
Mechanistic insights into positional and skeletal isomerization of cyclohexene in the H-BEA zeolite†
Peng
Liu
 a,
Qian
Liu
a,
Wei
Liu
*b,
Shaozhong
Peng
b and
Donghai
Mei
*ac
a,
Qian
Liu
a,
Wei
Liu
*b,
Shaozhong
Peng
b and
Donghai
Mei
*ac
aSchool of Chemical Engineering and Technology, Tiangong University, Tianjin 300387, P. R. China. E-mail: dhmei@tiangong.edu.cn
bSINOPEC Dalian Research Institute of Petroleum and Petrochemicals, Dalian, Liaoning Province 116045, P. R. China. E-mail: lw.fshy@sinopec.com
cSchool of Environmental Science and Engineering, Tiangong University, Tianjin, 300387, P. R. China
First published on 12th July 2022
Abstract
The isomerization of cycloalkenes via the formation of carbenium cations assisted by the Brønsted acid site (BAS) in zeolites is the vital reaction step in hydrocracking and hydroisomerization processes of the petrochemical industry. To understand the acid-catalyzed positional isomerization and skeletal isomerization of cycloalkenes via carbenium intermediates, a series of ab initio molecular dynamics (AIMD) simulations of cyclohexene within the H-BEA zeolite have been carried out. AIMD simulations combined with the enhanced sampling technique reveal that the half-chair conformer is the most stable conformation for cyclohexene within H-BEA. Free energy landscapes characterizing protonation/deprotonation, positional isomerization, and skeletal isomerization of cyclohexene have been mapped out at 413 K. The free energy barrier for the formation of carbenium is calculated to be 44 kJ mol−1. The skeletal isomerization of cyclohexene to methylcyclopentylium via the protonated cyclopropane transition state involves four stages with a total free energy barrier of 134 kJ mol−1. Further geometrical analysis provides additional information about the structural origin of free energy barriers.
1. Introduction
In the context of upgrading renewable biomass resources, natural gas, and/or crude oil into liquid fuels and high value-added chemicals,1 the major focus is on acid-catalyzed reactions over zeolite catalysts. Zeolites2 are microporous solids composed of tetrahedrally coordinated silicate units. Substitution of Si atoms by Al atoms and charge-compensating protons introduces Brønsted acid sites (BASs). The compensating protons possessing strong acid strength provide BASs within zeolites the catalytic capability of electrophilic attack to the electron-rich moieties,3 such as double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. The carbenium cations, which are formed by the protonation of these CC compounds with the BAS protons, then further transform into other chemical intermediates via proton/hydride transfer (positional isomerization) and/or carbon transfer (skeletal isomerization) processes. Gaining the fundamental understanding of the positional isomerization and skeletal isomerization of cycloalkenes will be greatly beneficial to improve the performance of hydrocracking and hydroisomerization reactions within zeolites.
C bonds. The carbenium cations, which are formed by the protonation of these CC compounds with the BAS protons, then further transform into other chemical intermediates via proton/hydride transfer (positional isomerization) and/or carbon transfer (skeletal isomerization) processes. Gaining the fundamental understanding of the positional isomerization and skeletal isomerization of cycloalkenes will be greatly beneficial to improve the performance of hydrocracking and hydroisomerization reactions within zeolites.
As one of the widely used and important model cycloalkene compounds in the petrochemical industry, cyclohexene obtained from the hydrogenation of benzene has been studied both experimentally and theoretically.4–7 For the acid-catalyzed positional isomerization of cycloalkenes within zeolites, the initial step is the protonation of the electron-rich π bond by the acidic proton (HB) of the BAS,8,9 generating one carbenium intermediate. Depending on the attached carbon atom (C1 or C2) by HB, there are two isomers for the carbenium cation. Isomerization of carbenium ions is achieved by the 1,2-hydride transfer process.10,11 The whole system can be stabilized by deprotonation via two pathways. For the first pathway, the HB within the carbenium ion, which originates from the BAS, returns to the framework oxygen at the BAS. This pathway involves three processes including the 1,2-hydride transfer, the protonation of cycloalkane, and deprotonation of carbenium. For the second pathway, an adjacent carbon (C3, see Scheme 1a) loses one proton to the framework oxygen and forms a new double C2C3 bond simultaneously. The combination of the last pathway with the protonation of the C1C2 double bond is the positional isomerization of the double CC bond,12 which is confirmed by the H/D isotope tracer study in conjunction with 1H NMR experiments13,14 in the dehydration process of cyclohexanol in various zeolite pores.15 Due to the lightest mass, hydrogen transfer is more feasible to achieve, which plays a significant role in the activation of reactants, transformation, and/or stabilization of intermediates toward products.
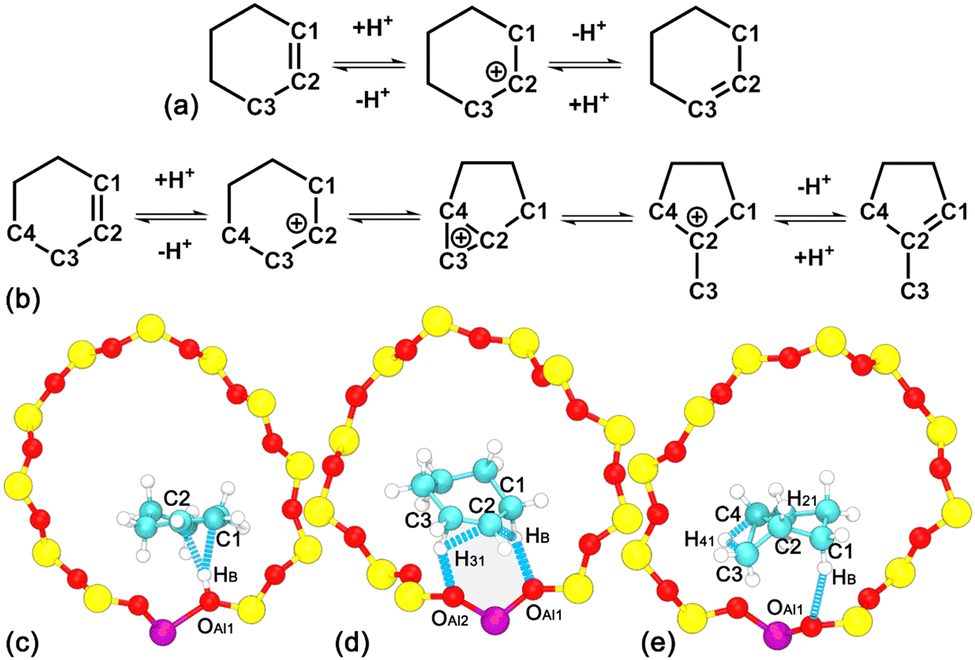 | ||
| Scheme 1 (a) The positional isomerization and (b) skeletal isomerization of cyclohexene via a carbenium intermediate. Atoms involved in the definition of collective variables characterizing (c) the protonation/deprotonation, (d) positional isomerization, and (e) skeletal isomerization of cyclohexene. | ||
For the carbenium cation, the carbon transfer changes the connectivity of heavy atoms causing skeletal isomerization of cycloalkenes. The skeleton of cyclohexylium can be rearranged via a protonated cyclopropane (PCP)-like structure16 connected edge-to-edge to a cyclopentane. The six-membered ring of cyclohexene shrinks into the five-membered ring of methylcyclopentene. A secondary carbenium is transformed into a tertiary one (see Scheme 1b). The ring contraction of cycloalkenes is the crucial industrial reaction for hydroisomerization of ethylbenzene,17 which is of importance in the production of paraxylene. Although enormous experimental efforts on the hydroisomerization processes of cycloalkenes in H-form zeolites have been made,18,19 how the positional and skeletal isomerization processes occur, in particular, at the atomic level, is still an open question. Due to the complexity of isomerization pathways, most of the available theoretical studies have primarily relied on the static density functional theory (DFT) calculation method and the transition-state theory,20 which cannot describe finite temperature effects with sufficient accuracy. Aimed at this problem, the enhanced sampling technique combined with ab initio molecular dynamics (AIMD) simulations is one of the best theoretical tools to tackle this question. Recently, this theoretical approach has been applied to investigate the isomerization of alkene in the cracking process of linear hydrocarbons within zeolites.16,21,22
In this contribution, the positional and skeletal isomerization processes of cyclohexene aided by the BAS were investigated. First, unconstrained AIMD simulations for one cyclohexene molecule and one 1-methylcyclopentene molecule in the vicinity of the BAS within the H-BEA zeolite have been performed to study their structural stability and preferred orientation. By combining AIMD simulations and the enhanced sampling technique, isomerization processes of cyclohexene were then systematically studied. High-dimensional free energy landscapes characterizing the conformational isomerization of cyclohexene, protonation of alkene/deprotonation of carbenium, positional isomerization of the double CC bond, and skeletal isomerization of cyclohexene were mapped out. The underlying mechanisms for the positional and skeletal isomerization of cyclohexene via the carbenium ion were clearly elucidated.
