
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A powder XRD, solid state NMR and calorimetric study of the phase evolution in mechanochemically synthesized dual cation (Csx(CH3NH3)1−x)PbX3 lead halide perovskite systems†‡
Sai S. H.
Dintakurti
 ab,
David
Walker
a,
Tobias A.
Bird
c,
Yanan
Fang
d,
Tim
White
d and
John V.
Hanna
*ad
ab,
David
Walker
a,
Tobias A.
Bird
c,
Yanan
Fang
d,
Tim
White
d and
John V.
Hanna
*ad
aDepartment of Physics, University of Warwick, Coventry, West Midlands CV4 7AL, UK. E-mail: j.v.hanna@warwick.ac.uk
bInterdisciplinary Graduate School, Nanyang Technological University, Singapore 639798, Singapore
cDepartment of Chemistry, University of Warwick, Coventry, West Midlands CV4 7AL, UK
dSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore
First published on 19th July 2022
Abstract
Methylammonium (MA+) lead halide perovskites (MAPbX3) have been widely investigated for photovoltaic applications, with the addition of Cs improving structural and thermal stability. This study reports the complete A site miscibility of Cs+ and MA+ cations in the lead chloride and lead bromide perovskites with nominal stoichiometric formulae (CsxMA1−x)Pb(Cl/Br)3 (x = 0, 0.13, 0.25, 0.37, 0.50, 0.63, 0.75, 0.87, 1). These suites of materials were synthesized mechanochemically as a simple, cost-effective synthesis technique to produce highly ordered, single phase particles. In contrast to previous studies using conventional synthetic routes that have reported significant solubility gaps, this solvent-free approach induces complete miscibility within the dual cation Cs+/MA+ system, with the resultant structures exhibiting high short-range and long-range atomic ordering across the entire compositional range that are devoid of solvent inclusions and disorder. The subtle structural evolution from cubic to orthorhombic symmetry reflecting PbX6 octahedral tilting was studied using complementary high resolution TEM, powder XRD, multinuclear 133Cs/207Pb/1H MAS NMR, DSC, XPS and UV/vis approaches. The phase purity and exceptional structural order were reflected in the very high resolution HRTEM images presented from particles with crystallite sizes in the ∼80–170 nm range, and the stability and long lifetimes of the Br series (10–20 min) and the Cl series (∼30 s–1 min) under the 200 kV/146 μA e− beam. Rietveld refinements associated with the room temperature PXRD study demonstrated that each system converged towards single phase compositions that were very close to the intended target stoichiometries, thus indicating the complete miscibility within these dual cation Cs+/MA+ solid solution systems. The multinuclear MAS NMR data showed a distinct sensitivity to the changing solid solution compositions across the MAPbX3–CsPbX3 partition. In particular, the 133Cs shifts demonstrated a sensitivity to the cubic–orthorhombic phase transition while the 133Cs T1s exhibited a pronounced sensitivity to the variable Cs+ cation mobility across the compositional range. Variable temperature PXRD studies facilitated the production of phase diagrams mapping the Cs+/MA+ compositional space for the (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 solid solution series, while Tauc plots of the UV/vis data exhibited reducing bandgaps with increasing MA+ incorporation through ranges of cubic phases where octahedral tilting was absent.
Introduction
Organic–inorganic hybrid Lead Halide Perovskites (LHPs) have been widely investigated for photovoltaic applications resulting in solar cells with a record power conversion efficiency (PCE) of >25%,1,2 and light emitting diodes (LEDs) with an external quantum efficiency (EQE) reaching 23.4%.3 The prototype material for solar cells is methyl ammonium lead iodide (CH3NH3PbI3). The key advantages of this class of materials are solution processability, inexpensive starting materials, good hole and electron conductivity and a high tolerance to defects.4 Apart from solar cells and light emitting diodes LEDs, LHPs show promise as semiconductor lasers with an ultra-low threshold as lasing media.5 Moisture and thermal sensitivity remain major challenges to the lifetime of perovskite photovoltaic devices and have delayed widespread adoption. Multiple strategies such as encapsulation, polymer coatings, fluoride additives and the incorporation of hydrophobic cations can inhibit moisture sensitivity.6–8 Saliba et al. were the first to report the advantages of a Cs additive in solar cell materials by using CsI to enhance the structural and thermal stability of photovoltaic devices; this produced smoother and more reproducible films and superior phase stability and moisture resistance.9 The presence of Cs+ cation incorporation in perovskite films was indirectly observed via changes in the X-ray diffraction (XRD) data (and the measured lattice parameters) and increases in the material band gap, however the short-range nature of Cs+ and Rb+ incorporation in lead halide perovskite powders was unambiguously identified using solid state MAS NMR techniques.10,11LHP nanoparticles have been successfully employed to fabricate the active layer in LEDs and are widely investigated for better efficiency and stability of these devices.12 This class of materials show excellent tunability of emission properties when prepared from facile, yet robust, synthetic methods employing ultrasonic mixing, hot injection and anti-solvent addition for low cost photovoltaics. Organic salts such as methylammonium (MA) and formamidinium (FA) are hygroscopic and have poor thermal stability as they decompose above 180 °C. Partial replacement of organic cations with Cs+ improves both moisture and thermal stability. Zhang et al. reported the then brightest and most efficient LEDs (EQE ∼ 10.4%) having an active layer composition of Cs0.87MA0.13PbBr3 demonstrating that the mixed cation varieties have better optoelectronic properties than single A site ion equivalents.13 Although the reported composition is precise, it was indirectly estimated from the valence band edge shift. Alternatively, others have assumed the precursor ratio as the nominal and actual composition.14 Without direct analysis of the phase purity and composition, sample reproducibility based on target nominal compositions alone can be difficult as experimental conditions can vary subtly between syntheses. This study demonstrates that the solid state NMR technique is a reliable, non-destructive method of analysis to ensure the targeted composition was obtained and verifying phase purity.
Optimising precipitation parameters including precursor ratios realized composition-tunable and morphology-controlled nanoparticles with high photoluminescence quantum yield (PLQY) of >90%.13 The feasibility of targeted compositions can be limited by the relative solubility of precursors, especially that of Cs salts in the common solvents dimethylformamide (DMF) or dimethylsulfoxide (DMSO). The low solubility of Cs salts in DMF restricts the quantities that can be added to the precursor solution, limiting the final Cs content to ∼16 atom%. Alternatively, thermal annealing has successfully synthesised many inorganic halide perovskites with large grain size and precise compositions, including those of mixed halide compositions.15 This involved heating the precursor salts to high temperatures in a sealed ampoule, followed by a slow cooling. The temperatures needed to achieve homogeneity are close to the melting points of the precursor salts (>400 °C); consequently, this approach is unsuitable for organic–inorganic hybrid perovskite systems containing MA+ and FA+ cations that decompose above 200 °C. Hence, low temperature, solvent-free approaches are far more advantageous for these syntheses. Karmakar et al. explored the mechanochemical fabrication of limited suites hybrid lead and tin halide perovskite systems to achieve better compositional control.16–19 Other studies by Mozur et al. attempted hand grinding followed by thermal annealing for 5 days at 150 °C for the synthesis of mixed cation hybrid perovskites; however, this was only partially successful as Cs+ cation incorporation levels of >40 mol% could not be achieved.20
This study demonstrates that mechanochemical synthesis techniques represent a simple, cost-effective approach for the production of wide-ranging compositional suites of dual-cation lead halide perovskites of nominal stoichiometry (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 (x = 0.13, 0.25, 0.37, 0.5, 0.63, 0.75, 0.87). Mechanochemical synthesis routes avoid the use of organic solvents (thus representing a more environmentally compatible option) and facilitate ultra-clean reactions that proceed to completeness subsequently rendering homogeneous and highly-ordered products.21–24 Mechanochemical reaction syntheses can be achieved using hand grinding methods (using mortar and pestle),21–24 however more recent studies have reported that improvements in reaction efficiency and time can be achieved using ball milling techniques.16–19 Ball milling efficiently combines the effects of mixing and thermal annealing by exploiting the conversion of kinetic energy to heat at the precise point of reaction, thus leading to extremely high yields and homogeneity.21–24 Through the use of characterisation techniques such as powder XRD, high resolution TEM (HRTEM) and multinuclear 133Cs/207Pb/1H MAS NMR, this study aims to demonstrate the success of mechanochemical ‘alloying’ to achieve complete miscibility within these hybrid perovskites series. The exceptional quality, single phase reaction products permit the unambiguous observation of the phase evolution from cubic MA-lead halides to lower symmetry hettotypes to be demonstrated.
Experimental
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. CsPbCl3 and CsPbBr3 were prepared from 0.5 M precursor salt solution in DMSO. The perovskite solutions passed through a 0.2 μm PVDF filter and heated to 80 °C. The crystalline precipitates were washed with diethyl ether and dried at 60 °C for 20 minutes then powdered prior to synthesis of mixed cation samples.
