
Pore size effects on surface charges and interfacial electrostatics of mesoporous silicas†
Kento
Murota
 *ab and
Takumi
Saito
c
*ab and
Takumi
Saito
c
aDepartment of Nuclear Engineering and Management, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: murota@radw.t.u-tokyo.ac.jp
bRegulatory Standard and Research Department, Secretariat of Nuclear Regulation Authority, 1-9-9 Roppongi, Minato-ku, Tokyo 106-8450, Japan
cNuclear Professional School, School of Engineering, The University of Tokyo, 2-22 Shirakata Shirane, Tokai-mura, Naka-gun, Ibaraki 319-1188, Japan
First published on 18th July 2022
Abstract
Water in confinement becomes more structured than bulk water, and its properties, such as the dielectric constant, change. It remains unclear, however, how the interfacial reactions in confinement, such as the adsorption of ions on the surfaces of small pores, differ from those in larger spaces. We focused on the deprotonation reaction of hydroxyl groups, a fundamental surface reaction, and investigated the dependence of the surface charge density on pore size by determining the surface charge densities of six types of mesoporous silicas with micropores and mesopores at different ionic strengths and pH levels from batch titration tests. The surface complexation model assuming a potential distribution based on the Poisson–Boltzmann equation in cylindrical coordinates was fitted to the obtained surface charge densities to relate the electrostatics near the surface to the surface reaction. The results showed that the absolute values of the surface charge densities decreased with decreasing pore diameter due to the overlap of the electrical double layers. Furthermore, the capacitance of the Stern layer optimized by fitting decreased with decreasing pore diameter, especially in pores smaller than 4 nm in diameter, which suggested that the dielectric constants of water decreased near the surfaces of small pores.
Introduction
Materials with mesopores (pores with widths ranging between about 2 and 50 nm) and micropores (those not exceeding about 2 nm)1 are related to a wide range of fields, from industrially used materials such as zeolites and carbon nanotubes, to proteins such as Aquaporin, to list a few.2 Rocks in underground environments, another example of such materials, have network structures of pores and fractures of diverse sizes.3 For example, pores in shale rocks sampled from about 250 m below ground level were reported to be mainly a few nm in size, and many of these pores are accessible to water or gas.4 Contaminants in subsurface environments such as radioactive materials and heavy metals resulting from buried wastes or contaminated soil are dissolved in groundwater and migrate with groundwater through pores and fractures. Cationic contaminants in groundwater tend to be adsorbed on rock surfaces, which retards their migration. Therefore, the interaction between contaminants and the surfaces of small pores, where the surface area to pore volume ratio is high,3 is important for their migration in subsurface environments.The behavior of ions and molecules in mesopores and micropores has been actively studied in the last decades with improved analytical and simulation techniques, and in particular many of the studies have focused on water.2,5,6 A number of studies have reported that, compared to bulk water, confined water is more similar to ice with restricted motion. It has been reported, for example, that the dielectric constant of confined water is lower than that of bulk water. The dielectric constant is important, affecting the electrostatics, hydration of ions, and acid–base reactions of pore surfaces, which has mainly been investigated through simulations,7,8 but recently there have also been studied by direct measurements of the dielectric constant of water in confinement.9–11 It remains unclear, however, whether the difference in the properties between bulk water and confined water also applies to interfacial water near the surfaces in pores, which can directly affect the surface reaction in small pores.
Materials containing mesopores or micropores have advantageous properties as adsorbents and are often used for separation or scavenging.12,13 Few studies, however, have examined the special characteristics of surface reactions in confinement and their size dependence. For example, changes in the hydration state of uranium adsorbed in mesopores and its resistance to reduction have been reported.14,15 These confinement effects have often been explained by changes in the hydration state of ions in small spaces. The confinement effects on ions or molecular adsorption remain controversial. Some reports have shown that confinement leads to stronger adsorption per unit area;16,17 another has demonstrated a decrease in the adsorption constants with decreasing pore sizes.18
While experimental studies have also been done to investigate the effects of confinement on the surface charges originated from protonation or deprotonation of surface hydroxyl groups, the processes underlying the surface reactions of oxide surfaces remain unclear. First, some studies have shown conflicting effects. Wang et al.19 measured the surface charge density of mesoporous alumina and non-porous alumina by titration tests and found that the former had much higher surface charge densities. Other studies using similar methods, meanwhile, have shown only small effects of confinement on the surface charge density.18,20 Further, these investigations on surface charge density lacked sufficient theoretical interpretations to supplement their findings, and the factors behind the peculiarities of surface reactions in confined space were only superficially clarified, if at all. Recent simulation studies on the surface charge density in mesoporous silica, on the other hand, have shown that the absolute value of the surface charge density decreases as the pore diameter decreases, due to an overlap of the electrical double layers (EDLs).21,22 These studies, however, considered the surface charge in simplified systems, which may have led to differences in the surface charges of actual mesoporous silica. Conceivably, the silica surface modeled in these studies was flat, and the effect of curvature of the pore surface was not considered. By contrast, the surface charge density of a nanoparticle has been shown to change with its size.23 In addition, Prelot et al.24 showed that the enthalpies of adsorption of cadmium on nonporous and mesoporous silicas were different. The difference, they explained, was due to a change in the distance between surface hydroxyl groups caused by the curvature of the mesopores. Furthermore, it was reported that migration of ions in mesoporous silicon was retarded due to high viscosity of water near hydrophilic surface and surface roughness.25 From these literatures, a study on the dependence of surface charge density on pore diameter will be required to link the experimental and simulation studies and provide direct evidence on the effects of confinement on interfacial reactions.
In this study we experimentally investigate the surface charge density in pores of different sizes to clarify how the sizes affect the surface deprotonation. The pore geometry is difficult to be determined in natural rocks with broad size distributions, complex shapes, and network structures. For this reason, mesoporous silicas are chosen as their simplified analogues, considering their apparent similarities with rock-forming minerals, as well as their relatively uniform pores. The surface charge densities of six mesoporous silicas with different pore-size distributions are determined by batch titration tests at different pH levels and ionic strengths. Furthermore, a surface complexation model is fitted to the obtained surface charge densities to analyze the relationship between the sizes of small pores and the surface reaction in more detail. The model adopts the Stern layer description of the interfacial region. The Stern layer capacitances are optimized by the fitting, which makes it possible to link the changes in water structures near the surface with those in the surface reaction, depending on the pore size. The model is combined with numerical calculations of the Poisson–Boltzmann (PB) equation in cylindrical coordinates to account for the effect of the overlap of EDLs. The confinement effects on the potential distribution and water structures near the interface in small pores found in this study may affect various surface reactions and cannot be obtained by studying the adsorption of individual ions.
