
Unravelling the origin of long-term stability for Cs+ and Sr2+ solidification inside sodalite†
Wenzhi
Luo‡
 a,
Xiaoqiang
Yang‡
a,
Hailin
Cao
b,
Luqian
Weng
c,
Gang
Feng
d,
Xian-Zhu
Fu
a,
Jing-Li
Luo
a and
Jianwen
Liu
*a
a,
Xiaoqiang
Yang‡
a,
Hailin
Cao
b,
Luqian
Weng
c,
Gang
Feng
d,
Xian-Zhu
Fu
a,
Jing-Li
Luo
a and
Jianwen
Liu
*a
aCollege of Materials Science and Engineering, Shenzhen University, Shenzhen, 518055, China. E-mail: jwliu@szu.edu.cn
bCollege of Materials Science and Engineering, Harbin Institute of Technology, Shenzhen, 518000, P. R. China
cShenzhen Aerospace New Materials Technology Cooperation, Shenzhen, P. R. China
dInstitute of Applied Chemistry, College of Chemistry, Nanchang University, No. 999 Xuefu Road, Nanchang 330031, P. R. China
First published on 18th July 2022
Abstract
Cesium (Cs+) and strontium (Sr2+) ions are the main fission byproducts in the reprocessing of spent nuclear fuels for nuclear power plants. Their long half-live period (30.17 years for 137Cs and 28.80 years for 90Sr) makes them very dangerous radionuclides. Hence the solidification of Cs+ and Sr2+ is of paramount importance for preventing them from entering the human food chain through water. Despite tremendous efforts for solidification, the long-term stability remains a great challenge due to the experimental limitation and lack of good evaluation indicators for such long half-life radionuclides. Using density functional theory (DFT), we investigate the origin of long-term stability for the solidification of Cs+ and Sr2+ inside sodalite and establish that the exchange energy and the diffusion barrier play an important role in gaining the long-term stability both thermodynamically and kinetically. The acidity/basicity, solvation, temperature, and diffusion effect are comprehensively studied. It is found that solidification of Cs+ and Sr2+ is mainly attributed to the solvation effect, zeolitic adsorption ability, and diffusion barriers. The present study provides theoretical evidence to use geopolymers to adsorb Cs+ and Sr2+ and convert the adsorbed geopolymers to zeolites to achieve solidification of Cs+ and Sr2+ with long-term stability.
1. Introduction
With the development of the nuclear industry, cheap and clean nuclear energy can supply about 13% of electricity worldwide and alleviate the pressure of petroleum resources.1–3 However, the disposal of the spent nuclear liquid has been one of most concerning issues for the international community.4,5 The Fukushima nuclear contamination accident in Japan3 and the Chernobyl Nuclear Power Plant accident in Ukraine6 have resulted in irreparable consequences for the natural environment and aroused the concern of people all over the world.7–10137Cs and 90Sr are the main fission byproducts in the raffinate of the traditional PUREX (Plutonium and Uranium Extraction) process, which contribute to most radioactivity and toxicities.11 The half-lives for 137Cs and 90Sr are 30.17 and 28.80 years, respectively, which are considered to be very dangerous radionuclides when entering the human food chain through the water.12 Moreover, the chemical properties of Cs and Sr are similar to K and Ca, respectively, which are essential elements of the human body.13–15 To this end, the aqueous Cs+ and Sr2+ are potential risks for the living organisms because of their high solubility. Thus the removal is highly desired.The chemical precipitation,12 solidification,12 adsorption,16 ion exchange,17 and membrane technologies18 have been widely used to remove the radioactive Cs+ and Sr2+ from wastewater. Among these methods, solidification has been considered as one of the most promising methods to deal with all kinds of radioactive elements since it is efficient, low-cost, and feasible.19 For the sake of economic and health benefits, it is highly desired for the trapping and solidification of the toxic and radioactive Cs+ and Sr2+ ions using these low-cost adsorbents from nuclear post-processing. It has been explored for decades.20 In the earlier studies, there were a lot of organic molecules experimentally used to capture Cs+ and Sr2+ with their chelating ability, such as crown,21 calix[4]-arene-crown-[6], and calix crown receptors.22–26 It was found that the type of counter-ion affects not only the extraction efficiency in solvent extractions but also the solidification process. For instance, Sessler et al. studied varieties of ion-pair receptors and found that the counter ion increased the Cs+ binding efficiency to the receptor.27 Although early work found that it was not efficient to extract harmful metal ions from the liquid phase when nitrate and chloride exist,28 the studies based on quantum chemical calculations suggested that counter ions such as nitrate indeed help in assisting metal binding at the crown cavity for both Cs+ and Sr2+,29,30 which indicates the counter ion might tune the solidification process for these radionuclides due to the favorable electrostatic interaction. This can also be inferred from the recent experimental observations for oligoether-strapped calyx [4] pyrrole.31
Recently, different materials were reported to be used in extracting Cs+ and Sr2+ successfully from an aqueous medium. For instance, Zhang and co-workers found that a clay-based hydrogel showed excellent Cs+ removal performance at an optimal adsorption amount of ∼173 mg g−1.32 Making use of the synergistic effect based on bentonite and chitosan, Wang and co-workers synthesized bio-sorbent-based composite bentonite-chitosan beads for removing the radioactive Cs+ in aqueous solution selectively.33 Dawson and co-workers reported sulfonated hyper-cross-linked polymers (SHCPs) possessing high BET surface areas for selective environmental remediation of Cs+ and Sr2+.17 In addition, aluminosilicate-based geopolymers and zeolites show a good removal performance for Cs+ and Sr2+. These aluminosilicate-based materials have been extensively investigated due to their inexpensive production cost, a wide range of commercial uses, a variety of structures and properties, and great compatibility with geopolymers that can be converted to zeolites for the solidification of harmful waste.34,35 He and co-workers found that Metakaolin-based geopolymers showed a better solidification performance than Portland cement, ceteris paribus.36 Shan and co-workers reported a CHA-type zeolite CHA-3 which could remove 95% Sr2+ ions and remains intact after 5 regeneration cycles. Li and co-workers used aspartic acid-modified zeolite Y to remove nuclide cation and it had great performance for the removal of Sr2+ and Cs+.37,38
Notably, studies have shown that the geopolymers can be converted to zeolites, such as sodalite,39 faujasite,40 zeolite X,41 zeolite Y,42 and zeolite NaA.41 Based on the characteristics of geopolymers and zeolites, a strategy can be proposed to make full use of their advantages, that is, use geopolymers to adsorb Cs+ and Sr2+ through ion exchange, and then in situ convert them into zeolite to immobilize the Cs+ and Sr2+ ions. For this kind of manipulation, the most concerning issue is the long-term stability of immobilized zeolites. Although there have been a number of studies on the zeolitic materials with improved performance to immobilize Cs+ and Sr2+,35,43 the understanding of the origin of long-term stability of these radioactive remains a challenge. The main reason lies in the fact that the radioactive experiments are harmful to human health and the long term experiments are not only time-consuming but also expensive. As a result, the long-term experiments are very limited. Furthermore, there is a lack of indicators to evaluate the long term stability. Nevertheless, the fast developed computational tool is an alternative way, which can not only save the experimental expense, but can also simulate the reaction by accelerating the reactions using theoretical virtual experiments. In this study, we perform a comprehensive study to unravel the origin of long term-stability for the solidification of Cs+ and Sr2+ at the molecular level thermodynamically and kinetically using these computational tools. The acidity/basicity, solvation, temperature and diffusion effect are systematically studied to uncover the long-term stability for the Cs+ and Sr2+ solidification.
2. Computational details
All the density function theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP).44,45 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional was employed46 with projected augmented wave (PAW) describing the electron–ion interaction.47,48 Additionally, the D3 dispersion correction49,50 were used for all the calculations based on the previous results that the dispersion correction can significantly enhance the calculation accuracy for the interaction for silica51,52 and aluminum silicate sodalite.53,54 The crystal structure was obtained from experimental data with a cubic cell, i.e. a = b = c = 8.848 Å.55 The plane-wave cut-off energy was set as 500 eV whereas the k-points were generated using the Monkhorst–Pack method with a grid size of 4 × 4 × 4. The convergence threshold for the energies and forces was set as 1.0 × 10−5 eV and 0.02 eV Å−1, respectively.The sodalite consists of sod (t-toc) cages,56 which are connected by corner-sharing SiO4 and AlO4 tetrahedra. The vertices of the cage are Si or Al atoms and the edges are the bridging oxygen atoms connecting Si and Al atoms. The truncated octahedron has six four-member rings (accounting Si and Al only) at the face centers and eight six-member rings at the corners, as shown in Fig. 1a. To model the radioactive waste Cs+ and Sr2+ solidification, both the acidic model H6[Si6Al6O24](H2O)8 and alkaline model Na8[Si6Al6O24] (OH)2(H2O)6 are built as shown in Fig. 1b.
