
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantifying the ion coordination strength in polymer electrolytes†
Rassmus
Andersson
 ,
Guiomar
Hernández
and
Jonas
Mindemark
*
,
Guiomar
Hernández
and
Jonas
Mindemark
*
Department of Chemistry – Ångström Laboratory, Uppsala University, Box 538, SE-751 21 Uppsala, Sweden. E-mail: jonas.mindemark@kemi.uu.se
First published on 28th June 2022
Abstract
In the progress of implementing solid polymer electrolytes (SPEs) into batteries, fundamental understanding of the processes occurring within and in the vicinity of the SPE are required. An important but so far relatively unexplored parameter influencing the ion transport properties is the ion coordination strength. Our understanding of the coordination chemistry and its role for the ion transport is partly hampered by the scarcity of suitable methods to measure this phenomenon. Herein, two qualitative methods and one quantitative method to assess the ion coordination strength are presented, contrasted and discussed for TFSI-based salts of Li+, Na+ and Mg2+ in polyethylene oxide (PEO), poly(ε-caprolactone) (PCL) and poly(trimethylene carbonate) (PTMC). For the qualitative methods, the coordination strength is probed by studying the equilibrium between cation coordination to polymer ligands or solvent molecules, whereas the quantitative method studies the ion dissociation equilibrium of salts in solvent-free polymers. All methods are in agreement that regardless of cation, the strongest coordination strength is observed for PEO, while PTMC exhibits the weakest coordination strength. Considering the cations, the weakest coordination is observed for Mg2+ in all polymers, indicative of the strong ion–ion interactions in Mg(TFSI)2, whilst the coordination strength for Li+ and Na+ seems to be more influenced by the interplay between the cation charge/radius and the polymer structure. The trends observed are in excellent agreement with previously observed transference numbers, confirming the importance and its connection to the ion transport in SPEs.
Introduction
The implementation of solid polymer electrolytes (SPEs) into batteries has rendered great interest due to their inherently advantageous mechanical, thermal and electrochemical stabilities.1–4 However, the relatively low ionic conductivity compared to liquid electrolytes, resulting from the slower transport mechanism, is an obstacle to overcome for full realization. Fundamental understanding of the transport properties and cation mobility in the systems is therefore required in the SPE development process. In SPEs, the cation is transported by segmental motions of the polymer chains. With the movement of the chains, new energetically favored coordination sites become available allowing for cation transport by continually exchanging ligands in the solvation shell of the ion. The cation transport can thus be described as a series of intra- and interchain solvation and desolvation events between the cation and the ligands in the polymer structure.4 In the prevailing theories on ion transport in such systems, the attention is primarily aimed at the polymer dynamics as the main contributor to cation transport. However, recent studies have uncovered that it is more of a “trifecta” that governs the ion transport, consisting of the chain dynamics together with effects of the polymer architecture and the ion coordination strength.5–9 The influence of the polymer architecture on the transport properties was revealed by Ebadi et al. as well as by the Balsara group, where significant structural effects were observed by incorporation of side chains and restructuring of the polymer backbone, respectively.6–8 It has furthermore been revealed that the ion coordination strength has a direct influence on the cation transport where weaker coordination bonds allows for easier ligand exchange, resulting in higher cation mobility and a higher cation transference number (T+), while stronger coordination bonds promote dissociation from the anion, leading to less ion–ion association in the system.5,10 The ion coordination thus has a very direct effect on the ion transport properties and, ultimately, the performance of an SPE in an operating battery. Still, the full effect of the coordination strength on the ion transport properties in polymers and its relation to polymer structure is relatively unknown. Clearly, a complete understanding of these phenomena is essential in order to create a unifying model of ion transport in these materials, enabling directed design of next-generation SPE host polymers.The coordination strength in a system can be divided into two contributions. Firstly, the ion–dipole interactions between the cation and the coordinating ligands and secondly, the coordination number (CN), i.e. the number of coordinating groups to a single cation. The ion–dipole interactions between the cation and the ligands is highly dependent on the intrinsic properties of the coordinating functional group and the structure of the polymer, which influences the bond distances and the coordination enthalpy of the coordinating bonds.9,11,12 The CN for any given ion is not a static value, since the coordination environment is dynamic and constantly evolving, but can be expressed as a number average of coordinating ligands to one cation in a system. The cation coordination strength is also influenced by the cation itself. The size and charge of the ion will influence its polarizability, the coordination distance and sterically limit the number of coordinating ligands, ultimately affecting the coordination strength.12,13
In earlier studies, the CN has mainly been determined for different systems, often by computational methods, but also experimentally by both scattering and spectroscopic methods, although frequently resulting in rather broad CN intervals.14–20 Only the CN itself, however, does not provide the whole picture of the coordination strength effect on the ion transport in a system, since the ion–dipole interactions between the cation and the ligand are excluded. The scarcity of coordination strength studies, hampered by a lack of suitable methods to quantify this phenomenon, currently limits our understanding of the role of the coordination chemistry on ion transport in SPEs. To fully understand the ion transport, it is desirable to explore the effects of the size and charge of the cations as well as the functional group of the coordinating ligand on ion coordination strength in systems. In this study, three methods based on conventional NMR and FTIR measurements are presented to determine the coordination strength properties for different salt and polymer hosts to enable a thorough understanding of the influence of coordination chemistry on ion transport characteristics in SPEs.
