
Predicted crystal structures of xenon and alkali metals under high pressures†
Yifan
Tian
a,
John S.
Tse
ab,
Guangtao
Liu
*a and
Hanyu
Liu
 a
a
aState Key Laboratory of Superhard Materials and International Center for Computational Method & Software, College of Physics, Jilin University, Changchun 130012, China. E-mail: liuguangtao@jlu.edu.cn
bPhysics and Engineering Physics Department, University of Saskatchewan, S7N 5E2, Canada
First published on 19th July 2022
Abstract
The pressure-induced reaction between xenon (Xe) and other non-inert gas elements and the resultant crystal structures have attracted great interest. In this work, we carried out extensive simulations on the crystal structures of Xe–alkali metal (Xe–AM) systems under high pressures. Among all predicted compounds, KXe and RbXe are found to become stable at a pressure of ∼16 GPa by adopting a cubic symmetry of space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The stabilization of KXe and RbXe requires slightly lower pressure compared with that of previously reported CsXe (25 GPa), interestingly, which is in contrast to the electronegativity order of the AMs and unexpected. Our simulations also indicate that all predicted Xe compounds contain negatively charged Xe. Moreover, our in-depth analysis indicates that the occupation of AM d-orbitals plays a critical role in stabilizing these Xe-bearing compounds. These results shed light on the understanding of the reaction between Xe and AMs and the formation mechanism of the resultant crystal structures.
m. The stabilization of KXe and RbXe requires slightly lower pressure compared with that of previously reported CsXe (25 GPa), interestingly, which is in contrast to the electronegativity order of the AMs and unexpected. Our simulations also indicate that all predicted Xe compounds contain negatively charged Xe. Moreover, our in-depth analysis indicates that the occupation of AM d-orbitals plays a critical role in stabilizing these Xe-bearing compounds. These results shed light on the understanding of the reaction between Xe and AMs and the formation mechanism of the resultant crystal structures.
Introduction
The seminal work on the synthesis of the first noble gas (NG) compound XePtF6 by Bartlett1 opens a new area of the field of NG chemistry. Xenon (Xe) has been accepted to form compounds with non-metals2–29 and transition metals30–36 in theory and experiment, where it exhibits a cationic feature at ambient and high pressure. Recently, theoretical work also demonstrated the anionic feature of Xe in some compressed compounds due to the fact that simulations indicate that it might attract electrons from electropositive elements, such as magnesium (Mg),37 while these reactions require a much higher pressure of 125 GPa. In addition, based on first-principles structural searches, Xe was also predicted to form ionic compounds with lithium (Li)38 and cesium (Cs)39 at 50 and 25 GPa, respectively. The pressure required to stabilize the compounds between Xe and other electropositive elements decreases with the decreasing electronegativity of the elements.40It is well known that alkali metals (AMs) are electropositive elements that often act as electron donors.41,42 Under compression, the valence electrons of the AM atoms are prompted to occupy the higher energy orbitals.43 As demonstrated previously,38–40 rare gas atoms, such as Xe, with a full outer shell of electrons, can form compounds with the alkali atoms under high pressure. However, the structure and chemical bonding of Xe–Li and Xe–Cs compounds are quite different. In one case, the chemical bond between Xe and Li is metallic, while that in Xe–Cs compounds is ionic. The differences in the electronic properties stem from the distinct electronic configurations of the AMs. The delocalized s-electrons transfer to empty orbitals of higher azimuthal quantum number increasing the spatial flexibility of the electrons and help to alleviate the Pauli repulsion. The two AMs that have s-orbital to p-orbital electron transition are Li and sodium (Na).43 Heavier AM atoms, such as potassium (K), rubidium (Rb) and Cs, have complex electronic configurations with mixed low-lying d-orbitals, which have an important effect on the structure and phase transition sequence of compressed elemental solids.44–46 On the other hand, the electronic structures of the alkali atoms can influence dramatically their binary alloys. For instance, Na becomes an electron donor once combined with Li,47 but it is an electron acceptor when alloyed with K.48
In this work, we systematically explored the compounds of AMxXey (AM = Na, K and Rb, x = 1–2, y = 1–4; x = 3, y = 1–2) under high pressures. As a result of extensive simulations, we find that two cubic compounds, KXe and RbXe, could be energetically stable under an unexpected low pressure of ∼16 GPa. By comparing projected density of states (PDOS) and the enthalpies of formation in the Xe–K system, the occupation of AM d-orbitals at the Fermi surface plays a critical role in stabilizing the compounds. These findings are analyzed to try and establish an overall trend of reactions between Xe and AMs.