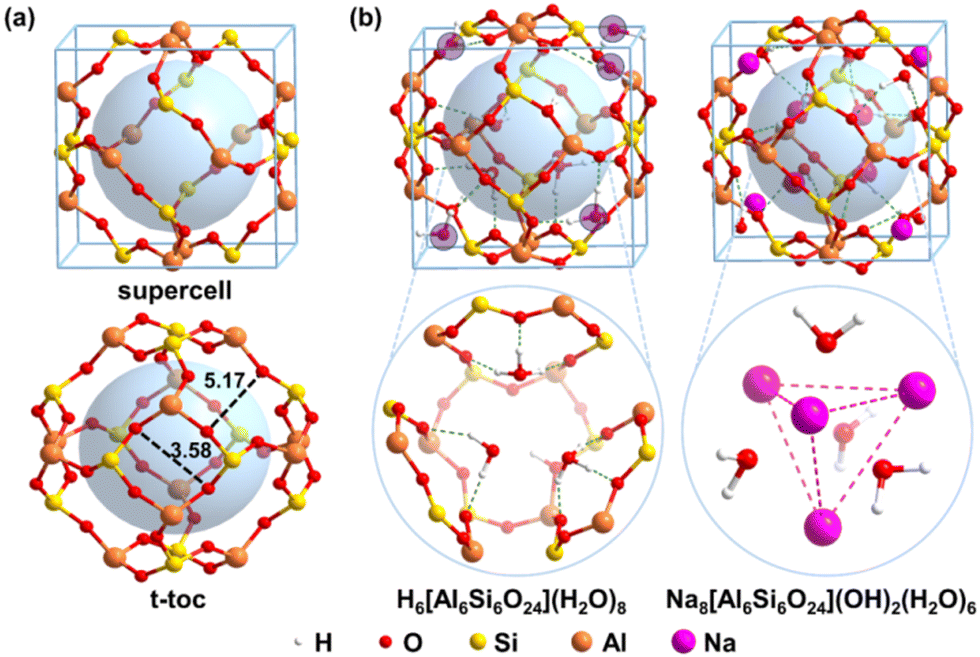 | ||
| Fig. 1 Solidification models for Cs+ and Sr2+. (a) The supercell and sodalite cage; (b) acidic model H6[Si6Al6O24](H2O)8 and alkaline model Na8[Si6Al6O24](OH)2(H2O)6. Bond lengths are in Å. The virtual pale blue is shown inside the cage of sodalite to better exhibit the hole. | ||
Because sodalite contains water molecules, various water molecules can combine to produce hydrated ions with Cs+ and Sr2+. There are numerous combinations, resulting in numerous configurations. As a result, finding the most stable configuration among them is quite challenging. Towards that end, all of the initial structures were first relaxed using ab initio molecular dynamics at 298 K for 1 picosecond with a step-size of 0.5 femtoseconds, generating 2000 configurations. The structures with the lowest energies were selected and optimized for use as the initial structures in future studies of the calculation of exchange and diffusion energies.
The solidification process can be considered as an ion exchange process which has been stated in detail previously.53 The exchange energies for H-type and Na-type sodalite are defined for a Mx+ (x = 1, 2) ion as follows:
| H-type: EEX = x × E(HNO3) + E(M@zeolite) − E(Hx@zeolite) − E(M(NO3)x) |
| Na-type: EEX = x × E(NaNO3) + E(M@zeolite) − E(Nax@zeolite) − E(M(NO3)x) |
The energies of M@zeolite and Hx@zeolite may be calculated directly from structural optimization. Energy calculations for H2O, HNO3, NaNO3, and M(NO3)x, on the other hand, are challenging since direct energy calculations for the interaction system are meaningless without taking the interaction into consideration. For example, because hydrogen bonding is so important in HNO3, ignoring it would render the calculations nonsensical. We used experimental crystalline structures for our calculations. The experimental lattice parameters for crystalline H2O,57 HNO3·3H2O,58 NaNO3,59 CsNO3,60 and Sr(NO3)261 are presented in Table S1 (ESI†). The H-type and Na-type sodalite models were used in the calculations. For H2O, HNO3, NaNO3, CsNO3, and Sr(NO3)2, the unit energy (Eunit) was derived by dividing the total energies by the number of unit components in a primitive cell. For example, the unit energy of a primitive cell of H2O is equal to the total energy divided by four, i.e. Eunit(H2O) = ETotal/4. Furthermore, because HNO3 crystalline structures are solvated as HNO3·3H2O, the energy from H2O molecules must be eliminated. Eunit(H2O), the unit energy for crystalline H2O, is utilized for subtraction.
The diffusion energy is critical for determining solidification. A diffusion channel from the center of one sodalite cage to the center of the neighboring one was proposed since it is the shortest and has the least steric barrier. The supercell (Fig. 1a) is made up of two cages, one with the center in the center of the supercell and the other with the center in the corner of the supercell, due to periodic boundary conditions. As a result, the actual reaction pathway for the diffusion of the Cs+/Sr2+ ions for the calculations is from the supercell's center to its corner.
We employed two distinct approaches to determine the energy barrier for diffusion due to the system's complexity. The climbing image nudged elastic band (CI-NEB)62 approach was used to compute the diffusion energy barrier for CsH5[Al6Si6O24](H2O)n and SrH4[Al6Si6O24](H2O)n (n = 0–4). In addition, we attempted another way, which is to choose a structure every 0.01 Å in the proposed reaction pathway for optimization, and then estimate the energy difference between the structure with the highest energy and the structure with the lowest energy in the reaction path as the energy barrier. Notably, each structure was initially relaxed at 298 K using ab initio molecular dynamics to try to get the low lying structure as the initial guess. The water molecules interacting with Cs+ and Sr2+ were then optimized to ensure the most stable configuration at each point along the diffusion path, while the Cs+ and Sr2+ were fixed to ensure that they did not depart from the proposed pathway. A unified explicit solvation technique was employed for all structures of CsH5[Al6Si6O24](H2O)n and SrH4[Al6Si6O24](H2O)n (n = 0–4). The gas phase calculation was performed specifically for the absence of water molecules (n = 0). The initial structures were obtained from the associated solvation effect studies of CsH5[Al6Si6O24](H2O)n and SrH4[Al6Si6O24](H2O)n (n = 0–4) for the diffusion energy calculations.