Methods
Materials
Both MCM-41 and SBA-15 are mesoporous silicas consisting of uniformly isotropic cylinders with hexagonal pores. Both silicas can be produced with different pore diameters by adjusting the type of a template and the chemical conditions during the synthesis. MCM-41 silicas with nominal pore sizes of 3, 6, and 8 nm and SBA-15 silicas with those of 4, 6, and 8 nm were purchased from Glantreo Limited. All six of the mesoporous silicas had a nominal particle size of 100 μm. The silicas were washed by dispersing 1 g of each silica in 200–300 ml of Milli-Q® water and solid–liquid separation three times to remove acid remaining after the manufacturing processes. The washed silicas were then dried in an oven at 45 °C for at least two days before the analyses and titrations.Characterization of silicas
The specific surface area and pore-size distribution of the silica samples were analyzed by nitrogen (N2) gas adsorption (3Flex Analyzer, Micromeritics Instrument Corporation). Details of the N2 gas adsorption method are shown in the ESI.† The density functional theory (DFT) method optimized for MCM-41 and similar mesoporous silicas was used to analyze the specific surface area and pore-size distribution.26 In addition, the specific surface area of external surface was evaluated by the t-plot method, which is known to be effective for samples containing micropores,27,28 using standard nitrogen adsorption data as reference of the thickness curve.29A thermogravimetric analysis (TGA) and differential thermal analysis (DTA) (Thermo plus EV02, Rigaku Corporation) were performed to estimate the site density of surface hydroxyl groups on the silica samples.30 The temperature was raised to 1000 °C at 10 °C min−1 under a continuous flow of argon at a rate of 200 ml min−1, and held for 10 min after reaching 1000 °C.
Measurement of the dissolved silicic acid
The structure of the mesoporous silica used in this study was an amorphous silica that could partly dissolve in the solution and release silicic acid in the liquid phase. To estimate the time required for the dissolution equilibrium and the effect of released silicic acid on the surface charge densities, the amounts of released silicic acid were measured at different immersion time and pH levels by the batch tests shown in the ESI.†Batch titration
We investigated the pH dependence of the negative surface charge density from the amounts of deprotonation of the hydroxyl groups on the surfaces at different ionic strengths. Titration tests are often performed by continuously adding small amounts of acid or base solutions, but in this study targeting mesoporous silica, titration tests were performed using a batch method, considering the presence of slow processes involving migration of ions through mesopores and micropores. Different concentrations and amounts of a NaOH solution were added to a polypropylene container containing 20 mg of each silica sample, the ionic strength was adjusted to 1, 10, and 50 mM using a NaNO3 solution, and Milli-Q® water was added to adjust the sample volume to 5 ml. The pH of the liquid phase was measured using the combination of a pH electrode (ROSS 8102BNUWP, Thermo Scientific) and a pH meter (VSTAR40A2, Thermo Scientific) after 5 days. This 5 day period corresponds to the period required for the dissolved silica to reach equilibrium, as described below. All batch experiments were conducted in ambient atmosphere at room temperature (20–25 °C) and the containers were continuously agitated with a shaker.Experimental results
Pore structure of the mesoporous silicas
To evaluate the pore size dependence of the surface charge density among different porous materials, it is necessary to properly estimate both their pore-size distribution and specific surface area. Particular attention should be paid to the specific surface area, as it directly affects the surface charge density and can vary when different evaluation methods are used. We used the adsorption branches of the isotherms of N2 adsorption to evaluate the pore-size distributions and specific surface areas, considering the shapes of the adsorption and desorption isotherms (see the ESI†).The pore-size distributions of the mesoporous silicas are presented in Fig. S2 in the ESI,† and the mesopore diameter of each silica based on the DFT method is shown in Table 1. We compared the DFT method with the BJH method and adopted the former because micropores were present (see the ESI†). The mesopore diameters of the different silica samples range from 3.4 to 12.4 nm, although the values tend to be larger than the corresponding nominal values. Henceforth, each mesoporous silica will be denoted by an acronym combining “M” or “S” (the first letters of MCM-41 or SBA-15) with the corresponding mesopore diameter shown in Table 1. Table 1 also shows the volumes per unit mass of pores smaller and larger than 2 nm in diameter, respectively, as determined by the DFT method. All the samples had micropores, especially M6.8 and the three types of SBA-15. In all the mesoporous silicas except M3.4, pore volumes larger than 2 nm in diameter increased with an increase in the mesopore diameters.
| MCM-41 | SBA-15 | |||||
|---|---|---|---|---|---|---|
| M3.4 | M6.8 | M12.4 | S7.1 | S7.9 | S8.2 | |
| a The values are determined with the DFT method. b The values are determined with the t-plot method. c The values are determined by the eqn (S1) in the ESI based on the results of TGA. | ||||||
| Mesopore diametera (nm) | 3.4 | 6.8 | 12.4 | 7.1 | 7.9 | 8.2 |
| Volume of pores smaller than 2 nm in diametera (cm3 g−1) | 0.010 | 0.104 | 0.038 | 0.089 | 0.095 | 0.101 |
| Volume of pores larger than 2 nm in diametera (cm3 g−1) | 0.506 | 0.279 | 0.928 | 0.312 | 0.352 | 0.449 |
| Specific surface areaa (m2 g−1) | 622 | 628 | 431 | 456 | 481 | 535 |
| <2 nm in diametera (m2 g−1) | 32 | 414 | 94 | 232 | 246 | 252 |
| >2 nm in diametera (m2 g−1) | 589 | 214 | 337 | 224 | 236 | 284 |
| External surface areab (m2 g−1) | 35 | 39 | 119 | 31 | 33 | 47 |
| Surface hydroxyl group densityc (sites per nm2) | 3.06 | 3.02 | 4.81 | 4.89 | 3.94 | 4.16 |
Fig. 1 shows the cumulative specific surface areas determined by the DFT method. Micropores are widely distributed in the range of 0.8 nm to 2 nm in diameter for M6.8 and the three types of SBA-15. In addition, M6.8 also exhibits pores as small as 0.56 nm in diameter, which is close to the lower limit of the measurement. Table 1 shows the specific surface areas of the total pores, pores smaller than 2 nm in diameter, and pores larger than 2 nm in diameter, as obtained based on Fig. 1. For M6.8 and three SBA-15, the mesopore diameters themselves are relatively close, but the specific surface areas of the micropores and mesopores are different. M6.8 has a much larger specific surface area of pores with diameters less than 2 nm, corresponding to micropores, than other mesoporous silicas. For SBA-15, the specific surface area increases as the mesopore diameter increases. For all the silicas, the percentages of pores smaller than 2 nm in diameter among the total pores are larger in the calculations based on the specific surface area than in those based on the volume. Although the pore-size distribution is often discussed in terms of pore volume, we need to consider it in terms of surface area when discussing the effect of the pore size on interfacial chemical reactions such as (de)protonation and ion adsorption. While the BET method is commonly used to derive a specific surface area, it is difficult to apply to the mesoporous silicas used in this study, as described in the ESI.†