Computational details
In this work, crystal structure predictions were performed by the CALYPSO method and software49,50 with a wide range of chemical compositions (AMxXey, AM = Na, K and Rb, x = 1–2, y = 1–4; x = 3, y = 1–2) at 50, 100 and 150 GPa. The structure prediction method previously helped in identifying novel NG compounds.51,52 All the structural relaxations and electronic property calculations are implemented by the Vienna ab initio simulation package (VASP)53 code within the framework of density functional theory (DFT).54,55 The Perdew–Burke–Ernzerhof (PBE) functional56 of the generalized gradient approximation (GGA)57 is adopted in these simulations. The all-electron projector augmented-wave (PAW)58 pseudopotentials are used to describe the ionic potentials, with 4s24p64d105s25p6, 2s22p63s1, 3s23p64s1 and 4s24p65s1 for Xe, Na, K and Rb treated as valence electrons, respectively. A plane wave basis energy cutoff is set to 1000 eV. A dense k-point sampling of the Brillouin zone with a resolution of 2π × 0.03 Å−1 was used to ensure that the enthalpy calculations converged within several meV per atom. To verify the dynamical stability of the predicted structures, phonon calculations were performed using the finite displacement approach with the supercell method as implemented in PHONOPY code.59 The harmonic interatomic force constants are calculated by a 1 × 1× 1 supercell for NaXe4, K2Xe3 and Rb2Xe3, 2 × 2 × 2 supercell for NaXe, 3 × 3 × 3 supercell for KXe and RbXe, 1 × 2 × 1 supercell for K3Xe, 2 × 2 × 1 supercell for K3Xe2, KXe4, KXe3, RbXe3 and RbXe4 and 3 × 3 × 1 supercell for KXe2, NaXe3 and RbXe2.Results and discussion