:1) mixture. CsPbCl3 and CsPbBr3 were prepared from 0.5 M precursor salt solution in DMSO. The perovskite solutions passed through a 0.2 μm PVDF filter and heated to 80 °C. The crystalline precipitates were washed with diethyl ether and dried at 60 °C for 20 minutes then powdered prior to synthesis of mixed cation samples.
 | ||
| Fig. 1 A comparison of the hand grinding (HG) and ball milling (BM) mechanochemical grinding procedures for the Cs0.5MA0.5PbCl3 sample preparation analysed by (a) 207Pb MAS NMR, (b) 133Cs MAS NMR, and (c) powder XRD techniques. These comparisons demonstrate that the ball milling route achieves complete solid solution character without the presence of impurity phases in approximately half the time that the hand grinding technique achieves. | ||
Laboratory source and synchrotron powder X-ray diffraction
High resolution laboratory source powder XRD (PXRD) data were measured at room temperature for the (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 series using a Bruker D8 Advance diffractometer fitted with a Cu Kα X-ray tube (λ = 1.54187 Å) operating at 40 kV and 40 mA. Each sample was mounted in a silicon single crystal zero background sample holder which was rotated at 40 rpm in the beam. The data was measured over the 10°–100° 2θ range using a 0.02° step size and a time duration of 1.5 s per step. Experimental fitting of the data was performed using TOPAS 6 employing a fundamental parameter approach with a simple axial model. The background was modelled by a third order Chebyshev function. The MA+ cation is known to be dynamically disordered in MAPbCl3 and MAPbBr3 over the Wyckoff position 24m.27–29 For Rietveld refinement of the powder XRD data, MA+ was modelled as a sphere with the same atomic scattering factor as isoelectronic K+ ion. The thermal parameters of all atoms were fixed to 1 as high angle data above 100° were not measured. Only the variable atomic fractional coordinates were refined. Average crystallite sizes were estimated by the Scherrer equation using the Lorentzian component of the diffraction peaks (see Table 3). This was implemented using the ‘crystallite size Lorentzian’ function in TOPAS 6, and it is noted that these values are semi-quantitative as microstrain contributions have not been deconvoluted.Variable temperature, high resolution laboratory source PXRD data were acquired for the (CsxMA1−x)PbCl3 series using a Rigaku Synergy S diffractometer fitted with a Cu Kα X-ray tube (λ = 1.54187 Å) and a large area Hypix-6000E detector. All samples were finely ground and packed in 0.3 mm sealed Mark tubes, rotated in the beam at 40 rpm during data acquisition, and cooled using an Oxford Cryostream Cobra Plus system with each measurement being undertaken over 5 °C intervals. The acquired images were integrated over 4°–66° 2θ range. As the sample absorption correction function was insufficient to account for significant absorption, Pawley refinement of all experimental data was performed using TOPAS 6 utilising pseudo-Voigt peakshapes, with peak and instrumental factors fixed using a LaB6 standard. Sequential batch refinements starting with the lowest temperature measurements, followed by successive higher temperatures, was adopted.
Variable temperature, high resolution synchrotron PXRD data were measured for the corresponding (CsxMA1−x)PbBr3 series at the I11 beamline at Diamond Light Source, UK using a wavelength of 0.82646 Å. A wide-angle Mythen microstrip position sensitive detector (PSD) was used to acquire the data in transmission geometry. All data was measured over the 2°–92° 2θ range with a resolution of 0.004° and an exposure time of 20 s per step. All samples were finely ground and packed in 0.3 mm sealed Mark tubes and rotated in the beam at 40 rpm during the data acquisition. The sample temperature was regulated using an Oxford Cryostream which enabled a heating/cooling rate of 6 °C min−1. For these variable temperature studies, the range of temperatures was limited to ±25 °C around the phase transition temperature as identified by DSC. All Rietveld refinement was undertaken using TOPAS 6 employing pseudo-Voigt peakshapes, with peak and instrumental factors fixed using a Si standard.
High resolution transmission electron microscopy (HRTEM)
Selected (CsxMA1−x)PbX3 specimens were prepared by ultrasonically dispersing a small quantity of powder in absolute acetone and placing a drop of suspension on a holey carbon-coated copper grid. These preparations were air dried. High-resolution transmission electron microscopy (HRTEM) was performed using a JEOL 2100F TEM with a field-emission gun operating at 200 kV and beam current of 146 μA. Samples were orientated with a double tilting holder and the selected area electron diffraction (SAD) patterns recorded. Observation times were limited to a few minutes due to electron amorphization, especially when a more converged beam was used for imaging.Solid state magic-angle-spinning NMR (MAS NMR)
Multinuclear solid state MAS NMR data were acquired at 14.1 T using a Bruker Avance 600WB Avance III spectrometer operating at 1H, 133Cs and 207Pb Larmor frequencies (νo) of 600.13, 78.7 and 125.5 MHz, respectively. Single pulse 133Cs MAS NMR data were obtained a using a Bruker 4 mm HX probe enabling a MAS rate (νr) of 12 kHz for all data acquisition. A CsNO3 0.1 M aqueous solution was used for pulse calibration and referencing. A non-selective (‘solution’) π/2 pulse duration of 6.0 μs was calibrated on solid CsCl, with a selective (‘solids’) π/2 pulse time 1.5 μs being used for acquisition of the I = 7/2 nucleus. A recycle delay of 300 s was used for all 133Cs measurements in the CsxMA1−xPbCl3 and CsxMA1−xPbBr3 (where x = 0.0, 0.13, 0.25, 0.37, 0.5, 0.63, 0.75, 0.87, 1.0) series which represented 5 × T1 to ensure complete relaxation, as determined on the slowest relaxing (CsPbCl3) system. The 133Cs T1 values were determined using a saturation-recovery experiment which implemented 100 pulses in the saturation pulse train with a 50 ms delay between pulses. All 133Cs chemical shifts were calibrated to the IUPAC recommended 0.1 M CsNO3 (in D2O) reference (δ 0.0 ppm) via a secondary solid CsCl reference (δiso 223.2 ppm) (representing a frequency ratio (Ξ) of 133Cs = 13.116142% to the 1H signal from 1% TMS in CDCl3).30 The corresponding 207Pb MAS NMR data were measured using a rotor-synchronized Hahn echo (90°–τ–180°) experiment on a Bruker 4 mm HX probe enabling MAS rate (νr) of 12 kHz. A π/2 pulse with a pulse width of 4 μs was calibrated using solid PbNO3 from which a π/2/π pulse pair of 4 μs/8 μs was used for all measurements. A recycle delay of 30 s was used for all 207Pb experiments in both series which represented 5 × T1 to ensure complete relaxation, as determined on the slowest relaxing (CsPbBr3) system. In similar fashion to the 133Cs measurements, the 207Pb T1 values were determined using a saturation-recovery experiment which implemented 100 pulses in the saturation pulse train with a 50 ms delay between pulses. All 207Pb chemical shifts were calibrated to the IUPAC recommended PbMe4 (neat) reference (δ 0.0 ppm) via a secondary solid PbNO3 reference (δ −3490 ppm) (representing a frequency ratio (Ξ) of 207Pb = 20.920599% to the 1H signal from 1% TMS in CDCl3).30 The 1H MAS NMR data were acquired using single pulse methods using a Bruker 4 mm HX probe enabling MAS rate (νr) of 12 kHz. A π/2 pulse length of 4.0 μs duration was calibrated solid using adamantane and π/4 pulse excitation pulses of 2 μs were sued throughout. A recycle delay of 120 s was used for all 1H measurements within both series which represented 5 × T1 to ensure complete relaxation, as determined on the slowest relaxing (MAPbBr3) system. In similar fashion to the 133Cs and 207Pb measurements above, the 207Pb T1 values were determined using a saturation-recovery experiment which implemented 100 pulses in the saturation pulse train with a 50 ms delay between pulses. All 1H chemical shifts were calibrated to the IUPAC recommended TMS (δ 0.0 ppm) reference via a secondary solid adamantane reference (δ 1.82 ppm).30X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) was performed using a Kratos AXIS Supra spectrometer with a monochromatic Al K-alpha source (15 mA, 15 kV) on solid powder samples pressed onto carbon tape. A 3.1 V bias was applied to neutralise surface charge. Individual core levels were measured with a pass energy of 20 eV.Differential scanning calorimetry
Differential Scanning Calorimetry (DSC) was performed using a Mettler Toledo DSC 1 Stare system. Standard aluminium pans with 40 μl volume were used to load the samples. The ramp rate for both the cooling and heating segments was maintained at 10 °C min−1.UV/Vis spectroscopy
The UV/Vis absorption data were measured on a PerkinElmer Lambda 850 spectrometer equipped with an integrating sphere. The acquired data is presented as Tauc plots with change in absorption coefficient as a function of photon energy. The band gap is estimated by extrapolating the linear change in absorption coefficient.Results and discussion
Mechanochemically prepared sample integrity, high resolution transition electron microscopy (HRTEM)
