
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntioxidant and copper-chelating power of new molecules suggested as multiple target agents against Alzheimer's disease. A theoretical comparative study†
Maciej
Spiegel
 a,
Tiziana
Marino
a,
Mario
Prejanò
b and
Nino
Russo
*a
a,
Tiziana
Marino
a,
Mario
Prejanò
b and
Nino
Russo
*a
aDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, I-87136 Rende, CS, Italy. E-mail: nrusso@unical.it
bDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm, SE-10691, Sweden
First published on 13th June 2022
Abstract
In this study, the scavenging activity against OOH radicals and the copper-chelating ability of two new synthesized molecules (named L1 and L2) that can act as multiple target agents against Alzheimer's disease have been investigated at the density functional theory level. The pKa and molar fractions at physiological pH have been predicted. The main antioxidant reaction mechanisms in lipid-like and water environments have been considered and the relative rate constants determined. The copper-chelating ability of the two compounds has also been explored at different coordination sites and computing the complexation kinetic constants. Results show the L1 compound is a more effective radical scavenging and copper-chelating agent than L2.
Introduction
Alzheimer's disease (AD), first described in 1906, is a serious neurodegenerative disorder that induces progressive memory loss, devastating the quality of life of patients, and is the leading cause of death worldwide. It is an age-dependent disease that occurs generally after the age of 65 years. With the increase in life expectancy in the most industrialized countries, it has become one of the most widespread diseases in the world. In 2015, AD affected nearly 50 million people, and the latest estimates predict that it will grow to around 135 million in 2050.1 The estimated economic cost of this disease is enormous and is set to increase dramatically. Suffice it to say that in the UK alone, the cost of caring for people with AD will reach £66 billion in 2050.2The pathology of AD is essentially characterized by the accumulation of senile plaques and neurofibrillary tangles and by abnormal levels of neurotransmitters.3–5 The senile plaques are formed by insoluble peptide segments composed of 39–43 amino acids (Aβ), and their aggregation (amyloid cascade hypothesis) and the possibility of the formation of small and soluble oligomers (oligomer hypothesis) are considered important phenomena and the basis of the disease.6,7 Despite the great amount of research carried out over the past twenty years, the exact cause of AD is still poorly understood and an effective cure has not yet been proposed.3,4,8,9 The few drugs approved by the US FDA and other European and Asian agencies essentially serve to alleviate the symptoms of the disease and improve the quality of life of patients, but they are ineffective in slowing down its evolution.10 For these reasons, the development of research in the field of AD is fundamental to both a better understanding of the mechanisms underlying its onset and the proposal of new, more effective and targeted drugs.
Autopsy examination of the brain tissues of patients who died due to AD has shown the presence of high and abnormal concentrations of metals such as copper, zinc and iron ions (about six and three times the normal quantity in the human brain, for Cu, and for Zn and Fe, respectively).11–13 It has been shown that this metal ion dyshomeostatis can play an important role since the formation of toxic metal–amyloid complexes promotes Aβ aggregation.13 Furthermore, owing to the redox nature of the metal ions involved, the presence of molecular oxygen in the brain tissue leads to the formation of reactive oxygen species (ROS) through Fenton-like reactions with a consequent increase in oxidative stress. In fact, due to the occurrence of high levels of unsaturated fatty acids (the preferred target of attack for ROS), the brain is particularly sensitive to oxidative stress, as has been proved in different neurodegenerative diseases.13–15
Among the three metal ions that undergo dyshomeostatis in the brains of AD patients, the most involved ions seem to be Cu2+ and Zn2+, which form stable complexes with the Aβ16 proteins, with different and much higher stability constants in the case of Cu2+.17,18
A strategy to limit ROS damage is based on the use of chelator agents that are able to control the increase in the concentration of metals, and to increase the level of antioxidant substances that are not usually present in the brain in sufficient quantities so as to counteract the effects caused by the rise in concentration of free radicals.19,20
On the basis of the observations reported above, the proposal of new drugs endowed with multiple actions in the latest research can contribute to increasing our knowledge not only about degenerative diseases, including Alzheimer's, but also to their treatment using multiple actions.21–23
Very recently, Cho et al.24 proposed some multifunctional molecular systems whose structure provided an antioxidant action together with a chelating action, to reduce the concentration of the copper ions in the brain (L1 and L2 of Scheme 1). The first action is due to the presence of a guaiacyl group, while some nitrogenous chelating groups, of different sizes that can coordinate the Cu2+ ion, express the second. Furthermore, some of their derivatives form stable Cu-radiolabeled complexes.25 These systems, which show values of lipophilicity such that their brain barrier penetration is possible, have been chemically characterized, and in vivo tests have suggested them as promising drugs against AD.
