
Ultrafast Förster resonance energy transfer between tyrosine and tryptophan: potential contributions to protein–water dynamics measurements†
Haoyang
Li
a,
Guanyu
Jiang
a,
Menghui
Jia
a,
Simin
Cao
a,
Sanjun
Zhang
 a,
Jinquan
Chen
a,
Haitao
Sun
a,
Jianhua
Xu
*a and
Jay R.
Knutson
*b
a,
Jinquan
Chen
a,
Haitao
Sun
a,
Jianhua
Xu
*a and
Jay R.
Knutson
*b
aState Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai 200062, China. E-mail: jhxu@phy.ecnu.edu.cn; Tel: +86 21 62233936
bLaboratory of Advanced Microscopy & Biophotonics, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, Maryland 20892, USA. E-mail: knutsonj@nhlbi.nih.gov; Tel: +1 3014962557
First published on 12th July 2022
Abstract
Ultrafast Förster Resonance Energy Transfer (FRET) between Tyrosine (Tyr, Y) and Tryptophan (Trp, W) in the model peptides Trp-(Pro)n-Tyr (WPnY) has been investigated using a femtosecond up-conversion spectrophotofluorometer. The ultrafast energy transfer process (<100 ps) in short peptides (WY, WPY and WP2Y) has been resolved. In fact, this FRET rate is found to be mixed with the rates of solvent relaxation (SR), ultrafast population decay (QSSQ) and other lifetime components. To further dissect and analyze the FRET, a spectral working model is constructed, and the contribution of a FRET lifetime is separated by reconciling the shapes of decay associated spectra (DAS). Surprisingly, FRET efficiency did not decrease monotonically with the growth of the peptide chain (as expected) but increased first and then decreased. The highest FRET efficiency occurred in peptide WPY. The kinetic results have been accompanied with molecular dynamics simulations that reconcile and explain this strange phenomenon: due to the strong interaction between amino acids, the distance between the donor and receptor in peptide WPY is actually closest, resulting in the fastest FRET. In addition, the FRET lifetimes (τcal) were estimated within the molecular dynamics simulations, and they were consistent with the lifetimes (τexp) separated out by the experimental measurements and the DAS working model. This benchmark study has implications for both previous and future studies of protein ultrafast dynamics. The approach taken can be generalized for the study of proximate tyrosine and tryptophan in proteins and it suggests spectral strategies for extracting mixed rates in other complex FRET problems.
1. Introduction
The intrinsic fluorescence of proteins is dominated by three aromatic amino acid residues: tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe). For convenience, the abbreviations or single-letter abbreviations of amino acids are often used in the following text, for example, tryptophan (Trp or W), tyrosine (Tyr or Y), phenylalanine (Phe or F), proline (Pro or P), or Leucine (Leu or L). Among all amino acids, W (Trp) has the highest extinction coefficient and quantum yield while remaining highly sensitive to the local environment. Compared with extrinsic dye molecules, as a naturally abundant biological molecule, it is an innocuous and faithful reporter of environment. Therefore, it has been widely used in the study of proteins.1,2 Previous studies have shown that even peptides or proteins containing a single W usually exhibit wavelength-dependent multi-exponential fluorescence decay, and each exponential term has a significant decay associated spectrum (DAS).3–6 There have been two different explanations for the complexity of W fluorescence decay in proteins, but there is still no consensus. One explanation is the loss of heterogeneous population, that is, the inhomogeneous decay of W experiencing differing environs. Ground state heterogeneity is the simplest explanation for the complex decay of W fluorescence lifetime. This heterogeneity can be caused by the “rotamer” positions of a W side chain in simple peptides and, in more complex chains, “conformers” should be used to refer to the different environs around the indole group in the proteins.7,8 In our previous work, through the analysis of quantum yield and lifetime data, “quasi-static self-quenching” (QSSQ) has been widely observed in peptides, proteins, and some other biomolecules (like NADH).9–13 In addition to nanosecond lifetimes, shorter lifetimes were observed by up-conversion technology. The observation of an ultrafast decay rate is believed due to the existence of some “ultrafast-quenched conformation”. An alternate view is that the complex decay is due to the excited state equilibrium produced by solvent or protein relaxation, which shifts spectra on the same timescale.14,15 The solvation correlation function developed by Zhong and colleagues has been widely used to explain the relaxation of W in peptides or proteins. Their research posits that there are two different forms of water molecules around peptides or proteins (e.g., bulk and “biological”), which lead to two different rates.16–19 It is also called the “general solvent relaxation” (SR) of free water and the “slow solvent relaxation” (SSR) of bound water. We have shown that when W is replaced with a non-natural amino acid W analog, the contribution of heterogeneity (through quasi-static self-quenching, QSSQ) can be made very weak, allowing the remaining solvent relaxation term to emerge from the mixture.20,21 Thus, for W, the two phenomena are seen to exist at the same time and often can be difficult to distinguish.For a long time, the interaction between Y and W in proteins or peptides has been studied,22–28 especially, energy transfer from Y to W,27,28 a very significant phenomena of protein fluorescence. As early as the 1940s, the rate equation of energy transfer was proposed by Förster:
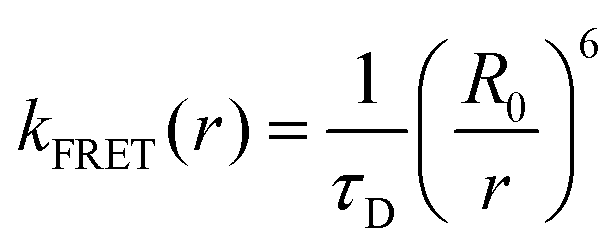 | (1) |
Polyproline exists in two forms, polyproline I (PPI) in which the peptide bonds are all cis, and polyproline II (PPII) in which the peptide bonds are all trans. In nonpolar or poor solvents, the more compact helix, PPI is the stable form; but in good solvents like water, the more extended helix, PPII is the stable form. In the cis(I) form the chain length is increased by 1.85 Å with each proline whereas in the trans form, the increment is 3.12 Å.30 The Y/W FRET pair is characterized by a very short Förster radius (R0 = 15 Å),31,32 which allowed distance determinations in such short peptides (WPnY, n < 5). There are many studies on “Spectroscopic Rulers” in short proline frames. Unfortunately, short proline stretches cannot be used as stable spacers, and the stability of spacing can be a very important thing in these studies. Comprehensive spectroscopic evidence for different model peptides demonstrates that the hexa-proline backbone forms a stable PPII helix in aqueous solution.24,33–37 For shorter polyprolines, the onset of the PPII secondary structure may vary; for example, peptides of the type Gly-(Pro)n-OH adopt the PPII structure only at n = 3 (onset),38 those of the type (Pro)n-OH39 and Trp-(Pro)n-Tyr-OH,30,40 only at n = 4. Thus, the mixing of the two structures (cis and trans) of proline in WPnY results in poor stability of the whole chain, i.e., the peptide chain can be partly folded (so the distance between donor and receptor will be closer than expected). According to Förster theory, the decrease of distance between donor and acceptor will lead to the sharp increase of energy transfer rate. Thus, for low n, these ultrafast energy transfer phenomena cannot be detected by traditional measurement methods, because they exceed the detection limit of the equipment. To study even shorter lifetimes, one abandons the photomultiplier transit time response limit by either resorting to “synchroscan” streak cameras or nonlinear mixing and/or sampling methods. The latter include pump/probe methods, stimulated emission tracing, or most directly, “up-conversion”. Up-conversion has been remarkably successful in identifying picosecond self-quenching and relaxation phenomena in protein fluorescence.9–11,21 We describe below the ultrafast femtosecond fluorescence dynamics of a series of peptide chains, Trp-(Pro)n-Tyr (WPnY, n = 0, 1, 2, 3, 5, 8), by up-conversion.