Ball milling involves a continuous process of particle fracturing, re-forming and annealing during collisions with the milling media. The average particle size reduces gradually to a limit where any further milling can induce amorphization. The presence of these amorphous phases and their compositions cannot be ascertained directly by PXRD, however the high resolution transmission electron microscopy (HRTEM) and solid state NMR techniques provide descriptions of the short range chemical environment about each atomic position rather than descriptions of averaged long range order. HRTEM data presented in Fig. 2 and Fig. S1 (ESI‡) depict very high resolution images of a number of the mechanochemically prepared dual cation lead halide perovskites comprising the (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 (x = 0.0, 0.13, 0.25, 0.37, 0.5, 0.63, 0.75, 0.87, 1.0) suites of samples. Previous attempts at HRTEM of MAPbBr3 only exhibited lattice fringe structure, with no evidence of atomic positions on the particle surface.31,32 This contrasts with the near-atomic resolution imaging demonstrated in Fig. 2 as measured from the (Cs0.50MA0.50)PbBr3, (Cs0.75MA0.25)PbBr3, (Cs0.5MA0.5)PbCl3 and (Cs0.75MA0.25)PbCl3 systems prepared under mechanochemical conditions. The incorporation of the Cs+ cation into the structure coupled with the high purity/high short range order products and the lack of solvent inclusions increases the stability under the electron beam facilitating near atomic resolution information. The (Cs0.5MA0.5)PbBr3 image in Fig. 2a shows equidistant (220) and (202) planes as expected from a [111]cubic zone axis. The Br series exhibits high electron beam stability of up to 10–20 minutes, whereas the analogous Cl series demonstrated a reduced persistence and degrades over a period of ∼30 s–1 min under the beam. In addition, an increased Cs+ cation content reduces the e-beam sensitivity as evidenced by comparisons of Fig. 2a and b (i.e. (Cs0.50MA0.50)PbBr3 and (Cs0.75MA0.25)PbBr3)), and comparisons of Fig. 2c and d (i.e. (Cs0.50MA0.50)PbCl3 and (Cs0.75MA0.25)PbCl3). High e− beam stability of the dual cation Cs+/Ma+ systems in comparison to pure MA+ analogues suggests that these materials may be more stable under high electric fields, which is critical for LED applications where the active layer experiences electric fields on the order of 2000 kV m−1.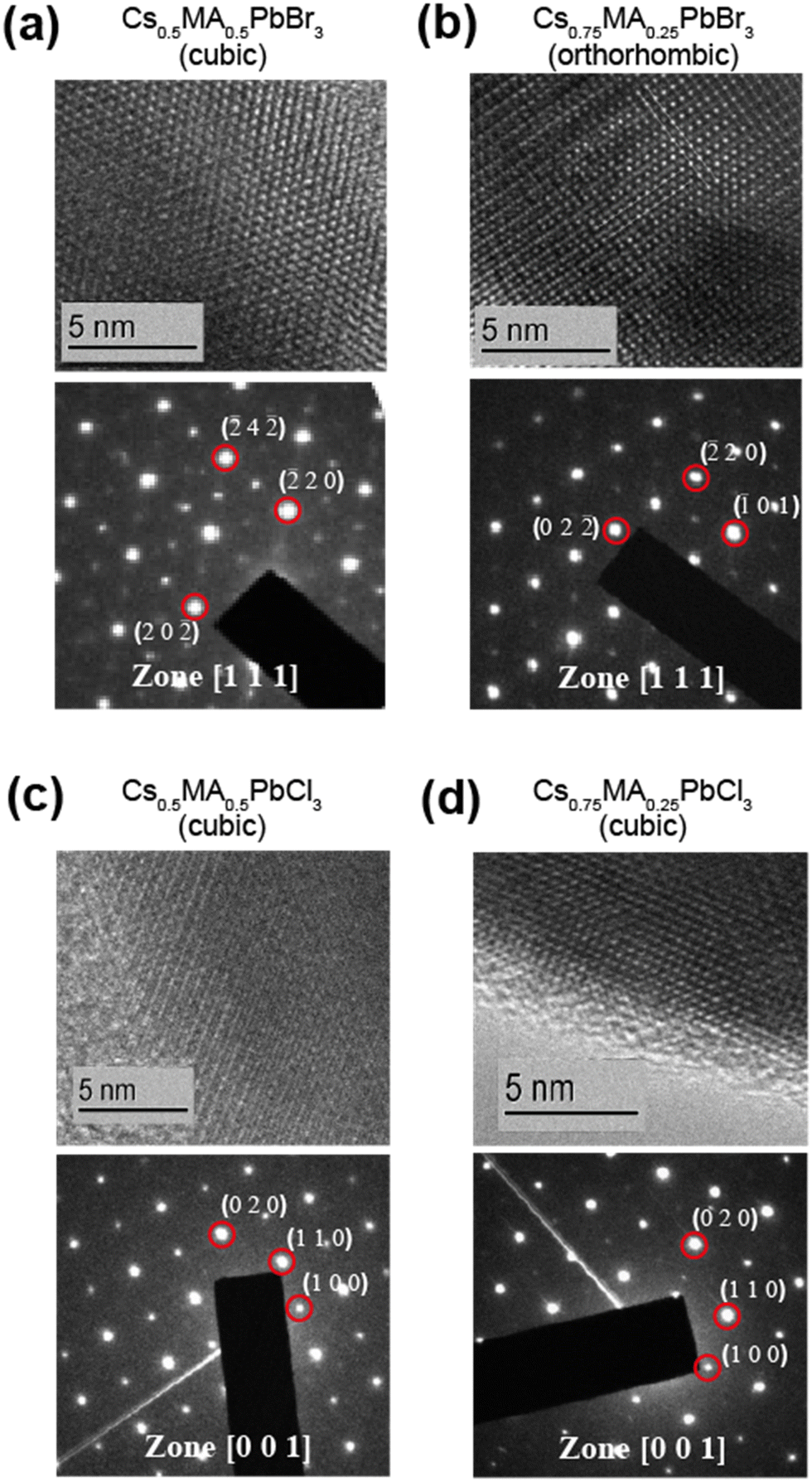 | ||
| Fig. 2 HRTEM images and selected area electron diffraction (SAD) patterns of powder samples prepared from mechanochemical synthesis. The indexed reflections confirm the lattice symmetry of Cs0.75MA0.25PbBr3 sample to be orthorhombic while the others are cubic. These images also indicate the exceptional stability of Cs–MA hybrid perovskite samples under an electron beam in high vacuum conditions. | ||
PXRD and solid state MAS NMR studies of the as-synthesized mechanochemically derived products
The perovskite ABX3 aristotype is cubic Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (space group no. 221) where A occupies the 1a Wyckoff position, B the 1b site and X the 3c site, permitting the crystallochemical formula to be expressed as [A][B][X3] with one formula unit per unit cell (Z = 1). MAPbCl3 and MAPbBr3 satisfy Pmm symmetry; however, because MA+ is a non-centrosymmetric cation it is necessarily disordered over the 24m Wyckoff position (rather than occupying the 1a position) to maintain Oh symmetry.29 The structural topology of perovskites consists of [BX6]4− octahedra that extends three dimensional connectivity by corner sharing such that the aristotype creates A-centred cuboctahedral interstices. As the octahedra are hinged, symmetry is readily modified by cooperative BX6 octahedral tilting that is mediated through varying temperature, pressure or composition. In addition, the ionic radius of MA+ cation (2.17 Å) is significantly larger than that of the Cs+ cation (1.88 Å).33,34
m (space group no. 221) where A occupies the 1a Wyckoff position, B the 1b site and X the 3c site, permitting the crystallochemical formula to be expressed as [A][B][X3] with one formula unit per unit cell (Z = 1). MAPbCl3 and MAPbBr3 satisfy Pmm symmetry; however, because MA+ is a non-centrosymmetric cation it is necessarily disordered over the 24m Wyckoff position (rather than occupying the 1a position) to maintain Oh symmetry.29 The structural topology of perovskites consists of [BX6]4− octahedra that extends three dimensional connectivity by corner sharing such that the aristotype creates A-centred cuboctahedral interstices. As the octahedra are hinged, symmetry is readily modified by cooperative BX6 octahedral tilting that is mediated through varying temperature, pressure or composition. In addition, the ionic radius of MA+ cation (2.17 Å) is significantly larger than that of the Cs+ cation (1.88 Å).33,34
Hettotypes arise when the A cation is too small to reside in an undistorted cuboctahedron, thus requiring a reduction of the coordination number from 12. To satisfy the bond valence criteria, the BX6 octahedra (to a first approximation considered as rigid bodies) tilt about the pseudocubic 100, 010 and 001 axes. Glazer proposed the commonly used notation to describe perovskite tilt systems using the rotation of octahedra in successive layers about each of the orthogonal Cartesian axis.35 In this nomenclature a, b and c represent rotations about the x, y and z axes, respectively, with the sense of rotation denoted by using 0/+/− superscripts. The aristotype in which the octahedra are not tilted is represented as a0a0a0, while in-phase tilts are denoted + and the out-of-phase tilts are denoted −. This approach was reanalysed by Howard et al.; Scheme 1 below illustrates the octahedral tilt systems and resultant lowering of symmetry in accordance with the group–subgroup relationships from their analysis.36 Tilt angles can be calculated from the Rietveld refinement of diffraction data yielding atomic fractional coordinates, bond angles, cell dimensions and polyhedral volumes, although complications can arise when the octahedra distort.37,38 In this respect, it is advantageous to use synchrotron X-ray or neutron diffraction when acquiring data to enhance the observation of weak superlattice peaks, especially as the X-ray scattering is dominated by the Pb atoms. Typical peak splitting observed in CsPbCl3 and CsPbBr3 are provided in Table 1. At room temperature, CsPbCl3 was found to be orthorhombic (Pnma), but upon heating above 37 °C it transforms into a tetragonal (P4/mbm) phase which upon further heating >47 °C, transforms into a cubic (Pmm) structure.39,40 From Scheme 1 and Table 1 these transformations are characterized by octahedral tilting as described by a+b−b− (orthorhombic) and a0a0c+ (tetragonal) tilt systems, respectively.
 | ||
| Scheme 1 Schematic representation of octahedral tilting modes and their group–subgroup relationships. The cubic aristotype (Pmm) transforms into tetragonal P4/mbm or I4/mcm with tilting in one direction while tilts in more than one direction give rise to lower symmetry space group settings. This schematic relationship shows that P4/mbm is an intermediate pathway between Pnma and Pmm. This relationship allows second order phase transformations according to Landau theory. At low temperatures MAPbCl3 transforms into P4/mmm which doesn’t occur in the above relationship confirming that it occurs via octahedral distortion rather than tilting. | ||
| Cubic Pmm |
Tetragonal P4/mbm | Tetragonal I4/mcm | Orthorhombic Cmcm | Pseudo-orthorhombic Pnma |
|---|---|---|---|---|
| a a is cubic unit cell length. | ||||
| a |

|

|
2a, 2a, 2a |

|
| a 0 a 0 a 0 | a 0 a 0 c + | a 0 a 0 c − | a 0 b + c − | a + b − b − |
| 100 | 001 | 002 | 020 | 110 |
| 110 | 110 | 002 | 002 | |
| 200 | ||||
| 110 | 200 | 200 | 022 | 020 |
| 111 | 112 | 200 | 112 | |
| 200 | ||||
| 111 | 201 | 202 | 222 | 202 |
| 022 | ||||
| 200 | 002 | 004 | 040 | 220 |
| 220 | 220 | 004 | 004 | |
| 400 | ||||
Fig. 3 and 4 illustrate the evolution of crystallographic parameters obtained from Rietveld refinement of laboratory PXRD data (see Fig. S3, S4, ESI,‡ and Tables 2, 3) measured from (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 series measured at room temperature. From Table 3 the estimated average crystallite sizes of the samples in these series resulting from mechanochemical synthesis fall within the ∼80–170 nm range. The a and b lattice parameters of CsPbCl3 sample differ from each other by <0.005 Å indicating that the room temperature structure is close to a phase transition. While in the PbCl6 octahedra displacements of the Cl would be characteristic of octahedral tilting, the relatively weak scattering of Cl− compared to Pb2+ weakens the sensitivity to superlattice peak asymmetry and splitting. However, lattice dilation through chemical substitution of Cs+ cations with MA+ is unequivocal and a transition to cubic symmetry, as indicated by the converging a and b lattice parameters (see Fig. 3c) in Cs0.87MA0.13PbCl3. Although (CsxMA1−x)PbCl3 compositions in the range x ≤ 0.87 exist in a cubic structure, the orthorhombic equivalent cell was used for comparison with CsPbCl3. Substitution of Cs+ with the larger MA+ cation causes a predictable monotonic change in the size of the unit cell and cell volume as seen in Fig. 3d. The difference in the X-ray scattering factors of Cs and MA ions causes a monotonic change in the relative intensity of 100 reflection at 15.9° and 110 reflection at 22.2° and permits a compositional refinement of Cs content that shows excellent agreement with the nominal composition (Fig. 2d). These results validate the viability of the mechanochemical synthesis in obtaining homogenous products with targeted compositions.