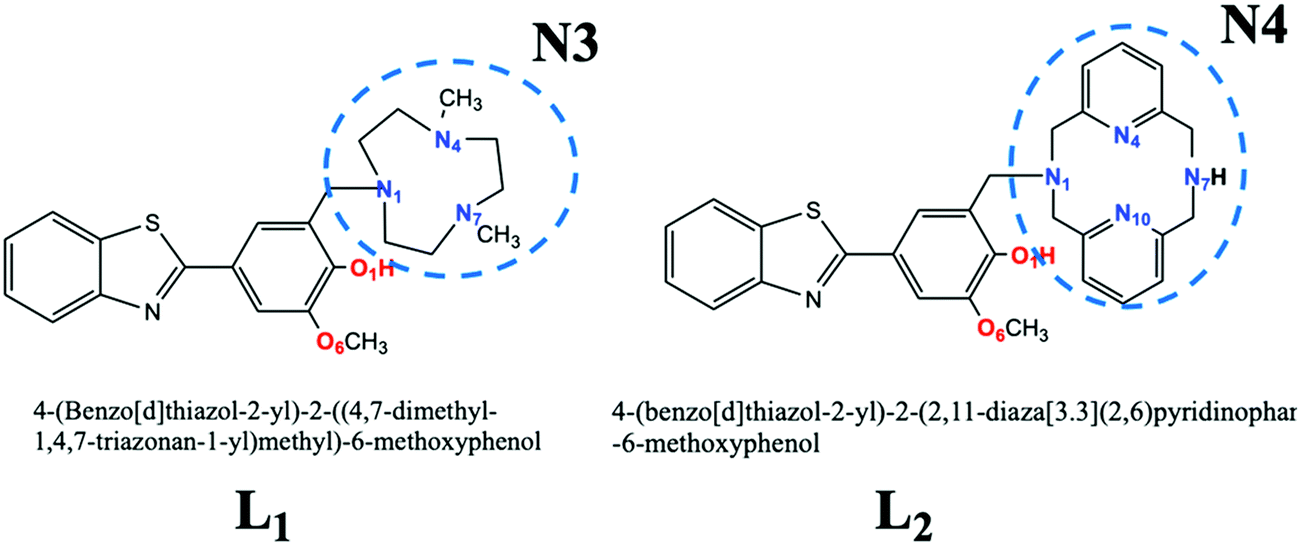 | ||
| Scheme 1 Chemical structure of the bifunctional chelators investigated. The atoms coloured represent the metal chelating sites considered in the present work. | ||
A better knowledge of their structural and electronic properties, as well as of their biological mechanisms, is very useful for their proposition as lead compounds for the development of new therapies in the treatment of AD. For these reasons, we have undertaken a detailed theoretical study using the density functional theory (DFT), which has proved to be particularly useful in the study and prediction of various molecular properties and reaction mechanisms, even in systems containing transition metals.26 In particular, we have considered different antioxidant reaction paths to establish what kind of scavenging mechanism these molecules follow and the possible chelating sites for copper-ion coordination.
Computational details
Geometry optimizations and frequency calculations were performed using the Gaussian 0927 package in the framework of DFT. The M05 exchange–correlation functional28 in conjunction with the 6-31+G(d,p) basis set and the SMD solvation model,29 to mimic lipid-like (ε = 4.7, pentyl ethanoate (PE)) and water environments (ε = 78.48), has been used. In order to improve the electronic energies, single-point computations on previously optimized geometries have been recalculated using the 6-311+G(d,p) basis sets. The unrestricted formalism has been employed for the treatment of radical species. The intrinsic reaction coordinate (IRC) procedure has been applied to verify that the intercepted transition state is properly connected to the relative minima (reactant and product) in the minimum-energy reaction path. The quantum mechanism-based method QM-ORSA,30,31 which is based on conventional transition state theory32,33 and Marcus theory,34 has been used. The computed rate constants, within or close to the diffusion-limited regime, have been corrected using Collins–Kimball theory.35The computational procedure previously described36 has been applied to obtain the molar fraction and pKa values. Numerous previous studies have demonstrated, in fact, that this computational protocol gives reliable results for thermochemistry and kinetics parameters relative to reactions involving free radicals and metals.37–40
Results and discussion
Primary antioxidant properties
From experimental studies on the protonation constants of 1,4,7-triazacyclononane and its derivatives, a high pK1 value related to protonation of the amino group (10.44) indicates that the neutral form is the preferred one.41 This evidence prompted us to consider the amino group of the macrocycle (N3; Scheme 1) in the neutral form and, by analogy, the macrocycle (N4; Scheme 1) is also considered to be deprotonated. Due to the lack of any experimental information on the pKa of the 2-phenylbenzothiazole fragment considered in both