2. Materials and methods
2.1 Samples
All peptides (WP, PY, WPnY) were synthesized by us in cooperation with Apeptide Co. Ltd (Shanghai, China), and the purity of each peptide was over 98% by high performance liquid chromatography (HPLC) and mass spectrometry (MS). The chemical structures were shown in Fig. 1A. Na2HPO4 and NaH2PO4 were purchased from Aladdin (Shanghai, China), dissolved in ultrapure Millipore water (18.2 MΩ cm−1), and then mixed in a certain proportion to obtain 10 mM phosphate buffer (pH = 7.2). All samples were not purified again before use. To eliminate the influence of internal filtration and the formation of aggregates, a typical concentration of peptides (WP, PY, WPnY) in 10 mM phosphate buffer was 0.2 mM for all the measurements. Before each up-conversion measurement, the fresh sample solution was prepared at room temperature (25 °C) and stored in a dark sample bottle. And the absorption spectra of the measured samples were carefully checked to ensure that there was no obvious photodamage/photodegradation during the whole measurement.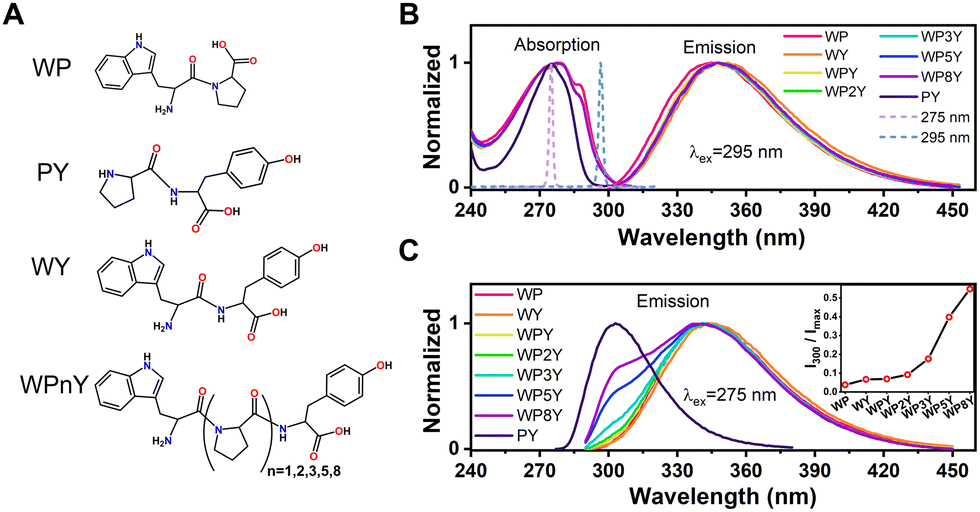 | ||
| Fig. 1 Chemical structure of all sample (WP, PY, WY and WPnY) molecules (A). Normalized steady-state absorption (B) and emission spectra with excitation wavelength at 295 nm (B) and 275 nm (C) of WP, WY, WPnY and PY. It is indicated by solid lines of different colors. Normalized 275 nm laser pulse and 295 nm laser pulse are represented by dotted lines (B). The parameter I300/Imax of all samples are given in the inset (C). | ||
2.2 Steady-state absorption and fluorescence spectra
Steady-state absorption and fluorescence spectra were obtained with TU1901 UV-Vis spectrophotometer (Beijing Purkinje General Instrument Co. Ltd) and FluoroMax-4 fluorescence spectrophotometer (Horiba, Jobin Yvon), respectively. For all samples, the steady-state absorption spectra were recorded between 240 nm and 320 nm in step of 1 nm. The steady-state fluorescence spectra were recorded from 300 nm to 460 nm (λex = 295 nm) and 290 nm to 460 nm (λex = 275 nm) with a step of 1 nm. For all measurements, samples were placed in the fluorescent quartz cells with optical path of 1 or 2 mm, and data were collected at 25 °C. All data were averages of at least three independent measurements. In the same environment, the baseline was measured in a phosphate buffer, and all data were corrected by background and instrument calibration files.2.3 Up-conversion spectrophotofluorometer
The detail of this experimental system was described in the previous work.12 In short, the seeded ultrafast Ti:sapphire regenerative CPA amplifier (Astrella, Coherent Inc.) produced a fundamental ultrashort laser pulse (∼90 fs) with an average power of 7 W, a central wavelength of 800 nm and a repetition rate of 1 kHz. 40% of the fundamental pulse was used as gate pulse, which was laterally shrunk after passing through a telescope system, and then passed through the delay line stage. To avoid damaging the mixing crystal, the average pulse power was reduced to less than 20 mW before the gate pulse was focused in the 0.2 mm BBO crystal. Leftover 60 percent of the pulse energy was used to pump an optical parametric amplifier (OperA solo, Coherent Inc.). The excitation pulse was generated by harmonic generation of the optical parametric amplifier and the wavelength was adjustable in the range of 260–295 nm with an average power of 4–10 mW. To further purify the excitation pulse, a simple spectrometer composed of a pair of UV prisms and a suitable filter were used. To avoid photodegradation and hole burning effects, the pulse energy was attenuated to less than 200 μW before the excitation pulse was focused on the sample. The sample was injected into the interior of a cuvette comprising a UV quartz sample disk with a diameter of 80 mm, which could rotate continuously with an angular velocity of not less than 120 rad s−1. After the sample was excited by the excitation pulse, the fluorescence was collected through a pair of parabolic focusing mirrors. Before the fluorescence was focused in the BBO mixed crystal, a suitable filter was used to remove the excitation pulse and select the fluorescence signal. Finally, the pure fluorescence was focused in BBO crystal (with a small angle to the gate pulse) and overlapped spatially with the gate pulse. The fluorescence and gate pulse generated up-conversion signal through type-I sum frequency. Up-converted fluorescence was resolved by a monochromator (Omnik500, Zolix) and then detected by a photomultiplier tube (CR317, Hamamatsu). The signal was then integrated/amplified by a fast-gating integrator (BOXCAR SR250, Stanford Instrument) and recorded by a 16-bit analog-to-digital converter (BNC2120, National Instruments). The magic angle (54.7°) polarization arrangement was used in most experiments to avoid the influence of fluorophore rotation rate contributions. The instrument response function (IRF) was obtained by measuring the up-conversion signal of water Raman scattering, and the IRF corresponding to each excitation wavelength (275 and 295 nm) was similar (∼350 fs) in width. All sub picosecond fluorescence decay curves were measured by this experimental system. According to the previously proposed method,3–6 all data were globally fitted and used to build DAS. And IGOR Pro program version 6.37 (Wavemetrics Inc., Portland, OR) was used to analyze the up-conversion data.2.4 Molecular dynamics (MD) simulations
The ground-state structure of each molecule was optimized at the PCM (water)41,42-B3LYP43,44-GD3(BJ)/6-311G(d) level using Gaussian 16 program,45 and then restrained electrostatic potential charges (RESP)46 were assigned for the optimized structure. The general amber force file (GAFF)47 was used for MD simulations. The initial structure was immersed in the center of a truncated octahedral box of TIP3P48 water model, and each of the molecules were immersed in a cubic water box (60 × 60 × 60 Å). Then dynamic simulations were performed as following: (i) used conjugated gradient (CG) method49 for energy minimization, simulation time of 10 ps; (ii) equilibrium phase simulations with the velocity-rescale thermostat50 and Berendsen51 pressure coupling, and the system reached equilibrium and stability, and eliminated the irrationality of structures, simulation time of 1 ns; (iii) production phase simulations with the velocity-rescale thermostat and Parrinello-Rahman52 pressure coupling, simulation time of 50 ns. All the MD simulations were performed by GROMACS software.53 To give an intuitive understanding of the non-covalent interactions, an independent gradient model (IGM)54,55 analysis based on the electron density gradient was performed using the Multiwfn code.56 Roughly 7500 structures taken from last 15 ns trajectories were clustering for IGM analysis. The representative conformations for WPY and WY with the largest percentages were selected, followed by geometries optimization using PCM model in water.3. Results and discussion
3.1 Steady-state absorption and fluorescence spectra
The normalized steady-state absorption and fluorescence spectra of WP, PY, WY, WPY, WP2Y, WP3Y, WP5Y and WP8Y are shown in Fig. 1B and C. According to Fig. 1B, WP showed the characteristic absorption band from indole (∼280 nm) and PY showed that from phenol (∼275 nm). Other peptides (WPnY) showed the same characteristic absorption bands (∼280 nm), but the spectral shape was slightly different from WP. Obviously, this was only due to the superposition of Y and W. Thus, the absorption spectra of all peptides showed that the insertion of proline did not disrupt the energy level structure of the two fluorophores (W and Y). The fluorescence spectral shapes of all peptides (except PY) with excitation wavelength at 295 nm were similar (Fig. 1B). Only the spectral peak wavelength values were slightly different. The reddest emission peak was generated by WY (∼352 nm). Moreover, with the insertion of P links, the peak value gradually blue shifted (WP was the bluest, ∼345 nm). Like the previous work,10,12 this is mainly due to the different properties of peptides, including the difference of charge distribution and hydrophobic properties. In conclusion, the photophysical properties of W alone in the peptides (WPnY) did not change significantly.For WPnY, W and Y will be excited at the same time, when the excitation pulse is 275 nm (Fig. 1C). With the increase of the number of P, the remanent fluorescence peak of Y (∼300 nm) gradually appears. It suggests that when the W is close to Y, the fluorescence of Y will be quenched by W through energy transfer. This phenomenon is common in proteins containing both W and Y. The distance distribution of W and Y in proteins is diverse. The contribution of Y to total fluorescence (I300/Imax) was taken as a simple parameter of energy transfer efficiency (Fig. 1C inset). Obviously, with an increased length of the spacer (P) between W and Y, the energy transfer efficiency decreases gradually. Among them, WY and WPY have the highest efficiency. This indicates that proteins with the same fluorescence spectral shape as WY or WPY will have ultrafast energy transfer lifetime 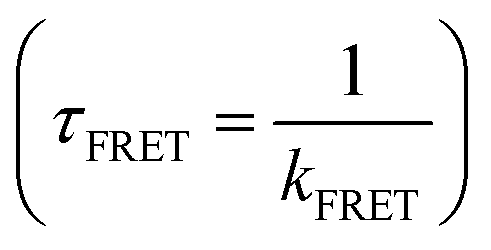 between Y and W. Traditional time-resolved measurement systems (phase or TCSPC) may not capture it, if τFRET ∼ <200 ps. Therefore, a series of studies were carried out by using femtosecond time-resolved fluorescence frequency up-conversion technology. To obtain the pure energy transfer signal from Y to W, the excitation pulse wavelengths were strictly controlled to be 275 nm and 295 nm (dotted line in Fig. 1B). A 295 nm pulse only excites W, and 275 nm pulses excite W and Y.