2. Computational details
2.1 Zeolite model
A periodic three-dimensional all siliceous BEA zeolite structure of Si64O128 is used in this work.23 The cell parameters are as follows: a = 12.6614 Å, b = 12.6614 Å, c = 26.4061 Å, and α = β = γ = 90°. One Si (T9) atom positioned at the twelve-membered ring is chosen. By replacing the chosen Si atom with one Al atom and adding one proton compensating the negative charge of the zeolite framework, a typical BAS is formed. The constructed H-BEA zeolite has a Si/Al ratio of 63. In the present work, two systems, viz., H-BEA–Chene and H-BEA–mCpene, have been constructed. The H-BEA–Chene and H-BEA–mCpene systems contain one cyclohexene and one 1-methylcyclopentene molecule adsorbed at the BAS of the H-BEA zeolite, respectively.2.2 Ab initio molecular dynamics simulations
All periodic AIMD simulations are carried out using the CP2K 6.1 package.24,25 The hybrid Gaussian and the plane wave density functional scheme with pseudopotential approximation are employed.26 All AIMD simulations are performed using the Perdew–Burke–Ernzerhof exchange/correlation functional27 with norm-conserving Goedecker–Teter–Hutter pseudopotentials28 for the core electrons. The double-ζ basis sets (DZVP-MOLOPT-GTH) are used to describe all atoms of the H-BEA–Chene and the H-BEA–mCpene systems. A 360 Ry cutoff energy is applied in all AIMD simulations. The dispersion force is taken into account using the DFT-D3 scheme.29 All AIMD simulations are performed in the canonical NVT ensemble. The simulation temperature is maintained at 413 K employing the Nosè–Hoover thermostat.30,31 The equations of motion in the AIMD simulations are integrated using the velocity Verlet method with a time step of 0.6 fs. The H-BEA–Chene and the H-BEA–mCpene systems are equilibrated for 4 ps, followed by a production run of 20 ps. Trajectory frames are captured every 4 steps. AIMD trajectories are visualized and analyzed using VMD 1.9.4.322.3 Free energy calculations
The multiple-walker well-tempered metadynamics method33,34 applied in the PLUMED 2.4.535,36 plug-in in combination with CP2K 6.1 has been used to perform enhanced sampling in the phase space. Four processes, i.e., the conformational isomerization, the protonation of alkene/deprotonation of carbenium, and the positional and skeletal isomerization of cyclohexene, have been systematically investigated. The free energy landscape characterizing each process has been mapped out. Four reaction steps are described by collective variables (CVs). Cremer–Pople puckering37 coordinates, viz., θ and ϕ, have been applied to describe the conformational isomerization of cyclohexene. For other processes, CVs are defined as the difference in coordination numbers (cn). cn(A–B) is functionalized as a switch function as follows, cn(A–B) = (1 − (dAB/d0)p)/(1 − (dAB/d0)p+q). dAB is the distance between atoms A and B. Detailed parameters for each cn(A–B) can be found in the ESI.† Atoms involved in the definition of CVs are labeled in Scheme 1. CVs, dcn,11 and dcn,12, are used to describe the protonation of alkene/deprotonation of carbenium. CVs, dcn,21 and dcn,22, are used to describe the positional isomerization of the double bond within cyclohexene. CVs, dcn,31, dcn,32, dcn,33, and dcn,34, are applied to monitor the ring contraction of cyclohexene. The above mentioned CVs are defined as follows:| dcn,11 = cn(C1–HB) − cn(C2–HB) | (1) |
| dcn,12 = cn(C1–HB) + cn(C2–HB) − cn(OAl1–HB) | (2) |
| dcn,21 = cn(OAl2–H31) − cn(C3–H31) | (3) |
| dcn,22 = cn(OAl1–HB) − cn(C1–HB) | (4) |
| dcn,31 = cn(C1–HB) − cn(OAl1–HB) | (5) |
| dcn,32 = cn(C2–C4) − cn(C3–C4) | (6) |
| dcn,33 = cn(C3–H41) − cn(C4–H41) | (7) |
| dcn,34 = cn(C4–H21) − cn(C2–H21) | (8) |
The Gaussian bias potentials are spawned every 30 steps. The height and width of the bias potential can be found in the ESI.† The bias factor is equal to 8. In the metadynamics simulation, the temperature is maintained at 413 K by applying the canonical sampling velocity rescaling thermostat.38 The least free energy pathways connecting minima are found with the aid of LFEP33 and NMFEP39 algorithms. The function of the position, s, is an order parameter applied in the description of free energy pathways. The detailed definition can be found in the publication by Branduardi et al.40 Local maxima within least free energy pathways correspond to transition states. The relative value between the transition state and the reactant/product state is the height of the free energy barrier in the forward/backward reaction step. In this work, four processes, viz., conformational isomerization, protonation/deprotonation, positional isomerization, and the ring contraction of cyclohexene, have been investigated. For each process, simulation time is accumulated up to at least 120 ps. Order parameters s0, s1, s2, and s3 are applied to plot the change of free energy and geometrical properties along the least free energy pathways.
3. Results and discussion
3.1. Thermal stability and structural features of H-BEA–Chene and H-BEA–mCpene systems
AIMD simulations have been performed to investigate the thermal stability of the H-BEA–Chene system. To locate HB of the BAS in the presence of an adsorbed cyclohexene molecule, the minimum distance between HB and oxygen atoms bonded with Al (dOAl–HB,min) is determined. Labels for atoms involved in the structural analysis are given in Fig. 1a. As shown in Fig. 1b, dOAl–HB,min fluctuates around 1.0 Å, indicating that HB predominantly resides within the BAS in the presence of an adsorbed cyclohexene.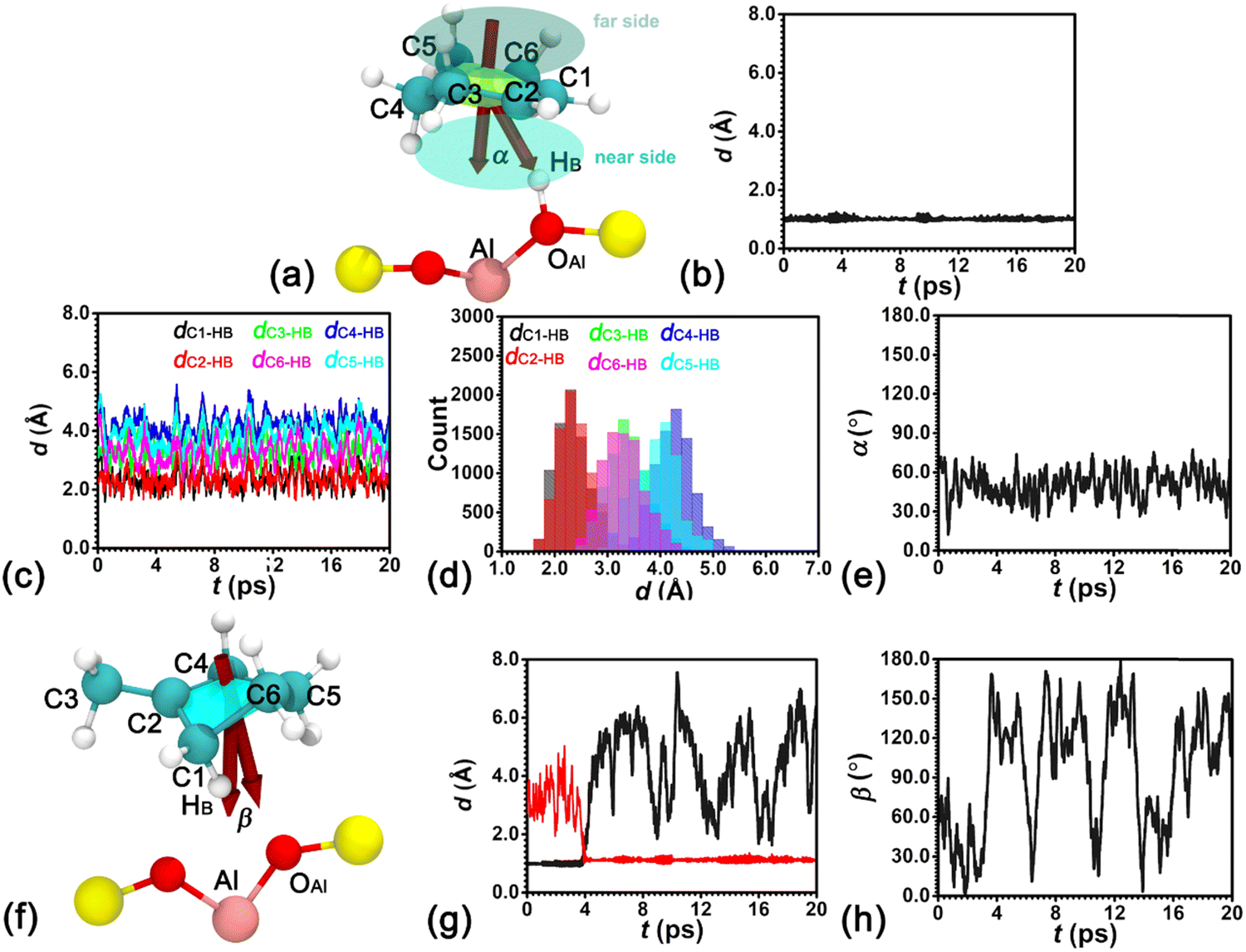 | ||
| Fig. 1 (a) Atoms involved in the structural analysis and the definition of the cross angle, α, between the normal vector of cyclohexene and the vector connecting the center of mass of cyclohexene and the acidic proton in the H-BEA–Chene system. The variation of (b) the minimum distance, dOAl–HB,min, between the acidic proton and oxygen atoms connecting Al. Evolutions (c) and distributions (d) of dCn-HB distances between the acidic proton and carbon atoms within cyclohexene. (e) The evolution of the cross angle, α. (f) Atoms involved in the structural analysis and the definition of the cross angle, β, between the normal vector of methylcyclopentylium and the vector connecting the center of mass of methylcyclopentylium and Al in the H-BEA–mCpene system. The variation of (g) the minimum distance, dOAl–HB,min, (black curve) between the acidic proton and oxygen atoms connecting Al and the minimum distance, dCs–HB,min (red curve), between the acidic proton and carbon atoms of methylcyclopentylium. (h) The evolution of the cross angle, β. | ||
Structural features of the adsorbed cyclohexene at the BAS in the H-BEA pores are explored. Distances between HB with every carbon atom of cyclohexene, dCn-HB (n = 1–6), have been measured. Results are shown in Fig. 1c. dCn-HB distances fluctuate around stable values in the production run of 20 ps. Distributions of these distances featuring Gaussian shapes are grouped into three categories. Distributions of (dC1–HB and dC2–HB), (dC3–HB and dC6–HB), and (dC4–HB and dC5–HB) are centered at ca. 2.3, 3.3, and 4.3 Å, respectively (see Fig. 1d). The shorter distances of dC1–HB and dC2–HB clearly indicate a strong interaction between the C1C2 double bond of the adsorbed cyclohexene and HB of the BAS.
To identify the orientation of cyclohexene relative to the BAS, the cross angle, α, between the normal vector of cyclohexene and the vector connecting the center of mass of cyclohexene and HB has been measured. As shown in Fig. 1e, the cross angle varies in the range of 30–60°, implying the attractive interaction between the double bond of cyclohexene and HB imposes a strong restraint on the orientation of cyclohexene. One side of cyclohexene is attached to the BAS. No flip-flop has been detected for the adsorbed cyclohexene in the production run of 20 ps.
AIMD simulations have been carried out to investigate the thermal stability of the H-BEA–mCpene system. To locate the acidic proton HB, dOAl–HB,min and the minimum distance between HB and carbon atoms of methylcyclopentene (dCs–HB,min) are measured. Labels for atoms involved in the structural analysis are shown in Fig. 1f. As shown in Fig. 1g, dOAl–HB,min fluctuates around 1.0 Å, while dCs–HB,min varies in the range of 2.5–5.0 Å in the first 4 ps run. This indicates that the acid proton HB resides on the oxygen atom of the BAS. At 4 ps, dOAl–HB,min abruptly increases, while dCs–HB,min decreases. In the remaining 16 ps, dOAl–HB,min and dCs–HB,min vary in the range larger than 2.0 Å and around 1.0 Å, respectively. Clearly, the acidic proton HB is detached from the BAS and bonded with methylcyclopentene. The stable species of methylcyclopentene in the vicinity of the BAS is the methylcyclopentylium cation. One ionic pair is formed, which is very similar to the previous observation by Cnudde et al.21
The orientation of methylcyclopentylium relative to the BAS has been studied by measuring the cross angle, β, between the normal vector of methylcyclopentylium and the vector connecting the center of mass of methylcyclopentylium and Al of the BAS. As shown in Fig. 1h, the cross angle varies in the full range of 0–180°, implying that methylcyclopentylium frequently flip-flops in the production run of 20 ps. The undirectional electrostatic interaction within the ionic pair imposes no restraint on the orientation of the positively charged methylcyclopentylium.