 | ||
| Fig. 3 High resolution laboratory source PXRD data and structural analyses from the (CsxMA1−x)PbCl3 (x = 0–1) solid solution series highlighting, (a) the measured PXRD data, (b) an expansion of the ∼15–23° 2θ region showing monotonic intensity changes intensity due to different scattering factors of the Cs+ and MA+ cations, (c) variation in the (CsxMA1−x)PbCl3 (x = 0–1) series lattice parameters obtained from Rietveld refinement using an orthorhombic lattice demonstrating that the lattice parameters gradually converge indicating an increase in symmetry, and (d) cell volume and Cs+ occupancy from the Rietveld refinements showing excellent agreement between the targeted (red line) and experimentally obtained compositions produced under mechanochemical conditions. | ||
 | ||
| Fig. 4 High resolution synchrotron PXRD data and structural analyses from the (CsxMA1−x)PbBr3 (x = 0–1) solid solution series highlighting, (a) the measured PXRD data, (b) an expansion of the ∼14–22° 2θ region showing monotonic intensity changes intensity due to different scattering factors of the Cs+ and MA+ cations, (c) variation in the (CsxMA1−x)PbCl3 (x = 0–1) series lattice parameters obtained from Rietveld refinement using an orthorhombic lattice demonstrating that the lattice parameters that gradually converge indicating an increase in symmetry from Cs0.25MAPbBr3 to Cs0.37MA0.63PbBr3, and (d) cell volume and Cs+ occupancy from the Rietveld refinements showing excellent agreement between the targeted (red line) and experimentally obtained compositions produced under mechanochemical conditions. | ||
| Space group | Pnma | a = 8.236(4) Å | b = 8.254(5) Å | c = 11.776(8) Å | R Bragg = 3.2 | B eq |
|---|---|---|---|---|---|---|
| Site | Multiplicity | x | y | z | Occupancy | |
| Cs1 | 4 | 0.990 | 0.971 | 0.25 | 0.89 | 1 |
| MA2 | 4 | 0.990 | 0.971 | 0.25 | 0.11 | 1 |
| Pb3 | 4 | 0.5 | 0.0 | 0.0 | 1 | 1 |
| Br4 | 4 | 0.046 | 0.505 | 0.25 | 1 | 1 |
| Br5 | 8 | 0.793 | 0.205 | 0.025 | 1 | 1 |
m and Pnma space groups, respectively
| Nominal composition | Lattice symmetry | R Bragg | a, b, c (Å) | Refined Cs occupancy (±0.03) | Cell volume | Bond length Pb–Xa (Å) | Bond length Cs/MA–Xb (Å) | Crystallite size L (nm) | Measured tilt angles |
|---|---|---|---|---|---|---|---|---|---|
| a Average bond lengths from the PbX6 octahedra (CN = 6). b Average bond lengths from the cuboctahedral Cs+ or MA+ cation containing interstices (CN = 12). | |||||||||
| (CsxMA1−x)PbBr3 series | |||||||||
| MAPbBr3 | Cubic | 6.7 | 5.9325(7) | 0 | 208.79(9) | 2.966(2) | 5.137(6) | 17(2) | |
| Cs0.13MA0.87 | Cubic | 4.0 | 5.9193(3) | 0.11 | 207.40(4) | 2.959(6) | 5.126(1) | 13(9) | |
| Cs0.25MA0.75 | Cubic | 4.1 | 5.9052(9) | 0.22 | 205.93(2) | 2.952(6) | 5.114(0) | 8(3) | |
| Cs0.37MA0.63 | Cubic | 6.6 | 5.8946(7) | 0.37 | 204.82(9) | 2.947(3) | 5.104(8) | 11(5) | |
| Cs0.50MA0.50 | Cubic | 4.9 | 5.8838(5) | 0.52 | 203.69(8) | 2.941(9) | 5.095(4) | 16(1) | |
| Cs0.63MA0.37 | Cubic | 5.9 | 5.8747(2) | 0.65 | 202.75(1) | 2.937(3) | 5.087(5) | 15(4) | |
| Cs0.75MA0.25 | Orthorhombic | 3.7 | 8.261(6) | 0.75 | 805.79(9) | Equatorial 2.965(7) axial 2.970(0) | Average 4.14(7) | 12(0) | In-phase 159.5° out-of-phase 169.2° |
| 8.272(1) | |||||||||
| 11.790(9) | |||||||||
| Cs0.87MA0.13 | Orthorhombic | 3.5 | 8.236(4) | 0.89 | 800.67(3) | Equatorial 2.959(5) axial 2.960(6) | Average 4.15(3) | 9(6) | In-phase 158.4° out-of-phase 167.8° |
| 8.254(5) | |||||||||
| 11.776(8) | |||||||||
| CsPbBr3 | Orthorhombic | 8.207(3) | 1 | 796.73(3) | Equatorial 2.952(4) axial 2.960(6) | Average 4.08(3) | 9(1) | In-phase 157.2° out-of-phase 165.9° | |
| 8.255(9) | |||||||||
| 11.758(4) | |||||||||
| (CsxMA1−x)PbCl3 series | |||||||||
| MAPbCl3 | Cubic | 12.2 | 5.6849(0) | 0 | 183.72(5) | 2.842(4) | 4.923(1) | 12(9) | |
| Cs0.13MA0.87 | Cubic | 6.8 | 5.6761(9) | 0.14 | 182.88(1) | 2.838(1) | 4.915(6) | 9(5) | |
| Cs0.25MA0.75 | Cubic | 4.6 | 5.6597(6) | 0.23 | 181.29(8) | 2.829(9) | 4.901(4) | 9(6) | |
| Cs0.37MA0.63 | Cubic | 4.9 | 5.6529(1) | 0.34 | 180.64(0) | 2.826(4) | 4.895(4) | 8(4) | |
| Cs0.50MA0.50 | Cubic | 6.9 | 5.6393(3) | 0.46 | 179.34(2) | 2.819(7) | 4.883(6) | 11(6) | |
| Cs0.63MA0.37 | Cubic | 8.2 | 5.6258(6) | 0.62 | 178.06(0) | 2.812(9) | 4.872(0) | 11(5) | |
| Cs0.75MA0.25 | Cubic | 7.5 | 5.6172(0) | 0.77 | 177.23(9) | 2.808(6) | 4.864(4) | 11(8) | |
| Cs0.87MA0.13 | Cubic | 5.3 | 5.6090(7) | 0.89 | 176.47(0) | 2.804(5) | 4.857(4) | 9(4) | |
| CsPbCl3 | Orthorhombic | 6.5 | 7.903(2) | 1 | 703.25(1) | Equatorial 2.812(8) | Average 3.97(8) | 11(7) | |
| 7.908(0) | Axial | ||||||||
| 11.252(3) | 2.833(5) | ||||||||
The room temperature laboratory XRD data is presented in Fig. 4a and b. As evident from Fig. 4c, CsPbBr3 is orthorhombic (Pnma) with the a and b lattice parameters differing by >0.05 Å. This larger difference in comparison to that of CsPbCl3 indicates that the CsPbBr3 structure is further removed from the cubic phase transition. This is also supported by the fact that CsPbBr3 transforms into a cubic phase above 130 °C, which is ∼105 °C above room temperature in comparison to ∼23 °C above room temperature for CsPbCl3.41 The superlattice peaks in the bromide samples show a higher intensity and are easily detected as Br has a higher electron density than Cl. The emergence of these peaks is observed for the bromide series of samples with <25% MA+ composition as the incorporated Cs+ destabilizes the cubic perovskite structure thus tilting the octahedra. From Fig. 4b and Table 1 the CsPbBr3 data exhibits clear peak splitting with the (100) reflection at 15° splitting into (110) and (002) in the orthorhombic setting, and these progressively converging when the composition evolves from Cs0.25MA0.75PbBr3 to Cs0.37MA0.63PbBr3 indicating an additional orthorhombic to cubic phase transition (see Fig. 4c and d). It can be inferred that PbBr6 octahedral tilting is also eliminated to satisfy the change in symmetry. Following the hierarchy of space groups proposed by Howard et al. (see Scheme 1 and Table 1) it can be inferred that during thermal expansion of the orthorhombic CsPbBr3 lattice the formation of a tetragonal intermediate at >88 °C occur; i.e. before transformation into the cubic aristotype. While not explicitly detected as a discrete tetragonal phase transition, this phase may exist over a narrow temperature and compositional range as indicated by the discontinuity in cell volume and lattice parameter variation in Fig. 4c and d, respectively.
Solid state MAS NMR analysis
The 133Cs and 207Pb MAS NMR data measured from the mechanochemically prepared (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 series (x = 0.0, 0.13, 0.25, 0.37, 0.5, 0.63, 0.75, 0.87, 1.0) shown in Fig. 5 and 6. They illustrate the acquired 133Cs MAS NMR and 207Pb MAS NMR spectra (see Fig. 5a and b, and Fig. 6a and b, respectively), the observed 133Cs and 207Pb chemical shift trends (see Fig. 5c and 6c, respectively), and the variation in the characteristic T1 relaxation times (see Fig. 5d and 6d, respectively) as function of composition for each series. | ||
| Fig. 5 The 133Cs MAS NMR study showing, (a) the 133Cs data for the (CsxMA1−x)PbCl3 series, (b) the 133Cs data for the (CsxMA1−x)PbBr3 series, (c) the variation in the 133Cs apparent chemical shifts with MA+ incorporation and their sensitivity to the orthorhombic–cubic phase transition, and (d) the sensitivity of the 133Cs T1s to MA+ incorporation and the pronounced T1 minimum exhibited at the orthorhombic–cubic phase transition. | ||
 | ||
| Fig. 6 The 207Pb MAS NMR study showing, (a) the 207Pb data for the (CsxMA1−x)PbCl3 series, (b) the 207Pb data for the (CsxMA1−x)PbBr3 series, (c) the variation in the 207Pb isotropic chemical shifts with MA+ incorporation and their insensitivity to the orthorhombic–cubic phase transition, and (d) the minimal decrease in the 207Pb T1s with increasing MA+ incorporation associated with increasing Pb–X bond lengths and cell volumes rather than sensitivity to the orthorhombic–cubic phase transition. | ||
The 133Cs MAS NMR spectra shown in Fig. 5a and b exhibit one resonance highlighting the phase purity of the mechanochemically prepared dual cation Cs+/MA+ solid solutions. This result corroborates the high quality HRTEM data presented in Fig. 2. As evidenced by the PXRD data of Fig. 3c and 4c, the lattice parameter (and hence the cubic phase bond length) increases proportionally with increasing mol% MA+ content, which is consistent with the larger ionic radius of the MA+ cation in comparison to that of its Cs+ counterpart. The expanding PbBr6 and PbCl6 frameworks reduce the electronic/covalent contact with the Cs+ positions, subsequently contracting the electron rich Cs+ cation valence electron clouds inducing a comparatively more ionic position with higher electron density. As a consequence, shifts trending to higher shielding (i.e. to lower ppm) are observed (see Fig. 5c). It is interesting to note that the 133Cs chemical shift is linearly proportional to mol% MA+ concentration within the same compositional range. As indicated in Fig. 5c, the cubic–orthorhombic phase transition can be observed as an inflection point in the gradient characterizing the rate of change of the 133Cs chemical shift with mol% MA+ incorporation. This linear δ(133Cs)/mol% MA+ trend can be used to predict the achieved nominal compositions and phase transition regions across these compositional series.