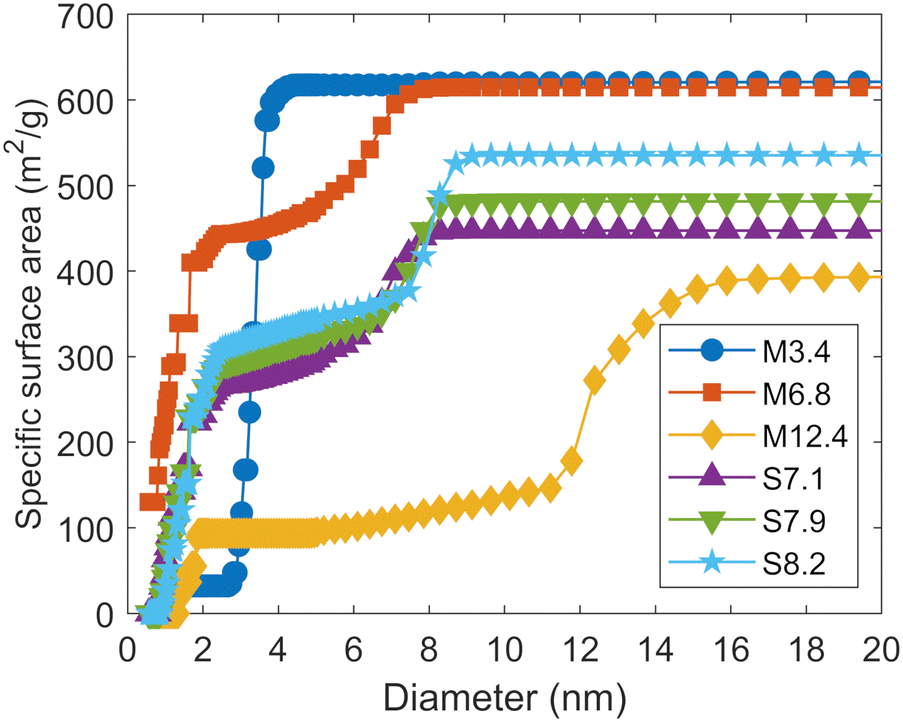 | ||
| Fig. 1 Cumulative distributions of the specific surface areas of the mesoporous silicas. | ||
The specific surface area of external surface of each silica determined with the t-plot method is also in Table 1. The external surface areas are sufficiently small compared to the surface areas of mesopores and micropores determined by the DFT method. Therefore, it can be assumed that (de)protonation and adsorption of ions on mesoporous silica surfaces occurs mainly inside the pores. Only M12.4 has a relatively large specific surface area on the external surface. However, as Fig. 1 shows, the mesopores of this silica are distributed over a wide range of sizes, and the t-plot method may not properly estimate the specific surface area of the external surface of M12.4 (see the ESI†).
Surface hydroxyl groups of the mesoporous silicas
The surface hydroxyl group densities of the silicas are calculated based on the amount of released structural water determined by TGA (see the ESI†). Fig. S5 in the ESI† shows the mass change of the mesoporous silicas as a function of temperature, expressed as a mass ratio to that before the temperature increase. The DTA curve of S7.1 is also shown as an example. The temperature at which the structural water begins to vaporize for each mesoporous silica was determined based on the temperature at which the large downward peak in the DTA curve ends and the mass loss occurs again in the TGA curve. The density of the surface hydroxyl groups of each mesoporous silica is shown in Table 1. The densities are distributed between 3 and 5 sites per nm2. The relatively small values for M3.4 and M6.8 are presumably due to the scarcity of the micropores in M3.4 and the presence of so small micropores that dehydration does not occur in M6.8, respectively (see the ESI†).Dissolution of the silicas
Silica partially dissolves in aqueous solutions, releasing silicic acid. Silicic acid is a weak acid that affects the solution pH. To quantify this effect on pH, we first examined the time required for the dissolution equilibrium (see the ESI†). As a result, the dissolution reached equilibrium after 5 days. Therefore, the silicic acid concentration and pH were measured more than 5 days after the silica was dispersed in the solution in the subsequent tests.Next, we investigated the effect of pH on the dissolution of silica. As a function of pH, the dissolution amount of silicic acid from both MCM-41 and SBA-15 remained constant at about 2 mM up to pH 9, but increased at pH > 9 (see the ESI†). As a greater degree of silica dissolution at higher pH not only increased proton release by the dissolved silicic acid, but also may have changed the microporous structure of mesoporous silica, the pH range will be limited to pH < 9 in subsequent discussions of surface charge density.
Surface charge density of the mesoporous silicas
When M3.4 and S7.1 were dispersed in a 10 mM NaNO3 solution, the pH of the liquid phases after 5 days were 4.76 and 4.84, respectively. Titration curves for each mesoporous silica at ionic strengths of 1, 10, and 50 mM are shown in the ESI.† To discuss the reactivity of the surface, we need to normalize the titration curves by the specific surface areas. Therefore, the surface charge densities were calculated based on measured pH values and the amounts of NaOH added (see the ESI†). It is noted that the surface charge density in this paper refers to the net proton surface charge density generated by the charges of deprotonated surface hydroxyl groups.20 The ESI† also shows how the correction was made for the measured H+ balance from deprotonation of silicic acid released from the silica dissolution. As the pH increases and the ionic strength decreases, the deprotonation reaction of the dissolved silicic acid affects the value of the calculated surface charge density.Fig. 2 shows the surface charge densities of the silicas at different ionic strengths as a function of pH. Note that the vertical scale is different for each ionic strength. The surface charge densities at the different ionic strengths around two different pH levels (7 and 8.5) are shown in Table S3 in the ESI.† The magnitude of the negative surface charge density increased with increasing ionic strength, which was consistent with findings in the literature investigating the surface charge densities of amorphous silica particles and quartz.31–33 This is because a higher ionic strength corresponds with a stronger screening of the negative charge on the silica surface by the EDL, leading to more deprotonation on the surface.32 Similar to the trend observed in the titration curve, the absolute value of the surface charge density of M12.4 was larger than those of the other mesoporous silicas at any ionic strength. The M12.4 silica had the smaller fraction of micropores and the mesopore diameter of M12.4 is the largest among all the silicas. This suggests that pores smaller than about 10 nm in diameter have smaller absolute surface charge densities than larger pores. While this trend is inconsistent with Wang et al.,19 who found that mesoporous alumina had a much higher surface charge density than non-porous alumina, it does concur with the results from Prélot et al.,34 who reported that the absolute value of the surface charge density of non-porous silica exceeded that of any SBA-15. Their finding supports the claim that confinement leads to a decrease in the absolute value of the surface charge density, which has also been shown by recent numerical calculations with the classical density functional theory (CDFT) or Poisson–Nernst–Plank (PNP) equations.21,22 As shown by their calculations, the overlap of EDLs increases as the pore gets smaller, which can affect the surface charge density. As the diameter of the pore decreases, the potential difference between the region near the surface and the center of the pore decrease due to the effect of EDL on the opposite surface, and the screening by electrolyte ions may be weaken.21,22 In addition, as the pore size becomes smaller, the effect of the curvature of the pore surface cannot be ignored. Note that the absolute value of the surface charge density of nanoparticles is known to increase as their size decreases,23,32 which may imply that the relationship between the size and surface charge density is opposite between pores and particles. The relationship in nanoparticles has been explained by the fact that the screening of the charge on the particle surface by the EDL becomes more efficient when the particle becomes smaller, as the screening atmosphere goes from a single half space to a spherically surrounding form.23 Judging from this, the effect of the surface curvature works for small pores in the opposite way as it does for nanoparticles, and the charge screening by EDL becomes inefficient as the diameter of the pores decreases.