3.2. Conformational isomerization of cyclohexene in the H-BEA zeolite
In our previous contribution,41 the confinement environment of pores within the zeolite affects the preferred conformation of cyclohexanol. Compared with cyclohexanol, cyclohexene exhibits more rigidity. To determine the preferred conformation of cyclohexene in the H-BEA zeolite, the two-dimensional free energy surface spanned by Cremer–Pople coordinates, θ and ϕ, has been calculated. As depicted in Fig. 2a, the free energy surface features two basins centered around (θ = 120°, ϕ = 90°) and (θ = 60°, ϕ = 270°) connected by a sinusoidal strip. The two basin regions correspond to the 5H4 and 4H5 half-chair conformers of cyclohexene, respectively. Two additional shallow basins centered around (θ = 90°, ϕ = 180°) and (θ = 90°, ϕ = 360°/0°) correspond to the B3,6 and 3,6B boat conformers, respectively. Further geometrical optimization validates that B3,6 and 3,6B conformers represent stable states. The least free energy pathway connecting four conformers can be identified. The free energy profile along the least free energy pathway is shown in Fig. 2b. The corresponding structures of reaction intermediates and transition states are given in the right panel of Fig. 2. As shown in Fig. 2a and b, the most possible conformers for cyclohexene at the BAS in H-BEA are 4H5 and 5H4. There is no preference between these two conformers. The transition from one conformer to another needs to cross an intermediate state (3,6B or B3,6) and two transition states with free energy barriers as high as 19 kJ mol−1. The free energy difference between transition states and intermediates is marginal (∼ 1 kJ mol−1 < kBT), which can be easily overcome by thermal fluctuations. Therefore, combined with our previous results of the adsorbed cyclohexanol in the H-BEA zeolite,41 it can be concluded that the cyclohexanol dehydration reaction converts the most stable conformer of the six-membered ring from 4C1 and/or 1C4 (chair conformers) for cyclohexanol to 4H5 and/or 5H4 for cyclohexene.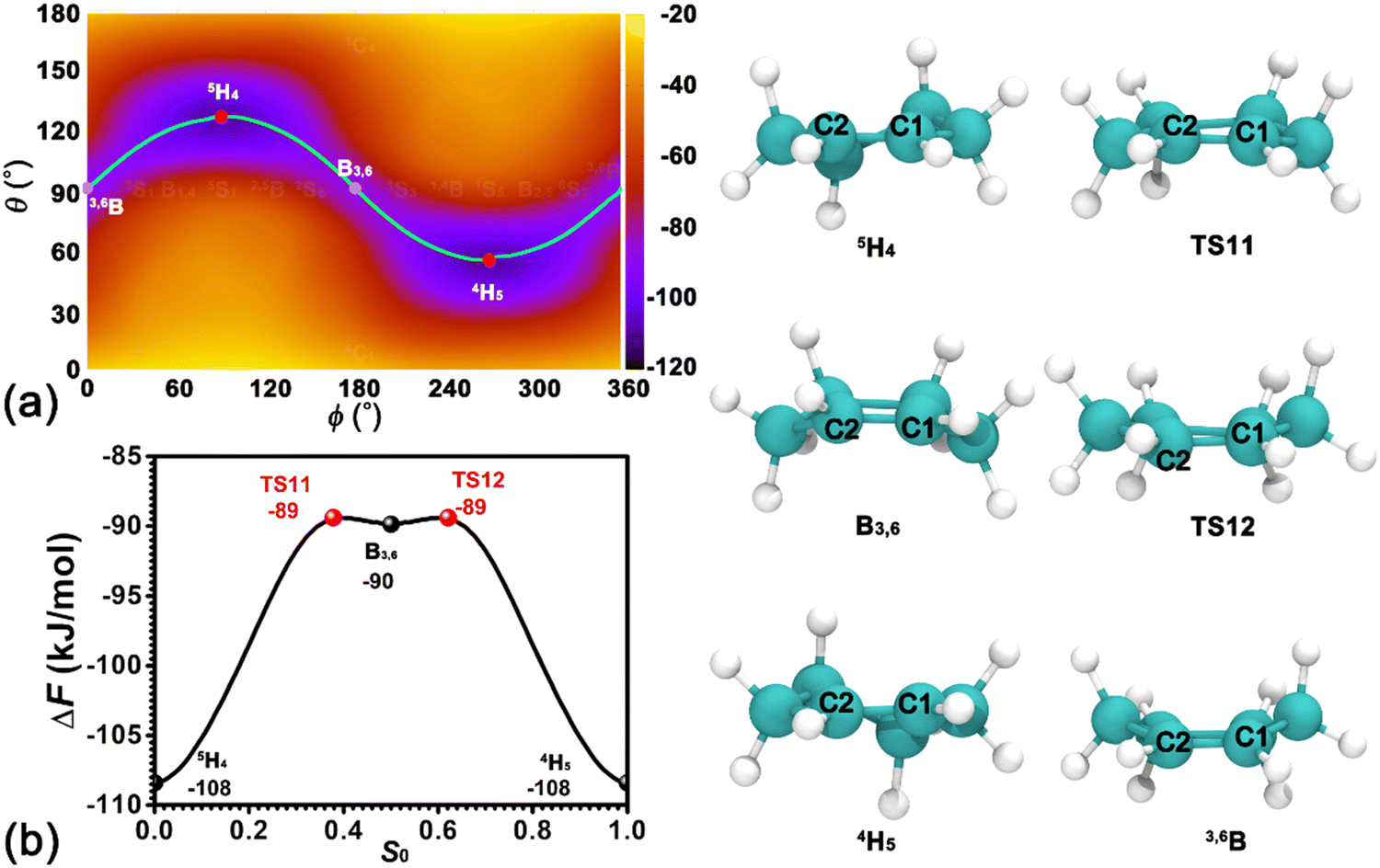 | ||
| Fig. 2 (a) Free energy landscape spanned on Cremer–Pople puckering angles, θ and ϕ, characterizing the conformational isomerization of cyclohexene in the H-BEA zeolite at 413 K. Regions corresponding to the boat (B) and the half-chair (H) conformers are indicated. (b) The identified least free energy pathway for the transition of conformers. Representative structures of local minima and saddle points are also depicted on the right. | ||
3.3. Protonation of cyclohexene/deprotonation of cyclohexylium
The orientation of adsorbed cyclohexene is restrained because of the strong interaction between the cyclohexene molecule and the BAS. The adsorbed cyclohexene relative to the BAS has two sides, viz., the near side and the far side (see Fig. 1a). Herein, the protonation of the C1C2 double bond/deprotonation of carbenium in the near side has been investigated. The calculated two-dimensional free energy surface is shown in Fig. 3a. Three local minima, viz., Chene(12) + H+, Ch+(ax,C1), Ch+(ax,C2), can be found at (dcn,1 = −0.01, dcn,2 = −0.56), (dcn,1 = −0.64, dcn,2 = 0.99), and (dcn,1 = 0.62, dcn,2 = 0.99), respectively. The least free energy pathway connecting three minima is shown in Fig. 3b. The corresponding structures of reaction intermediates and transition states are shown in Fig. 4. The ground state of the whole system is the neutral cyclohexene. The two intermediate states represent the cyclohexylium cations.