Similarly, the 207Pb MAS NMR spectra shown in Fig. 6a and b also exhibit one resonance mirroring the phase purity indicated by the 133Cs MAS NMR and HRTEM data. In contrast to the 133Cs MAS NMR data, the 207Pb chemical shift variation trends monotonically to higher deshielding (i.e. to higher ppm) with increasing mol% MA+ incorporation (see Fig. 6c). As reflected in Table 3, this behaviour is influenced by the increasing average 〈Pb–Cl〉 and 〈Pb–Br〉 bond distances comprising the PbCl6 and PbBr6 frameworks, respectively, as they accommodate the increasing proportions of the MA+ cation which possesses a larger ionic radius. Furthermore, to within the resolution afforded by the 207Pb linewidths (∼7–16 kHz) these data are insensitive to the cubic–orthorhombic phase transition suggesting that Cs+ cation positions are more responsive to the octahedral tilting defining this phase transition. As reported in previous 207Pb MAS NMR studies on the CsPbBr3 system, a heteronuclear 1J(Pb, Br) coupling has been observed and measured, although the ability to observe this phenomenon is intrinsically associated with the sample quality and the integrity of the short range order.42–44 From simulation of the 207Pb MAS NMR data from the CsPbBr3 endmember shown in Fig. 6b and Fig. S2a (ESI‡) 1J(Pb, Br) coupling constant of 2360 ± 50 Hz has been measured. Although this is consistent with previous studies, it is not detected in subsequent data reflecting increasing MA+ incorporation as the substitutional and positional Cs+/MA+ cation disorder influencing the PbBr6 octahedra precludes observation of this fine structure.42–44
From Fig. 5d and 6d it can be observed that the 133Cs and 207Pb T1 values measured across the mechanochemically synthesized (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 compositional series exhibit contrasting behavioral trends. Although the 133Cs nucleus (I = 7/2) is a quadrupolar nucleus it possesses a very small quadrupole moment (Q ∼ −0.343 × 10−30 m2), and coupled with the high point symmetry of the cuboctahedral Cs+ cation environment which induces small or zero electric field gradients, the overall 133Cs quadrupole coupling interaction within these systems is negligible. Hence, contributions from the quadrupole interaction to the 133Cs T1 relaxation rates/times will be small, and they will be thus dominated by a dipolar relaxation mechanism. Variation in the in dipolar-dominated 133Cs T1s have been previously used as diagnostic probes of mobility, dynamics and phase transitions in halide perovskite materials.18,43–46 The measured reduction in 133Cs spin-lattice relaxation rates have been correlated to phase transitions and associated increase in electrical conductivity due to increase in halide mobility.44,47Fig. 5d indicates marked variation in the 133Cs T1 with respect to sample composition. For the (CsxMA1−x)PbCl3 series the T1 decreases from 51 s in the CsPbCl3 end member to a T1 minimum of 8 s for (Cs0.87MA0.13)PbCl3 which represents an orthorhombic–cubic phase transition. As the mol% of the MA+ cation increases past this point the T1 increases to 44 s in the final dual cation (Cs0.13MA0.87)PbCl3 composition. Similarly for the (CsxMA1−x)PbBr3 series, the T1 decreases from 54 s in the CsPbBr3 end member to a T1 minimum of 17 s for the (Cs0.75MA0.25)PbBr3 system which also represents an orthorhombic–cubic phase transition for this series; this monotonically increases to 47 s for the final (Cs0.13MA0.87)PbBr3 dual cation composition.
The 133Cs T1 minima corresponding to the orthorhombic–cubic phase transition for each series represent compositions where the local (or short range) disorder is maximized, thus facilitating optimal conditions for Cs+ cation and halide anion mobility. As observed in Fig. 5d, these minima are represented by pronounced reductions in T1 by factors of ∼6–7 for the (CsxMA1−x)PbCl3 series and ∼3 for the (CsxMA1−x)PbBr3 series at this transition point. However, the subsequent increase in T1s back to values approaching the starting (orthorhombic) CsPbCl3 and CsPbBr3 endmembers occurs throughout cubic compositions where no change in lattice symmetry is observed. This phenomenon suggests that these T1 increases can be attributed primarily to Cs+ cation and halide anion mobility. Indeed, other studies of similar lead iodide perovskite analogues have shown that the electrical conductivity is dominated by halide mobility rather than cation mobility in these materials.48–50 Notably, the compositions associated with these phase transitions in Fig. 5d are coincident with the points on inflection (i.e. change in the slopes) of the δ(133Cs)/mol% MA+ trends for each system in Fig. 5c, indicating that the 133Cs T1 relaxation times and chemical shifts δ data are both sensitive to the precise compositions where phase transitions occur within the dual cation (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 series. Sensitivity to the orthorhombic–cubic phase transition is not mirrored by the accompanying 207Pb and 1H chemical shift δ and T1 data of Fig. 6a–d and 7a–c, respectively. Fig. 6a–d show that an increasing MA+ content induces an increase in the 207Pb chemical shifts δ that are correlated with the increasing average 〈Pb–X〉 bond lengths, while the 207Pb T1 data exhibit small monotonic decreases which correlate with increasing average octahedral 〈Pb–X〉 bond lengths, average cuboctahedral 〈Cs/MA–X〉 distances and cell volumes (see Table 3). In contrast, Fig. 7a–c show that the 1H chemical shifts δ are completely invariant with composition, while the associated T1 data also exhibits a similar invariance except for those compositions approaching (and including) the MAPbCl3 endmembers. This phenomenon is attributed to the local disorder introduced throughout the solid solution part of the series; however, an abrupt increase in T1 is observed for the ordered MAPbCl3 endmembers where optimal structural order is coupled with the maximised cell volume.
 | ||
| Fig. 7 The 1H MAS NMR data exhibiting methyl and ammonium resonances at δ ∼3 ppm and δ ∼6.5 ppm, respectively, for (a) the (CsxMA1−x)PbCl3 series, (b) the (CsxMA1−x)PbBr3 series, together with (c) the largely invariant 1H T1 data across the Cs+/MA+ compositional range (except for the MAPbX3 endmembers), indicating that the MA+ dynamics are relatively unaffected by the orthorhombic–cubic phase transition. | ||
PXRD and thermal analyses of the temperature induced phase transitions
Room temperature TEM, PXRD and solid state NMR studies of the as-synthesized mechanochemically derived (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 (x = 0.0, 0.13, 0.25, 0.37, 0.5, 0.63, 0.75, 0.87, 1.0) solid solution series all demonstrate the superior homogeneity, and long and short range structural order that emanates from this preparative method. These properties can be exploited to accurately map the more extensive temperature induced phase transitions enabling phase diagrams for these entire solid solution compositional ranges to be constructed.The CsPbCl3 and MAPbCl3 systems have been extensively studied. For CsPbCl3 the low temperature orthorhombic (Pnma, a−b+a−) phase transforms at 310 K to a tetragonal (P4/mbm, a0a0c+) variant through the elimination of out-of-phase PbCl6 octahedral tilting,39 with conversion to a cubic (Pmm, a0a0a0) phase following closely above 320 K as a second order transformation.15,40 Poglish et al. determined that cubic MAPbCl3 transforms into a pseudo-cubic tetragonal (P4/mmm, a0a0c0) structure below 177 K with disordered methylammonium ions,51 while Songvilay et al. established that phase transitions from orthorhombic to tetragonal and cubic forms are driven by thermally activated framework vibrations.52 The tetragonal P4/mmm polymorph possesses non-tilted BX6 octahedra similar to the cubic Pmm aristotype, but the PbCl6 octahedra are elongated to accommodate the rigid methylammonium ions leading to a reduction in symmetry, and consequently is not captured in Scheme 1. Kawamura et al. reported that the MA+ cations are incommensurately modulated along the c-axis of the tetragonal cell,53 with the tetragonal to cubic transition occurring at 177–178 K appearing second order in nature.54 Despite the large body of work that exists characterising the CsPbCl3 and MAPbCl3 systems, no study has attempted to map the phase transitions characterising Cs+/MA+ solid solution compositional series linking these endmembers.