3. Results and discussion
To uncover the long-term stability of solidification of Cs+ and Sr2+ in sodalite, not only the thermodynamic factors arising from the acidity/basicity effect, solvation effect, and temperature effect, but also the kinetic factors from the diffusion effect are comprehensively studied.3.1 Acidity/basicity effect
The acidity/basicity of zeolites is an essential factor to determine the ability of adsorption.63 For the solidification of hazardous radionuclides Cs+ and Sr2+, it is extremely important because it may lead to the leakage of Cs+ and Sr2+. To evaluate the acidity/basicity effect for the solidification of Cs+ and Sr2+, the exchange energies are introduced, which were successfully applied for the solidification of hazardous metals from fly ashes previously.53 The more negative the exchange energy is, the easier the reaction is.As shown in Fig. 2, the exchange energies are proposed to evaluate the stability of radioactive ions Cs+ and Sr2+ inside the acidic and alkaline model thermodynamically with various Si/Al ratios ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 5:1 based on the experimental lattice parameters (Table S1, ESI†). It is shown that the exchange energies for both Cs+ and Sr2+ in acidic and alkaline models increase with the Si/Al ratio, indicating the low Si/Al is favorable for solidification. The main reason is attributed to more Brønsted acid sites produced from the low Si/Al ratio zeolite compared to that from the high Si/Al ratio zeolite.51,64 These Brønsted acid sites subsequently enhance the adsorption. For Si/Al = 1, the exchange energies for Cs+ and Sr2+ inside acidic sodalite are exothermic at −1.04 and −0.83 eV, respectively. When the Si/Al ratio increases to two, the critical point for the solidification of Cs+ and Sr2+ appears. The exchange energies are −0.11 eV and 0.41 eV for Cs+ and Sr2+, respectively. Notably, the exchange energies of Cs+ and Sr2+ are both positive for alkaline sodalite, indicating that this process is not spontaneous. Additionally, it is found that the calculated exchange energies are inversely correlated with the ionic radius: Cs+ (1.67 Å) > Sr2+ (1.18 Å) (as listed in Table S2, ESI†) for the acidic model. Such properties arise from the excellent structure containing a six-member ring which is analogous to crown ethers and exhibits the great performance for alkali ion size-selective adsorption.65,66
:1 to 5:1 based on the experimental lattice parameters (Table S1, ESI†). It is shown that the exchange energies for both Cs+ and Sr2+ in acidic and alkaline models increase with the Si/Al ratio, indicating the low Si/Al is favorable for solidification. The main reason is attributed to more Brønsted acid sites produced from the low Si/Al ratio zeolite compared to that from the high Si/Al ratio zeolite.51,64 These Brønsted acid sites subsequently enhance the adsorption. For Si/Al = 1, the exchange energies for Cs+ and Sr2+ inside acidic sodalite are exothermic at −1.04 and −0.83 eV, respectively. When the Si/Al ratio increases to two, the critical point for the solidification of Cs+ and Sr2+ appears. The exchange energies are −0.11 eV and 0.41 eV for Cs+ and Sr2+, respectively. Notably, the exchange energies of Cs+ and Sr2+ are both positive for alkaline sodalite, indicating that this process is not spontaneous. Additionally, it is found that the calculated exchange energies are inversely correlated with the ionic radius: Cs+ (1.67 Å) > Sr2+ (1.18 Å) (as listed in Table S2, ESI†) for the acidic model. Such properties arise from the excellent structure containing a six-member ring which is analogous to crown ethers and exhibits the great performance for alkali ion size-selective adsorption.65,66
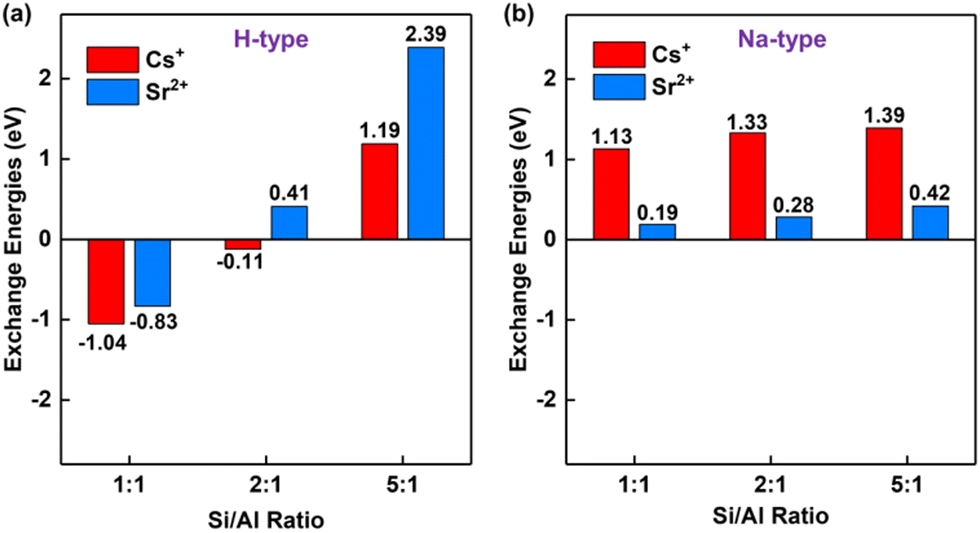 | ||
| Fig. 2 Exchange energies for Cs+ and Sr2+ in (a) acidic and (b) alkaline sodalite. | ||
The detailed structures are listed in Fig. 3 to reveal the essence of the solidification of the Cs+ and Sr2+ for the acidic and alkaline models, respectively. It is found that the distribution of crystalline water is essential for the stability of the structure. The most stable configure involves hexa-coordinations for Cs+, in which three are from water and the other three are from the framework. The Cs–O bond lengths from H2O molecules are 2.85, 3.14, and 3.15 Å, respectively (Fig. 3a). And the Cs–O bond lengths from the framework are 3.04, 3.11, and 3.14 Å, respectively. The average length for the coordinated Cs–O bonds is around 2.96 Å, which is much smaller than the previously reported average Cs–O bond length in the 8-member ring of CHA-2.05 (3.37Å) and MOR-6.53 (3.48Å).65 It indicates a crystalline structure instead of adsorption. Comparatively, the Sr–O lengths from the three water molecules for SrH4[Si6Al6O24](H2O)8 are 2.55, 3.22, and 3.39 Å, respectively (Fig. 3a). Due to the smaller ionic radius, the Sr–O lengths from the framework are 2.57, 2.61, and 2.68 Å, respectively. The distances of hexa-coordinated bonds for Sr2+ are uneven compared to that of Cs+. In the model CsNa7[Si6Al6O24](OH)2(H2O)6, one of the Na+ ions is substituted by one Cs+ whereas two Na+ ions are replaced by one Sr2+ as shown in Fig. 3. As the ionic radius follows the order of Cs+ (1.67 Å) > Sr2+ (1.18 Å) > Na+ (1.02 Å) shown in Table S2 (ESI†), introducing the larger ion Cs+ significantly make the original limited cage more crowded compared to that of Sr2+, resulting in the more endothermic for the solidification.
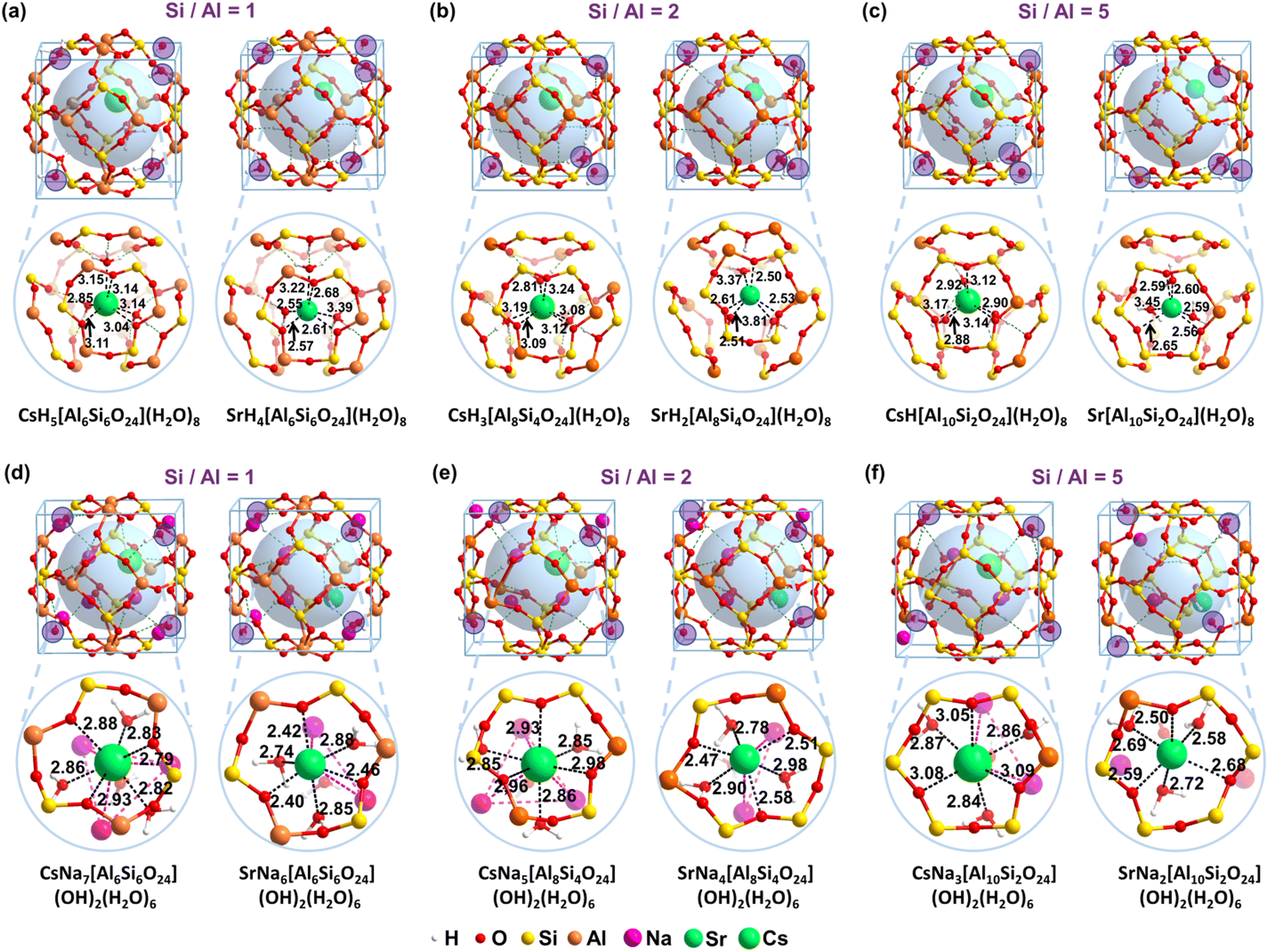 | ||
| Fig. 3 Structures for Cs+ and Sr2+ solidification in acidic and alkaline models. (a) Si/Al = 1 for the acidic model. (b) Si/Al = 2 for the acidic model. (c) Si/Al = 5 for the acidic model. (d) Si/Al = 1 for the alkaline model. (e) Si/Al = 2 for the alkaline model. (f) Si/Al = 5 for the alkaline model. To better display the framework, the critical bond lengths are given in Å. | ||
Collectively, the low Si/Al ratio is favorable for the solidification of Cs+ and Sr2+ due to the enhanced adsorption by Brønsted acid sites. The acid H-type sodalite is more favorable for the solidification of Cs+ and Sr2+ than the alkaline Na-type sodalite due to the strong coordination interaction between the oxygen atoms of the framework and the water molecules.