systems, as a first step of this work we calculated the pKa and the molar fractions at physiological pH (7.4). The results depicted in Fig. 1 show that the pKa values of the two systems are similar, although that of L1 is slightly lower than L2 (8.57 versus 9.22). For both the equilibria, at physiological pH the dominant form is the neutral one with the monoanion species assuming percentages of 6.3 and 1.5 for L1 and L2, respectively. Although these are smaller values for the dissociated form, they have been taken into account in the computation of the antioxidant properties.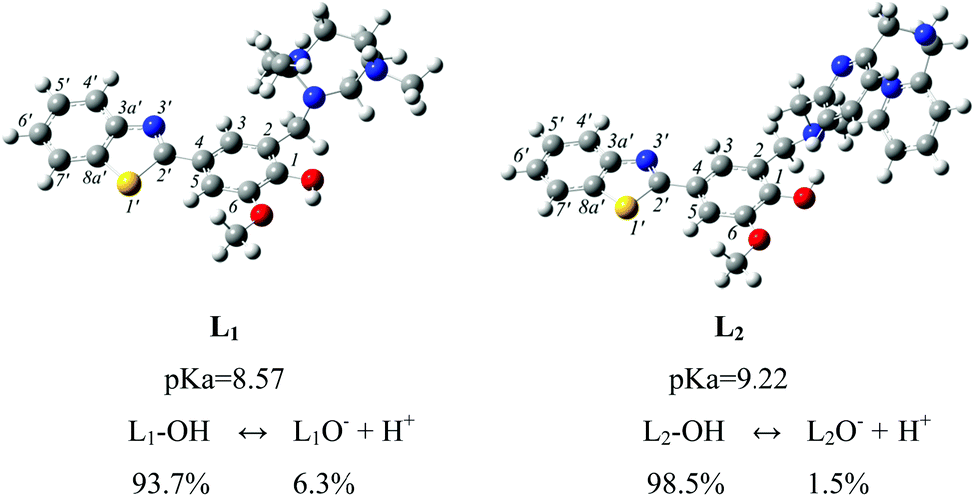 | ||
| Fig. 1 Optimized structures, dissociation constants and molar distribution at pH = 7.4 for L1 and L2 molecules. | ||
Based on our previous experience42,43 and that of other research groups,39,40,44–46 the following redox and non-redox pathways were explored:
| Hydrogen atom transfer (HAT): HX + R˙ → X˙ + RH |
| Single electron transfer (SET): HX + R˙ → HX+˙ + R– |
| Radical adduct formation (RAF): HX + R˙ → [HX−R]˙ |
The obtained Gibbs free energies of reaction are collected in Table 1. In both the considered environments and for both the considered species, the RAF mechanism generates endergonic reactions with Gibbs free energies higher than 10 kcal mol−1, suggesting that they do not take place. In addition, the SET mechanism in a lipid-like environment appears to be highly unlikely given the high endothermic values of the reaction Gibbs free energies. In the aqueous phase, calculations for both neutral and deprotonated forms confirm the feasibility of the SET mechanism. In fact, for the former we find ΔG values of 9.6 and 7.5 kcal mol−1 for L1 and L2, respectively, while for the latter they become −0.6 kcal mol−1 for L1 and 0.1 kcal mol−1 for L2.
| Mechanism | L 1(PE) (L2(PE)) | L 1(W) (L2(W)) | L 1 − (W) (L2−(W)) |
|---|---|---|---|
| HAT | −4.1 (−3.5) | −9.6 (−6.9) | — |
| RAF-C1 | 18.2 (19.0) | 14.2 (16.9) | 18.3 (17.5) |
| RAF-C2 | 24.8 (31.0) | 24.7 (30.3) | 21.5 (21.5) |
| RAF-C3 | 18.0 (14.9) | 16.7 (17.5) | 18.9 (18.5) |
| RAF-C4 | 25.5 (24.8) | 24.0 (24.0) | 21.9 (21.2) |
| RAF-C5 | 22.2 (22.5) | 20.3 (21.6) | 19.4 (19.5) |
| RAF-C6 | 20.0 (22.8) | 18.8 (22.0) | 11.7 (16.4) |
| RAF-C2′ | 20.6 (18.2) | 15.9 (17.3) | 17.9 (19.9) |
| RAF-C3a′ | 32.9 (33.8) | 29.2 (30.5) | 27.6 (29.5) |
| RAF-C4′ | 17.9 (17.8) | 15.3 (16.4) | 15.2 (13.9) |
| RAF-C5′ | 22.3 (22.2) | 20.6 (21.2) | 20.4 (19.7) |
| RAF-C6′ | 18.3 (18.7) | 17.5 (18.4) | 16.8 (16.4) |
| RAF-C7′ | 18.3 (18.6) | 16.9 (17.9) | 17.5 (17.0) |
| RAF-C7a′ | 27.9 (27.7) | 25.9 (27.1) | 24.4 (25.1) |
| SET | 45.8 (67.3) | 9.6 (7.5) | −0.6 (0.1) |
Analyzing the data for the HAT mechanism, we note that, in both environments, the ΔG values indicate exergonic reactions for both the studied compounds. In particular, those related to the aqueous medium assume exothermic values (−9.6 and −6.9 kcal mol−1 for L1 and L2, respectively).