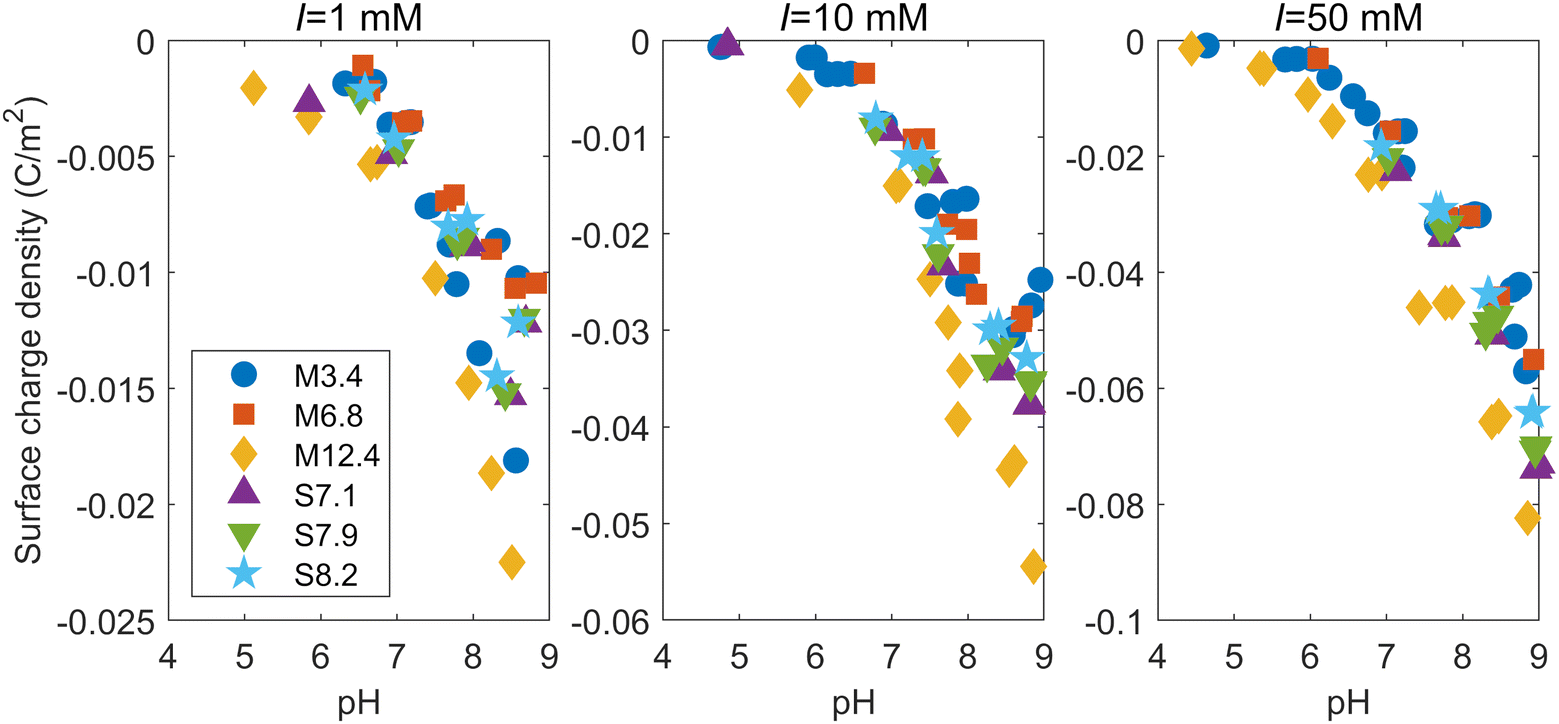 | ||
| Fig. 2 Surface charge densities of the mesoporous silicas at ionic strengths (I) of 1, 10, and 50 mM as a function of pH. | ||
In our study, the surface charge densities of the mesoporous silicas other than M12.4 shows no significant difference (Fig. 2). This result can be explained by the fact that the surface charge densities are the sums of those of both the mesopores and micropores of the mesoporous silicas. M3.4 has the smallest mesopores, but also a smaller percentage of micropores among its total pores compared to the other mesoporous silicas (Table 1). To study the effect of the different pore diameters in more detail, we used the surface complexation model to analyze the relationship between the pH and surface charge density for each ionic strength, reflecting the experimental pore-size distributions and the hydroxyl group densities (Table 1).
