzerhof (PBE) functional65 and Grimme's semiempirical DFT-D3 scheme66 of dispersion correction was employed to account for the weak van der Waals (vdW) interaction. A kinetic energy cut off 500 eV was used for the plane-wave basis. 10−7 eV was set as the convergence criteria for the energy and structure optimizations were performed until the Hellmann–Feynman force on each atom was less than 10−3 eV Å−1. A 2 × 2 C2N monolayer supercell with a vacuum space of 15 Å along the z-direction was applied to avoid the interaction between two periodic units. Further, a Monkhorst–Pack mesh67 of 3 × 3 × 1 for structural optimization and 6 × 6 × 1 for the calculation of electronic properties was used.The binding energy per atom (Eb) for the Ta based catalysts45 on the C2N monolayer and the N2 adsorption energy on the Tan@C2N catalysts were obtained using the given below equations
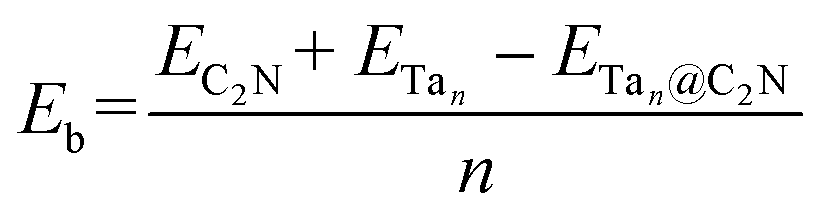
| Eads = EN2 + ETan@C2N − ETan@C2N–N2 |
The reaction free energy (ΔG) for each elementary step was calculated based on the computational hydrogen electrode (CHE) model proposed by Nørskov and co-workers68–70 by the following equation
| ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH |
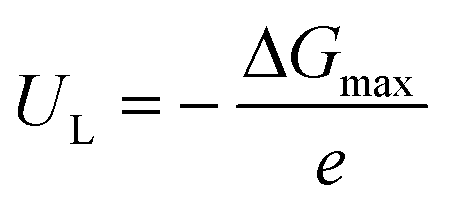
3. Results and discussion
We begin with a discussion on the structure, stability, and electronic properties of Tan (n = 1–3) species adsorbed on the C2N monolayer. To obtain the most stable structure of Tan@C2N systems, several possible adsorption patterns of Tan on C2N were considered. Fig. 1(a) represents the lowest energy structures of Tan@C2N catalysts with relevant geometrical parameters. The binding energy per atom of the Ta-monomer, dimer, and trimer species with C2N monolayer are also given in Fig. 1(a). As can be seen from the figure, the single Ta atom prefers to remain in the C2N plane with two Ta–N bonds and a significantly large binding energy of 8.50 eV. For the Ta2@C2N, one of the Ta resides inside the C2N plane and the second Ta atom is pushed slightly above the C2N plane while as for Ta3@C2N all the three Ta atoms reside above the C2N monolayer plane. The calculated binding energy per atom for the Ta2@C2N and Ta3@C2N were found to be 10.06 and 9.87 eV, respectively. Interestingly, the calculated binding energy for the Ta monomer, dimer and trimer is higher than the reported cohesive energy per atom of 8.10 eV for bulk Ta. We also evaluated the thermal stabilities of Tan@C2N systems by carrying out ab initio molecular dynamics (AIMD) simulations at 900 K (see Fig. 1(b)) for a time of 10 picoseconds with the help of NVT ensemble and Nose–Hoover thermostat as implemented in the CP2K package.72 AIMD simulations reveal that all the atoms in the studied catalysts vibrate about their equilibrium positions with no significant structural