3.2 Solvation effect
For the solidification of heavy metals using aluminosilicates, crystalline water always exists in the framework. Therefore, the solvation effect is essential for the heavy metals according to the previous report.53,67 To understand the solvation effect, comprehensive studies were performed with the results summarized in Fig. 4.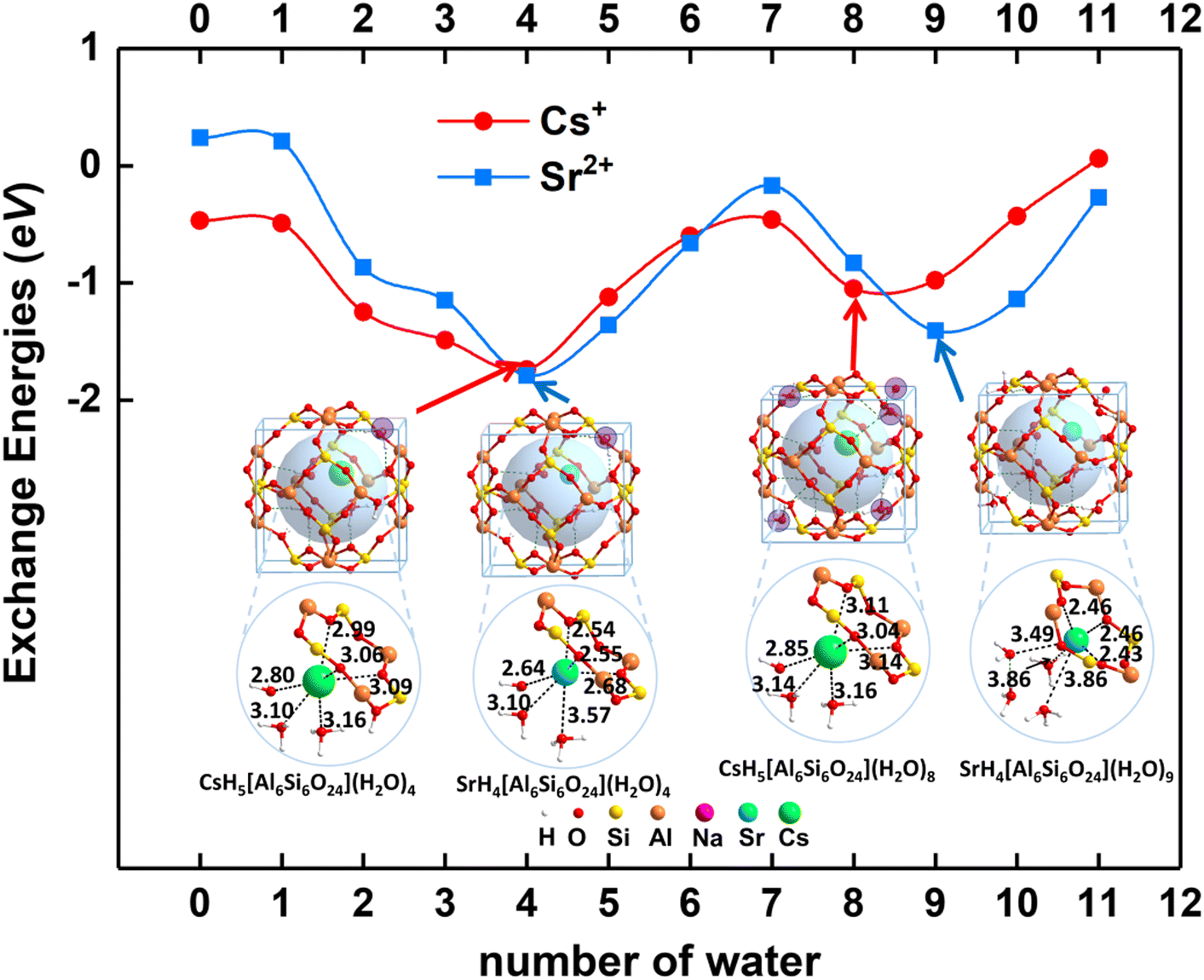 | ||
| Fig. 4 Solvation effect for the Cs+ and Sr2+ solidification in acidic sodalite. The associated exchange energies are listed in Table S4 (ESI†) and bond distances are listed in Table S5 in the ESI.† | ||
When n = 0, there are no water molecules in the sodalite cage. As shown in Fig. 4, the exchange energies for Cs+ and Sr2+ are −0.47 and 0.24 eV, respectively, suggesting Cs+ is easier to solidify than Sr2+ inside sodalite in the case of n = 0. The radioactive metal ions Cs+ and Sr2+ are chelated in the center of the six-member ring of the framework, which is similar to the crown organic molecules reported before.66,68,69 It is not as favorable as that for Cs+ (−1.04 eV) and Sr2+ (−0.83 eV) using the model H6[Si6Al6O24](H2O)8 as aforementioned, indicating that the solvation is vital for the solidification process. When n = 1–3, the water molecules are encapsulated in the framework. It is found that the exchange energies for Cs+ and Sr2+ become more and more exothermic when water molecules are introduced into the cage, indicating the solidification becomes more favorable. When n = 4–8, the most stable structure is the one with three water molecules located in the cage (denoted as waterin-cage) and the others located outside of the cage, i.e. in the adjacent cage (denoted as waterout-cage), showing the environmental effect. Especially, when the number of water molecules increases to four, the exchange energy reaches the minimum for both Cs+ and Sr2+. For this case, it can form the hexa-coordination with the oxygen atoms from water molecules and framework for both Cs+ and Sr2+. In addition, the fourth water molecule is attached to the six-member ring through hydrogen bonds to keep the structure stable. However, when more and more water molecules (n = 4 to 7) are introduced outside the cage of the framework (another cage in the adjacent), the exchange energies gradually become less negative because the more water molecules there are the more hydrogen bonds occur, which weaken the interaction between the water molecules and Cs+/Sr2+. When the number of water molecules is equal to eight, the exchange energy for Cs+ reaches a minimum, and that for Sr2+ reaches a minimum. The main reason may be due to the limited space for the two cages of a supercell, which can hold only 8 or 9 water molecules depending on the cation size. The further addition of water molecules in the cage leads to the increasing of exchange energy first (more negative) and then decreasing (less negative). The main reason may be due to the balanced effect arising from the increased hydrogen bonds which lower the system's energies and limit space confinement which increases the system's energies. For n > 9, the water molecules are added not only outside the cage but also in the cage, i.e. five in-case water molecules and the others are out-cage water molecules. This makes the cage very crowded and enhances the hydrogen bonds, weakening the interaction between the water molecules and Cs+/Sr2+.