Based on the ΔG values obtained for the various reaction mechanisms, we proceeded to determine the potential energy surfaces by characterizing the transition states and the products for those processes whose reaction energies result in being lower than 10 kcal mol−1. The results, reported in Table 2 and Fig. 2, indicate that in the lipid-like phase, for both systems, only the HAT mechanism is possible with energy barriers of 15.0 and 19.1 kcal mol−1, for L1 and L2, respectively. In the aqueous environment, for the neutral forms of both compounds, the reactions that occur with SET and HAT mechanisms are energetically feasible. In particular, the energy barriers for L1 turn out to be 11.9 (SET) and 15.4 kcal mol−1 (HAT). Those found for L2 are 11.2 and 22.4 kcal mol−1 for the SET and HAT channels, respectively. Finally, the only possible path for both the anionic compounds results in being the SET mechanism, for which a barrier of 3.3 kcal mol−1 for L1 (3.6 kcal mol−1 for L2) must be overcome. The transition-state structures for the HAT mechanism, reported in Fig. 2, show similar geometrical parameters for the H transfer from the OH–L1 (L2) to the OOH radical in both the considered environments. The analysis of the computed negative frequency accounts for this process satisfactorily.
| Mechanism | L 1(PE) (L2(PE)) | L 1(W) (L2(W)) | L 1 − (W) (L2−(W)) | |||
|---|---|---|---|---|---|---|
| ΔG | ΔG‡ | ΔG | ΔG‡ | ΔG | ΔG‡ | |
| HAT | −4.1 (−3.5) | 15.0 (19.1) | −9.6 (−6.9) | 15.4 (22.4) | — | |
| SET | 45.8 (67.3) | 52.3 (194.0) | 9.6 (7.5) | 11.9 (11.2) | −0.6 (0.1) | 3.3 (3.6) |
 | ||
| Fig. 2 L 1 and L2 transition-state structures for the HAT mechanism with the corresponding frequencies, main bond lengths (Å), and dihedral angles (degrees). | ||
The obtained kinetic rate constants (Table 3) in the PE solvent for the HAT path indicate that the L1 system is a better antioxidant than the L2 system, with their k values being 1.89 × 104 and 7.43 × 101 M−1 s−1, respectively. Comparison with the corresponding k value (3.40 × 103 M−1 s−1) of Trolox (generally used as reference antioxidant)31,47 suggests that L1 is a more efficient OOH scavenger than L2.
| Mechanism | k (M−1 s−1) | Γ (%) | k (M−1 s−1) | Γ (%) | k (M−1 s−1) | Γ (%) |
|---|---|---|---|---|---|---|
| L 1(PE) | L 1(W) | L 1 − (W) | ||||
| HAT | 1.89 × 104 | 100.00 | 1.02 × 105 | 89.87 | ||
| SET | 1.15 × 104 | 10.13 | 2.51 × 1010 | 100.00 | ||
| F | 100% | 93.7% | 6.3% | |||
| k total | 1.89 × 104 | 1.14 × 105 | 2.51 × 1010 | |||
| fk total | 1.07 × 105 | 1.58 × 109 | ||||
| L 2(PE) | L 2(W) | L 2 − (W) | ||||
| HAT | 7.43 × 101 | 100.00 | 1.22 × 102 | 3.39 | ||
| SET | 3.59 × 104 | 96.61 | 1.49 × 1010 | 100.00 | ||
| F | 100% | 98.5% | 1.5% | |||
| k total | 7.43 × 101 | 3.60 × 104 | 1.49 × 1010 | |||
| fk total | 3.55 × 104 | 2.24 × 108 | ||||
In water and at physiological pH (7.4) the situation is different. In particular, we underline that, in the neutral form, which has a higher molar fraction (93.7 and 98.5% for L1 and L2, respectively), the HAT mechanism in L1 (k = 1.02 × 105 M−1 s−1) is preferred, while the SET mechanism dominates for the L2 species (k = 3.59 × 104 M−1 s−1). Looking at the ktotal values, our data indicate that L1 is a better radical scavenger than L2 with a difference of one order of magnitude. The comparison with Trolox in the aqueous solvent and under physiological pH conditions (k = 8.96 × 104 M−1 s−1)46 shows that L1 is more efficient, while L2 has almost the same antioxidant power.