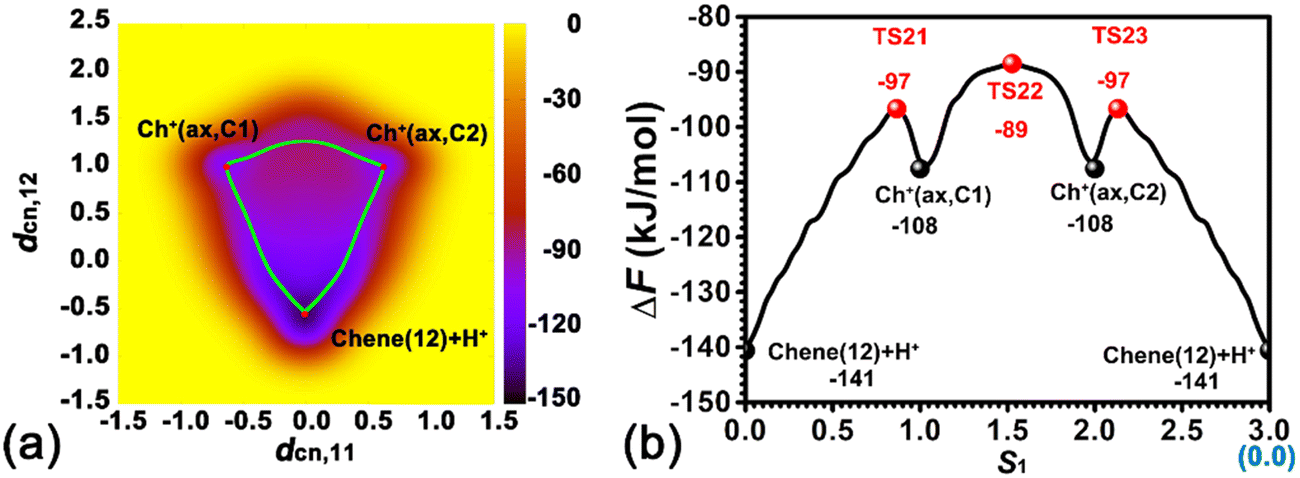 | ||
| Fig. 3 (a) Free energy landscape characterizing the protonation of cyclohexene/deprotonation of cyclohexylium within the H-BEA zeolite at 413 K. (b) The least free energy pathway for the protonation/deprotonation process. | ||
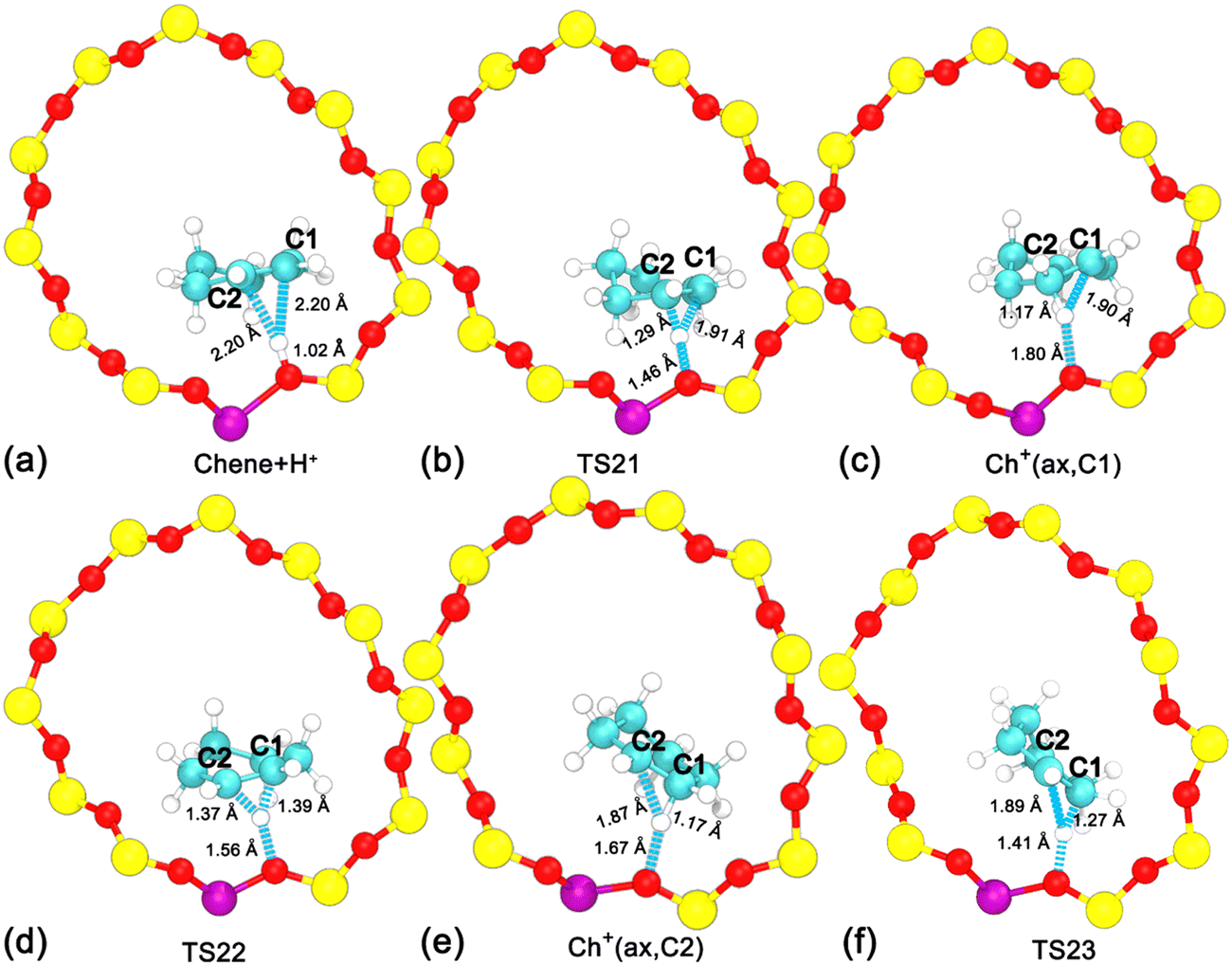 | ||
| Fig. 4 Structures of reaction intermediates and transition states shown in Fig. 3. | ||
Three minima are grouped into two categories, i.e., neutral cyclohexene and the carbenium cation. The free energy difference between cyclohexene and the carbenium ion is calculated to be 33 kJ mol−1. Transformations between two categories are the protonation/deprotonation process. The free energy barriers are estimated as 44 and 11 kJ mol−1 for the protonation of cyclohexene and deprotonation of cyclohexylium, respectively. This is in excellent agreement with our previous work where the free energy barriers for protonation of cyclohexene (by hydronium) and deprotonation of carbenium are calculated to be 47 kJ mol−1 and 12 kJ mol−1, respectively.35,37 Carbenium ions, viz., Ch+(ax,C1) and Ch+(ax,C2), are reciprocally symmetric structures. The difference is in the sequence of numbering of carbon atoms. The free energy difference between carbenium ions is marginal. The transformation between two carbenium ions involves the 1,2-hydride transfer, in which the hydride species transfers from C2 to C1 (or vice versa). The calculated free energy barrier for the 1,2-hydride transfer is 19 kJ mol−1, which is also in agreement with other theoretical calculations42,43 and experimental measurement (<23 kJ mol−1).44 For the carbenium ion, the deprotonation is more feasible than the 1,2-hydride transfer.
To further understand the effect of protonation/deprotonation on the strength of the CC double bond of cyclohexene, the length of the C1C2 bond (dC1–C2) has been measured. The evolution of the average value for dC1–C2 along the least free energy pathway is plotted in Fig. 5. For cyclohexene protonation, dC1–C2 increases from 1.35 Å to 1.44 Å. For the 1,2-hydride transfer, dC1–C2 firstly decreases from 1.44 Å to 1.40 Å and then regains to 1.44 Å. It has been well-documented that the single C–C and double CC bond lengths in the hydrocarbons are 1.54 Å and 1.34 Å, respectively.45 Therefore, the C1–C2 bond of the cyclohexene is the CC double bond in the beginning. As the protonation proceeds, the CC double bond is weakening with the increasing bond length. Please note that this C1–C2 bond length (1.44 Å) is still shorter than the C–C single bond length (1.54 Å), suggesting that the C1–C2 bond strength is more than the single C–C bond strength in the typical hydrocarbons. In the hydride transfer process, the C1–C2 bond is strengthened with the decreasing bond length and then recovers to the initial bond length.
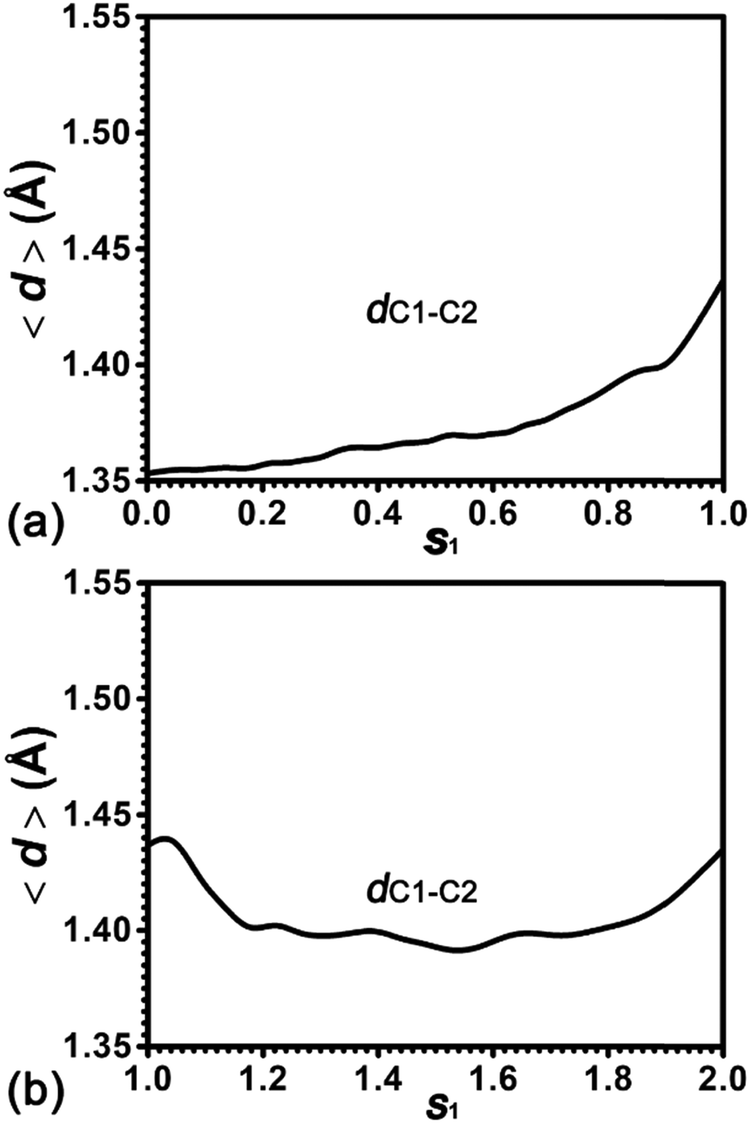 | ||
| Fig. 5 Evolution of the distance, dC1–C2, in the protonation process of alkene and the 1,2-hydride transfer within the carbenium ion. | ||
3.4. Positional isomerization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C double bond within cyclohexene
C double bond within cyclohexene
Using the H/D isotope tracer technique, Chen et al.15 have proposed a cyclohexanol dehydration reaction pathway via the carbenium intermediate in zeolites. They have identified two dehydration products of 1-D-cyclohexanol, i.e., 1-D-cyclohexene and 3-D-cyclohexene, suggesting an isomerization of cyclohexene via a carbenium intermediate. As shown in Scheme 2, the positional isomerization of the CC double bond within cyclohexene can be achieved by the combination of protonation, 1,2-hydride transfer, and deprotonation. While in Scheme 1a, which is discussed above, a simplified isomerization pathway involving only protonation and deprotonation steps was depicted. The 1,2-hydride transfer is circumvented in Scheme 1a. As indicated in the previous section, a high free energy barrier needs to be overcome for the 1,2-hydride transfer. Therefore, we will only discuss the positional isomerization of cyclohexene in terms of protonation of cyclohexene and deprotonation of cyclohexylium.
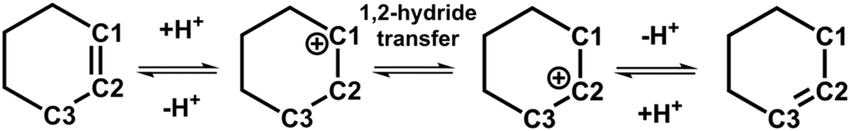 | ||
| Scheme 2 The possible pathway for the positional isomerization of cyclohexene. | ||
To disentangle the positional isomerization of cyclohexene assisted by the BAS in the H-BEA zeolite pore, the two-dimensional free energy surface has been mapped out (Fig. 6a). Three local minima can be located. Free energy pathways connecting these minima are determined using the LFEP algorithm, as shown in Fig. 6b. The corresponding structures of the reaction intermediates and transition states are shown in Fig. 7. Two minima are located at (dcn,21 = −0.87 and dcn,22 = 0.60) and (dcn,21 = 0.67 and dcn,22 = −0.87), which correspond to the initial state, Chene(12) + H+, and the final state, Chene(23) + H+. The free energy difference between the initial and final states is negligible. The third minimum, Ch+(ax,C2) (dcn,3 = −0.73 and dcn,4 = −0.80), is a carbenium intermediate. At this minimum, the positive charge is located at the C2 atom (trivalent carbon). The free energy difference between the carbenium ion and the initial/final state is calculated to be 31 kJ mol−1. The estimated free energy barriers for the protonation and deprotonation processes are 40 and 9 kJ mol−1, respectively. These results are consistent with the results discussed in the previous section. For the deprotonation process, a free energy barrier of 9 kJ mol−1 is required to overcome, indicating that the deprotonation process in the positional isomerization is more facile than the 1,2-hydride transfer, which has a higher free energy barrier of 19 kJ mol−1.