To reveal the solidification mechanisms for Cs+, the structural details of CsH5[Si6Al6O24](H2O)n (n = 0–11) are shown in Fig. 5. For comparison, the structures for H6[Si6Al6O24](H2O)n (n = 0–11) before solidification are listed in Fig. S3 (ESI†). For the structure CsH5[Si6Al6O24](H2O)n (n = 0–11), when n = 0, the distances from Cs+ to the framework are 3.05, 3.15 and 3.16 Å, respectively. When one water molecule is introduced, the distance from Cs+ to the water molecule is 3.06 Å, while those from Cs+ to the framework are 3.09, 3.12, and 3.12 Å, respectively. As the number of water molecules increases to two, the Cs–O bond lengths are 2.99 and 3.05 Å. And the Cs–O bond lengths from the framework are 3.00, 3.01, and 3.20 Å, respectively, showing enhanced adsorption. When n is equal to three as shown in model CsH5[Si6Al6O24](H2O)3, the lengths for the Cs–O bonds from the three water molecules are 3.05, 3.07, and 3.10 Å. In comparison, the lengths for the Cs–O bonds from the framework are 2.95, 3.04, and 3.09 Å, showing further enhanced adsorption. Notably, hexa-coordinated Cs+ in the first solvated layer is formed when n = 3. However, a water molecule of CsH5[Si6Al6O24](H2O)4 tends to be pushed out to the adjacent cage although hydrogen bonds are formed between the water molecules and the framework. As the number of water molecules continues to increase, the water molecules are located on each corner of the cage i.e. the adjacent cage for the structures CsH5[Si6Al6O24](H2O)n (n = 5–8). For CsH5[Si6Al6O24](H2O)9, five water molecules are located out of the cage while four molecules are located in the cage. As the water molecules increase to ten, the coordination number of Cs+ remains six with three oxygen atoms from water and three from the framework (structure CsH5[Si6Al6O24](H2O)10). The Cs–O bonds formed from the framework are slightly strengthened with distances of 3.00, 3.19, and 3.28 Å. In comparison, the Cs–O bonds formed from water are also strengthened with distances of 2.82, 2.95, and 3.00 Å. For n = 11, the Cs–O bonds formed from water are further strengthened with distances of 2.95, 2.97, and 2.98 Å. Correspondingly, the Cs–O bonds formed from the framework are also further strengthened with distances of 2.97, 3.01, and 3.02 Å. Notably, no matter how many water molecules are introduced into the cage, only three water molecules are found to interact with Cs+ to form the first coordination layer and the rest are used to form the second or the third layer through the interaction of hydrogen bonds.
 | ||
| Fig. 5 Structures for CsH5[Al6Si6O24](H2O)n (n = 0–11). The pale blue sphere is introduced to show the hole of the model. The H2O molecules adsorbed outside the framework are marked in pale pink. The Cs–O bond length units are in Å. | ||
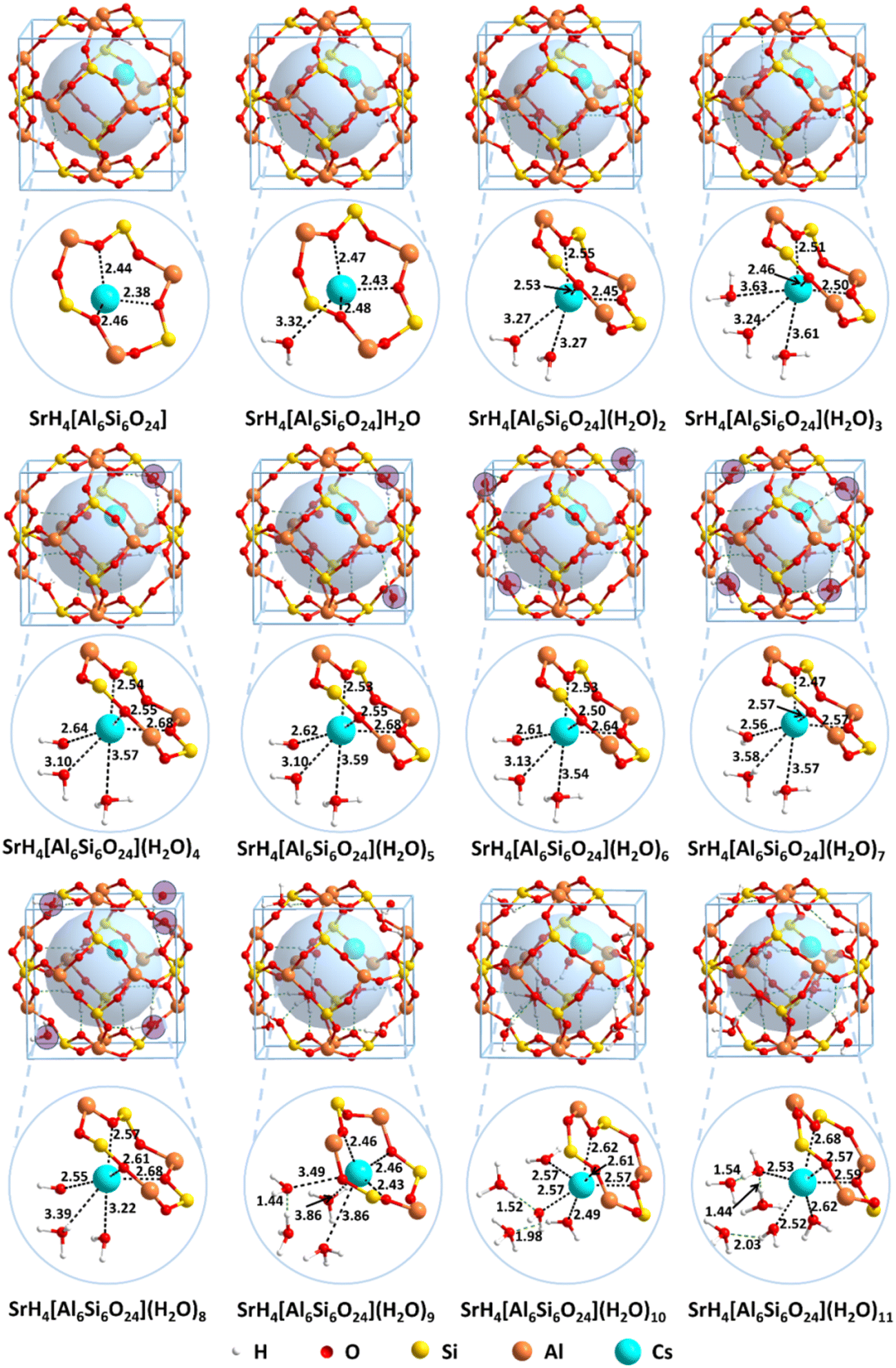
In contrast, the structural details for SrH4[Si6Al6O24](H2O)n (n = 0–11) are shown in Fig. 6. For n = 1–3, the distances of Sr–O bonds from water are larger than 3.00 Å, but the lengths of the Sr–O bonds from the framework are about 2.40–2.50 Å. For SrH4[Si6Al6O24](H2O)n (n = 4–8), the water molecules are evenly distributed out of the cage except for the three molecules encapsulated inside the cage. The distances of Sr–O bonds between Sr2+ and the framework remain in the range of 2.53–2.68 Å, indicating strong interactions. The situation diverges for the Sr–O bond lengths from water molecules. On one hand, two of the Sr–O bonds are larger than 3.0 Å as that for n = 1–3. On the other hand, the Sr–O bonds are enhanced to 2.55–2.65 Å, which are significantly stronger. The reason may be attributed to the increased water molecules, which enhance the interactions. Moreover, when the number of water molecules increases to nine, four water molecules are encapsulated in the inner cage while five water molecules are distributed in the adjacent cage. For the case that the four molecules are in the inner cage, three water molecules are located in the first solvated layer and the fourth water molecule is located in the second solvated layer. The distances between the three coordinated water molecules and Sr2+ are 3.49, 3.86, and 3.68 Å, respectively. As the value of n is equal to ten, the Sr2+ is still solvated by three water molecules with lengths of 2.49, 2.49, and 2.57 Å, which is significantly stronger than that for n = 9. Moreover, the extra two water molecules located in the second solvated layer further interact with coordinated water molecules through the hydrogen bonds. Similar to SrH4[Si6Al6O24](H2O)10, six water molecules are located in the cage with three in the first solvated shell and two in the second solvated shell when n = 11. The lengths of Sr–O bonds in the first coordinated layer are 2.52, 2.53, and 2.62 Å, respectively. Although the distances of Sr–O bonds are shortened with the value of n from nine to eleven, the crowded water molecules in the inner cage will result in the exchange energy increasing due to the limited space.