Surface complexation model for cylindrical pores of mesoporous silicas
Description of the model
The surface complexation model was applied to analyze the surface charge density of each mesoporous silica obtained from the batch titration tests, in order to relate the changes of surface equilibrium reactions such as the (de)protonation and adsorption of ions with pore size to the structure of the EDL or interfacial water. The mesopores and micropores of the mesoporous silicas were modeled as cylinders with infinite length, and the model considered the distribution of the radial one-dimensional potential, assuming that the internal electrostatic potential was uniform in the length direction. A schematic diagram of the distribution of ions and potential inside a pore are shown in Fig. 3. The model handles the radial potential distribution in a cylindrical pore by dividing it into two parts: the Stern layer and diffuse layer. While the hydrated counter ions (i.e., Na+) cannot directly adsorb on the charged surfaces of the silica pores, they can form outer-sphere complexes on a plane, being located slightly distant from the surface (called the outer Helmholtz plane (OHP)). The range where no charge exists is called the Stern layer, and the potential drop is assumed to be linear in this layer. The relationship between the surface charge density and the potential in the Stern layer can then be expressed by a simple capacitor model:| σ0 = C(ψ0 − ψOHP) | (1) |
 | ||
| Fig. 3 Schematic diagram of the distribution of ions (upper part) and the potential profile (lower part) inside a pore. | ||
The calculations of the surface complexation model, including the calculation of the potential distribution described above, were performed by formulating the mass action law and mass balance with MATLAB (MathWorks Inc.) software according to Tadanier and Eick.36 In this model, the activity of each ion pair was calculated from the activity of the free ions based on the mass action law considering the potential at the surface or OHP where the reaction occurs. Two surface reactions were considered: the deprotonation reaction of hydroxyl groups and the ion-pair formation of Na+ with a negative surface charge. The surface charge density was calculated based on the activity of ion pairs and the numerical solution of the PB equation calculated using the fourth-order method (bvp4c) function in MATLAB, with the arguments on the potential at the OHP, ionic strength, and pore size. During the above calculations, the mass balance of each ion and the site density (using the values in Table 1) and the charge balance at the surface and OHP were always checked to ensure that they were satisfied. By using the surface hydroxyl group density calculated from the TGA results as the site density considered in the mass balance, differences in surface hydroxyl group density between different silicas can be incorporated into the model. The detailed flow of the calculation is shown in the ESI.†
Fig. 4 shows the potential distributions of the diffuse layer in the pores with the different diameters calculated by the model shown above. The results for the ionic strength of 1 mM show that the potential at the center of the pore is not zero for all the pore diameters examined, which indicates an overlap of the EDLs. As the pore diameter decreases, the overlap of the EDLs becomes more pronounced and smaller difference can be found in the electric potential between the center of the pore and the head of the diffuse layer. As the ionic strength increases to 50 mM, the overlap of the EDLs occurs only when the pore diameter is less than 20 nm, due to the thin EDLs (the Debye length is about 1.4 nm). Fig. S10a in the ESI† shows the calculated surface charge densities for pores with different diameters at pH 7, with the capacitance of the Stern layer set at 1 C m−2. The absolute surface charge density increases with the ionic strength and with the pore diameter. These trends correspond to the changes of the potential gradient at the OHP in different pore diameters (Fig. 4) and can contribute to the small absolute value of the surface charge density in the small pores, as indicated by comparison of surface charge densities of the silicas with different pore sizes (Fig. 2).
 | ||
| Fig. 4 Potential distributions of the diffuse layer in pores with different diameters at pH 9 and ionic strengths of 1 mM (left) and 50 mM (right), based on the cylindrical one-dimensional PB equation. The capacitance of the Stern layer and surface hydroxyl group density were set at 1 C m−2 and 4 sites per nm2, respectively. The horizontal axis shows the radial position normalized by the radius of each pore. The dots on the vertical axis represent the potential at the surface of each pore. | ||
Fitting for experimentally determined surface charge densities
Since the mesoporous silicas used in this study have both micropores and mesopores, the surface complexation reaction on the micropore and mesopore surfaces needs to be calculated separately, reflecting the different potential distribution depending on the pore diameters. Based on the distribution of the specific surface areas (Fig. 1) and the relationship between calculated surface charge density and pore diameter (see the ESI†), all six of the mesoporous silicas in this study were modeled under the assumption that they had two different pore sizes: a micropore with a diameter of 1.36 nm and a mesopore with the diameters shown in Table 1. The surface charge density of each silica was calculated by determining the surface charge density of each pore when the two types of pores were present for their respective surface areas under each solution condition and their proportions were multiplied with respect to the surface areas. To minimize the difference between the calculated and experimental surface charge densities, the capacitance of the Stern layer was varied for seven types of pores, namely, six types of mesopores with different diameters for each mesoporous silica, and a micropore with a diameter of 1.36 nm (the micropore diameter shared by all samples). The fitting was performed using the Levenberg-Marquardt algorithm.The solid lines in Fig. 5 represent the fitting results of the surface charge densities obtained from the titration tests shown in Fig. 2. As Fig. 5 shows, the model adequately explains the surface charge densities derived from the experimental results for all the ionic strengths and all the samples. Fig. 5 also shows that the absolute values of the surface charge densities of M6.8, which has the largest proportion of micropores with respect to the specific surface area, and M3.4, which has the smallest mesopores, are relatively small. By contrast, the absolute values of the surface change densities of M12.4, which has the largest mesopores, are largest. The surface charge densities of SBA-15 fall in between those of MCM-41 and are also close to each other, as they share similar mesopore diameters and specific surface areas with similar size distributions.
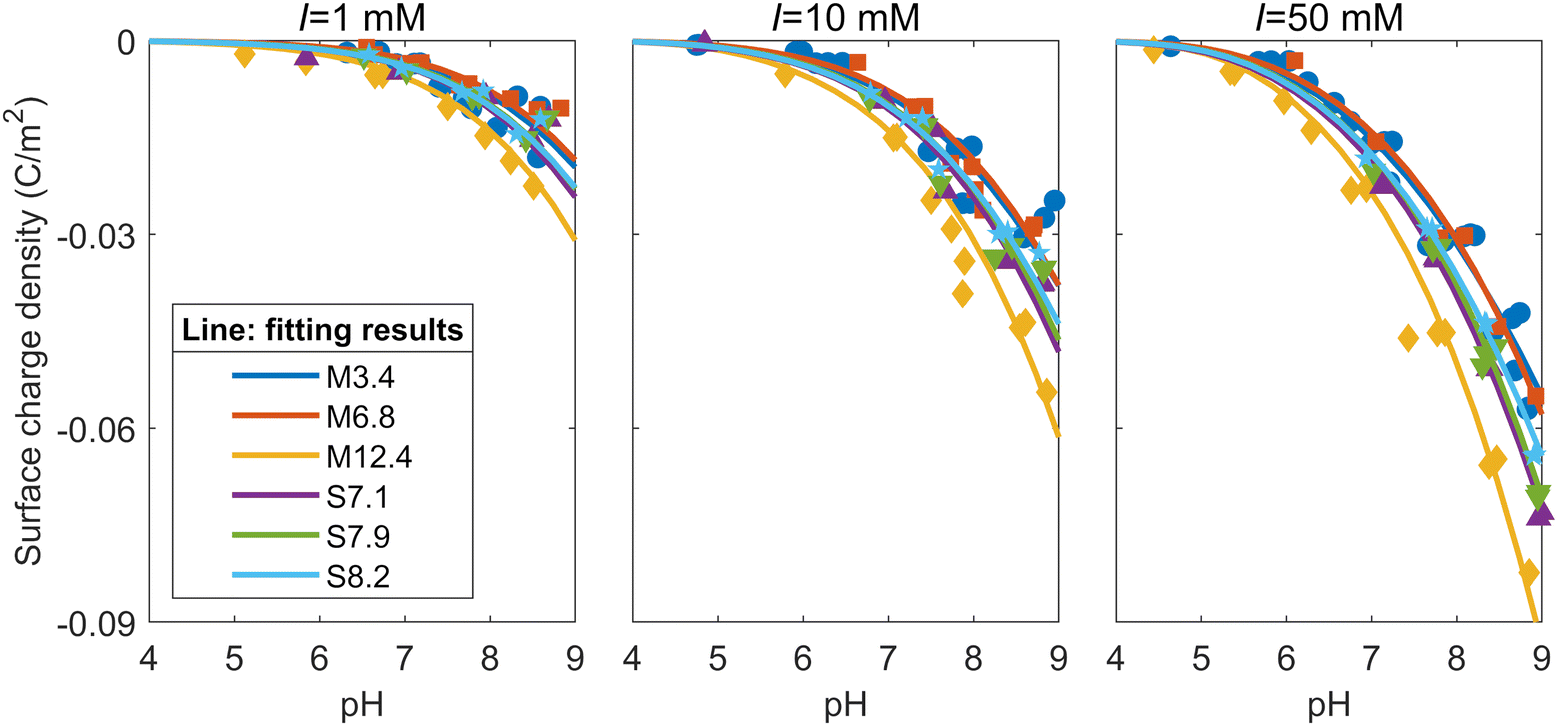 | ||
| Fig. 5 Fitting results to the surface charge densities of the mesoporous silicas at ionic strengths of 1, 10, and 50 mM. | ||
Fig. 6 shows the capacitances of the Stern layer of a 1.36 nm micropore and six different mesopores optimized by the fitting, as a function of the diameters of the pores. The 95% confidence intervals for the fitting are shown as error bars. The confidence interval of the capacitance of the 6.8 nm pore is relatively large. The reason for this large error is that as the capacitance of the Stern layer increases, the degree of change in surface charge density associated with the change in the capacitances becomes smaller (see the ESI†).