deformation. To further understand the stability of Ta-based catalysts, we carried out climbing image nudged elastic band (CI-NEB) calculations73 to evaluate the barrier for diffusion of tantalum atom from the nanopores of g-C2N to the hexagonal rings. The results (Fig. S1†) reveal that not only is the diffusion highly endergonic with an energy requirement of 6.12 eV but also entails a very large diffusion barrier of 6.57 eV. The significantly large binding energies compared to bulk Ta metal and negligible structural deformation around 900 K indicate the enhanced thermal stability of Tan@C2N catalysts.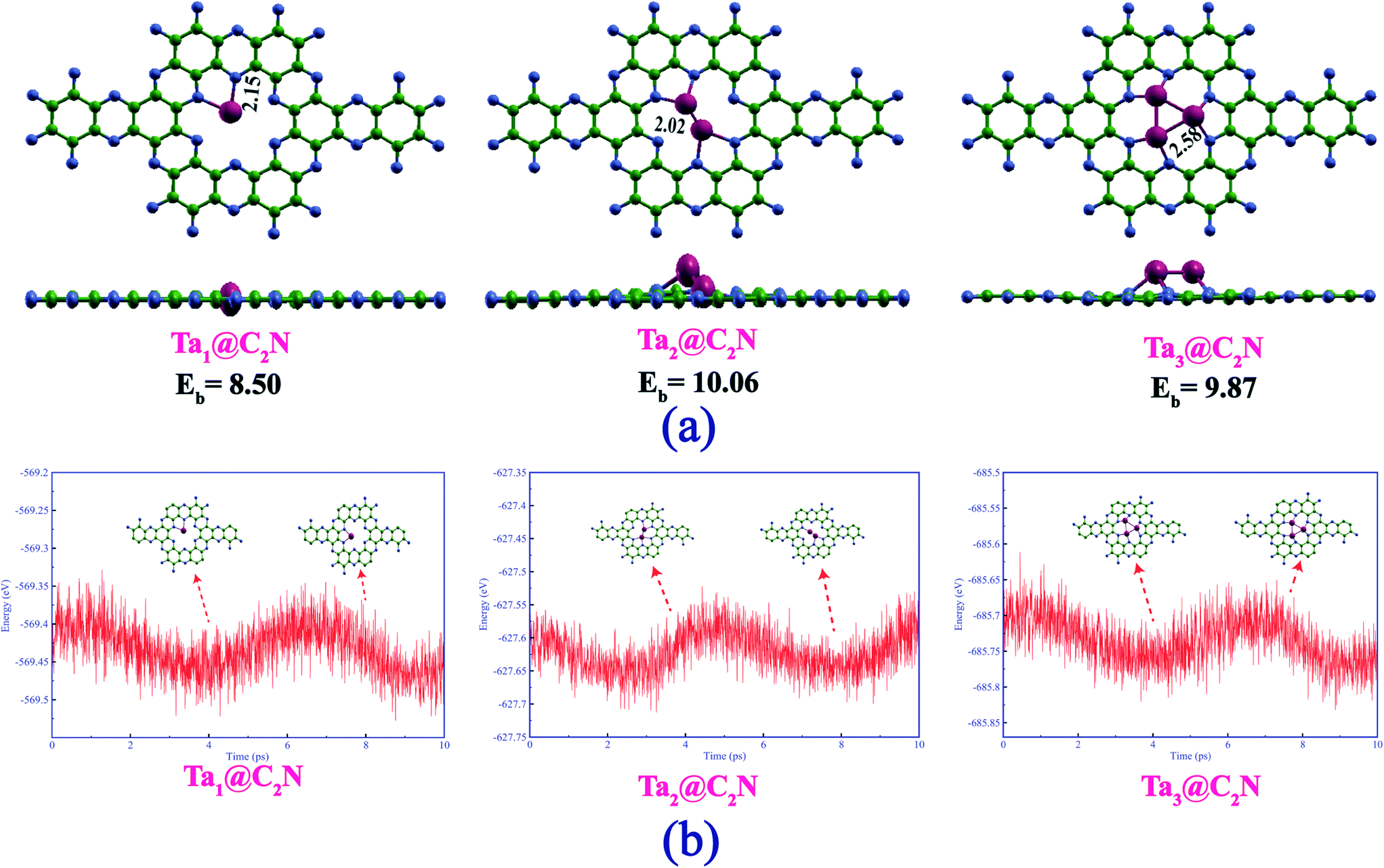 | ||
| Fig. 1 (a) Top and side view of Tan@C2N catalysts with relevant geometrical parameters. The numbers in black at the bottom are the calculated binding energy per atom in eV of the Tan (n = 1–3) species with C2N monolayer. (b) Variation of energy with respect to the AIMD simulation time in picoseconds for Tan@C2N catalysts. | ||
Since activation and adsorption of N2 is the first step towards N2 reduction process, we next looked at the N2 capturing ability of these catalysts by calculating the N2 adsorption energies in different possible adsorption modes. As can be seen from Fig. 2, nitrogen was adsorbed through side-on and end-on modes with two Ta–N and one Ta–N bonds, respectively. Table 1 highlights the computed nitrogen adsorption energies and N–N bond lengths of nitrogen molecule adsorbed on the single, double, and triple Ta atom catalysts. Our results indicate that the end-on mode of binding of N2 is preferred over the side-on mode in the Ta monomer whereas side-on mode is preferred over end-on mode for the Ta dimer and trimer catalysts. Further, there is a significant increase in the adsorption energy of N2 molecule in the side-on mode as we go from Ta1 to Ta3. A slight difference in N2 adsorption energy is noted in end-one mode of N2 adsorption. The calculated N2 adsorption energies for side-on and end-on modes were found to be 0.78, 1.88 and 3.37 eV, and 1.46, 1.35 and 1.43 eV, respectively for the single, double, and triple atom catalysts. Bader charge analysis (Table 1) demonstrates that the charge transfer to adsorbed N2 from the catalysts is larger in the side-on (0.47 to 1.65 e) mode than in the end-on (0.26 to 0.40 e) mode. The stronger binding and enhanced charge transfer with respect to the side-on N2 adsorption mode is also reflected by the more significant charge redistribution in charge density plots (Fig. 2) and increase in the population of p-states of N2 near the Fermi level (Fig. S3†). The electron transfer results in the occupation of the antibonding orbitals of the N2 molecule and subsequent N–N bond elongation. It can therefore be observed from Table 1 that the increase in N–N bond length up on adsorption follows the order Ta3@C2N > Ta2@C2N > Ta1@C2N in the side-on mode, whereas the end-on mode of N2 adsorption reveals almost similar N–N bond length.