Our results well agree with the experimental measurements that reveal L1 to be 1.6 times more efficient than Trolox as a radical-scavenging system.25
Secondary antioxidant properties
In this section, we report the results concerning the Cu2+ complexation abilities of L1 and L2 throughout the computation of Gibbs free energies (ΔGf) for the following reactions:| Cu(H2O)62+ + L → [Cu(H2O)6 L]2+ |
| Cu(H2O)62+ + L− → [Cu(H2O)6 L]+ |
For both the ligands we have considered three possible coordination sites of the Cu2+ ion (Scheme 1): coordination with the O1 and O6 oxygen atoms linked in the phenolic cycle, with the nitrogen atoms of the peripheral macrocycle (N3 or N4 for L1 and L2, respectively) and with both sites (the oxygen atoms of the phenyl ring and the nitrogen atoms of the macrocycle (N3 or N4 for L1 and L2)). This topology will hereafter be referred to as O,O, N3 (or N4), and N,O. The energetic values (ΔG and ΔΔG) and the main geometrical parameters around the copper ion are reported in Table 4 and Table S1 (ESI†), respectively. The optimized structures are given in Fig. 3 (for L1) and Fig. 4 (for L2).
| Coordination site | ΔGf (kcal mol−1) | ΔΔGf (kcal mol−1) | K f | ΣKf (KIIf) |
|---|---|---|---|---|
| L 1 (93.7%) | ||||
| N3 | −18.4 | 1.2 | 2.88 × 1013 | |
| N,O | −19.6 | 0.0 | 2.24 × 1014 | |
| O,O | −5.8 | 13.8 | 1.61 × 104 | |
| 2.53 × 1014 (2.37 × 1014) | ||||
| L 1 − (6.3%) | ||||
| N3 | −25.2 | 11.2 | 2.81 × 1018 | |
| N,O | −36.4 | 0.0 | 5.01 × 1026 | |
| O,O | −16.2 | 20.2 | 7.11 × 1011 | |
| 5.01 × 1026 (3.16 × 1025) | ||||
| K appf (L1) = 3.16 × 1025 | ||||
| L 2 (98.5%) | ||||
| N4 | −13.7 | 0.0 | 1.21 × 1010 | |
| O,O | 4.4 | 18.1 | 6.38 × 104 | |
| 1.21 × 1010 (1.19 × 1010) | ||||
| L 2 − (1.5%) | ||||
| N4 | −11.5 | 1.6 | 2.68 × 108 | |
| O,O | −13.1 | 0.0 | 4.58 × 109 | |
| 6.44 × 1018 (9.66 × 1016) | ||||
| K appf (L2) = 9.66 × 1016 | ||||
 | ||
| Fig. 3 Optimized structures for Cu–L1 (a) and Cu–L1− (b) complexes. Distances are in Å. | ||
For L1, Table 4 shows that the preferred coordination site is N,O followed by N3 at only 1.2 kcal mol−1. The O,O topology results in being at a higher energy (13.8 kcal mol−1). For N,O coordination, analysis of the Cu–X bond lengths (Fig. 3 and 4 and Table S1, ESI†) reveals that the Cu ion interacts with the three nitrogen atoms of the peripheral macrocycle, while the bond with the O1 oxygen is weak since Cu–O1 assumes a value of 2.812 Å. In addition, the Cu2+ interacts with the two water molecules at distances of 2.084 and 2.355 Å. The situation is slightly different for the interaction with the anionic ligand. In fact, although the preferred coordination continues to be N,O, the other two are at higher energies (11.2 and 20.2 kcal mol−1 for N3 and O,O, respectively). In the absolute minimum, the Cu–O1 bond is strong (Cu–O distance is 1.957 Å) due to the presence of a more negative charge on the interacting oxygen atom, and the solvation water molecules are away from the metal center (more than 3.1 Å).
 | ||
| Fig. 4 Optimized structures for Cu–L2 (a) and Cu–L2− (b) complexes. Distances are in Å. | ||
Analysis of the spin density maps reveals that in the N,O coordination mode, for both the Cu–L1 and Cu–L1− systems (see Fig. S1 and S2, ESI†), the spin density is almost distributed on the entire molecule, enhancing the stability of this conformation.
To discuss the complexation process for the L2 ligand, we note that for both the considered species, the N,O coordination is not a minimum in the potential energy surfaces. For the L2 neutral species the N4 topology is preferred over the O,O topology by 18.1 kcal mol−1 (see Table 4). From Table S1 (ESI†) and Fig. 4 it is possible to see that the Cu2+ ion is bonded with the N4 and N10 nitrogen atoms of the pyridinophane ring (2.085 and 2.073 Å for Cu–N4 and Cu–N10 bonds, respectively) and with the oxygen atoms of two water molecules with bond distances of about 2.0–2.1 Å. For the anionic form of L2, the preferred coordination site results in being the O,O topology while the N4 lies at only 1.6 kcal mol−1. In the O,O mode, the copper ion forms two bonds with the O1 and O6 atoms of the phenyl ring, assuming the values of 1.918 Å (Cu–O1) and 2.343 Å (Cu–O6). There are two other strong interactions with the oxygen atoms of two water molecules with bond distances of 2.031 and 2.244 Å.
For both anionic species (L1− and L2−) in the O,O topology the copper ion shows an octahedral coordination geometry.