The lattice parameter evolution from the Pawley refinement of the variable temperature laboratory PXRD data from the (CsxMA1−x)PbCl3 series, and the variable temperature synchrotron PXRD data from the (CsxMA1−x)PbBr3 series, are given in Fig. 8 and 9, respectively. All phase transitions detected using diffraction techniques are corroborated by DSC measurements shown in Fig. S5 and S6 (ESI‡), with the overall mapping of the temperature induced phase transitions determined from both techniques summarised in the phase diagrams of Fig. 10a and b. From Fig. 8, 10a and Fig. S5 (ESI‡) the tetragonal (P4/mbm)–cubic (Pmm) transition for the pure CsPbCl3 endmember measured at 320 K closely corroborates previous reports.40,55 The subsequent increase in MA+ content monotonically depresses the cubic–tetragonal transition temperature from 320 K in CsPbCl3 to 220 K in Cs0.63MA0.37PbCl3. Although the phase transitions for CsPbCl3 and Cs0.87MA0.13PbCl3 are closely mirrored by the DSC data of Fig. S5 (ESI‡), compositions from Cs0.63MA0.37PbCl3 to Cs0.13MA0.87PbCl3 undergo a slow and irreversible disproportionation or phase segregation below 210 K yielding the 2D layered CsPb2Cl5 structure and highly disordered MA+-containing MA/Cs/Pb/Cl phases of mixed compositions. At these reduced temperatures the solid solution enthalpy of formation (or activation) is not satisfied, and a more entropic state appears to be supported. However, pure MAPbCl3 alters to tetragonal (P4/mmm) perovskite below 177 K in agreement with earlier reports.53 In summary, the (CsxMA1−x)PbCl3 solid solution series demonstrates that the phase transition temperature can be tuned by replacing the smaller A site Cs+ cation with larger MA+ so that the cubic phase persists at room temperature.
 | ||
| Fig. 8 Refined lattice parameter evolution (pseudocubic axes a black, c blue) elucidated from Pawley refinement of laboratory XRD data collected on the (CsxMA1−x)PbCl3 solid solution series. The cubic (Pmm) to tetragonal (P4/mbm) phase transformations in Cs+-rich end of the series are dominated by second order effects. | ||
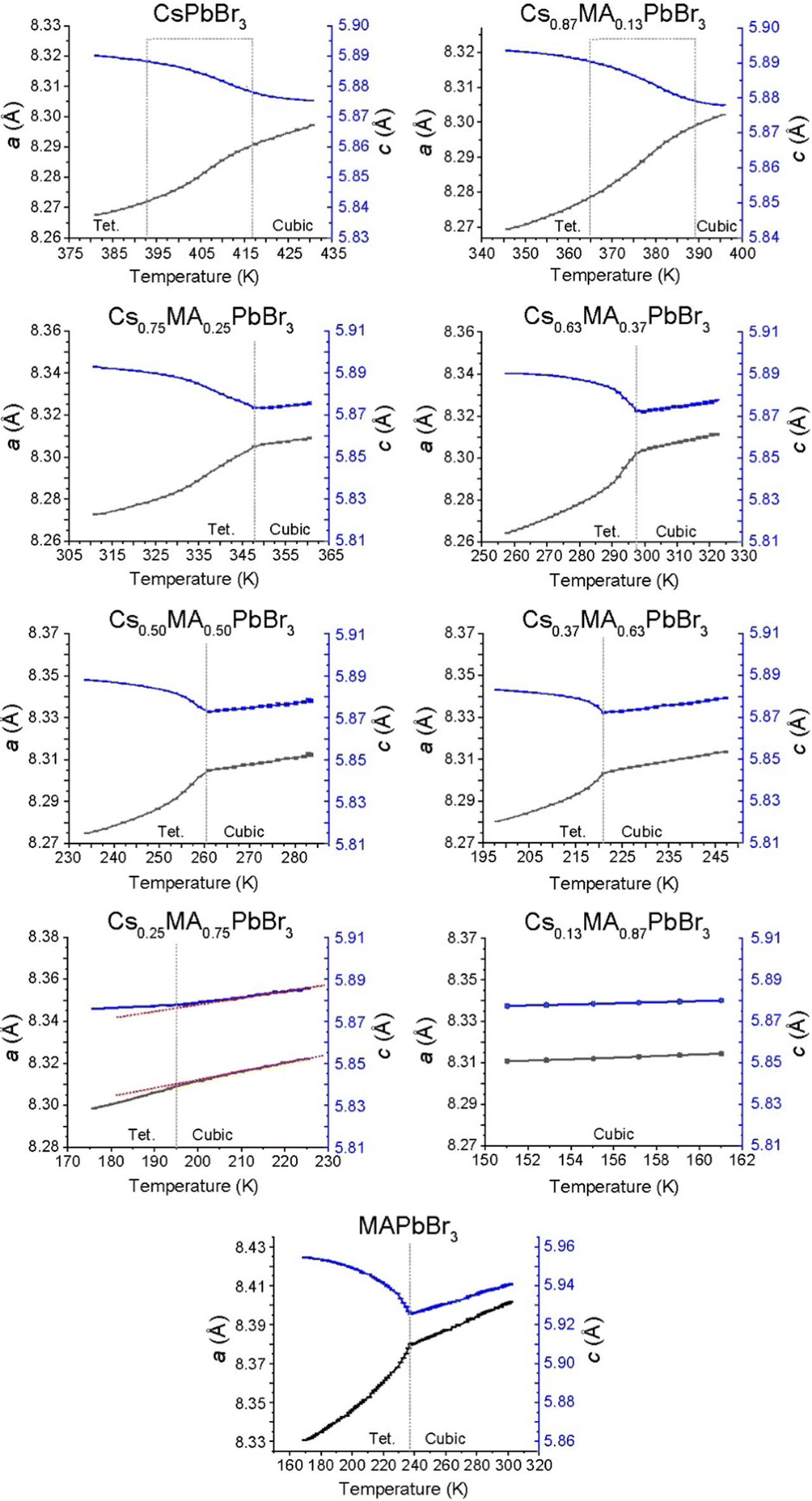 | ||
| Fig. 9 Refined lattice parameter evolution (pseudocubic axes a black, c blue) elucidated from Rietveld refinement of synchrotron XRD data collected on the (CsxMA1−x)PbBr3 solid solution series. The cubic (Pmm) to tetragonal (P4/mbm) phase transformations in Cs+-rich end of the series are dominated by second order effects. The second order effects gradually reduce in the MA+-rich compositions with MAPbBr3 resembling first order behavior. | ||
 | ||
| Fig. 10 Variable temperature phase diagrams for, (a) the (CsxMA1−x)PbCl3, and (b) the (CsxMA1−x)PbBr3 solid solution series highlighting the cubic–tetragonal transitions as determined by the combined DSC/laboratory source PXRD study (for the (CsxMA1−x)PbCl3 series) and the DSC/synchrotron source PXRD study (for the (CsxMA1−x)PbBr3 series). The additional lower symmetry phase transitions denoted with superscripts a, b and c corroborate previous reports in ref. 38, 39 and 59, respectively. The room temperature PXRD and solid state NMR studies correspond to the phase transitions indicated by the dotted lines in each series. | ||
The corresponding phase diagram generated from the diffraction and DSC data for the (CsxMA1−x)PbBr3 solid solution series (see Fig. 10b) exhibits a more homogeneous and less complex response to MA+ incorporation. As observed from Fig. 9, 10b and Fig. S6 (ESI‡) the tetragonal (P4/mbm)–cubic (Pmm) transition for the pure CsPbBr3 endmember identified at 403 K (130 °C) is consistent with previous studies,41 while the increasing MA+ content depresses the phase transition temperature monotonically to 195 K for the Cs0.25MA0.75PbBr3 composition. Thereafter, the Cs0.13MA0.87PbBr3 system represents an unusually stable cubic phase down to <150 K which is considerably lower than the adjacent MAPbBr3 and Cs0.25MA0.75PbBr3 phase transition temperatures at 177 and 195 K, respectively. Identifying this composition with exceptional phase stability could have potential implications for the fine tuning of photovoltaic device efficiency and stability. Furthermore, the PXRD data of Fig. 9 reveals that this monotonic behaviour is mirrored by a gradual change in the nature of the tetragonal–cubic phase transition. For CsPbBr3 this phase transition is distinctly second order and spread over a ∼20 K temperature range (∼395–415 K); however, this narrows and assumes greater first order character with increasing MA+ incorporation with MAPbBr3 exhibiting a completely first order phase transition with a temperature range of ∼3 K (∼236–239 K). As shown in Fig. 10b, the phase transitions detected from the DSC and PXRD measurements (see Fig. S6, ESI‡ and Fig. 9, respectively) are in excellent agreement.
In contrast to MAPbCl3 (P4/mbm), cubic MAPbBr3 cools into a tetragonal (I4/mcm) structure described by the tilt system a0a0c−.54 Tetragonal MAPbBr3 has been found to be incommensurately modulated similar to the chloride analogue,56,57 with the disorder of the MA+ cations in the tetragonal phase being previously characterised using neutron diffraction.58 The intrinsic structural difference between the tetragonal CsPbBr3 and MAPbBr3 structures centres around the octahedral tilting which is in-phase in the former system and out-of-phase in the latter. Since the PXRD data of Fig. S3 and S4 (ESI‡) alone cannot distinguish in-phase and out-of-phase tilts, reflections from space groups such and P4/mbm and I4/mcm cannot be resolved. While changes in the lattice parameters indicate variation in lattice symmetry, the lack of phase information from the reflections determines that precise space group information cannot be elucidated for the intermediate solid solution compositions. Hence, the reported tetragonal space groups shown in Fig. 10a and b are assumed directly from the parent endmembers.