between Y and W. Traditional time-resolved measurement systems (phase or TCSPC) may not capture it, if τFRET ∼ <200 ps. Therefore, a series of studies were carried out by using femtosecond time-resolved fluorescence frequency up-conversion technology. To obtain the pure energy transfer signal from Y to W, the excitation pulse wavelengths were strictly controlled to be 275 nm and 295 nm (dotted line in Fig. 1B). A 295 nm pulse only excites W, and 275 nm pulses excite W and Y.
3.2 Up-conversion fluorescence transients
The up-conversion results for control construct WP were shown in Fig. 2A. These were quite similar to transient decay of W (Trp) alone, as expected. A rapid decay at the blue edge, slow decay at peak emission, and a matching rapid rise at the red edge were all indicative of fast (<3 ps) solvent relaxation, as first described in Trp by Shen and Knutson.57,58 These sub-nanosecond components from W alone would mix with and confound recovery of FRET rates if Y (Tyr) was also excited. It was expected that both Y and W would be excited at 275 nm, in proportion to extinction coefficient, while 295 nm excitation (if sufficiently narrowband) should only excite W. Note that for WP, 275 and 295 nm produced essentially the same transients. WP would be partly excited to a higher excited state (1Lb) when excitation is at 275 nm.59 Previous work has shown that even if W was excited to that energy level, it would still reach the energy level 1La by ultrafast internal conversion (<100 fs).57,58 That exceeds the resolution of the up-conversion system and could not be captured. Therefore, solo W fluorescence decay kinetics excited at 275 and 295 nm measured by the up-conversion system were the same. This is helpful to analyze the effect of Y on W fluorescence decay by comparing the decay kinetics at 275 and 295 nm excitation.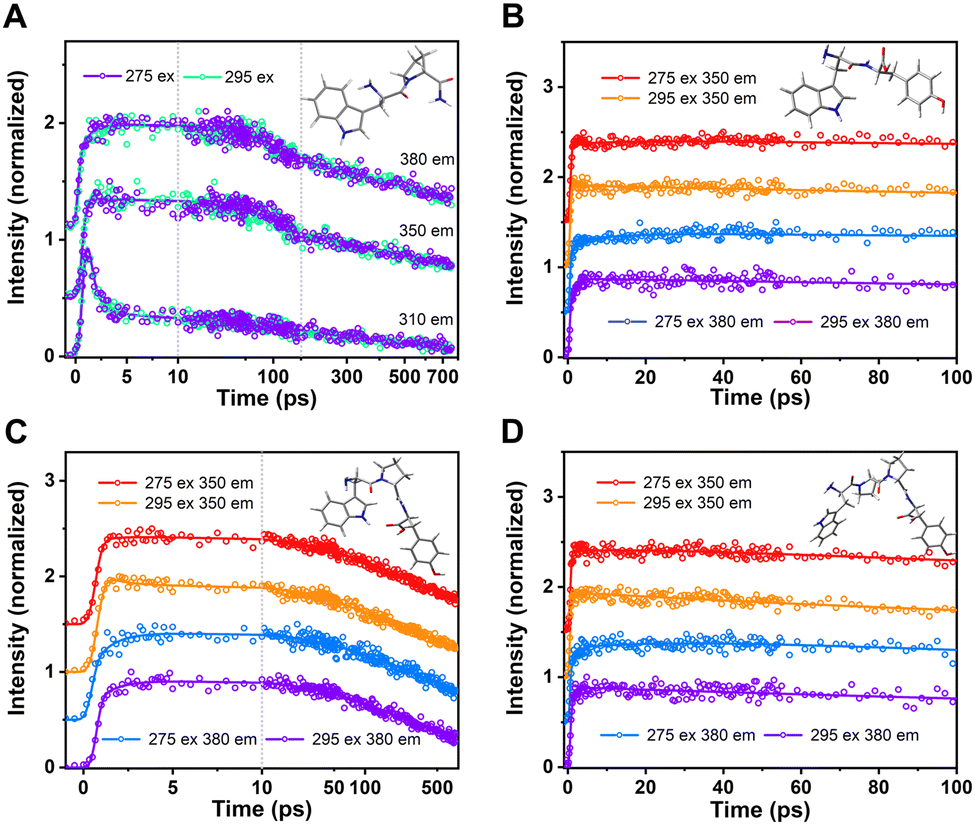 | ||
| Fig. 2 Normalized up-conversion fluorescence decay of WP (A), WY (B), WPY (C) and WP2Y (D) gated from typical emission wavelengths with excitation wavelength 275 nm and 295 nm. | ||
The compound expected a priori to yield the fastest FRET was WY (if the distance between indole and phenol is assumed to be the smallest). The up-conversion results for WY are shown in Fig. 2B. It was obvious that the decay curve had a distinct rising trend, when the 275 nm excitation occurred. The trend was more apparent when the detection wavelength was at 380 nm. This is mainly due to the weak contribution of QSSQ terms at the redder wavelength, so the negative amplitude (i.e., rising) signal from FRET dominated the picture in this time range. The addition of a fast FRET component at 275 nm would normally alter the total trace shape, unless that FRET rate was either below our 300 fs resolution or comparable to solvent relaxation of W. Thus, we saw the first evidence of a fast FRET (<100 ps) from Y to W in this peptide. To investigate the effect of distance on energy transfer between W and Y, multiple P were then inserted between the two amino acids as the spacer.
For peptide WPY, adding one “spacer” between W&Y, FRET slower than WY was expected. However, the rising trend like WY was not observed in the time range of >10 ps. In Fig. 2C, when comparing the two excitations detected at 350 nm, it was obvious that the ∼2 ps solvent relaxation signal, usually contributing at 350 nm only decaying or neutral kinetics, became a rising term, when excitation was at 275 nm. Similarly, with emission wavelength set at 380 nm, the normal rising trend of the ∼2 ps rate term strengthened more. This implied that the fast FRET in WPY was actually ultrafast (∼2 ps) and mixed/indistinguishable from solvent relaxation. Contrary to expectations, inserting a P between W and Y did not slow this FRET. This was due to unanticipated closer distances between W and Y, which may be related to the structure of the folded peptide. In the section describing molecular dynamics simulation, a more detailed explanation will be given.
For the peptide WP2Y, with two proline spacers, “normalcy” returns: it had longer energy transfer lifetimes and weaker energy transfer amplitude than WY and WPY, as expected. The transients of WP2Y were similar to WY (Fig. 2D), except that WP2Y had a weaker rise. Although WPY did not play the expected “ruler” role in increasing the distance between W and Y, WP2Y did. When the spacer number is increased further (WP3Y, WP5Y and WP8Y), the weaker efficiency and longer FRET lifetime becomes difficult to capture in the up-conversion time window. As shown in Fig. S1 (ESI†), the decays were almost indistinguishable when exciting at 295 vs. 275 nm. When spacer (proline) number was increased to more than 3, the energy transfer phenomenon could not be easily observed due to the sharp decrease of energy transfer efficiency and the increase of FRET linked lifetime. In addition, the fluorescence lifetime component of W alone is complex. The weak FRET signal was difficult to distinguish from normal “rotamer” (heterogeneous) fluorescence decay components.
Unfortunately, indistinguishability (or very subtle shape changes) in these single wavelength decay transient curves were not fully diagnostic; the emission observation wavelengths might be in a place where either the FRET rate was poorly manifested or was masked by other terms. Therefore, more in-depth data analysis and processing needed to be carried out.