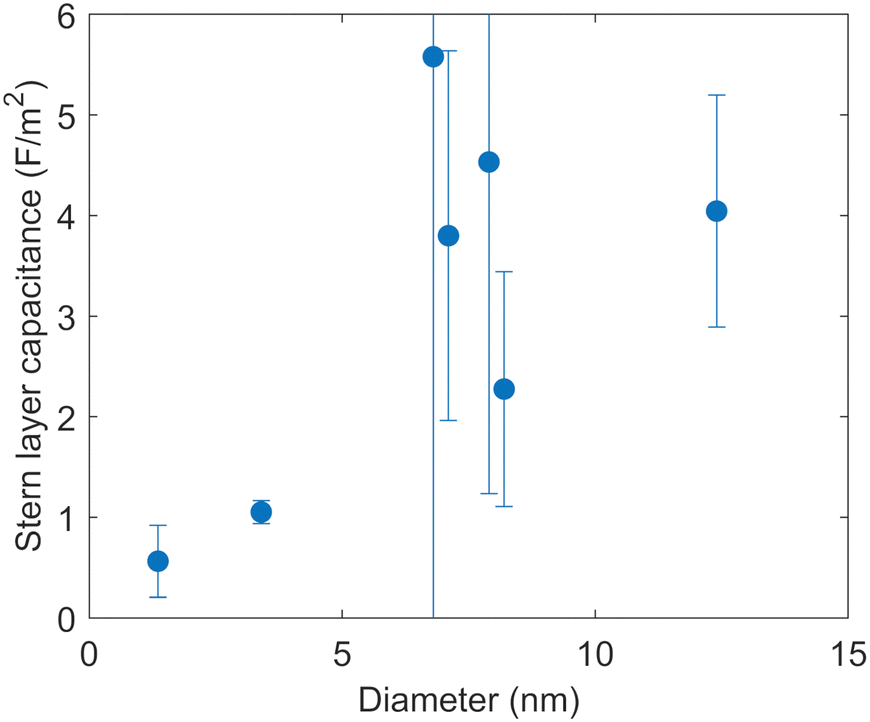 | ||
| Fig. 6 Capacitances of the Stern layer of pores with different diameters optimized by the fitting of the surface complexation model to the surface charge densities. The 95% confidence interval for the fitting is shown for each dot. | ||
The capacitances of the Stern layer of the pores with diameters of 1.36 nm and 3.4 nm in diameter are smaller than those of the larger pores in Fig. 6. While no clear trend can be seen for pores with diameters of more than 6 nm, the capacitance appears to decrease as the pore diameter decreases for pores with diameters of less than 6 nm. This indicates that there is a confinement effect due to something in addition to overlap of EDLs on the electrostatic reaction at the metal oxide surface. The capacitance of the Stern layer can be expressed as follows based on Gauss’ law by considering the hydroxyl groups on the solid surface and the electrolyte ions aligned at the OHP as a double cylindrical capacitor,37
 | (2) |
Fig. 6 shows that the decrease of the capacitance of the Stern layer with the decreasing pore diameter is bounded by a pore diameter of about 4–6 nm. There have been reports showing that confinement effects occur at similar scales for several chemical reactions. Knight et al.17 reported that SBA-15 with a pore diameter of 4.4 nm adsorbed more copper ions than SBA-15 with pore diameters of 5.2 nm and 7.0 nm. They found that the confinement effect, whereby the hydrogen bonding network between water molecules approached that of ice, was more pronounced in pore diameters of less than 5 nm.42 In addition, Baum et al.43 investigated the translational diffusion coefficient of water in different pore sizes and the electrolyte concentrations determined from quasi-elastic neutron scattering (QENS) for MCM-41 and SBA-15. As the pore size increased from 2.6 nm to 6.6 nm in diameter, they found that the effect of confinement on the water dynamics decreased and the contribution of electrolyte increased.
Meanwhile, the optimized capacitances of the Stern layer with pore diameters of 6 to 8 nm and 12.4 nm in diameter obtained in this study are higher than the value optimized to the surface charge densities of silica sol using a similar model by Hiemstra et al.44 There could be two possible reasons for the discrepancy as discussed in the ESI.† First, there may be a size effect on the surface charge densities that Hiemstra et al. used for the fitting. Another possible reason is the cut-off of the minimum pore size targeted in the fitting in our study.