The cubic polymorph stability can be displayed by plotting the minimum unit cell size determined by the PXRD data below which the tetragonal phase becomes measurable (see Fig. 11). These observations are supplemented by DSC measurements to assess the latent heat capacity near these transition temperatures (see Fig. S5 and S6, ESI‡) which are collated in the phase diagrams of Fig. 10a and b. All endmembers (i.e. CsPbCl3, CsPbBr3, MAPbCl3, MAPbBr3) show sharp, well-defined transitions, while only some intermediate compositions exhibit detectable changes in latent heat capacity. From the (CsxMA1−x)PbBr3 series the Cs0.87MA0.13PbBr3, Cs0.75MA0.25PbBr3 and Cs0.63MA0.37PbBr3 compositions the DSC data of Fig. S6 (ESI‡) demonstrate heat capacities corresponding to successively smaller changes in the unit cell volumes upon increasing MA+ content.
 | ||
| Fig. 11 Correlated structural, bandgap and XPS data from the mechanochemically prepared (CsxMA1−x)PbX3 (x = 0–1, X = Cs, Br) solid solution series highlighting, (a) refined lattice parameters immediately prior to the cubic (Pmm)–tetragonal (P4/mbm) phase transition, (b) estimation of the bandgap using Tauc plots based on the UV/vis absorption spectra shown in Fig. S7 and S8 (ESI‡), respectively, (c–f) XPS data of the Cl 2p, Br 3d, Pb 4f and Cs 4d electronic states, respectively. | ||
In addition to the detected cubic–tetragonal phase transitions characterising the (CsxMA1−x)PbCl3 solid solution series, Fig. 10a also indicates lower temperature transformations to reduced symmetry. In agreement with Linaberg et al.,39 the CsPbCl3 endmember shows a tetragonal–orthorhombic transition at ∼315 K close to its cubic–tetragonal counterpart, although the lower temperature orthorhombic-monoclinic transition previously reported by Hirotsu at ∼200 K was not detected under these experimental conditions.40 From Fig. 8 and 10a additional orthorhombic-monoclinic transitions can be observed for the Cs0.87MA0.13PbCl3 and Cs0.75MA0.25PbCl3 compositions at lower temperatures of ∼150 K with increasing MA+ incorporation, mirroring the cubic–tetragonal trend. As indicated above, compositions with higher MA content are unstable below 210 K, phase segregating to CsPb2Cl5 and other MA/Cs/Pb/Cl-containing phases. The room temperature XRD data (see Fig. 3 and 4) represent a slice of the phase diagram as marked with a dashed line in Fig. 10. When viewed along this line, the transition from orthorhombic to cubic is apparent as a crossover for compositions with MA+ contents of more than Cs0.87MA0.13PbCl3 and Cs0.63MA0.37PbBr3.
Octahedral tilting influence on the bandgap
The solid state NMR and diffraction data from the highly ordered dual cation (CsxMA1−x)PbX3 (x = 0–1, X = Cs, Br) solid solution series formed under mechanochemical conditions demonstrate that complete miscibility exists across the entire mixed A site cation compositional range. This contrasts with other mixed A site cation systems such as Cs+-formamidinium (FA+) which exhibits a pronounced miscibility gap. Some conjecture surrounds the size of this Cs+–FA+ miscibility gap as different studies by Ralf et al.60 and Prasanna et al.61 reported miscibility gaps of 15–100 mol% Cs+ and 33–100 mol% Cs+, respectively. The latter study employed materials modelling and DFT calculations to examine the band gap evolution with respect to lattice contraction for low-intermediate compositions, concluding that lattice contraction in Cs+-rich compositions correlated with smaller band gaps that were attributed to octahedral tilting. However, the study by Prasanna et al. did not consider the scenario of lattice contraction without tilting.61 While focusing on well-formed mechanochemically synthesized (CsxMA1−x)PbX3 (x = 0–1, X = Cs, Br) solid solution products, this study presents an opportunity to accurately test this hypothesis relating electronic structure and band gap size to the impact of bond length contraction and octahedral titling.As established in Table 3 and Fig. 3, 4, 8–10, this work shows that high proportions of the Cs+ cation can be introduced into the A site of the cubic phase at room temperature without any change in bond angle or octahedral tilting. This compositional range can be extended to higher Cs+ cation incorporation with increasing temperature (especially for the (CsxMA1−x)PbBr3 series). Fig. 11a emphasizes the uniformity of refined lattice parameters of cubic (CsxMA1−x)PbBr3 and (CsxMA1−x)PbCl3 solid solution series immediately prior to the cubic (Pmm)–tetragonal (P4/mbm) phase transition. For the Br series the PbBr6 octahedra are consistently compressed to assume unit cell dimensions of ∼5.872 Å, either through compositional (A site) or temperature variation, beyond which the PbBr6 octahedra tilt to avoid further bond length reduction. Similarly, for the Cl series the PbCl6 octahedra are consistently compressed to unit cell dimensions of ∼5.620 Å. The Tauc plots of the UV/vis absorption data of Fig. 11b and Fig. S7, S8 (ESI‡) display a gradual reduction in bandgap with increasing MA+ content, although the errors are large due to the nature of the measurement. Importantly, this bandgap trend exists over MA+ incorporation ranges (x = 0–0.87 for the Cl series, x = 0–0.63 for the Br series) where only cubic phases exist (i.e. phases not supporting octahedral tilting), suggesting that octahedral tilting exerts a comparatively smaller perturbation on the bandgap in comparison to other factors (i.e. A site cation size, unit cell size, Pb–X bond length) in this class of materials.
Further XPS data shown in Fig. 11c–f demonstrates that both series exhibit correlated trends of strengthening Cs 4d, Br 3d, Pb 4f and Cl 2p binding energies with increasing MA+ content despite the increasing unit cell sizes and bond lengths reported above (see Fig. 3d, 4d and 6c). These results emphasize that change of the A site cation size influences the Pb–X bond length and bond strength in the absence of octahedral tilting. Conventional electronic band structure modelling of metal halide perovskites is normally dominated by the electronic spin-orbit coupling and bonding hybridization schemes between the lead and halide positions.61 The increasing MA+ content through each solid solution series induces structural change and variable disorder that correlates empirically with these electronic factors and the resultant bandgap variation; however, the specific mechanisms are not well understood. This evidence highlights the need for further theoretical investigations of the structural influences on bandgaps in these materials, and presents implications and factors for the materials modelling of these systems that cannot be neglected.
Conclusion
Mechanochemical synthesis is demonstrated to be a viable method for the preparation of dual cation hybrid perovskites of the nominal stoichiometric form (CsxMA1−x)Pb(Cl/Br)3 (x = 0, 0.13, 0.25, 0.37, 0.50, 0.63, 0.75, 0.87, 1). The continuous compositional variation demonstrates that Cs+ and MA+ are completely miscible which allows the formation of homogeneous and high purity solid solution products. The phase purity was studied using HRTEM, XRD and solid state NMR techniques which established that room temperature cubic–orthorhombic phase transitions exists in the region of 0.63 < x < 0.75 for (CsxMA1−x)PbBr3 system, and in the region of 0.87 < x < 1 (CsxMA1−x)PbCl3 system. HRTEM studies showed the Br series exhibits higher electron beam stability of up to ∼10–20 min, whereas the analogous Cl series demonstrated a reduced persistence with degradation occurring over a period of ∼30 s–1 min under the beam, although lattice fringes were still able to be observed. High e− beam stability of the dual cation Cs+/Ma+ systems in comparison to pure MA+ analogues suggests that these materials may be more stable under high electric fields, which is critical for LED applications where the active layer experiences electric fields on the order of 2000 kV m−1.The 207Pb NMR chemical shift exhibits sensitivity to changes in the first coordination sphere of Pb2+ position promoted by Pb–X bond length variation upon A site compositional variation, and the halide composition. In contrast, the 133Cs resonance is sensitive to the Pb–X bond length change (upon A site compositional change and halide composition), and the phase transitions induced throughout the compositional range. Synchrotron and laboratory source PXRD data exhibit good agreement with the 133Cs MAS NMR study in identifying the phase transitions, indicating that the local (short range) structural changes observed from NMR are accurately reflected in the periodic (long range) structural transformation observed with XRD.
Phase diagrams mapping the Cs+/MA+ compositional space for the (CsxMA1−x)PbCl3 and (CsxMA1−x)PbBr3 solid solution series were undertaken using variable temperature PXRD. While the cubic polymorph in most compositions exhibited a stability range from 403–195 K, the Cs0.13MA0.87PbBr3 composition displayed an exceptionally wide stability field for the cubic polymorph that extended below <150 K. This unique phase stability has promising potential for photovoltaic devices as the phase stability minimises thermal degradation and phase segregation. The Cl series also shows increasing cubic phase stability down to 210 K, below which solid solution formation no longer occurs and phase segregation (or disproportionation) into a 2D layered CsPb2Cl5 and other disordered MA/Cs/Pb/Cl phases of mixed compositions are thermodynamically favoured. Tauc plots of the UV/vis data highlighted the occurrence of a bandgap reduction observed for increasing MA+ incorporation throughout the range of cubic phases where octahedral tilting was absent.
Author contributions
SSHD, TW and JVH designed the initial experiments relating to the mechanochemical synthesis of these materials. SSHD performed the materials syntheses, while SSHD, DW and YF undertook the initial PXRD/crystallographic analyses, TEM, UV/vis, XPS and DSC characterization. SSHD and JVH designed and performed all solid state NMR measurements, while SSHD and TAB performed the synchrotron PXRD/crystallographic analysis. SSHD and JVH wrote the manuscript with all authors viewing and contributing to its content.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
JVH and TJW gratefully acknowledge the Diamond Light Source at the Rutherford Appleton Laboratories for access to the I11 beamline through the Diamond Light Source Block Allocation Group award “Oxford/Warwick Solid State Chemistry BAG to Probe Composition–Structure–Property Relationships in Solids” (STFC grants EE18786 and CY25166). The authors thank Assoc Prof. Mark Senn and Dr Anna Herlihy for assistance in running the I11 beamline, acquisition of the diffraction data, and for data handling. JVH and DW thank the Diffraction Research Technology Platform at the University of Warwick for access to the laboratory source PXRD instrumentation. TJW, JVH and SSHD acknowledges financial support from the Singapore National Research Foundation, Prime Minister's Office, through the Competitive Research Program (CRP Award No. NRF-CRP14-2014-03). JVH and TJW thank Center of High Field NMR Spectroscopy and Imaging at NTU for access to the solid state MAS NMR capability.References
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. Il, Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- Best Research Cell Efficiency, 2021, https://www.nrel.gov/pv/cell-efficiency.html.
- Y.-H. Kim, S. Kim, A. Kakekhani, J. Park, J. Park, Y.-H. Lee, H. Xu, S. Nagane, R. B. Wexler, D.-H. Kim, S. H. Jo, L. Martínez-Sarti, P. Tan, A. Sadhanala, G.-S. Park, Y.-W. Kim, B. Hu, H. J. Bolink, S. Yoo, R. H. Friend, A. M. Rappe and T.-W. Lee, Nat. Photonics, 2021, 15, 148–155 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. V. Gustafsson, M. T. Trinh, S. Jin and X. Y. Zhu, Nat. Mater., 2015, 14, 636–642 CrossRef CAS PubMed.
- F. Bella, G. Griffini, J.-P. Correa-Baena, G. Saracco, M. Grätzel, A. Hagfeldt, S. Turri and C. Gerbaldi, Science, 2016, 354, 203 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312 CrossRef CAS PubMed.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Gratzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 14173–14180 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Gratzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 7232–7238 CrossRef CAS PubMed.