3.3 Decay associated spectra (DAS), time resolved emission spectra (TRES) and simulation
While individual transients have significant information content, the collection of a 3D surface (photons emitted vs. wavelength and time) allows one to recover much more information about underlying processes. A global analysis of an up-conversion 3D surface yielded DAS (decay associated spectra) and decay rates; the shapes of DAS add information to help interpret the mix of all terms: FRET, solvent relaxation (SR) and fast population decay (QSSQ). In addition, time-resolved emission spectral (TRES) can be obtained by analyzing the 3D surface. It intuitively presents the change of emission spectrum shape with time. Based on previous work,60 we constructed a simplified scheme for unraveling the amounts of FRET, SR and QSSQ (Fig. S2, ESI†). This working model can then reproduce the decay curves, DAS, and TRES.A working model leads to DAS that reflect the compartment kinetics and multiply a few basis spectra: Tyr (Y), a single spectrum ignoring SR around Y; unrelaxed Trp (W) and relaxed Trp (W′). For modeling purposes, these spectra are constructed from identical steady state spectra (assumes Pn and consequent Y–W distance changes do not significantly change the dielectric environment). In addition, the QSSQ term is associated with only one spectrum, neglecting SR for that species. The first-order model we impose is not a true “target analysis”, and we have not attempted to do full identifiability analysis on the compartment kinetics. The SNR (Signal to Noise Ratio) of the up-conversion traces (and therefore DAS) cannot support the recovery of more than 3 or 4 kinetic rates and spectra, especially when some of the rates are probably quite similar. Laplace inversion (the recovery of exponential lifetimes from decay transients) is an ill-conditioned mathematical problem even with exquisite SNR, so we kept the modeling basic. To separate the close mixed rates, we take advantage of a priori information (the known basis spectra). The adjustment of model rates causes spectral shifts – and consequent movement of “DAS crossings” = zero amplitude wavelength loci – in model DAS, and these can be adjusted to give the best match between experimental DAS and these model vectors. In the following, we will separate the lifetime components of FRET from the mixed system by adjusting the model rate and amplitude rationally.
When accompanied by model spectra, the ability to predict DAS from the rates became viable, and zero crossing locations become the best indicators of mixing unknown with known DAS. In Fig. 3A, we show the 275 nm excited DAS of WY (experimental). The SR contribution was clearly seen as a 1.1 ps DAS that crossed the axis near the peak of overall emission. This is an expected characteristic of a SR DAS, where small fast shifts of the entire envelope cause fast decline on the blue side and equivalent rising behavior (negative amplitude) on the red side. In contrast, the 74 ps DAS recovered crossings were very near 320 nm, where Y still dominates over W. Our simulation of this process (where FRET alone would cause a crossing near 326 nm) was shown in Fig. 3B. We therefore posit a mixing of FRET-induced DAS with a QSSQ component that blueshifts the crossing point and lengthens that average lifetime. The mixing of two lifetime components can be expressed by the following equation,
| a × DASQSSQ × τQSSQ + b × DASFRET × τFRET = DASMIX × τMIX | (2) |
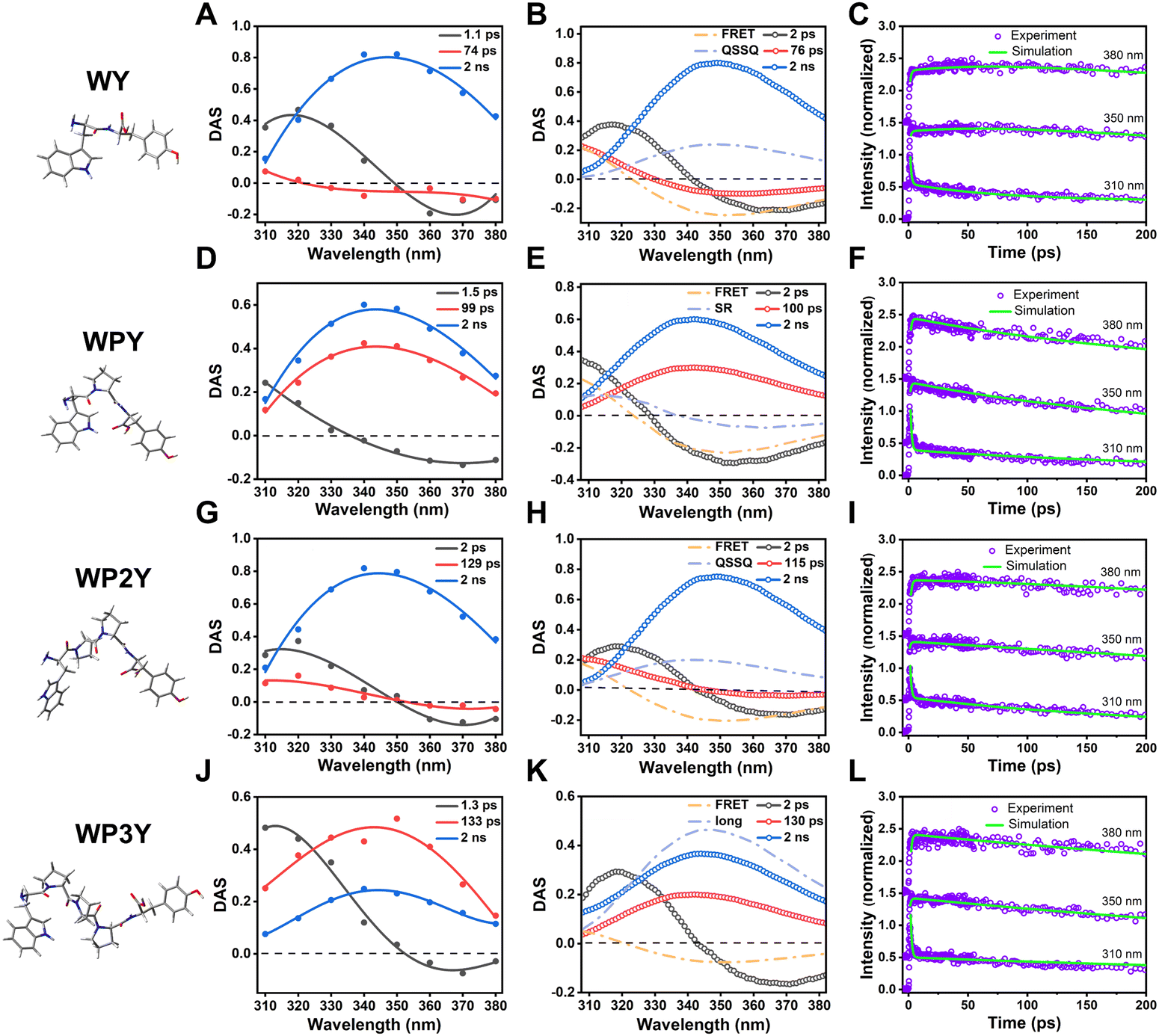 | ||
| Fig. 3 Decay associated spectrum (DAS) of WY (A), WPY (D), WP2Y (G) and WP3Y (J) with excitation wavelength 275 nm. Simulated DAS of WY (B), WPY (E), WP2Y (H) and WP3Y (K). The matching degree between the simulation results and the experimental results of WY (C), WPY (F), WP2Y (I) and WP3Y (L). | ||
Surprisingly, as mentioned before, WPY appears to exhibit faster FRET than WY; Fig. 3D shows that the zero crossing of the 1.5 ps DAS usually associated with SR alone is blue shifted. Usually, SR crosses zero near the fluorescence emission peak (∼350 nm). The strong blue shift (∼20 nm) is generally caused by the mixing of other components with the same lifetime. Thus, we assumed that FRET-induced DAS was mixed with SR components to blue shift the crossing point. Using a method similar to WY, the parameters DASQSSQ and τQSSQ in the eqn (2) were replaced by DASSR and τSR.
Reasonably adjusting the proportion of the two mixtures separated the FRET lifetime (τFRET = ∼2 ps). The simulated DAS are shown in Fig. 3E, and the details of parameters are in Table S1 (ESI†). In addition, the matching across time between the simulation results and the experimental results is checked at three typical wavelengths (Fig. 3F).
For WP2Y, similar to WY, the mixed lifetime component DAS of QSSQ and FRET crosses zero (Fig. 3G). Obviously, the zero-crossing location is near the emission peak (∼350 nm), which can imitate the slow solvent relaxation (SSR) signal. Our previous studies20,21 on proteins have shown that after shielding the signal of QSSQ, SSR will show a shape similar to SR (crossing zero near the emission peak). We will continue to discuss this point below. According to eqn (2), the lifetime of FRET was separated (τFRET = ∼50 ps). The simulated DAS is shown in Fig. 3H (see Table S1 for more parameter details, ESI†). Similarly, the matching in time plots between the simulation and the experimental results is checked to ensure the reliability of the simulation (Fig. 3I).