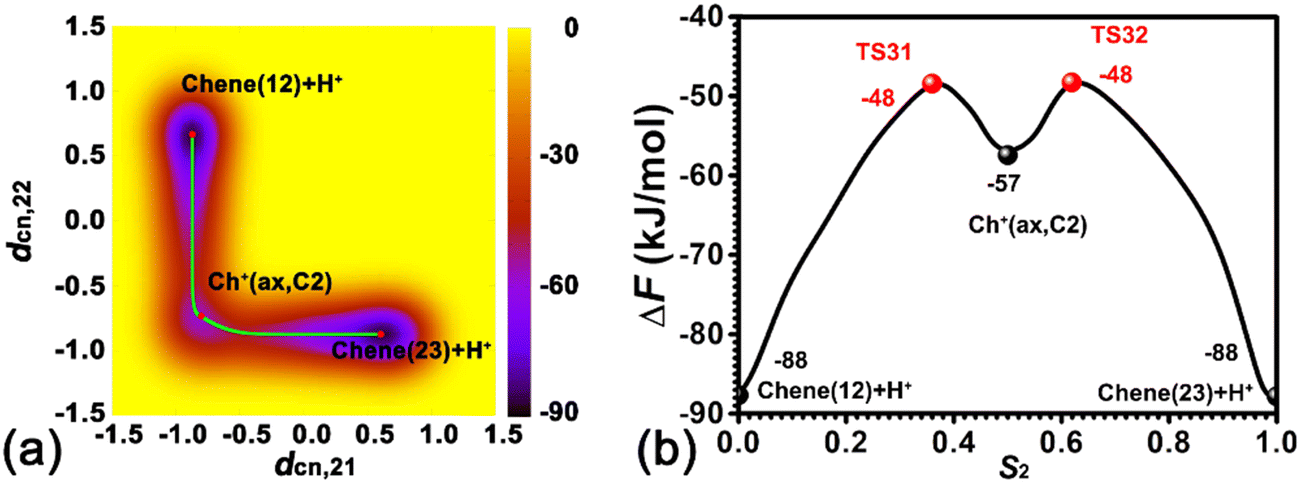 | ||
| Fig. 6 (a) Free energy landscape characterizing the positional isomerization of the CC double bond within cyclohexene assisted by the BAS in the H-BEA zeolite at 413 K. (b) The least free energy pathway for the positional isomerization of cyclohexene. | ||
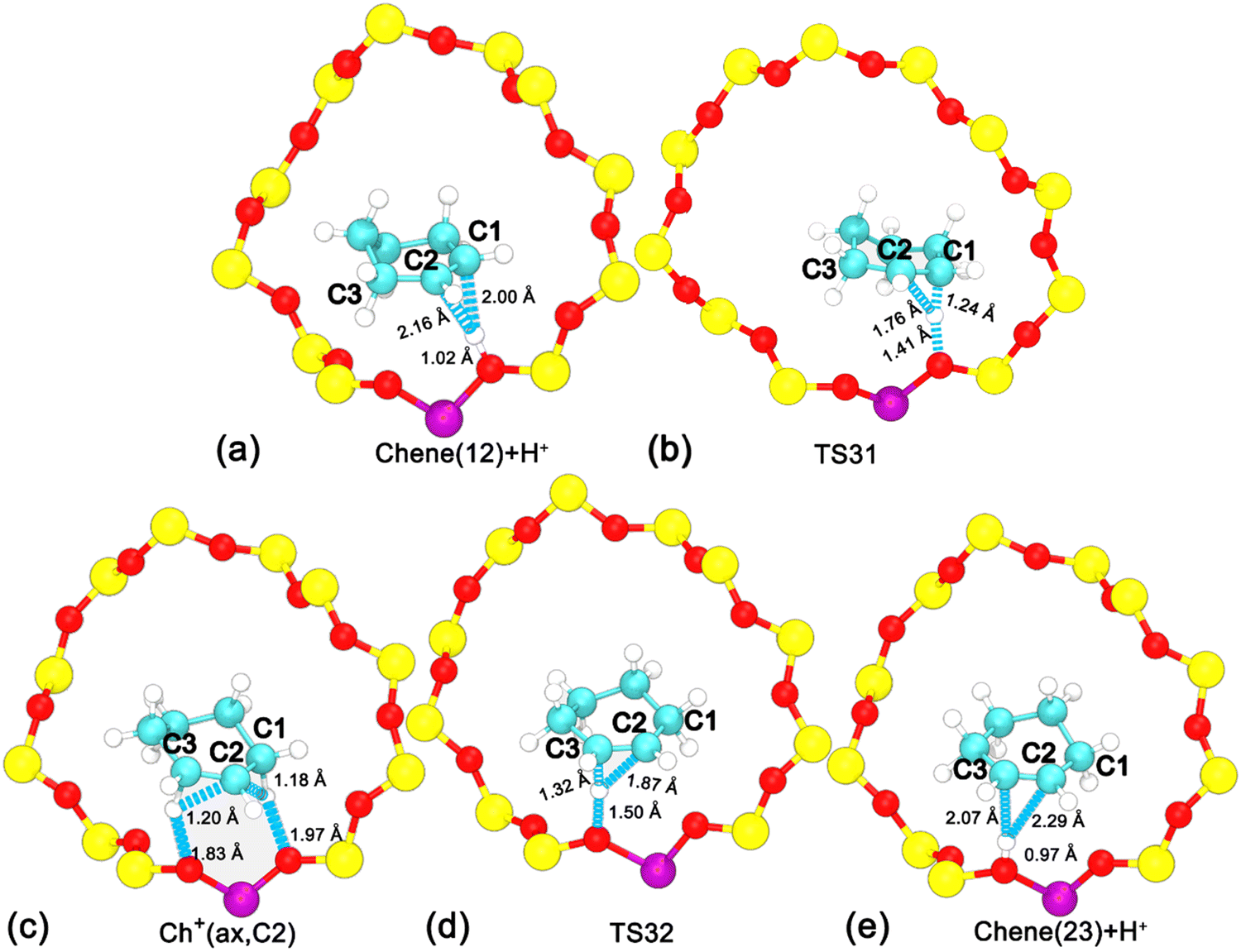 | ||
| Fig. 7 Structures of reaction intermediates and transition states shown in Fig. 6. | ||
To characterize the C–C bond strength in the positional isomerization of cyclohexene, two bond lengths, dC1–C2 and dC2–C3, have been measured. The evolution of average values for dC1–C2 and dC2–C3 along the least free energy pathway has been shown in Fig. 8. dC1–C2 increases from 1.35 to 1.52 Å, while dC2–C3 decreases from 1.52 Å to 1.35 Å. This indicates that the CC double bond shifts from the C1–C2 position to the C2–C3 position. At the point s2 = 0.5, which corresponds to the carbenium ion, Ch+(ax,C2), the C–C bond length is around 1.44 Å. This is also consistent with the measurement in the previous section.
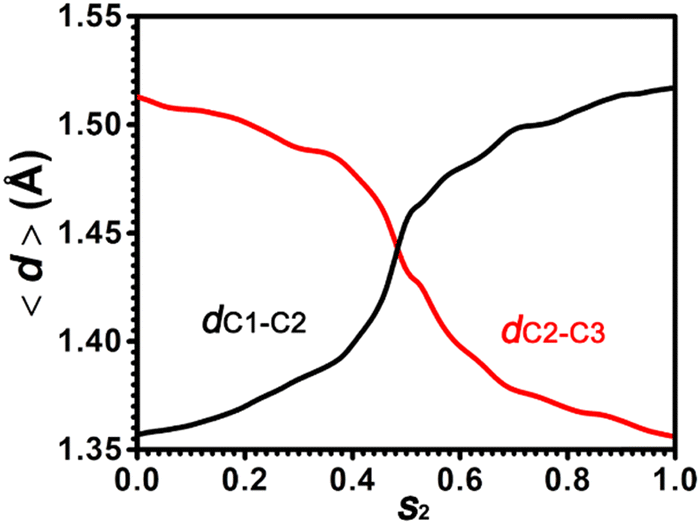 | ||
| Fig. 8 Evolution of dC1–C2 (black) and dC2–C3 (red) distances in the positional isomerization of the CC bond within cyclohexene. | ||
3.5. Skeletal isomerization of cyclohexene catalyzed by the BAS
To investigate the ring contraction of cyclohexene assisted by the BAS in the H-BEA zeolite pore, the skeletal isomerization of cyclohexene is examined by mapping out a high-dimensional free energy landscape (Fig. 9a, b). Five local minima have been identified. The free energy pathways connecting these minima are determined using the NMFEP algorithm, which are shown in Fig. 9c. The corresponding structures of reaction intermediates and transition states are shown in Fig. 10.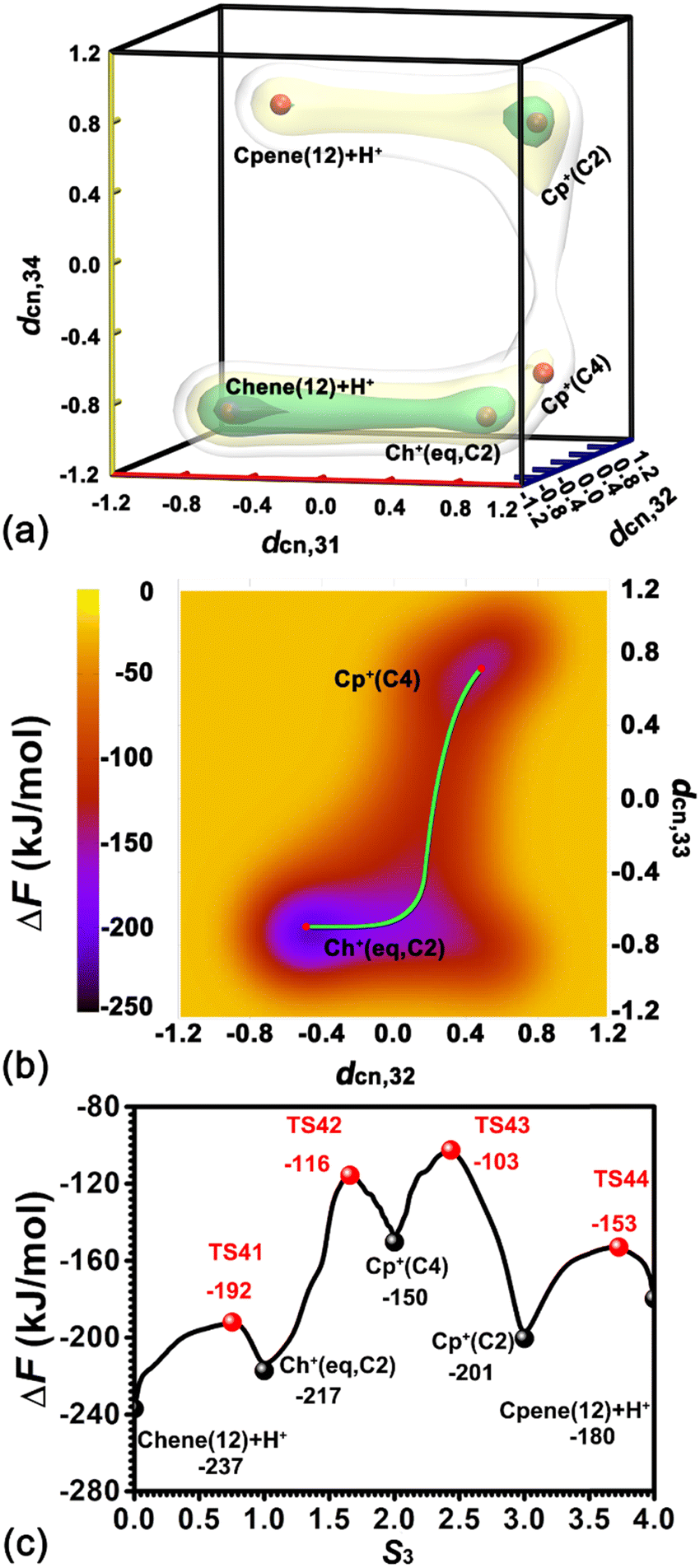 | ||
| Fig. 9 (a) Free energy landscape characterizing the skeletal isomerization of cyclohexene in the H-BEA zeolite at 413 K. (b) Free energy surface characterizing the transformation from Ch+(eq,C2) to Cp+(C4) at 413 K. (c) The least free energy pathway for the ring contraction of cyclohexene. | ||
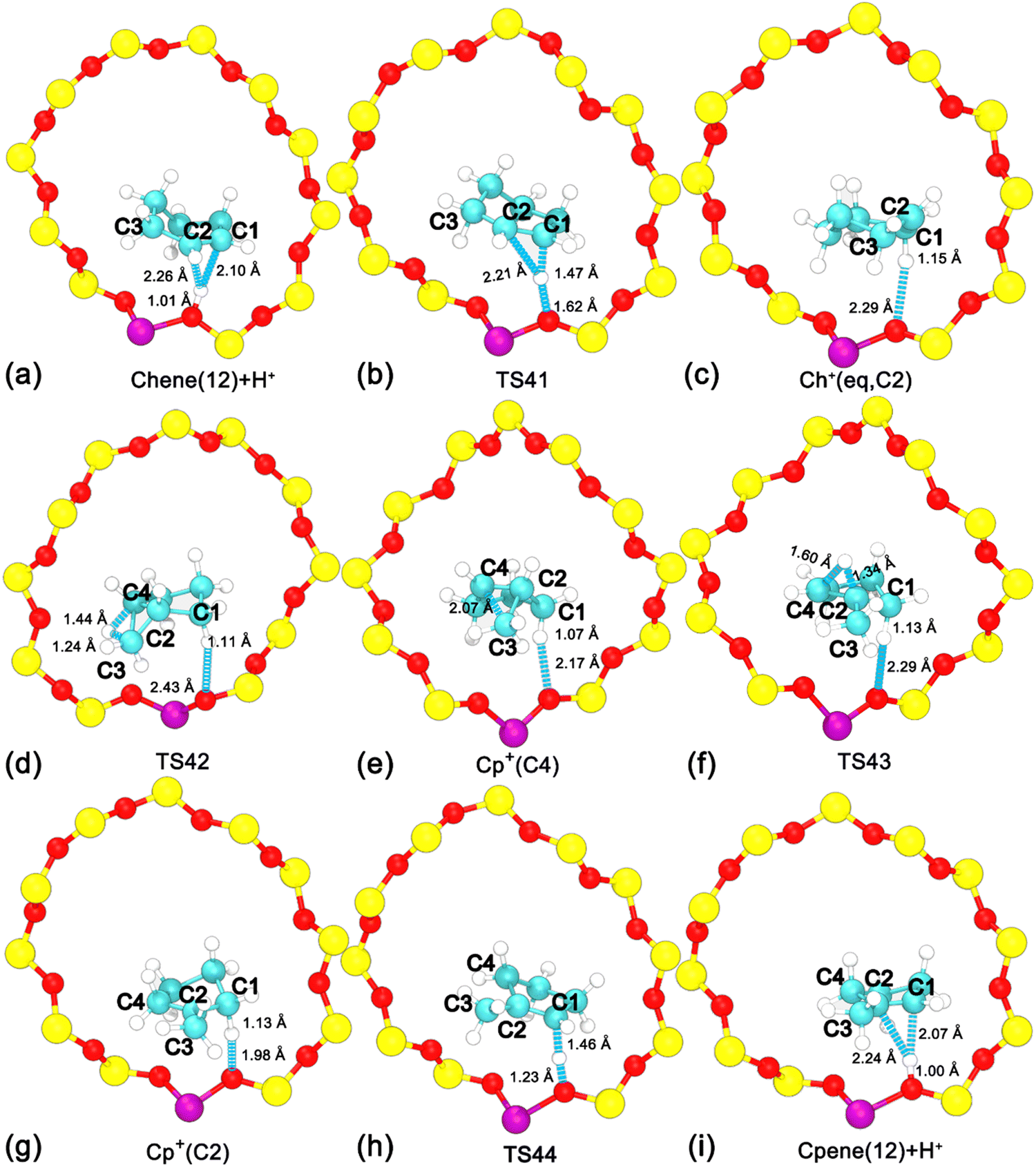 | ||
| Fig. 10 Structures of reaction intermediates and transition states shown in Fig. 9. | ||
Two minima are located at (dcn,31 = −0.68, dcn,32 = −0.55, dcn,33 = −0.71, and dcn,34 = −0.90) and (dcn,31 = 0.80, dcn,32 = 0.64, dcn,33 = 0.81, and dcn,34 = 0.65), which correspond to the initial state, Chene(12) + H+, and the final state, Cp+(C2). The free energy difference between the initial and final states is 36 kJ mol−1. The free energy barrier for the ring contraction process is 134 kJ mol−1. The ring contraction process consists of four stages. The first stage is the protonation of Chene(12) + H+ to Ch+(eq,C2) (dcn,31 = 0.80, dcn,32 = −0.48, dcn,33 = −0.71, and dcn,34 = −0.90), which is a carbenium intermediate. Within Ch+(eq,C2), the positive charge is located at C2 (trivalent carbon). Compared with the free energy difference between Ch+(ax,C2) and Chene(12) + H+ in the protonation of cyclohexene (31/33 kJ mol−1), the free energy difference between Ch+(eq,C2) and Chene(12) + H+ is decreased to be 20 kJ mol−1. This discrepancy can be ascribed to the difference between Ch+(ax) and Ch+(eq) isomers. In the protonation of cyclohexene/deprotonation of cyclohexylium and positional isomerization of