As a result of extensive structure searches for NaxXey, KxXey and RbxXey up to 150 GPa, we predicted several thermodynamically stable compositions for the Na–Xe, K–Xe and Rb–Xe systems at high pressures (Fig. 1 and Fig. S1, ESI†). Our further calculated phonon dispersions indicate the dynamical stability feature for all the predicted structures, where there are no imaginary frequencies in the whole Brillouin zone as shown in Fig. S2–S4 (ESI†). The predicted stable structures with respect to decompositions into the elements or other alloy compounds are also shown in Fig. S5–S7 (ESI†). Unlike many binary compounds, such as Mg–Ar compounds,37 adopting stacked hexagonal lattices, a majority of Xe–AM (Na, K, Rb and Cs) alloys consist of stacked square lattices with alternating Xe and AM layers. This structural type was previously mentioned in Mg–Xe and Mg–Kr.37 In comparison, the structures of Xe–Li38 compounds are predicted to adopt hexagonal symmetry. However, K3Xe, KXe4, and RbXe4 were predicted with space group of C2/m (Table S1, ESI†).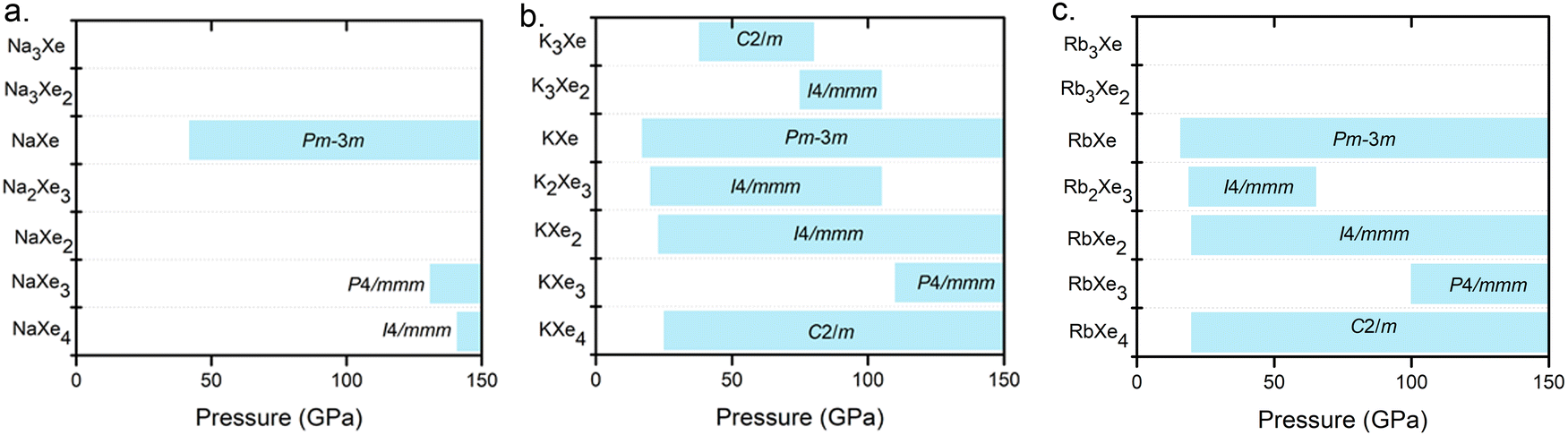 | ||
| Fig. 1 Pressure ranges of thermodynamically stable AMxXey compounds (AM = Na, K and Rb) from ambient pressure to 150 GPa. | ||
The pressures of stable KXe and RbXe are noticeably lower than those of the Xe–Li, Xe–Na and Xe–Cs, systems as shown in Fig. 2a. This observation is inconsistent with the electronegativity of AMs, which decreases along the group. This unexpected phenomenon motivated us to search for the origin of their stability. The enthalpy of the formation (ΔH) could be calculated by the sum of the internal energy (ΔE) and ΔpV terms by the following formula:
| ΔH = ΔE + ΔpV = (Ecompound − EAM − EXe) + (pVcompound − pVAM − pVXe). |
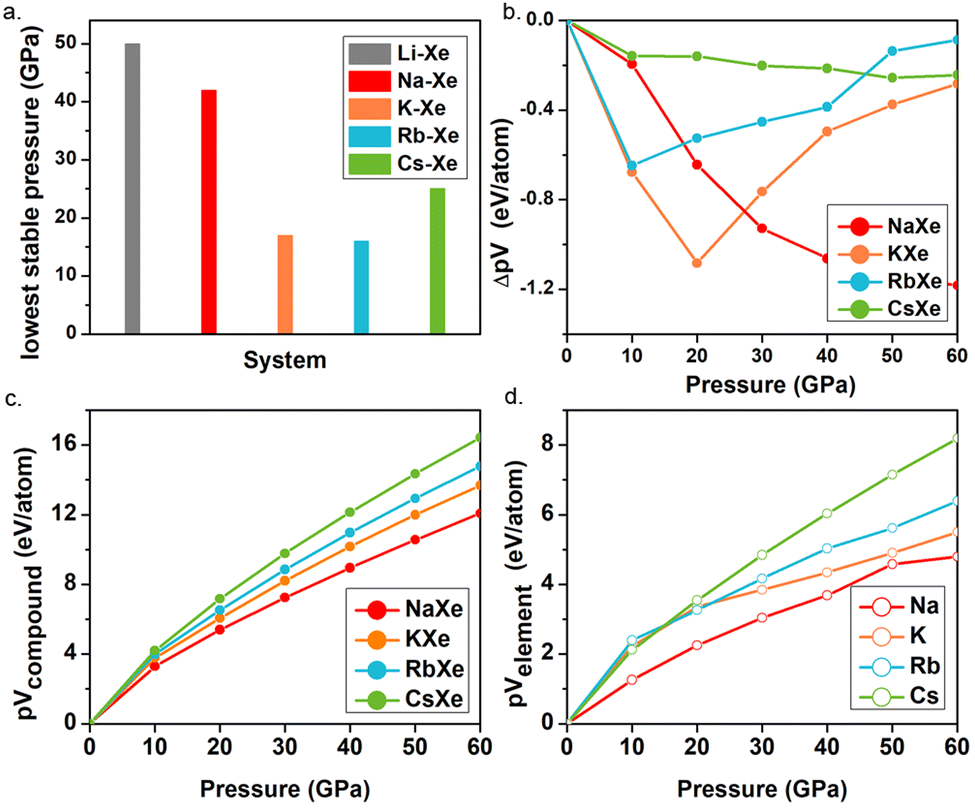 | ||
| Fig. 2 (a) The lowest stable pressures of the Li–Xe, Na–Xe, K–Xe, Rb–Xe and Cs–Xe systems. (b) ΔpV versus pressure for NaXe, KXe, RbXe and CsXe. (c) pV versus pressure for NaXe, KXe, RbXe and CsXe. (d) pV versus pressure for Na, K, Rb and Cs from ambient pressure to 60 GPa. | ||