Furthermore, the spin density behavior reveals in the neutral complex (Fig. S1, ESI†) that the resulting distribution with N4 coordination being shared in the two molecular fragments, whereas with O,O chelation it is more localized. In the charged complexes (Fig. S2, ESI†) spin delocalization is found, for both coordination modes, in only one segment, in agreement with the small energy difference between the complexes.
Comparing the molecular electrostatic potential of the L1 and L2 complexes (Fig. S3, ESI†) it is possible to note that in the L1 ligand the negative charge is mainly concentrated on the N3 site while in the L2 ligand, albeit to only a minor extent, the other fragment is also implicated. This distribution can be correlated with the fact that in L1 the preferred coordination site is the N,O.
According to the data gathered in Table 4, for the chelation processes predicted to be viable from a thermochemical point of view (exergonic or slightly endergonic reaction pathways) we computed the complexation kinetic constants according to a previously suggested procedure48 and taking into account the populations of the complexes estimated through the Maxwell–Boltzmann distribution (see Table 4).
The obtained apparent equilibrium constants (Kappf) for the L1 and L2 ligands were found to be 3.16 × 1025 and 9.66 × 1016 M−1, respectively, indicating that the former is predicted to be a stronger Cu(II)-chelating agent that can reduce the Cu(II) free ions produced by the Fenton reactions associated with Alzheimer's disease.
Conclusions
A careful quantum mechanical study on the antioxidant and copper-chelating power of two new molecules, proposed as combined multiple target agents against Alzheimer's disease, has been performed in the framework of density functional theory. From our results, the following conclusions can be drawn:The computed pKa values of the methoxyphenol group present in the 2-phenylbenzothiazole moiety show a higher value for the L2 species. The molar distribution at physiological pH indicates that for both molecules the neutral form is dominant. However, the anionic species, although present at a small concentration, should be considered for their contribution to both the antioxidant and Cu-chelating properties.
In lipid-like and water environments, the HAT mechanism results in being exergonic.
The obtained rate constants clearly indicate that L1 is much more efficient than L2 as an OOH scavenger in both the studied environments and that L1 is more efficient than Trolox, which is generally used as a reference antioxidant.
The Cu2+ preferred coordination sites have been determined. The computed apparent equilibrium constants clearly show that the L1 ligand is much more efficient as a chelating agent.
Our results well agree with the available experimental indications.
We hope that our results can further stimulate other investigations devoted to the possible use of these systems as multiple target drugs against Alzheimer's disease.
Author contributions
All authors contributed equally to this work and have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported in part by PLGrid Infrastructure (grant ID: plgantioxidants). The authors thank the Dipartimento di Chimica e Tecnologie Chimiche, Università della Calabria for the support.References
- T. P. C. Chierrito, S. P. Mantoani, C. Roca, C. Requena, V. Sebastian-Perez, W. O. Castillo, N. C. S. Moreira, C. Pérez, E. T. Sakamoto-Hojo, C. S. Takahashi, J. Jiménez-Barbero, F. J. Cañada, N. E. Campillo, A. Martinez and I. Carvalho, From dual binding site acetylcholinesterase inhibitors to allosteric modulators: A new avenue for disease-modifying drugs in Alzheimer's disease, Eur. J. Med. Chem., 2017, 139, 773–791 CrossRef CAS PubMed.
- F. Lewis, S. K. Schaffer and J. Sussex, Alzheimer's Research, UK, 2014, vol. 1, pp. 1–62 Search PubMed.
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors, Chem. Soc. Rev., 2017, 46, 310–323 RSC.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer's Disease, Parkinson's Disease, and Amyotrophic Lateral Sclerosis in the Last Decade, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS PubMed.
- C. Lane, J. Hardy and J. Schott, Alzheimer's disease, Eur. J. Neurol., 2018, 25, 59–70 CrossRef CAS PubMed.
- S. Tiwari, V. Atluri, A. Kaushik, A. Yndart and M. Nair, Alzheimer's disease: pathogenesis, diagnostics, and therapeutics, Int. J. Nanomed., 2019, 14, 5541–5554 CrossRef CAS PubMed.
- Z. He, J. L. Guo, J. D. McBride, S. Narasimhan, H. Kim, L. Changolkar, B. Zhang, R. J. Gathagan, C. Yue, C. Dengler, A. Stieber, M. Nitla, D. A. Coulter, T. Abel, K. R. Brunden, J. Q. Trojanowski and V. M. Y. Lee, Amyloid-β plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation, Nat. Med., 2018, 24, 29–34 CrossRef CAS PubMed.
- L. Blaikie, G. Kay and P. K. T. Lin, Current and emerging therapeutic targets of alzheimer's disease for the design of multi-target directed ligands, Med. Chem. Commun., 2019, 10, 2052–2072 RSC.