cyclohexene, the attached proton and the detached proton are located at the same side of cyclohexene. Only the carbenium ion, Ch+(ax), is involved. In the skeletal isomerization of cyclohexene, the attached proton and the following forming C2–C4 bond are on both sides of cyclohexene. Here, Ch+(eq) is involved. The energy difference between the two isomers of the cyclohexylium cation was estimated to be 8 kJ mol−1 in the vacuum,46 in which Ch+(eq) is more stable. Therefore, this energy difference between two isomers would partially compensate the free energy difference. Free energy barriers are calculated to be 45 and 25 kJ mol−1 for the protonation of cyclohexene and deprotonation of Ch+(eq) in the ring contraction process.
The second stage is the transformation from Ch+(eq,C2) to Cp+(C4) (dcn,31 = 0.87, dcn,32 = 0.50, dcn,33 = 0.74, and dcn,34 = −0.75), which is the intermediate state. Further geometrical optimization validates the stability of Cp+(C4). As shown in Fig. 10e, it features a methyl group sandwiched between a cyclopentene fragment and the deprotonated BAS. The methyl group is attached to C2 and stays in close proximity to C4. Transformation from Ch+(eq,C2) to Cp+(C4) experiences a PCP transition state increasing the free energy as high as 67 kJ mol−1. The calculated free energy barriers are 101 kJ mol−1 and 34 kJ mol−1 for the forward and backward processes, respectively. This transformation involves one carbon transfer process (formation of C2–C4 bonds and breakage of C3–C4 bonds) and one hydride transfer process (formation of C3–H41 bonds and breakage of C4–H41 bonds). In order to examine the correlation of two transfer processes, the free energy surface, characterizing the transformation from Ch+(eq,C2) to Cp+(C4) spanned on CVs, dcn,32 and dcn,33, has been drawn. As shown in Fig. 9b, the pathway initially moves from (dcn,32 = −0.49, dcn,33 = −0.70) to ca. (dcn,32 = 0.16, dcn,33 = −0.46) along the horizontal axis, dcn,32, corresponding to the carbon transfer. Then, the pathway moves from ca. (dcn,32 = 0.16, dcn,33 = −0.46) to (dcn,32 = 0.49, dcn,33 = 0.71) along the vertical axis, dcn,33, corresponding the hydride transfer. As a result, the transformation from Ch+(eq,C2) to Cp+(C4) undergoes a hydride transfer following a carbon transfer process. The free energy barrier in this stage appears in the hydride transfer process.
The third stage, the transformation from Cp+(C4) to Cp+(C2), involves another hydride transfer process (C4 → C2). The calculated free energy difference is 51 kJ mol−1. Free energy barriers of 47 kJ mol−1 and 98 kJ mol−1 are required to overcome the forward and backward processes, respectively. The highest free energy barrier in the ring contraction process appears in this stage.
The last stage is a deprotonation process of the methylcyclopentylium cation, which involves the transformation from Cp+(C2) to the local minimum Cpene(12) + H+ (dcn,31 = −0.68, dcn,32 = 0.57, dcn,33 = 0.81, and dcn,34 = 0.72). The calculated free energy difference is 21 kJ mol−1. The carbenium ion, Cp+(C2), is a more stable species. This is coincident with the previous result based on the analysis of unbiased molecular dynamics trajectories. For the deprotonation process of Cp+(C2) and the protonation of Cpene(12) + H+, free energy barriers of 48 kJ mol−1 and 27 kJ mol−1 are required to overcome, respectively.