 | ||
| Fig. 6 Structures for SrH4[Al6Si6O24](H2O)n (n = 0–11). The pale blue sphere is introduced to show the hole of the framework. The H2O molecules adsorbed outside the framework are marked in pale pink. The Sr–O bond length unit is in Å. | ||
Collectively, the solidification of Cs+ and Sr2+ is very susceptible to crystalline H2O with respect to the solvation effect based on the detailed structures and associated exchange energies analysis. It is shown that the synergistic effect between the adsorbed water molecules and the framework can make chelated Cs+ and Sr2+ stable.
3.3 Temperature effect
To evaluate the solidification, the temperature is also an important factor because the storage temperature for the immobilized materials may be quite high. Sometimes, high-temperature treatment is necessary to enhance stability. For instance, Kim et al.70 reported that CHA based composite adsorbent can be transformed to pollucite at 1000 °C thermal treatment for enhanced storage stability. It should be ensured that the immobilized Cs+ and Sr2+ will not leak even at very high temperatures.Inspired by the thermostability of zeolite and to understand whether the temperature will affect the solidification process of Cs+ and Sr2+ immobilized by acidic sodalite, the exchanged energy based on Gibbs free energies, i.e. exchange free energies were calculated for CsH5[Si6Al6O24](H2O)n and SrH4[Si6Al6O24](H2O)n (n = 0–11) at 0, 300, 600, 900 and 1200 K, as shown in Fig. 7. It is found that the temperature does not change the trend for both Cs+ and Sr2+, indicating the solidification shows good thermal stability. The exchange free energies decrease gradually as the water molecules are introduced to the cage of the framework, and the exchange free energies arrive at the minimum when n = 4. As the water molecules continue to increase, the exchange free energies increase until the value of n is equal to seven, which reaches another minimum. Subsequently, the exchange free energies start to decrease and reach the local minimum when the value of n is equal to eight. Moreover, the exchange free energies are rising due to the limited space of the modelled cage when more water molecules are introduced.
 | ||
| Fig. 7 Temperature effect for the (a) structure CsH5[Al6Si6O24](H2O)n (n = 0–11) and (b) structure SrH4[Al6Si6O24](H2O)n (n = 0–11) in 0, 300, 600, 900 and 1200 K, respectively. Exchange free energies are given in eV. The associated data are listed in Table S8 in the ESI.† | ||
In summary, the solidification process is not sensitive to the temperature for both Cs+ and Sr2+ as the number of water molecules increases, indicating that the solidification shows good thermal stability, which is consistent with the analogous zeolite that has been experimentally reported.70
3.4 Diffusion effect
It is important to evaluate the diffusion energy barrier for metal ions or molecules to be stuck in a confined space. Generally, the larger the barrier is, the more difficult the leaching of hazardous ions is, i.e., the more stable structure is associated with a larger diffusion energy.To reveal the long-term kinetic stability for Cs+ and Sr2+ encapsulated in the sod (toc) cage, the diffusion was studied as shown in Fig. 8 using two distinct methods as addressed in the computational details part. Based on the structure analysis, the most possible diffusion channel for Cs+ and Sr2+ are through the six-member ring of the sod (toc) cage to the adjacent cage as shown in Fig. 8. It was found that the diffusion energy barriers are 2.99 and 1.71 eV for Cs+ and Sr2+, respectively, in the case of four crystalline water molecules involved based on the CI-NEB calculations. However, when the crystalline water molecules decrease, it is found that the energy barriers decrease. The diffusion energy barrier for Cs+ is decreased to 2.27 eV whereas that for Sr2+ is decreased to 0.53 eV as shown in Fig. 9, indicating the crystalline water molecule contributes significantly for the solidification. For Cs+, the diffusion energy barrier is up to 2.27 eV in the case of the absence of water molecules, indicating it is very difficult for Cs+ to escape from the cage. When the number of water molecules increases from one to four, the diffusion energy barriers are rising continually, with values of 2.61, 2.64, 2.71, and 2.99 eV respectively. This indicates that it is not easy to leak once the hazardous Cs+ was immobilized in the sod cage. Thus it shows excellent long-term stability.
 | ||
| Fig. 8 Scheme of the diffusion process for (a) Cs+ and (b) Sr2+. | ||
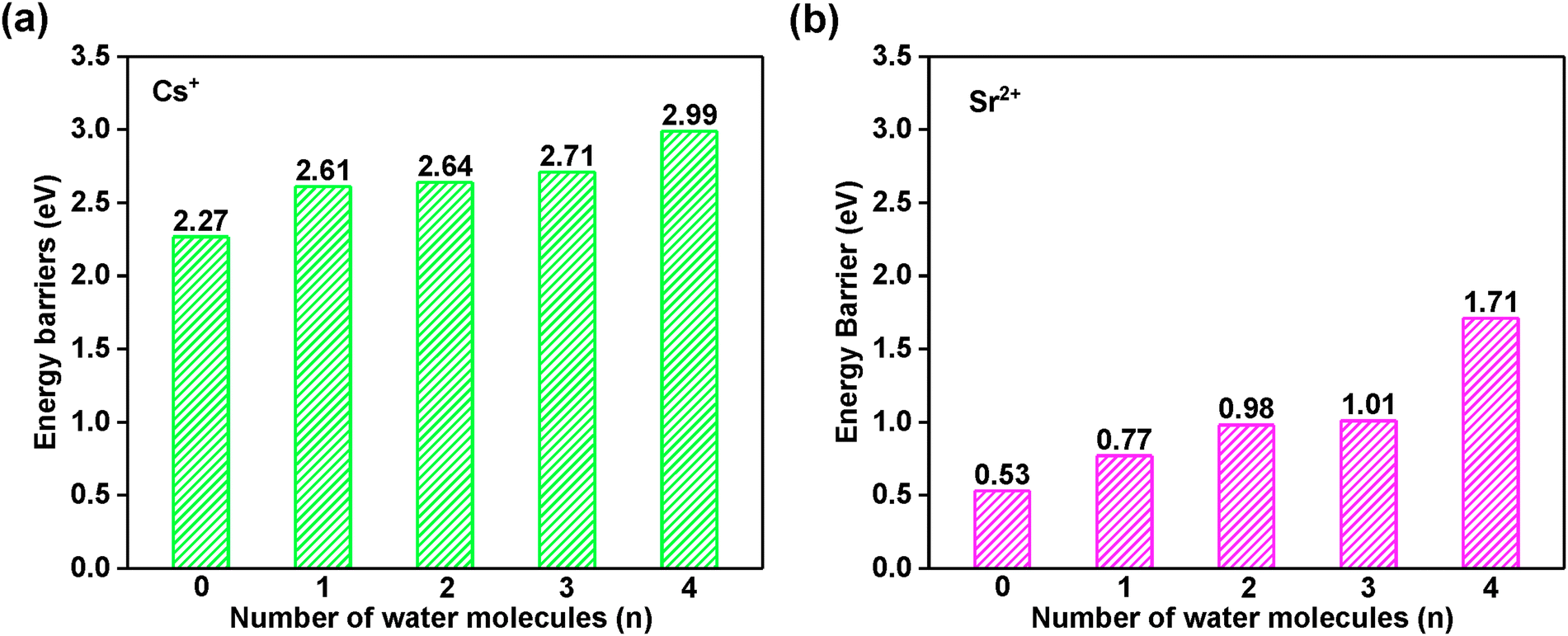 | ||
| Fig. 9 The diffusion Gibbs free energy barrier for (a) CsH5[Al6Si6O24](H2O)n and (b) SrH4[Al6Si6O24](H2O)n (n = 0–4) calculated by CI-NEB. The associated data are listed in Table S9 in the ESI.† | ||
Correspondingly, the diffusion energy barrier for Sr2+ is 0.53 eV in the absence of water, which is far smaller than that of Cs+. With the number of water molecules introduced to four, the diffusion energy barrier increases at values of 0.77, 0.98, 1.01, and 1.71 eV, respectively, as shown in Fig. 9b. It is also not easy to cross over with such a high barrier at 1.71 eV. Thus, the Sr2+ can also be immobilized in the cage of the framework with long-term stability.