For longer peptides (WP3Y), the effect of FRET on fluorescence decay curves and DAS is difficult to observe. WP3Y has low FRET efficiency and hence a longer FRET lifetime, exiting our ultrafast window. Obviously, for WP3Y, 275 nm DAS look the same as 295 nm DAS (Fig. 3J), showing three lifetime components (SR, QSSQ and 2 ns components). Longer FRET lifetime components may no longer be mixed with QSSQ. We assume that this lifetime component will instead be mixed with long-lifetime (∼2 ns) DAS. Finally, in Fig. 3K, one could see (by noticing reduced relative amplitude contributions) that the usual 2 ns DAS is now the one being mixed with the negative-going FRET DAS, while SR and QSSQ terms are in original W positions, unperturbed. According to eqn (2), replacing DASQSSQ and τQSSQ with DASlong and τlong, an apparent FRET lifetime can be separated (τFRET = ∼500 ps). Similarly, the matching and parameter details between the simulation and the experimental results are shown in Fig. 3L and Table S1 (ESI†). For peptides WP5Y and WP8Y, the experimental results and simulation methods were qualitatively similar to that of WP3Y.
In addition, since total intensity of fluorescence is simply the sum of amplitudes (i.e., DAS) multiplied by their individual lifetimes, we can easily construct models of steady state spectra. As seen in Fig. S3 (ESI†), the modeled and simulated DAS × τ sum does a good job of reproducing actual recorded steady state spectra. With a few individual transients, detecting these mixings would be impossible (or nearly so); the 3D data collection and extraction of global DAS is the key that permits one to further model and extract masked FRET rates. In each case, the Time-Resolved Emission Spectra (TRES) – a sum of all DAS – is also shown (Fig. 4), so the time-resolved spectral shape change can be visualized as a 3D surface.
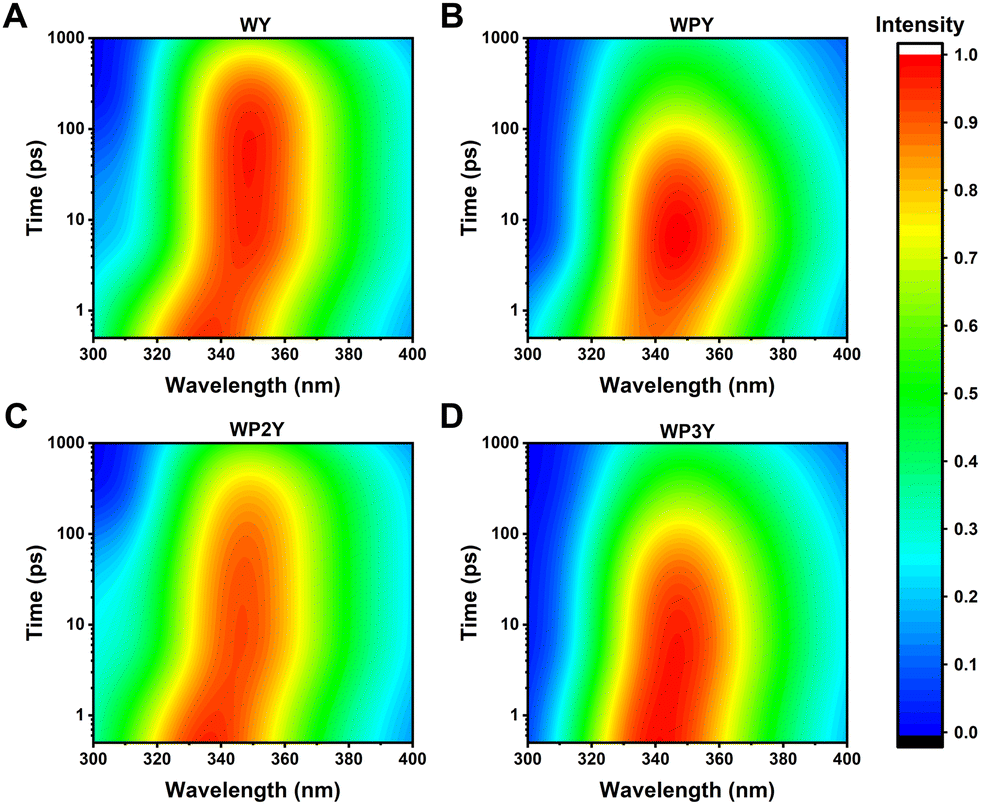 | ||
| Fig. 4 Time-Resolved Emission Spectra (TRES) of WY (A), WPY (B), WP2Y(C) and WP3Y (D) with excitation wavelength 275 nm. | ||
Obviously, all peptides (WY, WPY, WP2Y and WP3Y) showed a spectral red shift in the first 10 ps. This is a typical solvent relaxation process. However, due to the ultrafast FRET (∼2 ps), the peak of WPY gradually rises while red shifting (Fig. 4B). In addition, the peaks of WY and WP2Y also increased in varying degrees in ∼100 ps (Fig. 4A and C). WP3Y is always in decay, however, as there is no obvious rising trend (Fig. 4D). In short, from the above DAS, TRES, and simulation results, it can be clearly seen that the separated “FRET lifetime” extractions are WY (∼20 ps), WPY (∼2 ps), WP2Y (∼50 ps) and WP3Y (∼500 ps). These were not in line with a priori rigid ladder expectations. We surmised that this may be due to the folding of peptide WPY (shorter distance). Thus, we turned to molecular dynamics simulations.
3.4 MD simulations and calculations of energy transfer rate
The series of WPnY (n = 0, 1, 2, 3) “ruler” molecules, thought to be rigidly spaced, were studied with molecular dynamics (MD) simulations and quantum chemical calculations to better understand the true distance distribution and geometry between donor Y and acceptor W (Fig. 5). The strongest Förster resonance energy transfer (FRET) was observed for WPY, and the FRET intensity decreased with the increase of peptide length. The optimized geometries of the WPnY (n = 0, 1, 2, 3) are displayed in Fig. 5A and shown in real water box by MD simulation in Fig. 5B. To investigate the energy transfer between donor and acceptor as shown in Fig. 5C, the molecules were divided into three fragments, (F1, F2 and F3). And the centroid distances between fragments F1 and F3 were counted based on the trajectories of last 15 ns after the MD simulation reaches equilibrium (see Fig. S4 for details, ESI†).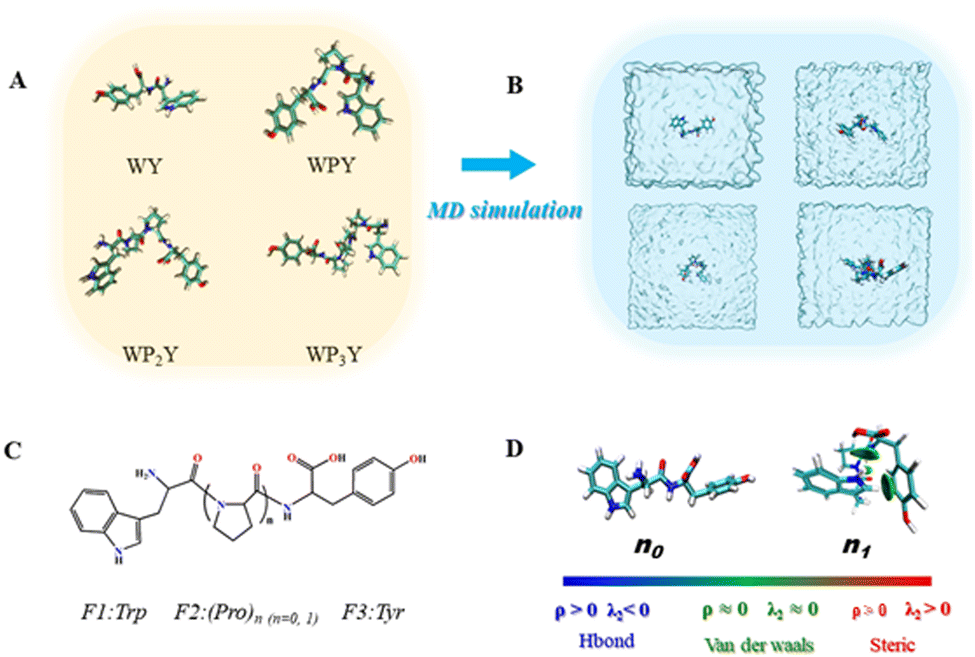 | ||
| Fig. 5 (A) Optimized geometries of the WPnY (n = 0, 1, 2, 3). (B) Diagrammatic sketch of the studied molecules in water box by MD simulation. (C) The molecules are divided into three fragments, F1(Trp), F2 (Pro)n (n = 0, 1) and F3 (Tyr). (D) The non-covalent interactions between F1(Trp) and F3(Tyr) through IGM method; the δginter = 0.01 a.u. iso-surfaces colored under the BGR scheme and the signed electron density range −0.05 a.u. < sign (λ2) ρ < 0.05 a.u. (The ρ denotes electron density, it is closely related to the strength of bond. The sign(λ2) denotes the sign of the second largest eigenvalue of Hessian of ρ.). | ||
The centroid distances r between the F1 and F3 are counted to be 0.58 nm for WY, 0.56 nm for WPY, 0.90 nm for WP2Y, 1.23 nm for WP3Y, 1.7 nm for WP5Y and 2.27 nm for WP8Y. The increasing r values can be attributed to the increasing numbers of (Pro)n units. Interestingly, the shortest centroid distance is observed for WPY instead of WY, which is also in agreement with the experimental observation of the maximum energy transfer efficiency for WPY. To explain this, non-covalent interaction analyses based on IGM method were performed for WPY and WY as shown in Fig. 5D. It can be seen that there are no significant interactions for WY (n0). But for WPY (n1), there are obviously H–π interactions between hydrogen (F1) and benzene (F3). Another dispersion interaction was also found between –NH(F1) and –COOH (F3). Due to the existence of the non-covalent interactions, the centroid distance of WPY is closer than that of WY. Therefore, for WPY, it is reasonable to observe the fastest FRET in the experiment. In addition, the centroid distance r increases as the number of the middle fragments (P) (n = 1, 2, 3) increases.