Results and discussion
In terms of polymer host materials, poly(ethylene oxide) (PEO) is by far the most well-studied material. PEO has ether groups in the polymer backbone acting as cation-coordinating moieties, which can be contrasted with, e.g., polyesters and polycarbonates, which coordinate by means of carbonyl groups. Intrinsically, PEO provides good ion solvation properties, one asset necessary for ion transport in SPEs, due to the chelating effect, originating from the “optimally” oxygen dense structure and the strong ion–dipole interaction between its ether oxygens and the cations.21,22 The strong interaction between the ligands in PEO and the cations has in recent studies been seen to impede the cation mobility, immobilizing the cation due to the high coordination strength, leading to a fairly low cation transference number (T+) in the SPE.23 In this perspective, the carbonyl-based polymers PCL and PTMC are interesting to investigate, since they demonstrate weaker coordination strength with the cations, leading to higher relative cation mobilities and thereby higher T+.5 In Fig. S1 (ESI†), the molecular structures of the polymers and salts investigated in this study are shown. Important to remember in this regard is that the coordination strength is influenced by both the individual interaction between the ligand and the cation, as well as the number of coordinating ligands to one cation, (the CN), as recently demonstrated by Eriksson et al. in copolymer systems where the composition of ester and carbonate groups was varied.24A technique that is sensitive to changes in the coordination environment is NMR. In a solution containing a salt as well as a cation-coordinating polymer, the following equilibrium is established:
| M+(solvent)x + y polymer ⇌ M+(polymer)y + x solvent | (1) |
The assessment of the coordination strength based on the equilibrium in eqn (1) is valid in a system where the salt concentration is sufficiently low to exclude a significant influence of ion–ion association. To minimize the ion–ion association, TFSI-based salts were used throughout this study, which provide good ion dissociation due to the delocalized charge distribution in the bulky TFSI anion. By observing the combined absorption band of the C–S and C–N stretching in TFSI− at 789 cm−1 with FTIR of dissolved LiTFSI in acetonitrile (ACN, Fig. 1), it could be confirmed that at 100 mM and 200 mM LiTFSI, the combination band remains unshifted, indicating little or no ion–ion association. In contrast, at 2 M and 4 M LiTFSI, the band shifts to 793 cm−1 and 798 cm−1, respectively, indicating the formation of ion pairs and ion clusters. Based on this, salt concentrations were kept at ≤100 mM for the solution-based NMR and FTIR measurements.
 | ||
| Fig. 1 FTIR absorption spectra demonstrating the ion–ion association effect through the red shift of the combination band of C–S and C–N stretching in the TFSI− at 800–785 cm−1 upon cation coordination.27,28 The absorbance is normalized by the concentration of the salt. | ||
NMR titration data with the 7Li chemical shift when adding PEO, PCL or PTMC to the solution is shown in Fig. 2.5 It is clear that the highest rate of chemical shift change as well as the lowest saturation value (plateau) is seen for PEO, indicating a stronger ion coordination strength for the polyether compared to the polycarbonate PTMC and polyester PCL. This can be explained by the chelating structures that PEO forms by wrapping its ether oxygens around the Li+ cation.29 Examples of the measured 7Li spectra and how the 7Li chemical shift changes upon addition of PEO can be seen in Fig. S2 (ESI†).