- J. Nasica-Labouze, P. H. Nguyen, F. Sterpone, O. Berthoumieu, N.-V. Buchete, S. Coté, A. De Simone, A. J. Doig, P. Faller, A. Garcia, A. Laio, M. S. Li, S. Melchionna, N. Mousseau, Y. Mu, A. Paravastu, S. Pasquali, D. J. Rosenman, B. Strodel, B. Tarus, J. H. Viles, T. Zhang, C. Wang and P. Derreumaux, Amyloid β Protein and Alzheimer's Disease: When Computer Simulations Complement Experimental Studies, Chem. Rev., 2015, 115, 3518–3563 CrossRef CAS PubMed.
- E. von Schaper, Everything but amyloid: new thinking prompts FDA revamp, Nat. Biotechnol., 2018, 36, 483–484 CrossRef CAS PubMed.
- Y. Liu, M. Nguyen, A. Robert and B. Meunier, Metal Ions in Alzheimer's Disease: A Key Role or Not?, Acc. Chem. Res., 2019, 52, 2026–2035 CrossRef CAS PubMed.
- K. P. Kepp, Bioinorganic chemistry of Alzheimer's disease, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Oxidative stress and the amyloid beta peptide in Alzheimer's disease, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- M. A. Greenough, J. Camakaris and A. I. Bush, Metal Dyshomeostasis and Oxidative Stress in Alzheimer's Disease, Neurochem. Int., 2013, 62, 540–555 CrossRef CAS PubMed.
- E. Atrian-Blasco, E. Cerrada, P. Faller, M. Laguna and C. Hureau, Role of PTA in the prevention of Cu(amyloid-b) induced ROS formation and amyloid-b oligomerisation in the presence of Zn, Metallomics, 2019, 11, 1154–1161 CrossRef CAS PubMed.
- B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Cu(II) Affinity for the Alzheimer's Peptide: Tyrosine Fluorescence Studies Revisited, Anal. Chem., 2013, 85, 1501–1508 CrossRef CAS PubMed.
- T. R. Young, A. Kirchner, A. G. Wedd and Z. Xiao, An integrated study of the affinities of the Aβ16 peptide for Cu(I) and Cu(II): implications for the catalytic production of reactive oxygen species, Metallomics, 2014, 6, 505–517 CrossRef CAS PubMed.
- J. T. Pedersen, K. Teilum, N. H. H. Heegaard, J. Østergaard, H.-W. Adolph and L. Hemmingsen, Rapid Formation of a Preoligomeric Peptide–Metal–Peptide Complex Following Copper(II) Binding to Amyloid β Peptides, Angew. Chem., Int. Ed., 2011, 50, 2532–2535 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, J. Kim and L. M. Mirica, The effect of Cu2+ and Zn2+ on the Aβ42 peptide aggregation and cellular toxicity, Metallomics, 2013, 5, 1529–1536 CrossRef CAS PubMed.
- M. W. Beck, J. S. Derrick, R. A. Kerr, S. B. Oh, W. J. Cho, S. J. C. Lee, Y. Ji, J. Han, Z. A. Tehrani, N. Suh, S. Kim, S. D. Larsen, K. S. Kim, J. Y. Lee, B. T. Ruotolo and M. H. Lim, Structure-mechanism-based engineering of chemical regulators targeting distinct pathological factors in Alzheimer's disease, Nat. Commun., 2016, 7, 13115 CrossRef CAS PubMed.
- A. K. Sharma, S. T. Pavlova, J. Kim, D. Finkelstein, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Bifunctional compounds for controlling metal-mediated aggregation of the Aβ42 peptide, J. Am. Chem. Soc., 2012, 134, 6625–6636 CrossRef CAS PubMed.
- A. K. Sharma, J. Kim, J. T. Prior, N. J. Hawco, N. P. Rath, J. Kim and L. M. Mirica, Small bifunctional chelators that do not disaggregate amyloid β fibrils exhibit reduced cellular toxicity, Inorg. Chem., 2014, 53, 11367–11376 CrossRef CAS PubMed.
- H.-J. Cho, A. K. Sharma, Y. Zhang, M. L. Gross and L. M. Mirica, A Multifunctional Chemical Agent as an Attenuator of Amyloid Burden and Neuroinflammation in Alzheimer's Disease, ACS Chem. Neurosci., 2020, 11, 1471–1481 CrossRef CAS PubMed.
- A. K. Sharma, J. W. Schultz, J. T. Prior, N. P. Rath and L. M. Mirica, Coordination chemistry of bifunctional chemical agents designed for applications in 64Cu PET imaging for Alzheimer's disease, Inorg. Chem., 2017, 56, 13801–13814 CrossRef CAS PubMed.