The term κ2 is orientation factor, eqn (3).
| κ2 = (eD·eA −3(eD·eR)(eA·eR))2 | (3) |
Further, the FRET lifetime can be evaluated by using eqn (4).
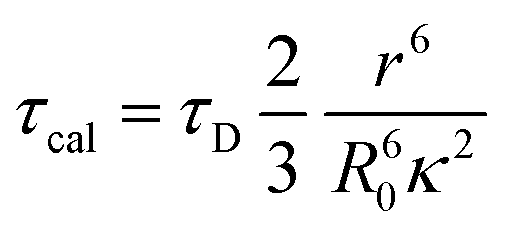 | (4) |
As shown in Table 1, τcal obtained by MD and calculation, τexp separated by up-conversion measurement and DAS simulation. Obviously, both τcal and τexp showed a trend of first decreasing and then increasing, with the increase of proline (n = 0, 1, 2, 3). And the lifetime values are also very consistent, except WY and WP3Y. This is not surprising. The reason for the deviation between the calculated and experimental values of WP3Y have been described earlier (free rotation leads to a smaller κ2 value). The calculation equation of τcal adopts the classical FRET theory. Previous work61 shows that the classical theory can fail for short peptides. Thus, for WY, there may be higher order deviation between the calculated results and the real results. Of course, other more complex behaviors are not excluded. Generally, the MD simulation and quantum chemical calculations are found to be in good agreement with experimental observation. Due to the interaction between amino acids, the distance between W and Y in WPY is the closest, and the FRET efficiency is the highest. With the increased extent of proline links, the efficiency decreased gradually. The distance information and dipole moment orientation information are obtained by MD, and the calculated lifetime τcal is consistent with the lifetime τexp separated from the experimental measurement. This shows that our proposed working model can successfully separate “pure” FRET lifetime, although it is usually masked by QSSQ.
| r (nm) | κ 2 | τ cal (ps) | τ exp (ps) | E (%) | |
|---|---|---|---|---|---|
| WY | 0.58 | 1.96 | 2.82 | ∼20 | 99.38 |
| WPY | 0.56 | 2.79 | 1.61 | ∼2 | 99.94 |
| WP2Y | 0.90 | 1.46 | 52.90 | ∼50 | 98.46 |
| WP3Y | 1.23 | 2.40 | 209.50 | ∼500 | 86.47 |
In addition, the FRET efficiencies were also calculated systematically for all the mentioned cases by using eqn (5).
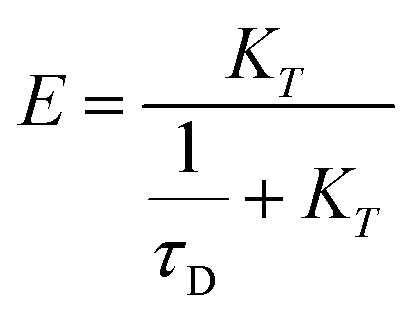 | (5) |
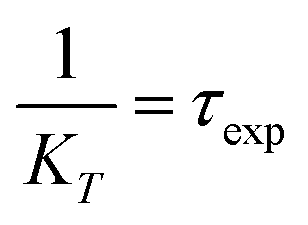 .
.
The FRET efficiency (E) shows that WPY has the highest efficiency (99.94%). According to the steady-state results (I300/Imax in Fig. 1C inset), we assume that the FRET efficiency of WP is 100%, then WY = 96.96%; WPY = 96.72%; WP2Y = 94.33%; WP3Y = 85.60%. It is worth noting that the FRET efficiency calculation only uses two parameters: the tyrosine lifetime and the FRET lifetime we separated. Although the parameters of the steady-state spectra are not used here, this is surprisingly consistent with the steady-state results. This shows that our “separate FRET model” is successful. The “pure” FRET lifetime is separated, and it can calculate the FRET efficiency independently. We also note that WP3Y results are the most similar. Using the equation from the FRET theory 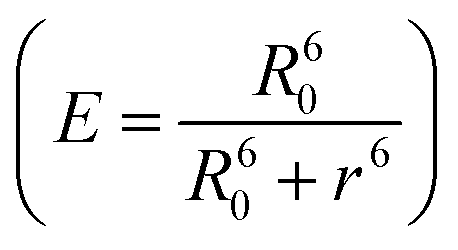 the distance between Y and W will be 11.6 Å which matches perfectly the distance calculated from our MD simulation. This actually shows the power of FRET as spectroscopic ruler. However, it is obvious that this power is weakened when n < 2. Steady state spectral parameters cannot accurately recovery the highest FRET efficiency of WPY. This indicates that the nearest W and Y FRET pairs in protein can be accurately described only by using femtosecond time resolution technology and our proposed working model.
the distance between Y and W will be 11.6 Å which matches perfectly the distance calculated from our MD simulation. This actually shows the power of FRET as spectroscopic ruler. However, it is obvious that this power is weakened when n < 2. Steady state spectral parameters cannot accurately recovery the highest FRET efficiency of WPY. This indicates that the nearest W and Y FRET pairs in protein can be accurately described only by using femtosecond time resolution technology and our proposed working model.
4. Conclusions
The importance of Tyrosine (Y) to Tryptophan (W) energy transfer lies in both its potential for uncovering structure and dynamics and in its possible interference with W studies. Y is about 3 times as abundant as W in proteins, and the fluorescence of Y in all but W-less proteins is usually efficiently quenched by FRET to W. That FRET process is usually so efficient that steady state emission spectra of proteins only rarely exhibit a “Y shoulder”. FRET between Y and W has been exploited over the years, but time-resolved FRET has been less often utilized, often because the Y → W rates in protein cores exceed the capabilities of most instruments. In this work, we designed and synthesized a series of peptide chains (WPnY) as classic energy transfer “spectroscopic rulers” (bridges of proline residues), bringing sub-picosecond resolution to bear on the ultrafast Y → W rate question.In doing so, we find that the WPnY construct does not follow classical expectations for distance-dependent FRET, and we reconcile that with MD simulation showing the breakdown of the n = 0, 1, 2 bridge order. Those simulations, when coupled with dipole–dipole interaction predictions (still classical), adequately explain our observations. We do not exclude the possibility of higher order (non-Förster) interaction; we simply find adequate agreement with dipole coupling. This study suggests that up-conversion (or equivalent stimulated emission probing) should provide a valuable tool for looking at the dynamics of protein cores, where Y and W often reside. Time resolution in the nanosecond regime has already been used to separate multiple W contributions,62 and the same approaches should be applicable to multiple Y – but now in the sub picosecond resolution world.