Expanding the comparison to the larger cation Na+, the same tendency is seen with a large 23Na chemical shift change observed for PEO and smaller for the polyester and polycarbonate (Fig. 2(b)). In accordance with the coordination strength order for Li+ of PEO > PCL > PTMC (Fig. 2(a)) observed by Rosenwinkel et al.,5,24 Na+ has a weaker coordination to the polycarbonate and polyester than to PEO.
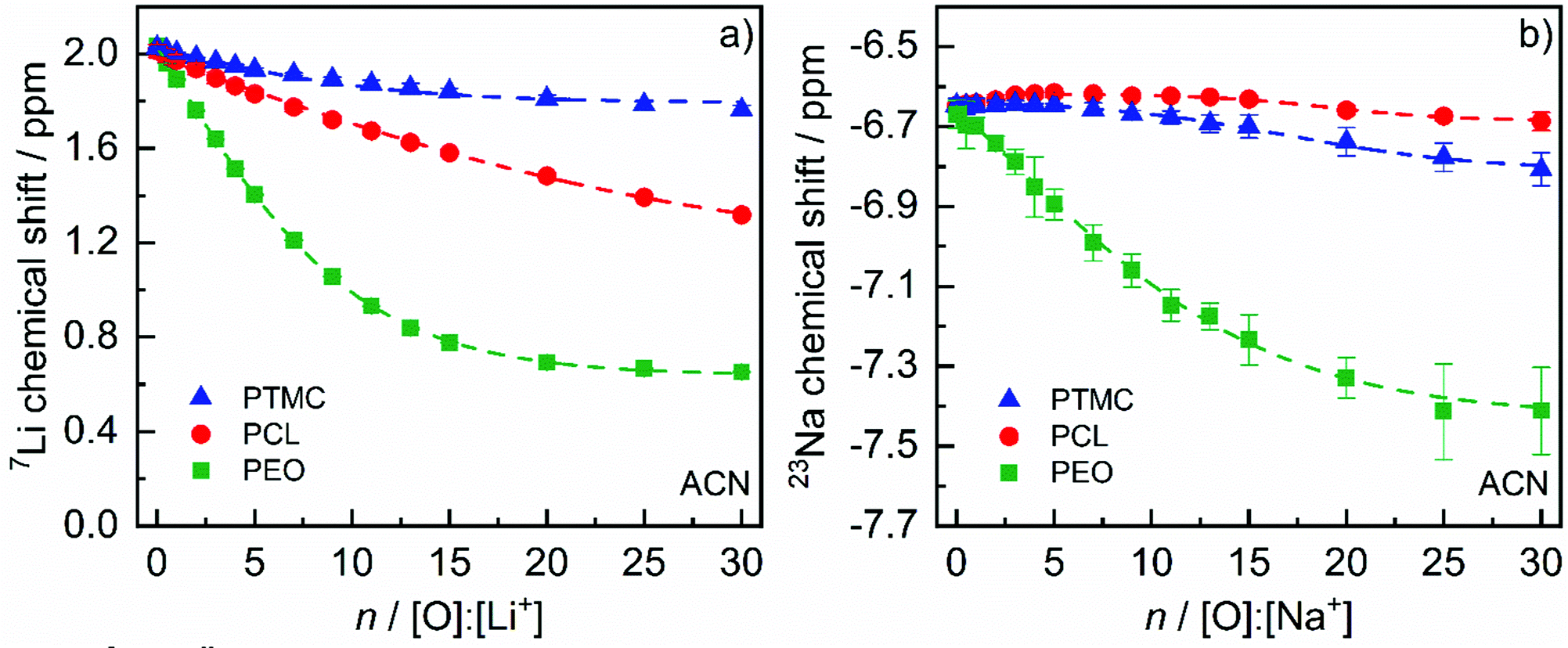 | ||
| Fig. 2 (a) 7Li and (b) 23Na chemical shift dependence of the polymer concentration, expressed as the ratio of coordinating polymer O to cations, in ACN solutions. 1 M LiCl or NaCl in D2O was used as an external reference for the measurements. The results are triplicates with the error bars representing the standard deviation. Dashed lines represent fits to either eqn (S1) (ESI†) (all Li+ systems as well as PEO:Na+) or S2 (PCL and PTMC with Na+). The data in (a) is reused from ref. 5. | ||
However, when small amounts of PCL and PTMC are added to the solutions with Na+, a different behavior is identified. Instead of observing an immediate exponential decline, a small initial increase of the chemical shift is observed before a decline commences. The increase is an indication of a weaker coordination strength for the polycarbonate and polyester to Na+ than to Li+. The weaker coordination strength for Na+ has been demonstrated by Johansson et al. by density-functional theory (DFT) methods to stem from the smaller charge/radius ratio for Na+, resulting in a reduced binding energy compared to Li+ with about 20%.13 The increase in chemical shift is still surprising, though, as it implies that small additions of polycarbonate and polyester promote a stronger interaction between Na+ and the acetonitrile solvent molecules. This can, however, be rationalized by considering the change in magnetic susceptibility in the solution on addition of the polymer to the solution, shifting the baseline to higher values.30 Indeed, the same phenomenon should be present in the case of Li+ and Na+ with PEO; however, since the effect of coordination to the polymer is so pronounced, the effect of the change in magnetic susceptibility becomes disguised. At sufficiently high polymer concentrations, the polymer–Na+ coordination dominates over the magnetic susceptibility effect and a declining chemical shift is indeed observed. Considering the coordination strength order for Na+ in Fig. 2(b), PTMC and PCL are switched compared to Li+ and is as follows for Na+: PEO > PTMC > PCL. The discrepancy in chemical shift between the polycarbonate and polyester is, however, modest, indicating a similar coordination strength to Na+ by these polymers. This difference has been observed in earlier studies,14 even though in general similar coordination structures are observed for Na+ and Li+.12,31