In addition, we used the pathway-approximation method to estimate the diffusion energy barrier. Although the computed absolute energy values differ, the energy barrier trend of CI-NEB can be successfully reproduced using the results of CsH5[Al6Si6O24](H2O)n and SrH4[Al6Si6O24](H2O)n (n = 1–4) (Fig. S5, ESI†). As a result, while this technique cannot capture aspects of bond break and formation, it is a fair estimate for calculating diffusion energy barriers for future investigations.
The previous experimental studies about the kinetics of ion-exchange show that the alkali cations were faster than those of alkali earth cations due to the enhanced hydration of the latter.71–73 The main reason is attributed to the solvation effect. The radius of dehydrated Sr2+ (1.18 Å) is smaller than that of Cs+ (1.67 Å), yet the hydrated Sr2+ is at a value of 4.12 Å, larger than that of Cs+ at a value of 3.29 Å.74 Hence, the ion-exchange for Sr2+ needs enough energy to break the hydration, which leads to the solidification being slightly difficult compared to Cs+. This is not the case in this system, which may be owing to the structure of sodalite, which contains oxygen atoms, which may also form hydrogen bonds with water. It weakens the connection between Cs+ and Sr2+ and the hydrated structure becomes more flexible in the confined spaces.
Based on the aforementioned thermodynamical and kinetic analysis, a promising strategy for the solidification of Cs+ and Sr2+ can be applied. Firstly, the Cs+ and Sr2+ions can be adsorbed using amorphous geopolymer, and then converted into zeolites with sod (t-toc) structural units since this process is facile to achieve with various products containing sod (t-toc) structural units such as sodalite,39 faujasite,40 zeolite X, zeolite Y, and zeolite NaA. Due to the long-term stability of this method of the solidification of Cs+ and Sr2+ ions, this method is expected to be an attractive safe method for the solidification of Cs+ and Sr2+ in the future.
4. Conclusions
The solidifications of Cs+ and Sr2+ inside sodalite were comprehensively studied based on the periodic DFT calculations. The exchange energy and diffusion barrier were introduced as indicators to evaluate the long-term stability thermodynamically and kinetically. Based on the studies on the acidity/basicity, solvation, temperature, and diffusion effect, the origin of long-term stability for the solidification of Cs+ and Sr2+ inside sodalite was revealed. It is found that the long-term stability of solidification is mainly determined by the solvation effect, zeolitic adsorption ability, and diffusion barriers. The solidification ability of sodalite for Cs+ and Sr2+ is mainly due to the enhanced solvation effect arising from the crystalline water. Moreover, the immobilized structures are found kinetically resistant to high temperatures. Additionally, it is found that the solidification of Cs+ and Sr2+ are more favorable in the acid environment with the exchange energies of −1.04 and −0.83 eV, respectively due to the synergistic effect between the inner water and the framework of zeolites. Further studies on the nature of the diffusion process show that the diffusion energy barrier increases as the number of water molecules increase, i.e., introducing the water molecules can strengthen the solidification of Cs+ and Sr2+ inside the sod (t-toc) cage preventing leaching out. For this reason, our studies provide theoretical evidence for a promising strategy of Cs+ and Sr2+ solidification, which is to first adsorb Cs+ and Sr2+ ions with amorphous geopolymer, and then convert the Cs+ and Sr2+ adsorbed geopolymers to zeolites. In this way, Cs+ and Sr2+ can be immobilized in the zeolite with long-term stability without dissolving out.Author contributions
Wenzhi Luo, Xiaoqiang Yang: data curation, investigation, formal analysis, methodology, software, validation, writing-original draft, writing-review and editing; Hailin Cao, Luqian Weng and Gang Feng: conceptualization, visualization, writing-review and editing. Xian-Zhu Fu and Jing-Li Luo: conceptualization, funding acquisition, project administration, resources, visualization; Jianwen Liu: conceptualization, investigation, funding acquisition, project administration, methodology, visualization, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the financial support from the National Natural Science Foundation of China (21975163), the Shenzhen Innovative Research Team Program (KQTD20190929173914967), and the Senior Talent Research Start-up Fund of Shenzhen University (000265).References
- C. Liu, P.-C. Hsu, J. Xie, J. Zhao, T. Wu, H. Wang, W. Liu, J. Zhang, S. Chu and Y. Cui, Nat. Energy, 2017, 2, 17007 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- R. M. Grossi, Science, 2021, 372, 1131 CrossRef CAS PubMed.
- J. W. Suh, S. Y. Sohn and B. K. Lee, Energy Policy, 2020, 146, 111794 CrossRef CAS.
- D. Alby, C. Charnay, M. Heran, B. Prelot and J. Zajac, J. Hazard. Mater., 2018, 344, 511–530 CrossRef CAS PubMed.
- R. Stone, Science, 2021, 372, 670 CrossRef CAS PubMed.
- X. Sun, H. Luo and S. Dai, Chem. Rev., 2012, 112, 2100–2128 CrossRef CAS PubMed.
- X. Guo, S. Gin, P. Lei, T. Yao, H. Liu, D. K. Schreiber, D. Ngo, G. Viswanathan, T. Li, S. H. Kim, J. D. Vienna, J. V. Ryan, J. Du, J. Lian and G. S. Frankel, Nat. Mater., 2020, 19, 962–963 CrossRef CAS PubMed.
- J. Liang, J. Li, X. Li, K. Liu, L. Wu and G. Shan, Sep. Purif. Technol., 2020, 230, 115874 CrossRef CAS.
- M. Aoyagi, Nat. Energy, 2021, 6, 326–328 CrossRef.
- P. Amesh, A. S. Suneesh, K. A. Venkatesan, R. Uma Maheswari and S. Vijayalakshmi, Sep. Purif. Technol., 2020, 238, 116393 CrossRef CAS.
- D. Alby, C. Charnay, M. Heran, B. Prelot and J. Zajac, J. Hazard. Mater., 2018, 344, 511–530 CrossRef CAS PubMed.
- A. Merceille, E. Weinzaepfel, Y. Barré and A. Grandjean, Sep. Purif. Technol., 2012, 96, 81–88 CrossRef CAS.
- M. R. Awual, S. Suzuki, T. Taguchi, H. Shiwaku, Y. Okamoto and T. Yaita, Chem. Eng. J., 2014, 242, 127–135 CrossRef CAS.
- J. M. Kaste, P. Volante and A. J. Elmore, Nat. Commun., 2021, 12, 1937 CrossRef CAS PubMed.
- E. S. Dragan, D. Humelnicu, M. Ignat and C. D. Varganici, ACS Appl. Mater. Interfaces, 2020, 12, 44622–44638 CrossRef CAS PubMed.
- A. M. James, S. Harding, T. Robshaw, N. Bramall, M. D. Ogden and R. Dawson, ACS Appl. Mater. Interfaces, 2019, 11, 22464–22473 CrossRef CAS PubMed.
- D. Ding, Z. Zhang, R. Chen and T. Cai, J. Hazard. Mater., 2017, 324, 753–761 CrossRef CAS PubMed.
- J. Wang and S. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- S. Chen, J. Hu, S. Han, Y. Guo, N. Belzile and T. Deng, Sep. Purif. Technol., 2020, 251, 117340 CrossRef CAS.
- S. Maleknia and J. Brodbelt, J. Am. Chem. Soc., 1992, 114, 4295–4298 CrossRef CAS.