Protein–water dynamics is an important tool to understand protein function, structure and even enzyme catalytic activity. However, the ultrafast energy transfer in protein may have a serious impact on it, resulting in the wrong judgment of protein–water dynamics. This has not been given enough attention. Thus, we add a cautionary note. Time Resolved Fluorescent Stokes Shifts (TDFSS) of W have been widely used to infer water dynamics and protein dynamics in the 1–200 ps window. As can be seen in the TRES of Fig. 4, the FRET process can easily mimic TDFSS. The key here is that for WP2Y one gets a negative amplitude (alpha) on a ∼50 ps term that one might think is “biological water” influence. Further, Y may play a role in any protein term with the range of 20–50 ps. In our previous work,20,21 we successfully isolated SSR signals from proteins very similar to the DAS of WY and WP2Y. Thus, the mixture of SSR and QSSQ that we have been concerned about may be further complicated by FRET from Y to W. This is usually not considered, as the excitation of W at 295 nm is thought to be in a place where Y has negligible absorbance. Several things can spoil that isolation:
– Y absorbance redshift
– Y abundance in proximity
– Broadband laser pulses from fs laser amplifiers, while centered at 295 nm, may have contributions even below 292 nm, especially if one seeks short pulses for better time resolution. Ironically, the time-bandwidth product of excitation pulses will thus set a limit on early time observations.
When observing proteins, if any of the above situations occur, Y will be excited. This will further yield energy transfer of Y to W, resulting in a negative amplitude lifetime component, which could be misunderstood as originating in W itself. Thus, this requires extra caution in Y-containing proteins, and all work should be examined with this in mind.
In addition, our model will provide an important feasible scheme for the separation of “pure” FRET from proteins. Tyrosine and tryptophan are native FRET pairs in proteins. As a spectroscopic ruler, they have great advantages in studying protein structure changes, compared with exogenous dyes. For a long time, this FRET pair has not been well developed due to the spectral overlap of donor/acceptor and the limitation of time resolution. With the help of up-conversion technology, our model successfully solved this problem. The “pure” FRET signal (Y to W) in protein will efficiently reveal the local structural changes of protein. Therefore, it will provide an important new scheme for the study of protein structure and dynamics.
In any case, model compounds for ultrafast Y to W FRET have been established and a scheme for “unmixing” ultrafast FRET in proteins was demonstrated. Model peptide data can, within limits, be a very favorable tool for protein research. In addition, future ultrafast studies of Y → W FRET should be useful in studying internal structure and dynamics of proteins, especially in cases where one should not perturb that native structure with labels.
Abbreviations
| FRET | Förster resonance energy transfer |
| Y | Tyrosine |
| W | Tryptophan |
| P | Proline |
| SR | Solvent relaxation |
| QSSQ | Quasi-static self-quenching |
| DAS | Decay associated spectra |
| SSR | Slow solvent relaxation |
| PPI | Polyproline I |
| PPII | Polyproline II |
| HPLC | High performance liquid chromatography |
| MS | Mass spectrometry |
| IRF | Instrument response function |
| RESP | Restrained electrostatic potential charges |
| GAFF | Reneral amber force file |
| MD | Molecular dynamics |
| CG | Conjugated gradient |
| IGM | Independent gradient model |
| TRES | Time resolved emission spectra |
| SNR | Signal to noise ratio |
| TDFSS | Time resolved fluorescent Stokes shifts. |
Author contributions
All authors have given approval to the final version of the manuscript.Funding sources
National Natural Science Foundation of China (No. 11674101), Intramural Funding of NHLBI, NIH (J. K.)Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by National Natural Science Foundation of China (No. 11674101). We also thank the Division of Intramural Research, NHLBI, NIH. We dedicate the manuscript to the memory of Ludwig Brand, who pioneered FRET in flexible polymer bridges, recognizing that small peptides might not form predicted ladders (R. Conrad and L. Brand, Biochemistry, 1968, 7(2), 777–787, DOI: 10.1021/bi00842a036).References
- C. P. Pan, P. R. Callis and M. D. Barkley, Dependence of Tryptophan Emission Wavelength on Conformation in Cyclic Hexapeptides, J. Phys. Chem. B, 2006, 110(13), 7009–7016 CrossRef CAS PubMed.
- S. D. Kamath, V. B. Kartha and K. K. Mahato, Dynamics of L-tryptophan in aqueous solution by simultaneous laser induced fluorescence (LIF) and photoacoustic spectroscopy (PAS). Spectrochimica Acta Part A Molecular & Biomolecular, Spectroscopy, 2008, 70(1), 187–194 Search PubMed.
- D. Rayner and A. Szabo, Time resolved fluorescence of aqueous tryptophan, Can. J. Chem., 1978, 56(5), 743–745 CrossRef CAS.
- A. Szabo and D. Rayner, Fluorescence decay of tryptophan conformers in aqueous solution, J. Am. Chem. Soc., 1980, 102(2), 554–563 CrossRef CAS.
- J. R. Knutson, D. G. Walbridge and L. Brand, Decay-associated fluorescence spectra and the heterogeneous emission of alcohol dehydrogenase, Biochemistry, 1982, 21(19), 4671–4679 CrossRef CAS PubMed.
- A. Stortelder, J. B. Buijs, J. Bulthuis, S. M. van der Vies, C. Gooijer and G. van der Zwan, Time-resolved fluorescence of the bacteriophage T4 capsid protein gp23, J. Photochem. Photobiol., B, 2005, 78(1), 53–60 CrossRef CAS PubMed.
- S. J. Kroes, G. W. Canters, G. Gilardi, A. van Hoek and A. J. Visser, Time-resolved fluorescence study of azurin variants: conformational heterogeneity and tryptophan mobility, Biophys. J., 1998, 75(5), 2441–2450 CrossRef CAS PubMed.
- M. Gonnelli and G. B. Strambini, Structure and dynamics of proteins encapsulated in silica hydrogels by Trp phosphorescence, Biophys. Chem., 2003, 104(1), 155–169 CrossRef CAS PubMed.
- X. Jianhua, T. Dmitri, K. J. Graver, R. A. Albertini, R. S. Savtchenko, N. D. Meadow, R. Saul, P. R. Callis, B. Ludwig and J. R. Knutson, Ultrafast fluorescence dynamics of tryptophan in the proteins monellin and IIAGlc, J. Am. Chem. Soc., 2006, 128(4), 1214–1221 CrossRef PubMed.
- X. Jianhua and J. R. Knutson, Quasi-static self-quenching of Trp-X and X-Trp dipeptides in water: ultrafast fluorescence decay, J. Phys. Chem. B, 2009, 113(35), 12084–12089 CrossRef PubMed.
- J. Xu, J. Chen, D. Toptygin, O. Tcherkasskaya, P. Callis, J. King, L. Brand and J. R. Knutson, Femtosecond fluorescence spectra of tryptophan in human γ-crystallin mutants: site-dependent ultrafast quenching, J. Am. Chem. Soc., 2009, 131(46), 16751–16757 CrossRef CAS PubMed.
- M. Jia, H. Yi, M. Chang, X. Cao, L. Li, Z. Zhou, H. Pan, Y. Chen, S. Zhang and J. Xu, Fluorescence kinetics of Trp-Trp dipeptide and its derivatives in water via ultrafast fluorescence spectroscopy, J. Photochem. Photobiol., B, 2015, 149, 243–248 CrossRef CAS PubMed.
- S. Cao, Z. Zhou, H. Li, M. Jia, Y. Liu, M. Wang, M. Zhang, S. Zhang, J. Chen and J. Xu, A fraction of NADH in solution is “dark”: implications for metabolic sensing via fluorescence lifetime, Chem. Phys. Lett., 2019, 726, 18–21 CrossRef CAS PubMed.
- M. Vincent, J. Gallay and A. P. Demchenko, Solvent relaxation around the excited state of indole: analysis of fluorescence lifetime distributions and time-dependence spectral shifts, J. Phys. Chem., 1995, 99(41), 14931–14941 CrossRef CAS.
- D. Toptygin, R. S. Savtchenko, N. D. Meadow and L. Brand, Homogeneous spectrally-and time-resolved fluorescence emission from single-tryptophan mutants of IIAGlc protein. The, J. Phys. Chem. B, 2001, 105(10), 2043–2055 CrossRef CAS.
- P. Jorge, Samir Kumar and P. Zewail, A. H., Hydration at the surface of the protein Monellin: dynamics with femtosecond resolution, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(17), 10964–10969 CrossRef PubMed.
- S. K. Pal, J. Peon and A. H. Zewail, Ultrafast Surface Hydration Dynamics and Expression of Protein Functionality: α-Chymotrypsin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(24), 15297–15302 CrossRef CAS PubMed.
- S. K. Pal, J. Peon and A. H. Zewail, Biological water at the protein surface: dynamical solvation probed directly with femtosecond resolution, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(4), 1763–1768 CrossRef CAS PubMed.