Conclusion
To investigate how the surface reaction at the interface between metal oxides and water in meso- to micro-scale spaces varies with the space size, the surface charge densities of six types of mesoporous silicas were determined by batch titration and analyzed using a surface complexation model with a potential distribution based on the PB equation and the Stern model to describe the interfacial charge distribution. The six types of mesoporous silicas used in this study had both micropores and mesopores. The six types included three SBA-15 silicas in which half of the specific surface areas were occupied by micropores, and M6.8 silica in which two-thirds of the specific surface area was occupied by micropores. As the presence of micropores led to an overestimation of specific surface area by the conventional BET method, the specific surface areas and pore-size distributions were derived from the DFT method in this study. The absolute values of the surface charge densities of M12.4 silica, which had the largest mesopore diameter and a relatively small proportion of micropores, were larger than those of the other mesoporous silicas, suggesting that the absolute values of the surface charge densities of the pores smaller than 12.4 nm in diameter were smaller than those of larger pores. The tendency for the absolute value of the surface charge density to be smaller for pores in smaller sizes is opposite to the size dependence of the surface charge density of nanoparticles due to the inward curvature effect.The model showed that the overlap of EDLs became significant with a decrease of the pore diameter, and that the surface charge density decreased accordingly. We simplified the pore-size distributions of the mesoporous silicas into two types, a micropore and mesopore, and fitted the model to the surface charge densities of the silicas to optimize the Stern-layer capacitance in each type of the pores. As a result, the capacitances of the pores with diameters below 6 nm were smaller than those of the pores with diameters above 6 nm. This suggests that the dielectric constant of water near the pore surface of silica decreased as the pore diameter decreased. The changes in the electrostatic potential and the dielectric constant of the water near the surface with the change in the pore size may have a significant effect not only on the surface charge density but also on the adsorption of ions on the pore surface or the migration of ions inside the pore. To evaluate the migration of contaminants in natural rocks, both better understanding of the size distributions of mesopores and micropores in porous media and development of a sorption/migration model that incorporates the size effects are necessary as demonstrated in this study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Kunihisa Nakajima for his help in performing the thermogravimetric analysis and differential thermal analysis. This work was done as a joint research program between the Secretariat of the Nuclear Regulation Authority and the University of Tokyo.References
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the ‘Gold Book’). Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 1997 Search PubMed.
- L. V. Belovolova and M. V. Glushkov, Porous Matrices and Specific Features of Water in Nanostructures, Phys. Wave Phenom., 2021, 29, 249–277 CrossRef.
- J. Zachara, S. Brantley, J. Chorover, R. Ewing, S. Kerisit, C. Liu, E. Perfect, G. Rother and A. G. Stack, Internal Domains of Natural Porous Media Revealed: Critical Locations for Transport, Storage, and Chemical Reaction, Environ. Sci. Technol., 2016, 50, 2811–2829 CrossRef CAS PubMed.
- X. Gu, D. R. Cole, G. Rother, D. F. R. Mildner and S. L. Brantley, Pores in marcellus shale: A neutron scattering and FIB-SEM study, Energy Fuels, 2015, 29, 1295–1308 CrossRef CAS.
- H. R. Corti, G. A. Appignanesi, M. C. Barbosa, J. R. Bordin, C. Calero, G. Camisasca, M. D. Elola, G. Franzese, P. Gallo, A. Hassanali, K. Huang, D. Laria, C. A. Menéndez, J. M. M. de Oca, M. P. Longinotti, J. Rodriguez, M. Rovere, D. Scherlis and I. Szleifer, Structure and dynamics of nanoconfined water and aqueous solutions, Eur. Phys. J. E: Soft Matter Biol. Phys., 2021, 44, 136 CrossRef CAS PubMed.
- W. H. Thompson, Perspective: Dynamics of confined liquids, J. Chem. Phys., 2018, 149, 170901 CrossRef PubMed.
- H. Itoh and H. Sakuma, Dielectric constant of water as a function of separation in a slab geometry: A molecular dynamics study, J. Chem. Phys., 2015, 142, 1–10 CrossRef PubMed.
- S. Senapati and A. Chandra, Dielectric constant of water confined in a nanocavity, J. Phys. Chem. B, 2001, 105, 5106–5109 CrossRef CAS.
- L. Fumagalli, A. Esfandiar, R. Fabregas, S. Hu, P. Ares, A. Janardanan, Q. Yang, B. Radha, T. Taniguchi, K. Watanabe, G. Gomila, K. S. Novoselov and A. K. Geim, Anomalously low dielectric constant of confined water, Science, 2018, 360, 1339–1342 CrossRef CAS PubMed.
- T. H. H. Le, A. Morita and T. Tanaka, Refractive index of nanoconfined water reveals its anomalous physical properties, Nanoscale Horiz., 2020, 5, 1016–1024 RSC.
- K. Morikawa, Y. Kazoe, K. Mawatari, T. Tsukahara and T. Kitamori, Dielectric constant of liquids confined in the extended nanospace measured by a streaming potential method, Anal. Chem., 2015, 87, 1475–1479 CrossRef CAS PubMed.
- H. Sepehrian, S. J. Ahmadi, S. Waqif-Husain, H. Faghihian and H. Alighanbari, Adsorption Studies of Heavy Metal Ions on Mesoporous Aluminosilicate, Novel Cation Exchanger, J. Hazard. Mater., 2010, 176, 252–256 CrossRef CAS PubMed.
- Y. Kim, C. Kim, I. Choi, S. Rengaraj and J. Yi, Arsenic Removal Using Mesoporous Alumina Prepared via a Templating Method, Environ. Sci. Technol., 2004, 38, 924–931 CrossRef CAS PubMed.
- Y. Sun, S. Yang, G. Sheng, Z. Guo, X. Tan, J. Xu and X. Wang, Comparison of U(VI) removal from contaminated groundwater by nanoporous alumina and non-nanoporous alumina, Sep. Purif. Technol., 2011, 83, 196–203 CrossRef.
- H. B. Jung, M. I. Boyanov, H. Konishi, Y. Sun, B. Mishra, K. M. Kemner, E. E. Roden and H. Xu, Redox Behavior of Uranium at the Nanoporous Aluminum Oxide-Water Interface: Implications for Uranium Remediation, Environ. Sci. Technol., 2012, 46, 7301–7309 CrossRef CAS PubMed.
- D. R. Ferreira and C. P. Schulthess, The Nanopore Inner Sphere Enhancement Effect on Cation Adsorption: Sodium, Potassium, and Calcium, Soil Sci. Soc. Am. J., 2011, 75, 389–396 CrossRef CAS.
- A. W. Knight, A. B. Tigges and A. G. Ilgen, Adsorption of copper(II) on mesoporous silica: The effect of nano-scale confinement, Geochem. Trans., 2018, 19, 1–13 CrossRef PubMed.
- M. Baca, X. Carrier and J. Blanchard, Confinement in nanopores at the oxide/water interface: Modification of alumina adsorption properties, Chem. – Eur. J., 2008, 14, 6142–6148 CrossRef CAS PubMed.
- Y. Wang, C. Bryan, H. Xu and H. Gao, Nanogeochemistry: Geochemical reactions and mass transfers in nanopores, Geology, 2003, 31, 387 CrossRef CAS.