While the NMR titrations allow for qualitative comparisons across a series of polymers, it is essentially limited in scope to Li+ and Na+ due to the lack of suitable NMR-active nuclei for other relevant cations such as K+, Mg2+ and Ca2+, due to low abundances and/or excessive peak broadening along with minimal peak shifts (Fig. S3, ESI†). For simple equilibria, the equilibrium constant for complex formation can be extracted from the titration curve, but for more complex systems, where there is a distribution of coordination environments with different CN, this quickly becomes complicated.25,32,33 The ligand exchange is also too fast compared to the NMR timescale to resolve the different coordination environments.
These limitations can be avoided by instead utilizing infrared spectroscopy. By utilizing FTIR, information about the cation coordination can be extracted, regardless of cation studied, by detecting molecular bond vibrations in the coordination ligands that are sensitive to ion coordination. To facilitate quantitative analysis of FTIR data, isolated absorption bands are preferred for the analysis. Considering PEO, the ether absorption band around 1200–1100 cm−1 is demanding to quantitatively analyze with confidence, due to many strong absorption bands in a narrow wavenumber range.34,35
The carbonyl stretch absorption band, on the other hand, is more accessible at 1800–1700 cm−1, a region with few other distinct vibrations.36 However, in order to reliably characterize any polymer, regardless of coordinating groups, it is better to shift the attention away from the polymer, and instead use the solvent as a probe. Assuming that the CN does not change as the ligands are exchanged, i.e., that the sum of the variables x and y in eqn (1) is constant, it is possible to deduce the average CN of the coordinating polymer by measuring the average CN of the coordinating solvent molecules. Considering the solvent, it is again preferential to use one with absorption bands that are isolated from the polymer and salt absorption bands. Nitrile solvents are advantageous for this purpose, with their C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N absorption band in the range of 2300–2200 cm−1.37,38
N absorption band in the range of 2300–2200 cm−1.37,38
Since the deconvoluted area of the coordinated and non-coordinated absorption bands in the FTIR spectra is proportional to the concentration of the functional group for each band, respectively, the apparent coordination number (CNapp) for the M+ can be calculated at any specific concentration of the polymer in the solvent, as described by Mindemark et al.39 Knowing CNapp with respect to the solvent, CNapp with respect to the polymer can be directly calculated, assuming that the total coordination number will be constant, i.e., remain the same as CNapp with respect to the solvent at the beginning of the titration, when there is yet no polymer present in the solution.