- W. Qiu, L. Zhang, Y.-T. Kao, W. Lu, T. Li, J. Kim, G. M. Sollenberger, L. Wang and D. Zhong, Ultrafast hydration dynamics in melittin folding and aggregation: helix formation and tetramer self-assembly, J. Phys. Chem. B, 2005, 109(35), 16901–16910 CrossRef CAS PubMed.
- A. Biesso, J. Xu, P. L. Muino, P. R. Callis and J. R. Knutson, Charge invariant protein–water relaxation in GB1 via ultrafast tryptophan fluorescence, J. Am. Chem. Soc., 2014, 136(7), 2739–2747 CrossRef CAS PubMed.
- J. Xu, B. Chen, P. Callis, P. L. Muino, H. Rozeboom, J. Broos, D. Toptygin, L. Brand and J. R. Knutson, Picosecond fluorescence dynamics of tryptophan and 5-fluorotryptophan in monellin: slow water–protein relaxation unmasked, J. Phys. Chem. B, 2015, 119(11), 4230–4239 CrossRef CAS PubMed.
- M. Faraggi, M. R. DeFelippis and M. H. Klapper, Long-range electron transfer between tyrosine and tryptophan in peptides, J. Am. Chem. Soc., 1989, 111(14), 5141–5145 CrossRef CAS.
- M. Weinstein, Z. B. Alfassi, M. R. DeFelippis, M. H. Klapper and M. Faraggi, Long range electron transfer between tyrosine and tryptophan in hen egg-white lysozyme, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1991, 1076(2), 173–178 CrossRef CAS.
- K. Bobrowski, J. Holcman, J. Poznanski, M. Ciurak and K. L. Wierzchowski, Pulse radiolysis studies of intramolecular electron transfer in model peptides and proteins. 5. Trp. bul. fwdarw. Tyr. bul. radical transformation in H-Trp-(Pro) n-Tyr-OH series of peptides, J. Phys. Chem., 1992, 96(24), 10036–10043 CrossRef CAS.
- K. Bobrowski, J. Poznański, J. Holcman and K. L. Wierzchowski, Pulse radiolysis studies of intramolecular electron transfer in model peptides and proteins. 8. Trp [NH˙ + ] → Tyr [O˙] radical transformation in H-Trp-(Pro) n-Tyr-OH, n = 3–5, series of peptides, J. Phys. Chem. B, 1999, 103(46), 10316–10324 CrossRef CAS.
- O. B. Morozova, R. Kaptein and A. V. Yurkovskaya, Changing the Direction of Intramolecular Electron Transfer in Oxidized Dipeptides Containing Tryptophan and Tyrosine, J. Phys. Chem. B, 2012, 116(40), 12221–12226 CrossRef CAS PubMed.
- G. Weber, Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds, Biochem. J., 1960, 75(2), 335 CrossRef CAS PubMed.
- G. Weber, Fluorescence-polarization spectrum and electronic-energy transfer in proteins, Biochem. J., 1960, 75(2), 345 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed.
- H. Chiu and R. Bersohn, Electronic energy transfer between tyrosine and tryptophan in the peptides trp-(pro) n-Tyr, Biopolymers, 1977, 16(2), 277–288 CrossRef CAS PubMed.
- G. Karreman, R. H. Steele and A. Szent-Gyorgyi, On Resonance Transfer of Excitation Energy Between Aromatic Aminoacids in Proteins, Proc. Natl. Acad. Sci. U. S. A., 1958, 44(2), 140–143 CrossRef CAS PubMed.
- R. F. Borkman and S. R. Phillips, Tyrosine-to-tryptophan energy transfer and the structure of calt gamma-II crystallin, Exp. Eye Res., 1985, 40(6), 819–826 CrossRef CAS PubMed.
- R. A. Malak, Z. Gao, J. F. Wishart and S. S. Isied, Long-range electron transfer across peptide bridges: the transition from electron superexchange to hopping, J. Am. Chem. Soc., 2004, 126(43), 13888–13889 CrossRef CAS PubMed.
- S. S. Isied, M. Y. Ogawa and J. F. Wishart, Peptide-mediated intramolecular electron transfer: long-range distance dependence, Chem. Rev., 1992, 92(3), 381–394 CrossRef CAS.
- A. K. Mishra, R. Chandrasekar, M. Faraggi and M. H. Klapper, Long-range electron transfer in peptides. Tyrosine reduction of the indolyl radical: reaction mechanism, modulation of reaction rate, and physiological considerations, J. Am. Chem. Soc., 1994, 116(4), 1414–1422 CrossRef CAS.
- M. Y. Ogawa, J. F. Wishart, Z. Young, J. R. Miller and S. S. Isied, Distance dependence of intramolecular electron transfer across oligoprolines in [(bpy) 2RuIIL. bul.-(Pro) n-CoIII (NH3)5]3+, n = 1–6: different effects for helical and nonhelical polyproline II structure, J. Phys. Chem., 1993, 97(44), 11456–11463 CrossRef CAS.
- A. Vassilian, J. F. Wishart, B. Van Hemelryck, H. Schwarz and S. S. Isied, Electron transfer across polypeptides. 6. Long-range electron transfer in osmium-ruthenium binuclear complexes bridged with oligoproline peptides, J. Am. Chem. Soc., 1990, 112(20), 7278–7286 CrossRef CAS.
- N. Helbecque and M. Loucheux-Lefebvre, Critical chain length for polyproline-II structure formation in H-Gly-(Pro) n-OH: circular dichroism approach, Int. J. Peptide Protein Res., 1982, 19(1), 94–101 CrossRef CAS PubMed.
- H. Okabayashi, T. Isemura and S. Sakakibara, Steric structure of L-proline oligopeptides. II. Far-ultraviolet absorption spectra and optical rotations of L-proline oligopeptides, Biopolymers, 1968, 6(3), 323–330 CrossRef CAS PubMed.
- K. L. Wierzchowski, K. Majcher and J. Poznański, CD investigations on conformation of HX-(Pro) nY-OH peptides (X = Trp, Tyr; Y = Tyr, Met); models for intramolecular long range electron transfer, Acta Biochim. Pol., 1995, 42(2), 259–268 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105(8), 2999–3094 CrossRef CAS PubMed.
- B. Mennucci and R. Cammi, Continuum solvation models in chemical physics: from theory to applications. John Wiley & Sons, 2008 Search PubMed.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98(45), 11623–11627 CrossRef CAS.
- M. Frisch; G. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci and G. Petersson, Gaussian 09, Revision d. 01, Gaussian. Inc., Wallingford CT, 2009, 201 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 1993, 97(40), 10269–10280 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- R. Fletcher and C. M. Reeves, Function minimization by conjugate gradients, Comput. J., 1964, 7(2), 149–154 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126(1), 014101 CrossRef PubMed.
- H. J. Berendsen, J. V. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81(8), 3684–3690 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: a new molecular dynamics method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem. Theory Comput., 2008, 4(3), 435–447 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132(18), 6498–6506 CrossRef CAS PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density, Phys. Chem. Chem. Phys., 2017, 19(27), 17928–17936 RSC.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- X. Shen and J. R. Knutson, Femtosecond internal conversion and reorientation of 5-methoxyindole in hexadecane, Chem. Phys. Lett., 2001, 339(3), 191–196 CrossRef CAS.
- X. Shen and J. R. Knutson, Subpicosecond Fluorescence Spectra of Tryptophan in Water, J. Phys. Chem. B, 2001, 105, 6260–6265 CrossRef CAS.
- M. R. Eftink, L. A. Selvidge, P. R. Callis and A. A. Rehms, Photophysics of indole derivatives: experimental resolution of La and Lb transitions and comparison with theory, J. Phys. Chem., 1990, 94(9), 3469–3479 CrossRef CAS.
- H. M. Watrob, C.-P. Pan and M. D. Barkley, Two-step FRET as a structural tool, J. Am. Chem. Soc., 2003, 125(24), 7336–7343 CrossRef CAS PubMed.
- E. Sobakinskaya, M. Schmidt am Busch and T. Renger, Theory of FRET “spectroscopic ruler” for short distances: application to polyproline, J. Phys. Chem. B, 2017, 122(1), 54–67 CrossRef PubMed.
- S. Verheyden, A. Sillen, A. Gils, P. J. Declerck and Y. Engelborghs, Tryptophan properties in fluorescence and functional stability of plasminogen activator inhibitor 1, Biophys. J., 2003, 85(1), 501–510 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional up-conversion fluorescence decay curve, energy level structure, simulated fluorescence spectra, calculation details of centroid distance, all parameters used in the working model, fragment transition dipole moments. See DOI: https://doi.org/10.1039/d2cp02139k |
| This journal is © the Owner Societies 2022 |