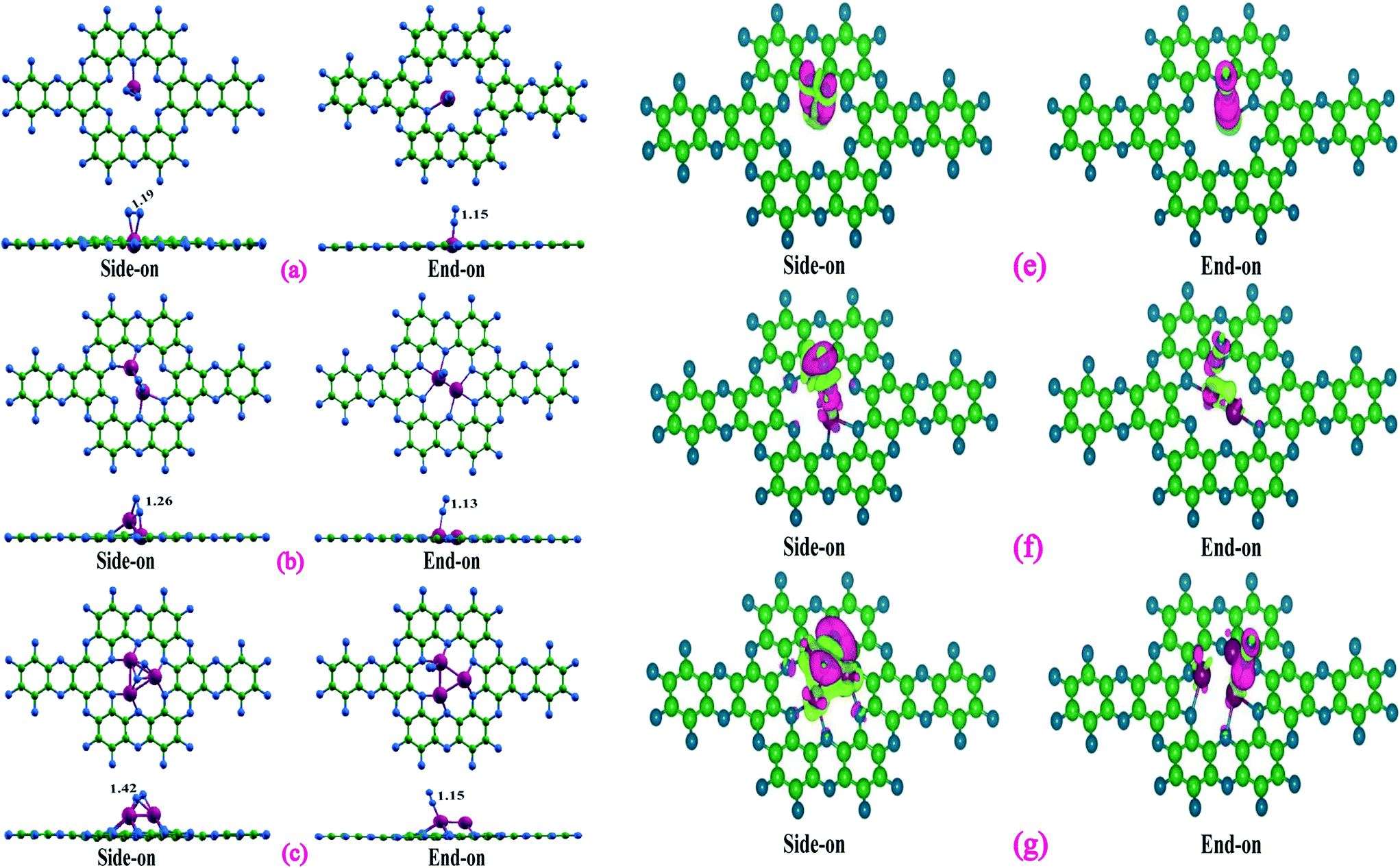 | ||
| Fig. 2 Top and side view of side-on and end-on N2 adsorption configurations on (a) Ta1@C2N (b) Ta2@C2N and (c) Ta3@C2N catalysts with relevant geometrical parameters. Charge density difference plots of side-on and end-in N2 adsorption configurations on (e) Ta1@C2N (f) Ta2@C2N and (g) Ta3@C2N catalysts. The isosurface level is set to 0.004 e Å−3. Pink indicates the electron density accumulation area, and light green indicates the electron density depletion area. | ||
| System | Binding mode | E ads (eV) | R N–N (Å) | Q b (e) |
|---|---|---|---|---|
| Ta@C2N | Side-on | 0.78 | 1.19 | −0.47 |
| End-on | 1.46 | 1.15 | −0.39 | |
| Ta2@C2N | Side-on | 1.88 | 1.26 | −1.02 |
| End-on | 1.35 | 1.13 | −0.26 | |
| Ta3@C2N | Side-on | 3.37 | 1.42 | −1.65 |
| End-on | 1.43 | 1.15 | −0.40 |
To further understand N2 binding energy trends, we performed detailed electronic structure analysis using projected density of state (PDOS) calculations. As can be seen from Fig. 3 that the PDOS of Ta1@C2N has an appreciable contribution of Ta d-states near the Fermi level in the spin-up channel. Moreover, the contribution of Ta d-states increases linearly from Ta1@C2N to Ta3@C2N (Fig. 3, cyan box). This increase in the electronic population of Ta d-states near the Fermi level facilitates the charge transfer and enables effective N2 adsorption. Indeed, a linear relationship (Fig. S3 and Table S1†) between the total integrated PDOS of Ta d-states near the Fermi level (in the range of −2.0 – 0.0 eV) versus the binding energies of N2 rationalizes the increase in corresponding N2 binding energies for side-on mode. To further quantify the N2 binding energy results, we plotted the IPDOS of N2 p-states near Fermi level (−2.0–0.0 eV) against N2 binding energy. Interestingly, a linear relationship between the N2 p-states IPDOS and binding energy was identified (Fig. S3†), suggesting that the binding energy of N2 in the side-on mode results due to strong hybridization between N2 p- and Ta d-states. In the end-on mode, N2 binds with catalysts through a single nitrogen atom, therefore, the overlap of N2 p-states with metal d-states is expected to be lower than the side-on mode. The visual inspection of PDOS (Fig. 4) clearly confirms that the extent of hybridization of N2 (p) with Ta (d) states near Fermi level follow the order Ta1@C2N > Ta3@C2N > Ta2@C2N, explaining the N2 binding energy trends qualitatively. Additionally, as the side-on and end-on modes of adsorption of N2 molecules involves different nature of N2 binding, the relationship between N2 binding energies and N2 p-states IPDOS is found to be mode dependent (Fig. S3c†).