- K. W. Goyne, A. R. Zimmerman, B. L. Newalkar, S. Komarneni, S. L. Brantley and J. Chorover, Surface charge of variable porosity Al2O3(s) and SiO2(s) adsorbents, J. Porous Mater., 2002, 9, 243–256 CrossRef CAS.
- J. Yang, H. Su, C. Lian, Y. Shang, H. Liu and J. Wu, Understanding surface charge regulation in silica nanopores, Phys. Chem. Chem. Phys., 2020, 22, 15373–15380 RSC.
- T. Sen and M. Barisik, Size dependent surface charge properties of silica nano-channels: Double layer overlap and inlet/outlet effects, Phys. Chem. Chem. Phys., 2018, 20, 16719–16728 RSC.
- Z. Abbas, C. Labbez, S. Nordholm and E. Ahlberg, Size-dependent surface charging of nanoparticles, J. Phys. Chem. C, 2008, 112, 5715–5723 CrossRef CAS.
- B. Prelot, S. Lantenois, C. Chorro, M. C. Charbonnel, J. Zajac and J. M. Douillard, Effect of nanoscale pore space confinement on cadmium adsorption from aqueous solution onto ordered mesoporous silica: A combined adsorption and flow calorimetry study, J. Phys. Chem. C, 2011, 115, 19686–19695 CrossRef CAS.
- M. Brinker and P. Huber, Wafer-Scale Electroactive Nanoporous Silicon: Large and Fully Reversible Electrochemo-Mechanical Actuation in Aqueous Electrolytes, Adv. Mater., 2022, 34, 2105923 CrossRef CAS PubMed.
- M. Jaroniec, M. Kruk, J. P. Olivier and S. Koch, A new method for the accurate pore size analysis of MCM-41 and other silica based mesoporous materials, Stud. Surf. Sci. Catal., 2000, 128, 71–80 CrossRef CAS.
- A. Galarneau, D. Mehlhorn, F. Guenneau, B. Coasne, F. Villemot, D. Minoux, C. Aquino and J. P. Dath, Specific Surface Area Determination for Microporous/Mesoporous Materials: The Case of Mesoporous FAU-Y Zeolites, Langmuir, 2018, 34, 14134–14142 CrossRef CAS PubMed.
- B. Coasne, A. Galarneau, R. J. M. Pellenq and F. Di, Renzo, Adsorption, intrusion and freezing in porous silica: The view from the nanoscale, Chem. Soc. Rev., 2013, 42, 4141–4171 RSC.
- M. Jaroniec, M. Kruk and J. P. Olivier, Standard nitrogen adsorption data for characterization of nanoporous silicas, Langmuir, 1999, 15, 5410–5413 CrossRef CAS.
- R. Mueller, H. K. Kammler, K. Wegner and S. E. Pratsinis, OH surface density of SiO2 and TiO2 by thermogravimetric analysis, Langmuir, 2003, 19, 160–165 CrossRef CAS.
- M. Kobayashi, F. Juillerat, P. Galletto, P. Bowen and M. Borkovec, Aggregation and charging of colloidal silica particles: Effect of particle size, Langmuir, 2005, 21, 5761–5769 CrossRef CAS PubMed.
- M. Barisik, S. Atalay, A. Beskok and S. Qian, Size dependent surface charge properties of silica nanoparticles, J. Phys. Chem. C, 2014, 118, 1836–1842 CrossRef CAS.
- G. H. Bolt, Determination of the charge density of silica sols, J. Phys. Chem., 1957, 61, 1166–1169 CrossRef CAS.
- B. Prélot, S. Lantenois, Y. Nedellec, M. Lindheimer, J. M. Douillard and J. Zajac, The difference between the surface reactivity of amorphous silica in the gas and liquid phase due to material porosity, Colloids Surf., A, 2010, 355, 67–74 CrossRef.
- W. Qu and D. Li, A model for overlapped EDL fields, J. Colloid Interface Sci., 2000, 224, 397–407 CrossRef CAS PubMed.
- C. Tadanier and M. Eick, Formulating the Charge-distribution Multisite Surface Complexation Model Using FITEQL, Soil Sci. Soc. Am. J., 2002, 66, 2040–2041 CrossRef CAS.
- M. Fujimoto, Physics of Classical Electromagnetism, Springer New York, New York, NY, 2007 Search PubMed.
- D. A. Sverjensky, Interpretation and prediction of triple-layer model capacitances and the structure of the oxide-electrolyte-water interface, Geochim. Cosmochim. Acta, 2001, 65, 3643–3655 CrossRef CAS.
- M. A. Brown, Z. Abbas, A. Kleibert, R. G. Green, A. Goel, S. May and T. M. Squires, Determination of surface potential and electrical double-layer structure at the aqueous electrolyte-nanoparticle interface, Phys. Rev. X, 2016, 6, 1–12 Search PubMed.
- D. L. Oatley, L. Llenas, N. H. M. Aljohani, P. M. Williams, X. Martínez-Lladó, M. Rovira and J. de Pablo, Investigation of the dielectric properties of nanofiltration membranes, Desalination, 2013, 315, 100–106 CrossRef CAS.
- R. Renou, A. Ghoufi, A. Szymczyk, H. Zhu, J. C. Neyt and P. Malfreyt, Nanoconfined electrolyte solutions in porous hydrophilic silica membranes, J. Phys. Chem. C, 2013, 117, 11017–11027 CrossRef CAS.
- A. W. Knight, N. G. Kalugin, E. Coker and A. G. Ilgen, Water properties under nano-scale confinement, Sci. Rep., 2019, 9, 8246 CrossRef PubMed.
- M. Baum, F. Rieutord, F. Juranyi, C. Rey and D. Rébiscoul, Dynamical and Structural Properties of Water in Silica Nanoconfinement: Impact of Pore Size, Ion Nature, and Electrolyte Concentration, Langmuir, 2019, 35, 10780–10794 CrossRef CAS PubMed.
- T. Hiemstra, J. C. M. De Wit and W. H. Van Riemsdijk, Multisite proton adsorption modeling at the solid/solution interface of (hydr)oxides: A new approach: II. Application to various important (hydr)oxides, J. Colloid Interface Sci., 1989, 133, 105–117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Method for nitrogen gas adsorption; method for measurement of the dissolved silicic acid; adsorption and desorption isotherms of nitrogen; comparison between the BJH and DFT methods; applicability of the BET method; relatively high specific surface area of external surface of M12.4; the surface hydroxyl group densities of the mesoporous silicas; dissolution of the silicas; titration curves; calculation of surface charge density; application of the PB equation to the model; calculation flow of the model; simplification of the size distribution of the specific surface areas of the silicas; possible reasons for the large confidence interval; comparison of the optimized capacitance of the Stern layer to that of a previous study. See DOI: https://doi.org/10.1039/d2cp02520e |
| This journal is © the Owner Societies 2022 |