ΔE of the KXe and RbXe compounds decreased significantly with increasing pressure compared to that of CsXe and NaXe, while it keeps positive below 30 GPa, suggesting that ΔE makes a lower contribution to the stability of the structures (Fig. S8, ESI†). As shown in Fig. 2b, ΔpV terms of the KXe and RbXe compounds show a tendency of falling first and then rising, but those of the compounds NaXe and CsXe are continuously decreasing upon compression. To identify the factors affecting the ΔpV term, pV terms with respect to the pressure of both compounds and elements are compared further, because ΔpV here is the difference between pVcompound and pVelement. It can be clearly seen that, as shown in Fig. 2c for predicted compounds, the trends of four polylines are well fitted from ambient pressure to 60 GPa. To be specific, the value of pV term gradually increases from NaXe to KXe to RbXe to CsXe. However, as shown in Fig. 2d below 20 GPa, this trend is not followed for elements K and Rb. This is mainly because the phase transition sequence including its pressure range are distinct for all AMs. For K, it adopts an oP8 structure below 20 GPa,60 in contrast to oP8-Na at ∼120 GPa for the convenience of its charge transfer from s to d orbitals under pressure. After oP8-K comes out, this high-pressure phase exhibits a short bond length of 2.8 Å at 30 GPa (Fig. S9, ESI†). This may provide an explanation for why the slope of the pV term becomes flatter after 20 GPa and further causes an increase of ΔpV in Fig. 2(b). Similarly, the bond length of high-pressure phase Cmca-Cs is shorter than that of fcc and I41/amd Rb under the corresponding pressure, leading to a relatively small volume of Cs. Thus, the large volumes of K and Rb make the KXe and RbXe compounds thermodynamically stable against decompositions at moderate pressures.