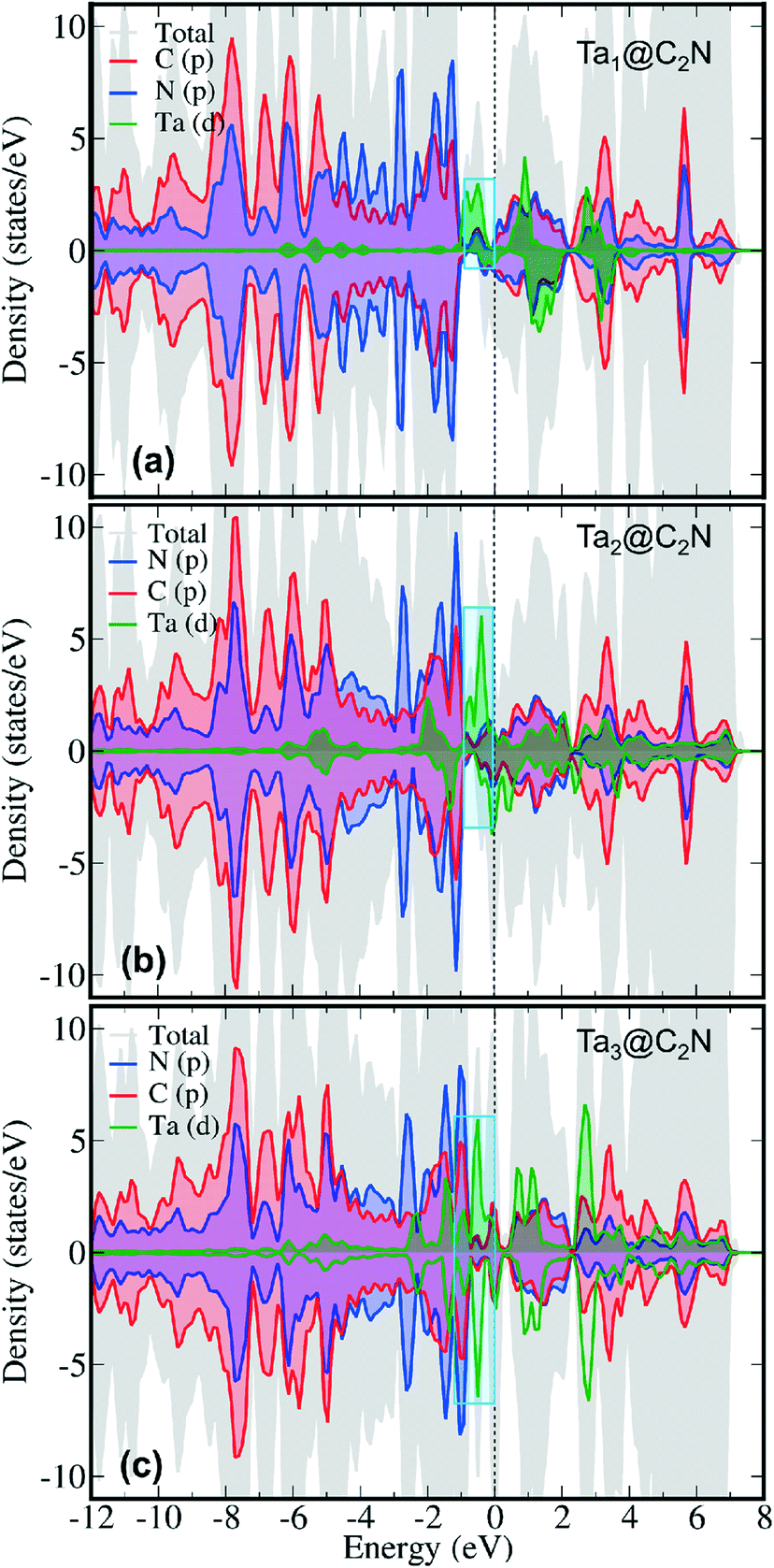 | ||
| Fig. 3 Spin-polarized orbital projected DOS (oPDOS) of (a) Ta1@C2N (b) Ta2@C2N and (c) Ta3@C2N catalysts. Colour codes for PDOS: grey – total, red – carbon (p), blue – nitrogen (p), and green – Ta (d). Fermi level is set has been zero. The PDOS is truncated within −12 to 8 eV for the sake of clarity. | ||
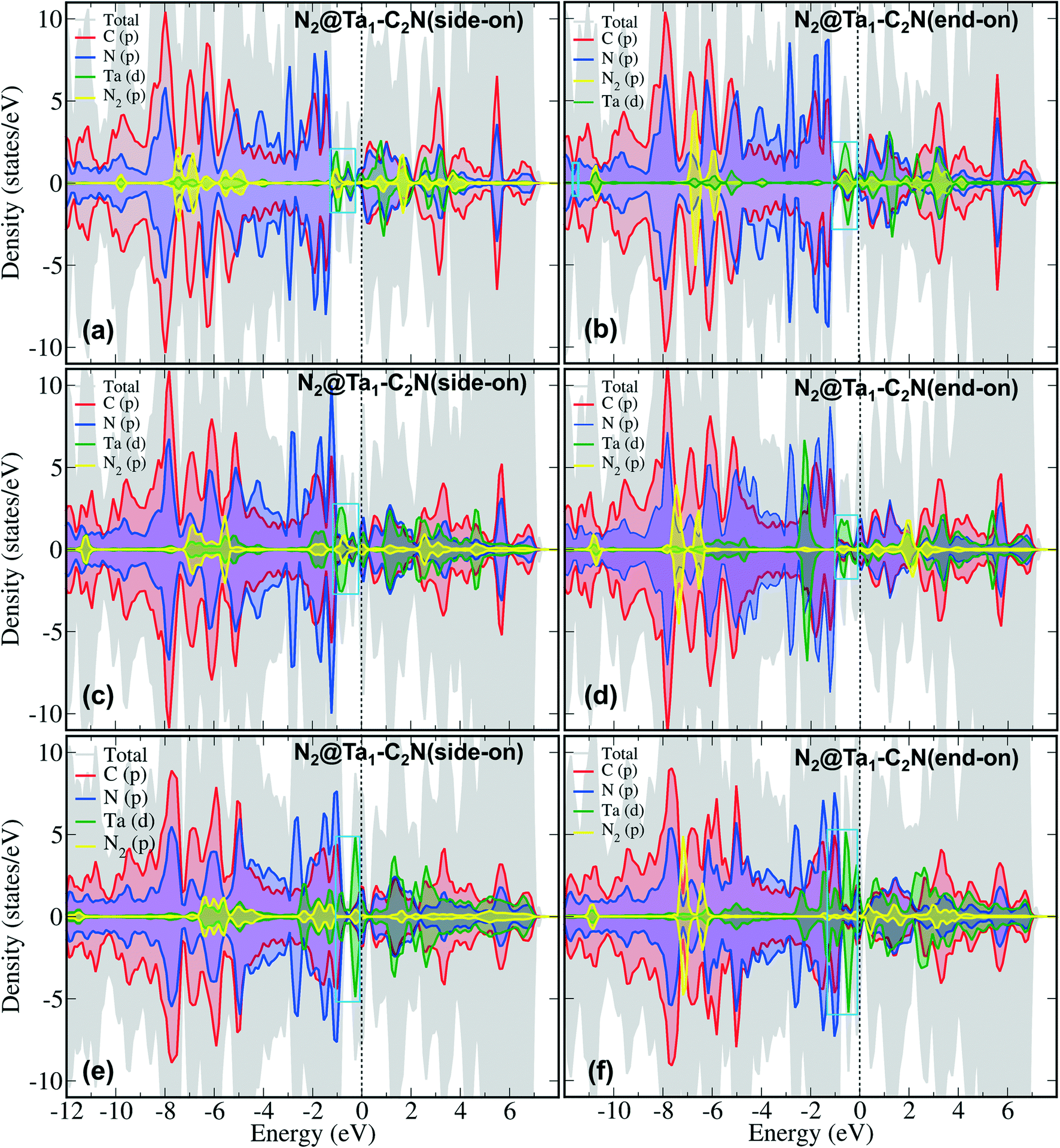 | ||
| Fig. 4 Spin-polarized oPDOS for the N2 adsorbed configurations on Tan@C2N in the side-on (a) Ta1@C2N (c) Ta2@C2N (e) Ta3@C2N and end-on (b) Ta1@C2N (d) Ta2@C2N (f) Ta3@C2N, mode. The population of Ta (d) states near Fermi level is highlighted with cyan boxes. Colour codes for PDOS: grey – total, red – carbon (p), blue – nitrogen (p), and green – Ta (d). Fermi level is set has been zero. The PDOS is truncated within −12 to 8 eV for the sake of clarity. | ||
We next explored the transformation of N2 to two NH3 molecules via the three already well-known mechanisms for NRR. According to earlier studies, the alternating and distal mechanisms are associated with end-on mode of adsorption whereas the enzymatic mechanism is associated with side-on mode of adsorption. In the distal mechanism, the proton–electron pairs initially attack the nitrogen atom not attached to the catalyst surface resulting in the formation of the first NH3 molecule and then the formation of the second NH3 molecule by attacking the left-over N-atom. In contrast, the alternating and enzymatic mechanisms involve the alternate attack of the proton–electron pairs on the two N atoms leading to the consecutive formation of two NH3 molecules. It is important to mention here that although N2 adsorbs strongly in the end-on mode in single atom tantalum catalyst and side-on mode on the double and triple atom tantalum catalysts, the side-on mode in single tantalum catalyst and the end-on modes in the double and triple tantalum catalysts are also considerably thermodynamically favorable. Thus, in an ensemble picture the side-on mode for single atom and end-on mode of N2 adsorption for double and triple atom catalysts will also be populated, thereby, making it imperative to evaluate all the three known pathways i.e., alternating, distal, and enzymatic for NRR on these catalysts. Before, we investigate the detailed NRR mechanism on the Tan@C2N catalysts, earlier studies have exclusively pointed that due to the inert nature of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond, the first proton–electron pair transfer step requires a large input of energy and is quite often the PDS. Therefore, we calculated the ΔG values for the
N triple bond, the first proton–electron pair transfer step requires a large input of energy and is quite often the PDS. Therefore, we calculated the ΔG values for the 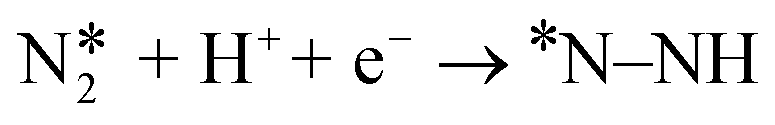 step on all the three catalysts for both side-on and end-on N2 modes and are presented in Table 2.
step on all the three catalysts for both side-on and end-on N2 modes and are presented in Table 2.
| Catalyst | Pathway | ΔG*NNH | PDS | U L (V) |
|---|---|---|---|---|
| Ta1@C2N | Alternating | 0.79 | *N–N + H+ + e− → *N–NH | −0.79 |
| Distal | 0.79 | *N–NH2 + H+ + e− → *N + NH3 | −1.39 | |
| Enzymatic | 0.21 | *NH–*NH2 + H+ + e− → *NH2–*NH2 | −0.97 | |