Starting with a neutral cyclohexene, the free energy barrier for the skeletal isomerization process is 134 kJ mol−1, which is higher than the barrier of 114 kJ mol−1 for the case starting with the positively charged cyclohexyl cation. In this work, the ring contraction process in the BEA zeolite pore proceeds via a protonated cyclopropane (PCP)-like intermediate state. While starting with an alkoxide species, two ring-contraction pathways through the PCP-like intermediate and the neutral bicycle[3.1.0]hexane intermediate in the MFI zeolite are proposed.20 The activation barriers for the two reaction routes are 115 and 65 kJ mol−1, respectively. Hu et al. reported a barrier of 108 kJ mol−1 for the contraction of cyclohexene encapsulated in MFI starting with cyclohexylium through bicycle[3.1.0]hexane.47 We have also noted that starting with ethylcyclohexylium, the lower activation barrier ranging from 50 to 77 kJ mol−1 for the ring contraction process of ethylcyclohexene within the EUO zeolite via a PCP-like intermediate was reported by Chizallet's group.17 Therefore, it can be presumed that the activation barrier for the ring contraction of cyclo-alkene is dependent on the location of the Brønsted acid site and the topology of the zeolite framework. The underlying mechanism for the ring contraction of cyclohexene is still an open question. The contraction via the PCP-like intermediate involves only one protonation process. The contraction via bicycle[3.1.0] requires two protonation and one deprotonation processes. Only a relatively simple pathway was examined in this work.
To characterize the strengths of C–C, C–H and O–H bonds in the ring contraction process of cyclohexene, the average lengths of four C–C bonds, viz., dC1–C2, dC2–C3, dC3–C4, and dC2–C4, five C–H bonds, viz., dC1–HB, dC3–H41, dC4–H41, dC2–H21, and dC4–H21, and one O–H bond, viz., dOAl1–HB, have been measured. The evolution of these distances along the least free energy pathway has been shown in Fig. 11. In the first stage (0.00 < s3 < 1.00), the decreasing dC1–HB and the increasing dOAl1–HB curves indicate the migration of the acidic proton HB of the BAS from OAl1 to C1. Curves of dC1–HB and dOAl1–HB intersect in the range 0.55 < s3 < 0.65. The location of the free energy barrier (pink vertical line) is within the intersection range. On the left side of the pink vertical line, four C–C distances fluctuate around their initial values. After the intersection range, dC1–C2 and dC2–C3 are converged to 1.45 Å from 1.35 Å and 1.50 Å, respectively. The cyclohexylium cation is formed. The C2 atom is positively charged. The C1–C2 bond and the C2–C3 bond are almost identical. dC2–C4 and dC3–C4 are around ca. 2.50 Å and 1.60 Å in the entire stage. This can be ascribed to the C4 atom that was not involved in the protonation of the C1C2 double bond.
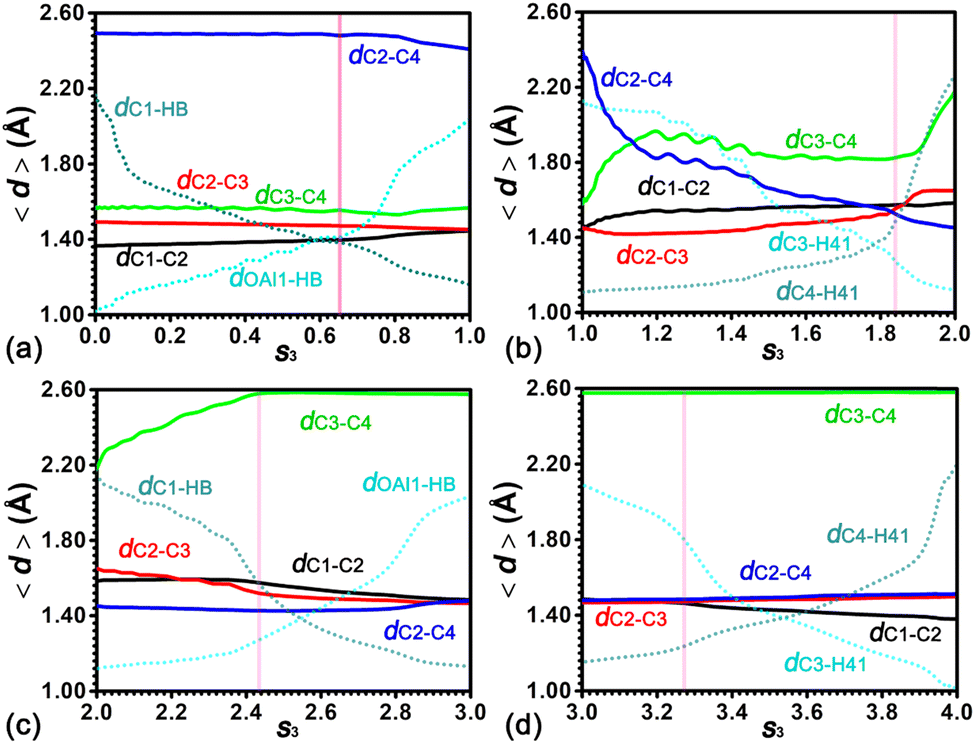 | ||
| Fig. 11 Evolution of C–C, C–O and C–H distances in the ring contraction of cyclohexene assisted by the BAS. The pink vertical line denotes the position of the free energy barrier in each stage. | ||
In the second stage (1.00 < s3 < 2.00), the most notable feature is the decrease of dC2–C4 in the entire stage. The curve of dC2–C4 intersects with the curve of dC3–C4 and dC2–C3 at the points s3 = 1.14 and s3 = 1.84, respectively. In the initial range (1.00 < s3 < 1.14), the dC2–C4 distance decreases to 1.90 Å. The dC3–C4 distance is stretched from 1.57 Å to 1.90 Å. At the intersection point (s3 = 1.14), a PCP fragment is formed. In the following range (1.14 < s3 < 1.84), dC2–C4 decreases from 1.90 Å to 1.53 Å, indicating the further strengthening of the C2–C4 bond. dC3–C4 varies in the range from 1.82 Å to 1.96 Å. The small increase of dC2–C3 from 1.42 Å to 1.53 Å reveals that the C2–C3 bond is slightly weakened. Within the same range, curves of dC3–H41 and dC4–H41 cross. The hydride, H41, migrates from C4 to C3. The intersection point (s3 = 1.84) is coincident with the location of the free energy barrier in the second stage. In the final range (1.84 < s3 < 2.00), dC2–C4 decreases from 1.53 Å to 1.45 Å, which is the representative length of the C–C bond involving the positively charged carbon atom within carbenium. dC1–C2 and dC2–C3 increase to 1.58 Å and 1.64 Å, respectively, featuring the neutral C–C single bond. dC3–C4 increases to 2.17 Å from 1.82 Å, revealing the breakage of the C3–C4 bond within the three-membered ring PCP.
In the third stage (2.00 < s3 < 3.00), dC3–C4 increases from 2.17 Å to 2.58 Å, suggesting that the favorable interaction between C3 and C4 completely disappears. In the following range, the same distance fluctuates around 2.58 Å. The turning point, s3 = 2.43, is coincident with the location of the free energy barrier in this stage. In the second range (2.43 < s3 < 3.00), curves of dC2–H21 and dC4–H21 cross, indicating the migration of the hydride, H21, from C2 to C4. Curves of dC1–C2, dC2–C3 and dC2–C4 converge to the same length (1.48 Å) featuring the C–C bond involving the tertiary carbenium cation.
In the final stage (3.00 < s3 < 4.00), the location of the free energy barrier is at s3 = 3.27. The point splits the stage into two ranges. In the first range (3.00 < s3 < 3.27), four C–C distances fluctuate around its initial values. dOAl1–HB and dC1–HB decrease and increase, respectively. In the second range (3.27 < s3 < 4.00), curves of dOAl1–HB and dC1–HB cross, indicating the migration of the proton, HB, from C1 to OAl1. The positively charged methylcyclopentylium is deprotonated. Meanwhile, dC1–C2, dC2–C3 and dC2–C4 diverge. dC1–C2 decreases to 1.38 Å, featuring the formation of the CC double bond. dC3–C4 fluctuates around its initial value in the entire stage.
4. Conclusions
In the present work, the underlying mechanisms for conformational isomerization, protonation/deprotonation, positional isomerization, and skeletal isomerization of cyclohexene have been elucidated using AIMD simulations combined with the enhanced sampling technique. With additional unbiased AIMD simulations, the attractive interaction of the CC double bond of cyclohexene with the BAS restrains the orientation of cyclohexene. For methylcyclopentene, the positively charged tertiary carbenium cation is the stable species. There is no restraint on the orientation of the methylcyclopentylium cation. By drawing the free energy surface spanned by Cremer–Pople coordinates, the most favored conformation of cyclohexene at the BAS has been quantitatively determined as the half-chair conformers. Free energy landscapes for protonation/deprotonation and the positional isomerization of cyclohexene are mapped out. The calculated free energy barriers are 44 kJ mol−1 and 11 kJ mol−1 for the protonation of cyclohexene and deprotonation of the cyclohexylium cation, respectively. While the free energy barrier for the 1,2-hydride transfer is estimated to be 19 kJ mol−1. For the carbenium ion, the deprotonation process is more feasible than the 1,2-hydride transfer. Positional isomerization of the CC double bond within cyclohexene can be considered as the combination of one protonation process (at C1) and one deprotonation process (at C3). Free energy barriers are calculated to be 40 kJ mol−1 and 9 kJ mol−1, respectively. Geometrical analysis confirms the transformation from the CC double bond to the C–C single bond (or vice versa) for the C1–C2 (or C2–C3) bond.
Catalyzed by the acidic zeolite, the neutral cyclohexene is transformed into a tertiary carbenium cation, methylcyclopentylium, via a PCP transition state. The ring contraction of cyclohexene involves four stages containing one free energy barrier in each stage. The first stage is the protonation of cyclohexene. The free energy barrier is in the intersection range of curves for dHB–C1 and dHB–OAl1. The PCP transition state composed of C2, C3 and C4 atoms is formed in the second stage. Among dC2–C3, dC2–C4, and dC3–C4, dC2–C4 decreases from the longest distance to the shortest one. In the just point where dC2–C4 becomes the shortest bond, the free energy barrier emerges. In the third stage, dC3–C4 increases to 2.58 Å and fluctuates in the following range. The turning point is coincident with the free energy barrier of 134 kJ mol−1. Deprotonation of methylcyclopentylium is the fourth stage. Within the tertiary carbenium cation, dC1–C2, dC2–C3, and dC2–C4 share the same length. The location of the free energy barrier is the just point where curves of three distances start to diverge. The present contribution sheds new insights into the isomerization of cycloalkenes in H-form zeolites by providing the fundamental understanding of the formation, stability and dynamic nature of the carbenium ion involved in zeolite acid-catalyzed reactions such as alkylation, hydrocarbon cracking, ring opening/closing, and C–H activation.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 91961119 and 21603160). This work is also financially supported by the SINOPEC Dalian Research Institute of Petroleum and Petrochemicals.References
- B. Browning, P. Alvarez, T. Jansen, M. Lacroix, C. Geantet and M. Tayakout-Fayolle, A Review of Thermal Cracking, Hydrocracking, and Slurry Phase Hydroconversion Kinetic Parameters in Lumped Models for Upgrading Heavy Oils, Energy Fuels, 2021, 35, 15360–15380 CrossRef CAS.