To further investigate the electronic properties of the Xe–AM compounds, the PDOS at 100 GPa are calculated as shown in Fig. 3. Among AMs, Na is the last AM atom without having the electron transition from s-orbital to d-orbital, while K is the first element with that electron transition. Thus, we here focus on the compounds of the Xe–Na and Xe–K systems. In order to understand the electronic behaviour, we constructed model systems in which all Xe atoms or AM atoms are removed from the predicted structures. Notably, after removing the AM atoms from the compounds, electrons of Xe would hardly occupy the 5d-orbital, suggesting that the 5d electrons of Xe in the compounds come from AM atoms instead of other Xe atoms. However, the occupation of the 3d-orbital of K is still reserved partially once the Xe atoms are removed. Therefore, as seen in Fig. 3, for Na, electron transfer occurs from the 3s-orbital to the 3p-orbital, while for K, occupied electrons for the 3d orbital come from 4s and 3p orbitals. This is in agreement with the previous analysis that the relative order of atomic orbital energies would change under pressure.61
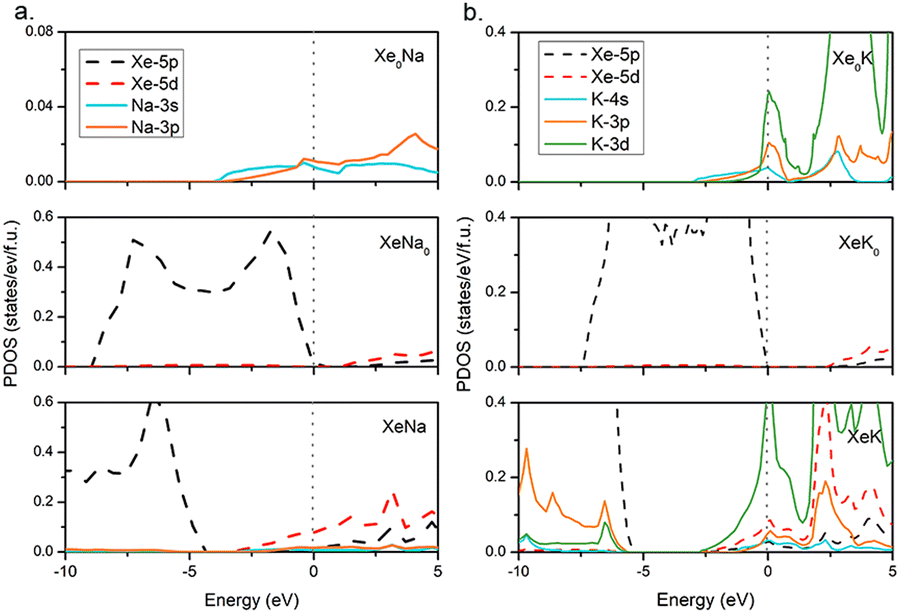 | ||
| Fig. 3 Projected densities of states (PDOS) for predicted compounds (a) Pmm–NaXe and (b) Pmm–KXe. The top and middle figures represent PDOS of the hypothetical structures, in which we remove all Xe atoms and alkali atoms from the compounds, respectively. The vertical dotted lines indicate the Fermi energy. | ||
Our further simulations suggest a peak of 3d orbital for K around the Fermi level in the KXe compound (Fig. S10, ESI†). As the Xe composition increased, we found that d-orbital electrons are no longer occupied at near the Fermi level. If the Xe composition is decreased, the PDOS of the 3d-orbital for K will gradually decrease. Thus, KXe, with the low enthalpy of formation in the Xe–K system, is also the compound with the most PDOS of the d orbital for K at the Fermi level. This may suggest that the occupation of the d orbital for AM elements at the Fermi level influences the stability of the compounds. Subsequently, we performed systematically Bader charge analysis62 for all the stable Xe–AM compounds based on atoms in molecules (AIM) theory 60 at 100 GPa, as shown in Fig. 4a. We found that Bader charge is more notable in the Xe–Li and Xe–Na systems than in the others. This is because parts of the s orbital electrons in K, Rb and Cs transferred to the d orbital, which reduced the quantity of Xe d orbital electrons. For a better understanding of the electronic structures, we calculated the electron localization function (ELF) as shown in Fig. 4b. Here, we take Pmm KXe as a representative example, the small ELF values between Xe and K atoms reflecting the ionic bonding nature. The positive Laplacian63 values of 3.63 a.u. between Xe and K and 1.67 a.u. between Xe and Xe, together with low values for electron density of 0.28 a.u between Xe and K and 0.25 a.u. between Xe and Xe, at critical points, indicate ionic bonds as shown in Fig. 4c. We further performed the chemical bond analysis for the compounds of NaXe and KXe as shown in Fig. S12 (ESI†). We found the existence of the lowest ELF values between the Xe atom and AM atom with 0.04 for NaXe, 0.07 for KXe, and 0.09 for RbXe, suggesting ionic bond features for the compounds of NaXe, KXe, and RbXe again.