- T. D. Chung, J. Inclusion Phenom., 1998, 32, 179–193 CrossRef CAS.
- H.-F. Ji, R. Dabestani, G. M. Brown and R. A. Sachleben, Chem. Commun., 2000, 833–834 RSC.
- A. Casnati, A. Pochini, R. Ungaro, F. Ugozzoli, F. Arnaud, S. Fanni, M.-J. Schwing, R. J. M. Egberink, F. de Jong and D. N. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 2767–2777 CrossRef CAS.
- E. Ghidini, F. Ugozzoli, R. Ungaro, S. Harkema, A. Abu El-Fadl and D. N. Reinhoudt, J. Am. Chem. Soc., 1990, 112, 6979–6985 CrossRef CAS.
- Z. Asfari, C. Bressot, J. Vicens, C. Hill, J.-F. Dozol, H. Rouquette, S. Eymard, V. Lamare and B. Tournois, Anal. Chem., 1995, 67, 3133–3139 CrossRef CAS.
- S. K. Kim, V. M. Lynch, N. J. Young, B. P. Hay, C.-H. Lee, J. S. Kim, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 20837–20843 CrossRef CAS PubMed.
- I. H. Gerow, J. E. Smith and M. W. Davis, Sep. Sci. Technol., 1981, 16, 519–548 CrossRef CAS.
- B. Sadhu, M. Sundararajan, G. Velmurugan and P. Venuvanalingam, Dalton Trans., 2015, 44, 15450–15462 RSC.
- B. Sadhu, M. Sundararajan, A. Pillai, R. Singh and T. Bandyopadhyay, Int. J. Quantum Chem., 2017, 117, e25418 CrossRef.
- I.-W. Park, J. Yoo, B. Kim, S. Adhikari, S. K. Kim, Y. Yeon, C. J. E. Haynes, J. L. Sutton, C. C. Tong, V. M. Lynch, J. L. Sessler, P. A. Gale and C.-H. Lee, Chem. – Eur. J., 2012, 18, 2514–2523 CrossRef CAS PubMed.
- H. Zhang, C. S. Hodges, P. K. Mishra, J. Y. Yoon, T. N. Hunter, J. W. Lee and D. Harbottle, ACS Appl. Mater. Interfaces, 2020, 12, 33173–33185 CrossRef CAS PubMed.
- K. Wang, H. Ma, S. Pu, C. Yan, M. Wang, J. Yu, X. Wang, W. Chu and A. Zinchenko, J. Hazard. Mater., 2019, 362, 160–169 CrossRef CAS PubMed.
- A. M. El-Kamash, M. R. El-Naggar and M. I. El-Dessouky, J. Hazard. Mater., 2006, 136, 310–316 CrossRef CAS PubMed.
- H. Y. Lee, H. S. Kim, H.-K. Jeong, M. Park, D.-Y. Chung, K.-Y. Lee, E.-H. Lee and W. T. Lim, J. Phys. Chem. C, 2017, 121, 10594–10608 CrossRef CAS.
- P. He, J. Cui, M. Wang, S. Fu, H. Yang, C. Sun, X. Duan, Z. Yang, D. Jia and Y. Zhou, J. Hazard. Mater., 2020, 384, 121377 CrossRef CAS PubMed.
- Y. Li, A. O. Simon, C. Jiao, M. Zhang, W. Yan, H. Rao, J. Liu and J. Zhang, Microporous Mesoporous Mater., 2020, 302, 110244 CrossRef CAS.
- K. Wang, F. Wang, F. Chen, X. Cui, Y. Wei, L. Shao and M. Yu, ACS Sustainable Chem. Eng., 2019, 7, 2459–2470 CrossRef CAS.
- Y.-b Zong, C.-y Zhao, W.-h Chen, Z.-b Liu and D.-q Cang, Int. J. Min. Met. Mater., 2020, 27, 55–62 CrossRef CAS.
- J. L. Provis, G. C. Lukey and J. S. J. van Deventer, Chem. Mater., 2005, 17, 3075–3085 CrossRef CAS.
- E. Ofer-Rozovsky, M. A. Haddad, G. Bar Nes and A. Katz, J. Mater. Sci., 2016, 51, 4795–4814 CrossRef CAS.
- P. Rozek, M. Krol and W. Mozgawa, J. Clean. Prod., 2019, 230, 557–579 CrossRef CAS.
- E. Han, Y.-G. Kim, H.-M. Yang, I.-H. Yoon and M. Choi, Chem. Mater., 2018, 30, 5777–5785 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- P. E. Blöchl, C. J. Först and J. Schimpl, Bull. Mater. Sci., 2003, 26, 33–41 CrossRef.
- T. Kerber, M. Sierka and J. Sauer, J. Comput. Chem., 2008, 29, 2088–2097 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Feng, D. Yang, D. Kong, J. Liu and Z.-H. Lu, RSC Adv., 2014, 4, 47906–47920 RSC.
- J. Liu, W. Luo, Y. Yin, X.-Z. Fu and J.-L. Luo, J. Catal., 2021, 396, 333–341 CrossRef CAS.
- J. Liu, W. Luo, H. Cao, L. Weng, G. Feng, X.-Z. Fu and J.-L. Luo, Microporous Mesoporous Mater., 2020, 306, 110409 CrossRef CAS.
- J. Liu, Y. Yin, X.-Z. Fu and J.-L. Luo, Appl. Surf. Sci., 2019, 503, 144148 CrossRef.
- J. Felsche, S. Luger and C. Baerlocher, Zeolites, 1986, 6, 367–372 CrossRef CAS.
- B. J. Campbell, J. M. Delgado, A. K. Cheetham, B. B. Iversen, N. P. Blake, S. R. Shannon, S. Latturner and G. D. Stucky, J. Chem. Phys., 2000, 113, 10226–10239 CrossRef CAS.
- A. Goto, T. Hondoh and S. J. Mae, J. Chem. Phys., 1990, 93, 1412–1417 CrossRef CAS.
- I. Taesler, R. G. Delaplane and I. Olovsson, Acta Crystallogr., Sect. B, 1975, b31, 1489–1492 CrossRef CAS.
- P. Cherin, W. C. Hamilton and B. Post, Acta Crystallogr., 1967, 23, 455–460 CrossRef CAS.
- M. S. Kalliomaki and V. P. J. Meisalo, Acta Crystallogr., Sect. B, 1979, 35, 2829–2835 CrossRef.
- H. Nowotny and G. Heger, Acta Crystallogr., Sect. C, 1983, 39, 952–956 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- B. Smit and T. L. M. Maesen, Chem. Rev., 2008, 108, 4125–4184 CrossRef CAS PubMed.
- F. Chen, L. Zhang, G. Feng, X. Wang, R. Zhang and J. Liu, Appl. Surf. Sci., 2018, 433, 627–638 CrossRef CAS.
- S. Kwon, C. Kim, E. Han, H. Lee, H. S. Cho and M. Choi, J. Hazard. Mater., 2021, 408, 124419 CrossRef CAS PubMed.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- F. Lolli, H. Manzano, J. L. Provis, M. C. Bignozzi and E. Masoero, ACS Appl. Mater. Interfaces, 2018, 10, 22809–22820 CrossRef CAS PubMed.
- M. R. Awual, T. Yaita, T. Taguchi, H. Shiwaku, S. Suzuki and Y. Okamoto, J. Hazard. Mater., 2014, 278, 227–235 CrossRef CAS PubMed.
- M. R. Awual, Chem. Eng. J., 2016, 303, 539–546 CrossRef CAS.
- J. Kim, K. Lee, B.-K. Seo and J.-H. Hyun, Environ. Res., 2020, 191, 110099 CrossRef CAS PubMed.
- R. M. Barrer, R. F. Bartholomew and L. V. C. Rees, J. Phys. Chem. Solids, 1963, 24, 51–62 CrossRef CAS.
- R. M. Barrer, J. A. Davies and L. V. C. Rees, J. Inorg. Nucl. Chem., 1969, 31, 219–232 CrossRef CAS.
- H. S. Hassan, O. A. Abdel Moamen and W. F. Zaher, Adv. Powder Technol., 2020, 31, 1125–1139 CrossRef CAS.
- E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1cp04164a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2022 |