- Y. Wang, C. Wang, L. Wang, L. Wang and F.-S. Xiao, Zeolite Fixed Metal Nanoparticles: New Perspective in Catalysis, Acc. Chem. Res., 2021, 54, 2579–2590 CrossRef CAS PubMed.
- J. J. Molloy, T. Morack and R. Gilmour, Positional and Geometrical Isomerisation of Alkenes: The Pinnacle of Atom Economy, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS PubMed.
- W. Chen, X. Yi, Z. Liu, X. Tang and A. Zheng, Carbocation chemistry confined in zeolites: spectroscopic and theoretical characterizations, Chem. Soc. Rev., 2022, 51, 4337–4385 RSC.
- H. Xu, Z. Li, S. Meng, J. Jarvis and H. Song, Highly Selective Skeletal Isomerization of Cyclohexene over Zeolite-Based Catalysts for High-Purity Methylcyclopentene Production, Commun. Chem., 2021, 4, 34 CrossRef CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Recent Trends and Fundamental Insights in the Methanol-to-Hydrocarbons Process, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- Z. Zhao, H. Shi, C. Wan, M. Y. Hu, Y. Liu, D. Mei, D. M. Camaioni, J. Z. Hu and J. A. Lercher, Mechanism of Phenol Alkylation in Zeolite H-BEA Using In Situ Solid-State NMR Spectroscopy, J. Am. Chem. Soc., 2017, 139, 9178–9185 CrossRef CAS PubMed.
- A. D. Chowdhury, K. Houben, G. T. Whiting, S.-H. Chung, M. Baldus and B. M. Weckhuysen, Electrophilic Aromatic Substitution over Zeolites Generates Wheland-Type Reaction Intermediates, Nat. Catal., 2018, 1, 23–31 CrossRef CAS.
- F. Liu, K. Huang, A. Zheng, F.-S. Xiao and S. Dai, Hydrophobic Solid Acids and Their Catalytic Applications in Green and Sustainable Chemistry, ACS Catal., 2018, 8, 372–391 CrossRef CAS.
- C. Liu, R. A. van Santen, A. Poursaeidesfahani, T. J. H. Vlugt, E. A. Pidko and E. J. M. Hensen, Hydride Transfer versus Deprotonation Kinetics in the Isobutane–Propene Alkylation Reaction: A Computational Study, ACS Catal., 2017, 7, 8613–8627 CrossRef CAS PubMed.
- A. Corma, P. J. Miguel and A. V. Orchillés, Product Selectivity Effects During Cracking of Alkanes at Very Short and Longer Times on Stream, Appl. Catal., A, 1996, 138, 57–73 CrossRef CAS.
- D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- X. Yi, Y.-K. Peng, W. Chen, Z. Liu and A. Zheng, Surface Fingerprinting of Faceted Metal Oxides and Porous Zeolite Catalysts by Probe-Assisted Solid-State NMR Approaches, Acc. Chem. Res., 2021, 54, 2421–2433 CrossRef CAS PubMed.
- A. Zheng, S. Li, S.-B. Liu and F. Deng, Acidic Properties and Structure–Activity Correlations of Solid Acid Catalysts Revealed by Solid-State NMR Spectroscopy, Acc. Chem. Res., 2016, 49, 655–663 CrossRef CAS PubMed.
- F. Chen, M. Shetty, M. Wang, H. Shi, Y. Liu, D. M. Camaioni, O. Y. Gutiérrez and J. A. Lercher, Differences in Mechanism and Rate of Zeolite-Catalyzed Cyclohexanol Dehydration in Apolar and Aqueous Phase, ACS Catal., 2021, 11, 2879–2888 CrossRef CAS.
- J. Rey, P. Raybaud, C. Chizallet and T. Bučko, Competition of Secondary versus Tertiary Carbenium Routes for the Type B Isomerization of Alkenes over Acid Zeolites Quantified by ab Initio Molecular Dynamics Simulations, ACS Catal., 2019, 9, 9813–9828 CrossRef CAS.
- E. Gutierrez-Acebo, J. Rey, C. Bouchy, Y. Schuurman and C. Chizallet, Location of the Active Sites for Ethylcyclohexane Hydroisomerization by Ring Contraction and Expansion in the EUO Zeolitic Framework, ACS Catal., 2019, 9, 1692–1704 CrossRef CAS.
- K. Cheng, L. I. van der Wal, H. Yoshida, J. Oenema, J. Harmel, Z. Zhang, G. Sunley, J. Zečević and K. P. de Jong, Impact of the Spatial Organization of Bifunctional Metal–Zeolite Catalysts on the Hydroisomerization of Light Alkanes, Angew. Chem., Int. Ed., 2020, 59, 3592–3600 CrossRef CAS PubMed.
- J. Kim, S. W. Han, J.-C. Kim and R. Ryoo, Supporting Nickel To Replace Platinum on Zeolite Nanosponges for Catalytic Hydroisomerization of n-Dodecane, ACS Catal., 2018, 8, 10545–10554 CrossRef CAS.
- Y. V. Joshi and K. T. Thomson, Embedded Cluster (QM/MM) Investigation of C6 Diene Cyclization in HZSM-5, J. Catal., 2005, 230, 440–463 CrossRef CAS.
- P. Cnudde, K. De Wispelaere, L. Vanduyfhuys, R. Demuynck, J. Van der Mynsbrugge, M. Waroquier and V. Van Speybroeck, How Chain Length and Branching Influence the Alkene Cracking Reactivity on H-ZSM-5, ACS Catal., 2018, 8, 9579–9595 CrossRef CAS PubMed.
- J.-M. Schweitzer, J. Rey, C. Bignaud, T. Bučko, P. Raybaud, M. Moscovici-Mirande, F. Portejoie, C. James, C. Bouchy and C. Chizallet, Multiscale Modeling as a Tool for the Prediction of Catalytic Performances: The Case of n-Heptane Hydroconversion in a Large-Pore Zeolite, ACS Catal., 2022, 12, 1068–1081 CrossRef CAS.
- C. Baerlocher and L. B. McCuskerDatabase of Zeolite Structures. https://www.iza-structure.org/databases/..
- T. D. Kühne, et al., CP2K: An Electronic Structure and Molecular Dynamics Software Package - Quickstep: Efficient and Accurate Electronic Structure Calculations, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, cp2k: Atomistic Simulations of Condensed Matter Systems, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- G. Lippert, J. Hutter and M. Parrinello, The Gaussian and Augmented-Plane-Wave Density Functional Method for ab initio Molecular Dynamics Simulations, Theor. Chem. Acc., 1999, 103, 124–140 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Goedecker, M. Teter and J. Hutter, Separable Dual-Space Gaussian Pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Nosé, A Unified Formulation of the Constant Temperature Molecular Dynamics Methods, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695–1697 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- A. Laio and M. Parrinello, Escaping Free-Energy Minima, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562 CrossRef CAS PubMed.
- A. Barducci, G. Bussi and M. Parrinello, Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- M. Bonomi, et al., Promoting Transparency and Reproducibility in Enhanced Molecular Simulations, Nat. Methods, 2019, 16, 670–673 CrossRef PubMed.
- M. Bonomi, et al., PLUMED: A Portable Plugin for Free-Energy Calculations with Molecular Dynamics, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- D. Cremer and J. A. Pople, General Definition of Ring Puckering Coordinates, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical Sampling through Velocity Rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Moradi, G. Enkavi and E. Tajkhorshid, Atomic-Level Characterization of Transport Cycle Thermodynamics in the Glycerol-3-phosphate:Phosphate Antiporter, Nat. Commun., 2015, 6, 8393 CrossRef CAS PubMed.
- D. Branduardi, F. L. Gervasio and M. Parrinello, From A to B in Free Energy Space, J. Chem. Phys., 2007, 126, 054103 CrossRef PubMed.
- P. Liu, W. Hao and D. Mei, Understanding the Effects of Water Molecules on Cyclohexanol Dehydration over Zeolitic Acid Sites, J. Phys. Chem. C, 2021, 125, 15283–15291 CrossRef CAS.
- X. Qian and X. Wei, Glucose Isomerization to Fructose from ab initio Molecular Dynamics Simulations, J. Phys. Chem. B, 2012, 116, 10898–10904 CrossRef CAS PubMed.
- Y.-P. Li, M. Head-Gordon and A. T. Bell, Analysis of the Reaction Mechanism and Catalytic Activity of Metal-Substituted Beta Zeolite for the Isomerization of Glucose to Fructose, ACS Catal., 2014, 4, 1537–1545 CrossRef CAS.
- D. M. Brouwer and H. Hogeveen, Electrophilic Substitutions at Alkanes and in Alkylcarbonium Ions, Progress in Physical Organic Chemistry, 1972, pp. 179–240 Search PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 95th edn, CRC Press, 2014–2015 Search PubMed.
- I. V. Alabugin and M. Manoharan, Effect of Double-Hyperconjugation on the Apparent Donor Ability of σ-Bonds:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Insights from the Relative Stability of δ-Substituted Cyclohexyl Cations, J. Org. Chem., 2004, 69, 9011–9024 CrossRef CAS PubMed.
Insights from the Relative Stability of δ-Substituted Cyclohexyl Cations, J. Org. Chem., 2004, 69, 9011–9024 CrossRef CAS PubMed. - M. Hu, C. Wang, X. Gao, Y. Chu, G. Qi, Q. Wang, G. Xu, J. Xu and F. Deng, Establishing a Link Between the Dual Cycles in Methanol-to-Olefins Conversion on H-ZSM-5: Aromatization of Cycloalkenes, ACS Catal., 2020, 10, 4299–4305 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed parameters for coordination numbers. An example for the cp2k input file in the AIMD simulations. Plumed input file for the conformational isomerization of cyclohexene, the protonation of cyclohexene/deprotonation of cyclohexylium, the positional isomerization of alkene within cyclohexene, and the ring contraction of cyclohexene. See DOI: https://doi.org/10.1039/d2cp02310e |
| This journal is © the Owner Societies 2022 |