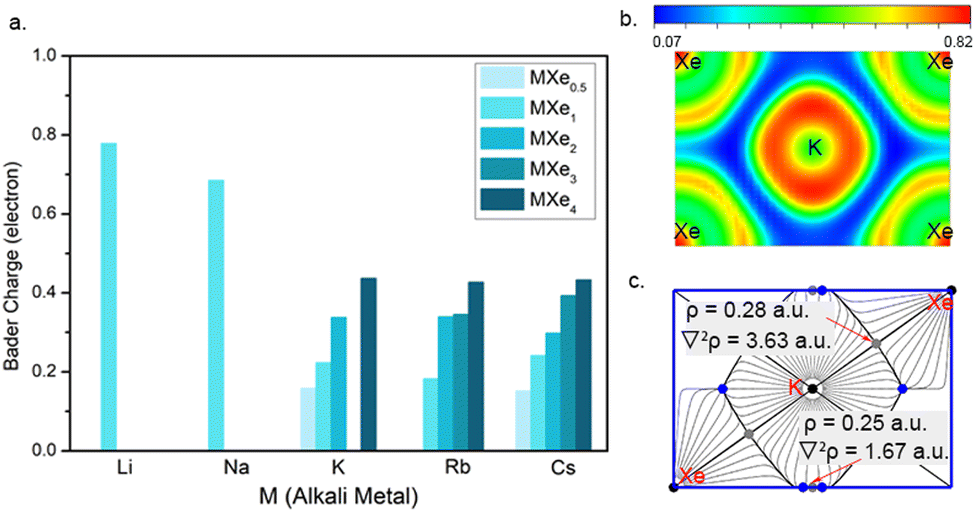 | ||
Fig. 4 (a) Calculated Bader charges for AM atoms in energetically stable compounds Xe–AM (AM = Li, Na, K, Rb and Cs) at 100 GPa. (b) Electron location function (ELF) of the compound KXe (space group Pmm) in the (01![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) plane at 100 GPa. (c) Gradient paths and critical points derived from a QTAIM analysis for Pmm KXe. ) plane at 100 GPa. (c) Gradient paths and critical points derived from a QTAIM analysis for Pmm KXe. | ||
Conclusions
In summary, we have performed extensive simulations on the high-pressure structures in the Xe–Na, Xe–K and Xe–Rb systems based on swarm intelligence structure simulations and globally explored the stability and electronic properties of the predicted structures. These compounds show both lattice dynamical stability and thermodynamic stability with respect to competing phases. Most of the stable compounds tend to adopt stacked square lattices with Xe layers and AM layers. Among these structures, Pmm KXe and RbXe could be stable at surprisingly low pressures as anionic Xe compounds predicted so far. Combining with the previous study, all the Xe–AM compounds are ionic bonded with negatively charged Xe, whereas the behaviors of electrons are different in these systems because of the change of orbital order under compression. We found that the occupation of d-orbitals for AMs at the Fermi level influences the formation enthalpy values of the compounds. Our results are expected to extend the understanding of the trend of reactions between AMs and Xe and the structural and electronic properties of anionic noble gas compounds at high pressures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 12074138), Jilin Province Science and Technology Development Program (grant no. YDZJ202102CXJD016), Program for Jilin University Science and Technology Innovative Research Team, the Program for Jilin University Computational Interdisciplinary Innovative Platform, and computing facilities at the High-Performance Computing Centre of Jilin University.References
- N. Bartlett, Proc. Chem. Soc., 1962, 6, 218 Search PubMed.
- F. Peng, J. Botana, Y. Wang, Y. Ma and M. Miao, J. Phys. Chem. Lett., 2016, 7, 4562–4567 CrossRef CAS PubMed.
- N. Zarifi, H. Liu, J. Tse and E. Zurek, J. Phys. Chem. C, 2018, 122, 2941–2950 CrossRef CAS.
- D. Schröder, J. N. Harvey, M. Aschi and H. Schwarz, J. Chem. Phys., 1998, 108, 8446–8455 CrossRef.