| Ta2@C2N | Alternating | 0.94 | *N–N + H+ + e− → *N–NH | −0.94 |
| Distal | 0.94 | *N–NH + H+ + e− → *N–NH2 | −1.15 | |
| Enzymatic | −0.51 | *NH2–*NH2 + H+ + e− → *NH2–NH3 | −1.04 | |
| Ta3@C2N | Alternating | 0.09 | *NH–NH2 + H+ + e− → *NH2–NH2 | −1.11 |
| Distal | 0.09 | *NH + H+ + e− → *NH2 | −0.72 | |
| Enzymatic | −0.42 | *NH2–*NH2 + H+ + e− → *NH2 + NH3 | −2.48 |
Intriguingly, the end-on mode of Ta-monomer along the alternating and dimer catalysts along the alternating and distal pathways require large energy for the first step of hydrogenation. In sharp contrast, for the Ta-trimer catalyst, a small amount of energy is required for the first step of hydrogenation along alternating/distal pathways, indicating the potential of these catalysts for high NRR activity. The most favourable reaction pathway for NRR on the Tan@C2N catalysts is presented in Fig. 5 and detailed reaction pathways with the images of various intermediates and corresponding limiting potential are given in ESI.†Table 2 depicts the potential-determining step with the limiting potential along the studied alternate, distal and enzymatic pathways on Ta-based single, double, and triple atom catalysts. From Fig. 5 and Table 2, one can see that the limiting potential value for the most favourable pathway for formation of two NH3 molecules on Ta2@C2N is −0.94 V. Contrastingly, the limiting potential values for the most favourable pathways on Ta1@C2N and Ta3@C2N are −0.79 and −0.72 V respectively. To simulate the solvent effect on the most favourable reaction pathways, we used VASPsol code74 with an implicit solvent model. The reaction free energy profiles for the most favourable reactions pathways after including the solvent corrections are presented in Fig. S7.† As can be seen from the figure, the inclusion of the solvent effect leads to an appreciable change in the relative stability of different intermediates along the NRR pathways for the Ta-single and double atom catalysts, whereas the NRR pathway for the triple atom catalyst reveals no significant change as compared to gas phase calculations. We note an increase in the limiting potential from −0.94 to −1.56 V for the Ta-double atom catalyst. However, importantly the single and triple atom catalysts reveal slightly lower limiting potentials of −0.76 and −0.70 V respectively as compared to the gas phase results. It is important to mention here that for the completion of the catalytic cycle, the second NH3 must undergo desorption from the single, double, and triple atom catalysts. The calculations reveal that this step requires 0.82, 1.20 and 1.42 eV of energy for single, double, and triple Ta-atom catalysts, which is comparable or lower than the recently reported results.46,75,76 Also, it is reported this obstacle can be overcome either by the energy released by the subsequent hydrogenation steps75 of NH3 formation or protonation of NH3 to NH4+ under acidic conditions.27,77 Finally, for comparison, the calculated limiting potential values for the Ta-monomer and trimer catalysts are in close proximity with already experimentally reported Ru SAC catalyst78,79 supported on nitrogen-doped carbon (UL = −0.73/−0.77 V) and much lower that of R(0001) stepped surface80 (UL = −0.98 V), which has the best NRR performance among bulk metal catalysts.