- Y. H. Kim, H. Cho, J. H. Heo, T. S. Kim, N. Myoung, C. L. Lee, S. H. Im and T. W. Lee, Adv. Mater., 2015, 27, 1248–1254 CrossRef CAS PubMed.
- L. Zhang, X. Yang, Q. Jiang, P. Wang, Z. Yin, X. Zhang, H. Tan, Y. M. Yang, M. Wei and B. R. Sutherland, Nat. Commun., 2017, 8, 15640 CrossRef CAS PubMed.
- M.-H. Park, S.-H. Jeong, H.-K. Seo, C. Wolf, Y.-H. Kim, H. Kim, J. Byun, J. S. Kim, H. Cho and T.-W. Lee, Nano Energy, 2017, 42, 157–165 CrossRef CAS.
- S. Hirotsu and S. Sawada, Phys. Lett. A, 1969, 28, 762–763 CrossRef CAS.
- A. Karmakar, A. M. Askar, G. M. Bernard, V. V. Terskikh, M. Ha, S. Patel, K. Shankar and V. K. Michaelis, Chem. Mater., 2018, 30, 2309–2321 CrossRef CAS.
- A. M. Askar, A. Karmakar, G. M. Bernard, M. Ha, V. V. Terskikh, B. D. Wiltshire, S. Patel, J. Fleet, K. Shankar and V. K. Michaelis, J. Phys. Chem. Lett., 2018, 9, 2671–2677 CrossRef CAS PubMed.
- A. Karmakar, A. Bhattacharya, D. Sarkar, G. M. Bernard, A. Mar and V. K. Michaelis, Chem. Sci., 2021, 12, 3253–3263 RSC.
- A. Karmakar, A. Bhattacharya, G. M. Bernard, A. Mar and V. K. Michaelis, ACS Mater. Lett., 2021, 3, 261–267 CrossRef CAS.
- E. M. Mozur, A. E. Maughan, Y. Cheng, A. Huq, N. Jalarvo, L. L. Daemen and J. R. Neilson, Chem. Mater., 2017, 29, 10168–10177 CrossRef CAS.
- G. A. Bowmaker, Effendy, J. V. Hanna, P. C. Healy, S. P. King, C. Pettinari, B. W. Skelton and A. H. White, Dalton Trans., 2011, 40, 7210–7218 RSC.
- G. A. Bowmaker, J. V. Hanna, R. D. Hart, B. W. Skelton and A. H. White, Dalton Trans., 2008, 5290–5292, 10.1039/B810659M.
- G. A. Bowmaker, J. V. Hanna, B. W. Skelton and A. H. White, Chem. Commun., 2009, 2168–2170, 10.1039/B822370J.
- G. A. Bowmaker, J. V. Hanna, R. D. Hart, P. C. Healy, S. P. King, F. Marchetti, C. Pettinari, B. W. Skelton, A. Tabacaru and A. H. White, Dalton Trans., 2012, 41, 7513–7525 RSC.
- G. Maculan, A. D. Sheikh, A. L. Abdelhady, M. I. Saidaminov, M. A. Haque, B. Murali, E. Alarousu, O. F. Mohammed, T. Wu and O. M. Bakr, J. Phys. Chem. Lett., 2015, 6, 3781–3786 CrossRef CAS PubMed.
- M. I. Saidaminov, A. L. Abdelhady, B. Murali, E. Alarousu, V. M. Burlakov, W. Peng, I. Dursun, L. Wang, Y. He and G. Maculan, Nat. Commun., 2015, 6, 7586 CrossRef PubMed.
- D. Weber, Z. Naturforsch. B, 1978, 33, 1443–1445 CrossRef.
- D. Weber, Z. Naturforsch. B, 1978, 33, 862–865 CrossRef.
- T. Baikie, Y. N. Fang, J. M. Kadro, M. Schreyer, F. X. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 RSC.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- S. S. Mali, C. S. Shim and C. K. Hong, NPG Asia Mater., 2015, 7, e208 CrossRef CAS.
- V. Malgras, J. Henzie, T. Takei and Y. Yamauchi, Chem. Commun., 2017, 53, 2359–2362 RSC.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr. B, 2008, 64, 702–707 CrossRef CAS PubMed.
- H. A. Maddah, V. Berry and S. K. Behura, Comput. Mater. Sci., 2020, 173, 109415 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Sci., 1972, 28, 3384–3392 CrossRef CAS.
- C. J. Howard and H. T. Stokes, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 782–789 CrossRef.
- Y. S. Zhao, D. J. Weidner, J. B. Parise and D. E. Cox, Phys. Earth Planet. Inter., 1993, 76, 1–16 CrossRef CAS.
- N. W. Thomas and A. Beitollahi, Acta Crystallogr., Sect. B: Struct. Sci., 1994, 50, 549–560 CrossRef.
- M. R. Linaburg, E. T. McClure, J. D. Majher and P. M. Woodward, Chem. Mater., 2017, 29, 3507–3514 CrossRef CAS.
- S. Hirotsu, J. Phys. Soc. Jpn., 1971, 31, 552–560 CrossRef CAS.
- S. Hirotsu, J. Harada, M. Iizumi and K. Gesi, J. Phys. Soc. Jpn., 1974, 37, 1393–1398 CrossRef CAS.
- M. Aebli, L. Piveteau, O. Nazarenko, B. M. Benin, F. Krieg, R. Verel and M. V. Kovalenko, Sci. Rep., 2020, 10, 1–9 CrossRef PubMed.
- L. Piveteau, V. Morad and M. V. Kovalenko, J. Am. Chem. Soc., 2020, 142, 19413–19437 CrossRef CAS PubMed.
- P. Vashishtha, S. A. Veldhuis, S. S. Dintakurti, N. L. Kelly, B. E. Griffith, A. A. Brown, M. S. Ansari, A. Bruno, N. Mathews, Y. Fang, T. J. White, S. G. Mhaisalkar and J. V. Hanna, J. Mater. Chem. C, 2020, 8, 11805–11821 RSC.
- P. Vashishtha, B. E. Griffith, A. A. M. Brown, T. J. N. Hooper, Y. Fang, M. S. Ansari, A. Bruno, S. H. Pu, S. G. Mhaisalkar, T. White and J. V. Hanna, ACS Appl. Electron. Materials, 2020, 2, 4002–4011 CrossRef CAS.
- P. Vashishtha, B. E. Griffith, Y. Fang, A. Jaiswal, G. V. Nutan, A. P. Bartók, T. J. White and J. V. Hanna, J. Mater. Chem. A, 2022, 10, 3562–3578 RSC.
- K. Yamada, Y. Kuranaga, K. Ueda, S. Goto, T. Okuda and Y. Furukawa, Bull. Chem. Soc. Jpn., 1998, 71, 127–134 CrossRef CAS.
- W. M. J. Franssen, S. G. D. van Es, R. Dervişoğlu, G. A. de Wijs and A. P. M. Kentgens, J. Phys. Chem. Lett., 2017, 8, 61–66 CrossRef CAS PubMed.
- A. Osherov, Y. Feldman, I. Kaplan-Ashiri, D. Cahen and G. Hodes, Chem. Mater., 2020, 32, 4223–4231 CrossRef CAS.
- W. Peng, C. Aranda, O. M. Bakr, G. Garcia-Belmonte, J. Bisquert and A. Guerrero, ACS Energy Lett., 2018, 3, 1477–1481 CrossRef CAS.
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef CAS.
- M. Songvilay, Z. Wang, V. G. Sakai, T. Guidi, M. Bari, Z. G. Ye, G. Xu, K. L. Brown, P. M. Gehring and C. Stock, Phys. Rev. Mater., 2019, 3, 125406 CrossRef CAS PubMed.
- Y. Kawamura and H. Mashiyama, J. Korean Phys. Soc., 1999, 35, S1437–S1440 Search PubMed.
- H. Mashiyama, Y. Kawamura, E. Magome and Y. Kubota, J. Korean Phys. Soc., 2003, 42, S1026–1029 CAS.
- M. Hidaka, Y. Okamoto and Y. Zikumaru, Phys. Status Solidi A, 1983, 79, 263–269 CrossRef CAS.
- D. Wiedemann, J. Breternitz, D. W. Paley and S. Schorr, J. Phys. Chem. Lett., 2021, 12, 2358–2362 CrossRef CAS PubMed.
- Y. Guo, O. Yaffe, D. W. Paley, A. N. Beecher, T. D. Hull, G. Szpak, J. S. Owen, L. E. Brus and M. A. Pimenta, Phys. Rev. Mater., 2017, 1, 042401 CrossRef.
- C. A. López, M. C. Álvarez-Galván, M. V. Martínez-Huerta, M. T. Fernández-Díaz and J. A. Alonso, Chem. – Eur. J., 2019, 25, 4496–4500 CrossRef PubMed.
- N. Onoda-Yamamuro, T. Matsuo and H. Suga, J. Phys. Chem. Solids, 1990, 51, 1383–1395 CrossRef CAS.
- R. G. Niemann, L. Gouda, J. G. Hu, S. Tirosh, R. Gottesman, P. J. Cameron and A. Zaban, J. Mater. Chem. A, 2016, 4, 17819–17827 RSC.
- R. Prasanna, A. Gold-Parker, T. Leijtens, B. Conings, A. Babayigit, H.-G. Boyen, M. F. Toney and M. D. McGehee, J. Am. Chem. Soc., 2017, 139, 11117–11124 CrossRef CAS PubMed.
Footnotes |
| † The experimental data for this study are provided as a supporting dataset from WRAP, the Warwick Research Archive Portal at https://wrap.warwick.ac.uk/160750/. |
| ‡ Electronic supplementary information (ESI) available: Supporting experimental and materials characterization including HRTEM, 207Pb MAS NMR data, high resolution laboratory source powder XRD data for the Cl series and Br series, DSC data, UV/vis data with Tauc plots, in addition to tables summarising the 133Cs, 207Pb and 1H T1 (spin-lattice) relaxation times and the variable temperature evolution of refined lattice parameters for each system. See DOI: https://doi.org/10.1039/d2cp02131e |
| This journal is © the Owner Societies 2022 |