- D. S. Brock and G. J. Schrobilgen, J. Am. Chem. Soc., 2011, 133, 6265–6269 CrossRef CAS PubMed.
- Q. Zhu, D. Y. Jung, A. R. Oganov, C. W. Glass, C. W. Gatti and A. O. Lyakhov, Nat. Chem., 2013, 5, 61–65 CrossRef CAS PubMed.
- X. Yan, Y. Chen, S. Xiang, X. Kuang, Y. Bi and H. Chen, Phys. Rev. B, 2016, 93, 214112 CrossRef.
- F. Peng, Y. Wang, H. Wang, Y. Zhang and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 094104 CrossRef.
- X. Yan, Y. Chen, X. Kuang and S. Xiang, J. Chem. Phys., 2015, 143, 124310 CrossRef PubMed.
- Y. Wang, J. Zhang, H. Liu and G. Yang, Chem. Phys. Lett., 2015, 640, 115–118 CrossRef CAS.
- J. F. Wacker and E. Anders, Geochim. Cosmochim. Acta, 1984, 48, 2373–2380 CrossRef CAS.
- J. I. Matsuda and K. Matsubara, Geophys. Res. Lett., 1989, 16, 81–84 CrossRef CAS.
- R. Hoppe, W. Dahne, H. Mattauch and K. Rodder, Angew. Chem., Int. Ed. Engl., 1962, 1, 599 CrossRef.
- R. M. Gavin Jr. and L. S. Bartell, J. Chem. Phys., 1968, 48, 2460 CrossRef.
- D. F. Smith, J. Am. Chem. Soc., 1963, 85, 816–817 CrossRef CAS.
- D. H. Templeton, A. Zalkin, J. Forrester and S. M. Williamson, J. Am. Chem. Soc., 1963, 85, 817 CrossRef CAS.
- H. H. Claassen, H. Selig and J. G. Malm, J. Am. Chem. Soc., 1962, 84, 3593 CrossRef CAS.
- H. Selig, H. H. Claassen, C. L. Chernick, J. G. Malm and J. L. Huston, Science, 1964, 143, 1322–1323 CrossRef CAS PubMed.
- C. Sanloup, H. K. Mao and R. J. Hemley, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 25–28 CrossRef CAS PubMed.
- A. Dewaele, N. Worth, C. J. Pickard, R. J. Needs, S. Pascarelli, O. Mathon, M. Mezouar and T. Rifune, Nat. Chem., 2016, 8, 784–790 CrossRef CAS PubMed.
- A. Dewaele, P. Loubeyre, P. Dumas and M. Mezouar, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014103 CrossRef.
- G. Weck, A. Dewaele and P. Loubeyre, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 014112 CrossRef.
- A. Hermann and P. Schwerdtfeger, J. Phys. Chem. Lett., 2014, 5, 4336–4342 CrossRef CAS PubMed.
- M. Somayazulu, P. Dera, A. F. Goncharov, S. A. Gramsch, P. Liermann, W. Yang, Z. Liu, H. K. Mao and R. J. Hemley, Nat. Chem., 2010, 2, 50–53 CrossRef CAS PubMed.
- C. Sanloup, B. C. Schmidt, E. M. C. Perez, A. Jambon, E. Gregoryanz and M. Mezouar, Science, 2005, 310, 1174–1177 CrossRef CAS PubMed.
- M. Somayazulu, P. Dera, J. Smith and R. J. Hemley, J. Chem. Phys., 2015, 142, 104503 CrossRef PubMed.
- C. L. Chernick, H. H. Claassen, P. R. Fields, H. H. Hyman, J. G. Malm, W. M. Manning, M. S. Matheson, L. A. Quarterman, F. Schreiner, H. H. Selig, I. Sheft, S. Siegel, E. N. Sloth, L. Stein, M. H. Studier, J. L. Week and M. H. Zirin, Science, 1962, 138, 136–138 CrossRef CAS PubMed.
- J. G. Malm, I. Sheft and C. L. Chernick, J. Am. Chem. Soc., 1963, 85, 110–111 CrossRef CAS.