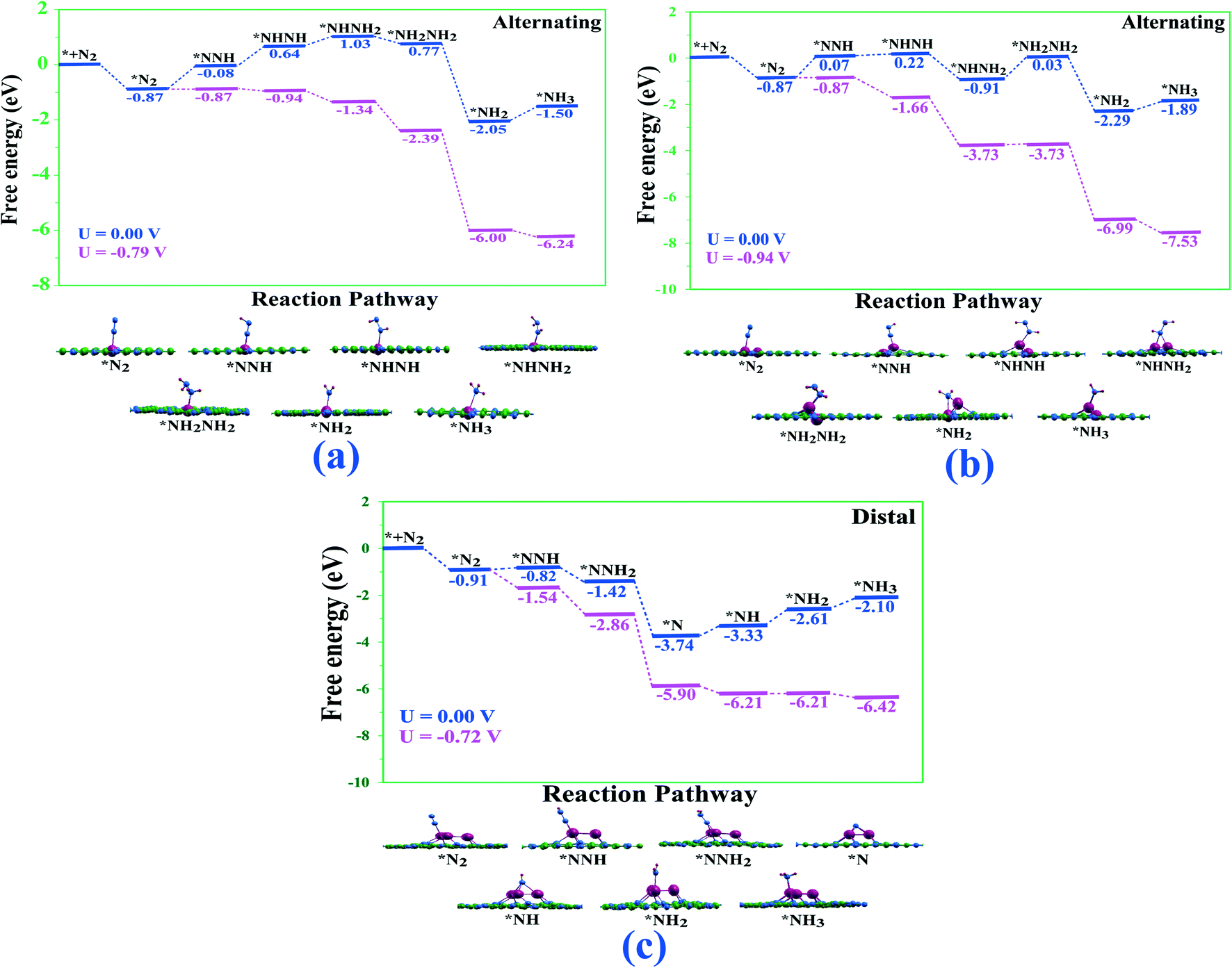 | ||
| Fig. 5 Most favourable reaction pathway for NRR on (a) Ta1@C2N (b) Ta2@C2N and (c) Ta3@C2N catalysts with corresponding limiting potentials. The sub-figures represent the optimized geometries of the different reaction intermediates involved in the reaction pathways. | ||
Further, hydrogen evolution reaction (HER) is the main competing reaction for NRR as the proton-electron pair required for N2 reduction can easily cover the catalyst surface, thereby reducing the selectivity for NRR. As per the CHE model, the free energy change for N2 and H adsorption respectively for NRR and HER provides a useful estimate of the reaction selectivity. Therefore, the adsorption free energies of *N2 and *H are calculated and plotted in Fig. 6. As can be seen from the figure, all the three Ta-catalysts preferentially adsorb N2 compared to H, indicating a higher selectivity for NRR than HER. Thus, from the above results, we conclude that the Ta-timer catalyst supported on a C2N monolayer acts as better catalysts for NRR than Ta-monomer and dimer catalysts.