- M. E. Alberto, T. Marino, N. Russo, M. Toscano and E. Sicilia, The performance of density functional based methods in the description of selected biological systems and processes, Phys. Chem. Chem. Phys., 2012, 14, 14943–14953 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone and G. A. Petersson, et al., Gaussian 09, revision D.01, 2014 Search PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. Galano and J. R. Alvarez-Idaboy, A computational methodology for accurate predictions of rate constants in solution: application to the assessment of primary antioxidant activity, J. Comput. Chem., 2013, 34, 2430–2445 CrossRef CAS PubMed.
- A. Galano and J. R. Alvarez-Idaboy, Computational strategies for predicting free radical scavengers’ protection against oxidative stress: where are we and what might follow?, Int. J. Quantum Chem., 2019, 119, 25665 CrossRef.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, Current status of transition-state theory, J. Phys. Chem., 1996, 100, 12771–12782 CrossRef CAS.
- M. G. Evans and M. Polanyi, Some applications of the transition state method to the calculation of reaction velocities, especially in solution, Trans. Faraday Soc., 1935, 31, 875–882 RSC.
- R. A. Marcus, Electron transfer reactions in chemistry: Theory and experiment (Nobel lecture), Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef.
- F. C. Collins and G. E. Kimball, Diffusion-controlled reaction rates, J. Colloid Sci., 1949, 4, 425–437 CrossRef CAS.
- A. Galano, A. Perez-Gonzalez, R. Castaneda-Arriaga, L. Munoz-Rugeles, G. Mendoza-Sarmiento, A. Romero-Silva, A. IbarraEscutia, A. M. Rebollar-Zepeda, J. R. Leon-Carmona, M. A. Hernandez-Olivares and J. R. Alvarez-Idaboy, Empirically Fitted Parameters for Calculating pKa Values with Small Deviations from Experiments Using a Simple Computational Strategy, J. Chem. Inf. Model., 2016, 56, 1714–1724 CrossRef CAS PubMed.
- S. Ahmadi, T. Marino, M. Prejanò, N. Russo and M. Toscano, Antioxidant Properties of the Vam3 Derivative of Resveratrol, Molecules, 2018, 23, 2446 CrossRef PubMed.
- A. Galano, G. Mazzone, R. Alvarez-Diduk, T. Marino, J. R. Alvarez-Idaboy and N. Russo, Food Antioxidants: Chemical Insights at the Molecular Level, Annu. Rev. Food Sci. Technol., 2016, 7, 335–352 CrossRef CAS PubMed.
- Z. Milanovic, J. Tosovic, S. Markovic and Z. Markovic, Comparison of the scavenging capacities of phloroglucinol and 2,4,6-trihydroxypyridine towards HO radical: a computational study, RSC Adv., 2020, 10, 43262–43272 RSC.
- T. C. Ngo, T. H. Nguyen and D. Q. Dao, Radical Scavenging Activity of Natural-Based Cassaine Diterpenoid Amides and Amines, J. Chem. Inf. Model., 2019, 59, 766–776 CrossRef CAS PubMed.
- R. Luckaya, R. D. Hancock, I. Cukrowskia and J. H. Reibenspiesb, Study of protonation of 1,4,7-tris(2_hydroxyethyl)- 1,4,7-triazacyclononane, and its complexes with metal ions, by crystallography, polarography, potentiometry, molecular mechanics and NMR, Inorg. Chim. Acta, 1996, 246, 159–169 CrossRef.
- M. Leopoldini, N. Russo and M. Toscano, The molecular basis of working mechanism of natural polyphenolic antioxidants, Food Chem., 2011, 125, 288–306 CrossRef CAS.
- A. Parise, B. C. De Simone, T. Marino, M. Toscano and N. Russo, Quantum Mechanical Predictions of the Antioxidant Capability of Moracin C Isomers, Front. Chem., 2021, 9, 666647 CrossRef CAS PubMed.
- A. Galano and A. Martínez, Capsaicin, a tasty free radical scavenger: Mechanism of action and kinetics, J. Phys. Chem. B, 2012, 116, 1200–1208 CrossRef CAS PubMed.
- M. Spiegel, T. Marino, M. Prejanò and N. Russo, On the scavenging ability of scutellarein against the OOH radical in water and lipid-like environments: A theoretical study, Antioxidants, 2022, 11, 224 CrossRef CAS PubMed.
- M. Spiegel, T. Andruniów and Z. Sroka, Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship. A Quantum Chemical Study, Antioxidants, 2020, 9, 461 CrossRef PubMed.
- M. E. Alberto, N. Russo, A. Grand and A. Galano, A physicochemical examination of the free radical scavenging activity of Trolox: mechanism, kinetics and influence of the environment, Phys. Chem. Chem. Phys., 2013, 15, 4642–4650 RSC.
- A. Pérez-González, M. Prejanò, N. Russo, T. Marino and A. Galano, Capsaicin, a powerful OH-inactivating ligand, Antioxidants, 2020, 9, 1247 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01918c |
| This journal is © the Owner Societies 2022 |