- E. E. Weaver, B. Weinstock and C. P. Knop, J. Am. Chem. Soc., 1963, 85, 111–112 CrossRef CAS.
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Nat. Chem., 2014, 6, 644–648 CrossRef CAS PubMed.
- D. Zhou, D. Szczęśniak, J. Yu, C. Pu and X. Tang, J. Phys. Chem. C, 2019, 123, 9323–9330 CrossRef CAS.
- K. K. Lee and G. Steinle-Neumann, J. Geophys. Res.: Solid Earth, 2006, 111, B02202 Search PubMed.
- D. Seoung, Y. Lee, H. Cynn, C. Park, K. Choi, D. A. Blom, W. J. Evans, C. Kao, T. Vogt and Y. Lee, Nat. Chem., 2014, 6, 835–839 CrossRef CAS PubMed.
- E. Stavrou, Y. Yao, A. F. Goncharov, S. S. Lobanov, J. M. Zaug, H. Liu, E. Greenberg and V. B. Prakapenka, Phys. Rev. Lett., 2018, 120, 096001 CrossRef CAS PubMed.
- A. Dewaele, C. M. Pépin, G. Geneste and G. Garbarino, High Pressure Res., 2017, 37, 137–146 CrossRef CAS.
- F. Peng, X. Song, C. Liu, Q. Li, M. Miao, C. Chen and Y. Ma, Nat. Commun., 2020, 11, 5227 CrossRef CAS PubMed.
- M. Miao, X. Wang, J. Brgoch, F. Spera, M. G. Jackson, G. Kresse and H. Lin, J. Am. Chem. Soc., 2015, 137, 14122–14128 CrossRef CAS PubMed.
- Z. Liu, J. Botana, M. Miao and D. Yan, EPL, 2017, 117, 26002 CrossRef.
- S. Zhang, H. Bi, S. Wei, J. Wang, Q. Li and Y. Ma, J. Phys. Chem. C, 2015, 119, 24996–25002 CrossRef CAS.
- M. Miao, Y. Sun, H. Liu and Y. Ma, Commun. Chem., 2022, 5, 15 CrossRef.
- J. C. Schon, Z. Cancarevic and M. Jansen, J. Chem. Phys., 2004, 121, 2289 CrossRef CAS PubMed.
- Z. Cancarevic, J. C. Schoen and M. Jansen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 224114 CrossRef.
- A. K. McMahan, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 5982(R) CrossRef.
- G. Fabbris, J. Lim, L. S. I. Veiga, D. Haskel and J. S. Schilling, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 085111 CrossRef.
- Y. Ma, A. R. Oganov and Y. Xie, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 014102 CrossRef.
- E. Gregoryanz, O. Degtyareva, M. Somayazulu, R. J. Hemley and H. K. Mao, Phys. Rev. Lett., 2005, 94, 185502 CrossRef PubMed.
- Y. Chen, H. Geng, X. Yan, Z. Wang, X. Chen and Q. Wu, Chin. Phys. B, 2017, 26, 056102 CrossRef.
- L. Yang, X. Qu, X. Zhong, D. Wang, Y. Chen, J. Yang, J. Lv and H. Liu, J. Phys. Chem. Lett., 2019, 10, 3006–3012 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- J. Zhang, H. Liu, Y. Ma and C. Chen, Natl. Sci. Rev., 2022, 9, nwab168 Search PubMed.
- J. Zhang, J. Lv, H. Li, X. Feng, S. A. T. Redfern, C. Lu, H. Liu, C. Chen and Y. Ma, Phys. Rev. Lett., 2018, 121, 255703 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- L. F. Lundegaard, M. Marques, G. Stinton, G. J. Ackland, R. J. Nelmes and M. I. McMahon, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 020101 CrossRef.
- M. Miao and R. Hoffmann, Acc. Chem. Res., 2014, 47, 1311–1317 CrossRef CAS PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- D. Vega and D. Almeida, J. Comput. Methods Sci. Eng., 2014, 14, 131–136 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02657k |
| This journal is © the Owner Societies 2022 |