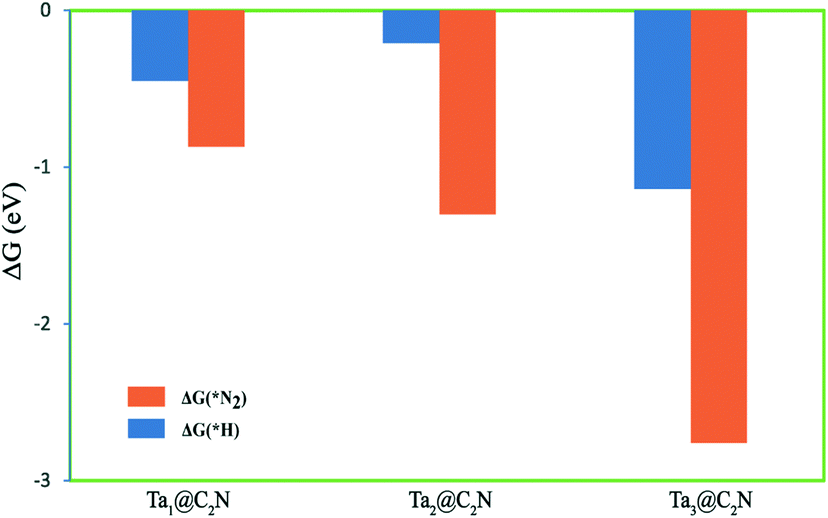 | ||
| Fig. 6 Calculated adsorption free energies of N2 molecule and H atom on the Tan@C2N catalysts. The free energy change for N2 adsorption has been calculated using end-on mode for Ta1@C2N and side-on mode for Ta2@C2N and Ta3@C2N catalysts. | ||
4. Conclusions
In summary, density functional theory calculations were carried on single, double, and triple Ta-atom catalysts anchored on the C2N monolayer for electrochemical reduction of nitrogen. Importantly, Ta3@C2N catalyst with appropriate electronic structure was found to be the most promising candidate for NRR with high thermal stability, strong N2 adsorption ability, a low limiting potential and good NRR selectivity. We expect the current work could lead to further investigations to stimulate the design of triple atom catalysts of varying metal atoms for efficient NRR and other electrocatalytic applications.Author contributions
A. R., I. A., and A. M. performed the theoretical calculations. M. A. D. and S. K. conceptualized the study and carried out the advising. M. D. performed and conceptualized the electronic structure analysis. All authors contributed to the preparation of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
M. A. D. acknowledges the Start-up Research Grant (SRG/2020/000654) of SERB, India and M. D. acknowledges the Core Research Grant (CRG/2020/005626) of SERB, India for financial support toward the completion of this work. We acknowledge National Supercomputing Mission (NSM) for providing computing resources of ‘PARAM Brahma’ at IISER Pune, which is implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India.References
- V. Rosca, M. Duca, M. T. de Groot and M. T. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS
.
- R. Schlogl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed
- C. J. van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35, 6964–7029 CrossRef CAS
- D. Erdemir and I. Dincer, Int. J. Energy Res., 2021, 45, 4827–4834 CrossRef
- Z. Yan, M. Ji, J. Xia and H. Zhu, Adv. Energy Mater., 2020, 10, 1902020 CrossRef CAS
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1055–1074 CrossRef CAS
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC
- L. Ma, Y. Li, Y. Xu, J. Sun, J. Liu, T. Wu, X. Ding and Y. Niu, Electrochem. Commun., 2021, 125, 107002 CrossRef CAS
- K. R. G. Lim, A. D. Handoko, S. K. Nemani, B. Wyatt, H.-Y. Jiang, J. Tang, B. Anasori and Z. W. Seh, ACS Nano, 2020, 14, 10834–10864 CrossRef CAS
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Phys. Chem. C, 2018, 122, 25268–25273 CrossRef CAS
- Z. Guo, S. Qiu, H. Li, Y. Xu, S. J. Langford and C. Sun, ChemCatChem, 2021, 13, 1239–1245 CrossRef CAS
- Q. Li, C. Liu, S. Qiu, F. Zhou, L. He, X. Zhang and C. Sun, J. Mater. Chem. A, 2019, 7, 21507–21513 RSC
- Y. Zhang, J. Hu, C. Zhang, Y. Liu, M. Xu, Y. Xue, L. Liu and M. K. H. Leung, J. Mater. Chem. A, 2020, 8, 9091–9098 RSC
- B. Yang, W. Ding, H. Zhang and S. Zhang, Energy Environ. Sci., 2021, 14, 672–687 RSC
- S. Vajda and M. G. White, ACS Catal., 2015, 5, 7152–7176 CrossRef CAS
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed
- M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed
- R. Gusmão, M. Veselý and Z. Sofer, ACS Catal., 2020, 10, 9634–9648 CrossRef
- L. Zhang, Y. Ren, W. Liu, A. Wang and T. Zhang, Natl. Sci. Rev., 2018, 5, 653–672 CrossRef CAS
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef
- L. Shi, Y. Yin, S. Wang and H. Sun, ACS Catal., 2020, 10, 6870–6899 CrossRef CAS
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS PubMed
- Q. Li, S. Qiu, C. Liu, M. Liu, L. He, X. Zhang and C. Sun, J. Phys. Chem. C, 2019, 123, 2347–2352 CrossRef CAS
- C. Choi, S. Back, N.-Y. Kim, J. Lim, Y.-H. Kim and Y. Jung, ACS Catal., 2018, 8, 7517–7525 CrossRef CAS
- H. Guo, L. Li, X. Wang, G. Yao, H. Yu, Z. Tian, B. Li and L. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 36506–36514 CrossRef CAS
- J. Qi, L. Gao, F. Wei, Q. Wan and S. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 47525–47534 CrossRef CAS
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2019, 3, 1800368 CrossRef
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, Small Methods, 2019, 3, 1800376 CrossRef
- L. Gao, F. Wang, M.-a. Yu, F. Wei, J. Qi, S. Lin and D. Xie, J. Mater. Chem. A, 2019, 7, 19838–19845 RSC
- L. Lin, L. Gao, K. Xie, R. Jiang and S. Lin, Phys. Chem. Chem. Phys., 2020, 22, 7234–7240 RSC
- K. Bhattacharyya and A. Datta, Phys. Chem. Chem. Phys., 2019, 21, 12346–12352 RSC
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Mater. Chem. A, 2019, 7, 4771–4776 RSC
- L. Xu, L.-M. Yang and E. Ganz, ACS Appl. Mater. Interfaces, 2021, 13, 14091–14101 CrossRef CAS
- C.-X. Huang, G. Li, L.-M. Yang and E. Ganz, ACS Appl. Mater. Interfaces, 2021, 13, 608–621 CrossRef CAS PubMed
- H. Niu, X. Wang, C. Shao, Z. Zhang and Y. Guo, ACS Sustainable Chem. Eng., 2020, 8, 13749–13758 CrossRef CAS
- M. Yao, Z. Shi, P. Zhang, W.-J. Ong, J. Jiang, W.-Y. Ching and N. Li, ACS Appl. Nano Mater., 2020, 3, 9870–9879 CrossRef CAS
- S. Tang, T. Liu, Q. Dang, X. Zhou, X. Li, T. Yang, Y. Luo, E. Sharman and J. Jiang, J. Phys. Chem. Lett., 2020, 11, 5051–5058 CrossRef CAS PubMed
- C. Ren, Q. Jiang, W. Lin, Y. Zhang, S. Huang and K. Ding, ACS Appl. Nano Mater., 2020, 3, 5149–5159 CrossRef CAS
- L. Cai, N. Zhang, B. Qiu and Y. Chai, ACS Appl. Mater. Interfaces, 2020, 12, 20448–20455 CrossRef CAS
- Z. W. Chen, L. X. Chen, C. C. Yang and Q. Jiang, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC
- X. Lv, W. Wei, B. Huang, Y. Dai and T. Frauenheim, Nano Lett., 2021, 21, 1871–1878 CrossRef CAS PubMed
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed
- T. Deng, C. Cen, H. Shen, S. Wang, J. Guo, S. Cai and M. Deng, J. Phys. Chem. Lett., 2020, 11, 6320–6329 CrossRef CAS PubMed
- S. Wang, L. Shi, X. Bai, Q. Li, C. Ling and J. Wang, ACS Cent. Sci., 2020, 6, 1762–1771 CrossRef CAS PubMed
- L. Jasin Arachchige, Y. Xu, Z. Dai, X. Zhang, F. Wang and C. Sun, J. Phys. Chem. C, 2020, 124, 15295–15301 CrossRef CAS
- Z. W. Chen, L. X. Chen, M. Jiang, D. Chen, Z. L. Wang, X. Yao, C. V. Singh and Q. Jiang, J. Mater. Chem. A, 2020, 8, 15086–15093 RSC
- G. Zheng, L. Li, Z. Tian, X. Zhang and L. Chen, J. Energy Chem., 2021, 54, 612–619 CrossRef
- C. Cui, H. Zhang and Z. Luo, Nano Res., 2020, 13, 2280–2288 CrossRef CAS
- M. Li, Y. Cui, X. Zhang, Y. Luo, Y. Dai and Y. Huang, J. Phys. Chem. Lett., 2020, 11, 8128–8137 CrossRef CAS PubMed
- D. Ma, Z. Zeng, L. Liu, X. Huang and Y. Jia, J. Phys. Chem. C, 2019, 123, 19066–19076 CrossRef CAS
- F. Studt, B. A. MacKay, M. D. Fryzuk and F. Tuczek, J. Am. Chem. Soc., 2004, 126, 280–290 CrossRef CAS PubMed
- C. Geng, J. Li, T. Weiske and H. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11680 CrossRef CAS
- C. Geng, J. Li, T. Weiske and H. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21416 CrossRef CAS
- Y. Zhao, J.-T. Cui, M. Wang, D. Y. Valdivielso, A. Fielicke, L.-R. Hu, X. Cheng, Q.-Y. Liu, Z.-Y. Li, S.-G. He and J.-B. Ma, J. Am. Chem. Soc., 2019, 141, 12592–12600 CrossRef CAS
- H. Sahin, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 085421 CrossRef
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, I.-Y. Jeon, S.-M. Jung, H.-J. Choi, J.-M. Seo, S.-Y. Bae, S.-D. Sohn, N. Park, J. H. Oh, H.-J. Shin and J.-B. Baek, Nat. Commun., 2015, 6, 6486 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, WIREs Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef
- L. Xu, L.-M. Yang and E. Ganz, ACS Appl. Mater. Interfaces, 2021, 13, 14091–14101 CrossRef CAS PubMed
- S. K. Sahoo, J. Heske, M. Antonietti, Q. Qin, M. Oschatz and T. D. Kühne, ACS Appl. Energy Mater., 2020, 3, 10061–10069 CrossRef CAS
- H.-J. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7, 3869–3882 CrossRef CAS
- L.-H. Zhang, F. Yu and N. R. Shiju, ACS Sustainable Chem. Eng., 2021, 9, 7687–7703 CrossRef CAS
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1870301 CrossRef
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Zero-point and entropic corrections to the free energy of the gas phase and the adsorbed species along the most favourable reaction pathways, diffusion energy barrier plot of Ta atom from the cavity of g-C2N to the hexagonal ring, IPDOS of Ta d-states and N2 p-states near the Fermi level, detailed free energy diagrams for the NRR pathways on the studied catalysts, solvation effect corrected most favourable free energy pathways, activation barriers for side-on to end-on transition for N2 adsorption on Ta2 and Ta3 catalysts, and coordinates of various adsorbed species along the most favourable pathways of NRR on Tan@C2N catalysts. See DOI: 10.1039/d1cy01292d |
| This journal is © The Royal Society of Chemistry 2022 |