As seen in Fig. 3(a), this method gives a similar response as the earlier NMR titrations, with the exception that the values more directly represent the coordination number. However, looking at the data in Fig. 3(a), it becomes immediately apparent why we have chosen to refer to the obtained CN as the apparent CN. The obtained values are obviously unrealistic for Li+ complexes in ACN, which have been reported as in the range of 4–6 for both Li+ and Na+.19,40–42 While the values are at least superficially reasonable in propionitrile (PPN, Fig. 3(b)), they become even more ridiculous for Mg2+ in ACN (Fig. 3(c)). It thus appears that the extinction coefficient of the CN stretch vibration changes on coordination and that this change is highly dependent on both the particular nitrile solvent used and the cation that is interacting with the coordinating group. Although CN up to 8 have been determined for Na+ by computational methods in the literature,15 which is similar to what is observed for Li+ in ACN (Fig. 3), the ridiculously high CN of Mg2+ in PPN in addition to the large CN variation for cations between the solvents, indicate that a reliable experimental determination of the extinction coefficient is required to correct CNapp. Considering that the extinction coefficients seem to be dependent on all species in the system (cation, polymer and solvent),43 it may be beneficial to determine the absolute CN for the cation in the pure solvent by other methods and adjust the data accordingly. Without independently measuring the true CN with a different technique, it is not possible to correct for this discrepancy and obtain the CN instead of the CNapp.
 | ||
| Fig. 3 FTIR results of the derived nitrile CNapp of Li+, Na+ and Mg2+ as a function of n = [O]/[M+] showing the effect of (a) polymer (PEO, PCL, PTMC) with Li+ dissolved in ACN, (b) solvent (ACN, PPN) with Li+ and PEO/PCL and (c) cation (Li+, Na+, Mg2+) with PEO dissolved in PPN. All results are triplicates with the standard deviation represented by error bars. Dashed lines are fits to the empirical function in eqn (S1) (PEO systems) and (S2) (PCL and PTMC systems) (ESI†). In Fig. S4 and S5 (ESI†), deconvolution examples of the results in (b) and (c) are shown. | ||
Alas, while enabling measurements of a much wider array of cations, this method thus becomes limited to the same type of qualitative comparisons as the NMR measurements. Since these FTIR titrations essentially measure the same thing as the NMR titrations, it is also possible to fit the CNapp decrease for PEO to the same empirical functions (eqn (S1) and (S2), ESI†). Considering the polycarbonate and polyester, a more pronounced sigmoidal behavior is seen, recognized as an initial “plateau” at low polymer concentrations before the CNapp declines, which differs from the NMR results with Li+ and instead is similar to the observation for Na+ with NMR. In the same manner, CNapp was fitted to the empirical function in eqn (S2) (ESI†) for PTMC and PCL. The same behavior was observed for both PPN and ACN solvents (Fig. 3(b)), with a small difference in fitting parameters to the empirical fit (eqn (S2), ESI†) for PCL between the solvents.
Furthermore, the CNapp for Li+, Na+ and Mg2+ in PEO was explored in Fig. 3(c), where a large decline of the CNapp is observed in all cases, although it is more pronounced for Mg2+, suggesting a strong coordination strength to PEO. Based on the observations by Johansson et al., stronger binding to Mg2+ is expected since this divalent ion has a similar radius to Li+ resulting in a larger charge/radius ratio compared to both Li+ and Na+.13 However, since the extinction coefficient for the observed vibrations of interest are unknown, normalizations cannot be performed, making the cation comparison more speculative than concrete, only strengthened by previous observations.
Just as for the NMR titrations, based on FTIR data it should be relatively straightforward to determine the equilibrium constant for simple equilibria involving formation of complexes with well-defined stoichiometry.32,33 This should in principle be possible to do also for these much more complex systems, but in the absence of reliable CN values, this becomes exceedingly difficult.
This prompts a shift of attention to an equilibrium that is not reliant on the CN at all. The dissolution of a simple binary salt MX in a polymeric (or low-molecular-weight) solvent involves the dissociation of ion pairs according to the following equilibrium:
| MX ⇌ M+ + X− | (2) |
In the case of divalent cations, such as Mg2+, two anions are involved in the dissociation equilibrium. Compared to the previous methods, this method is liberated from the assumption of no significant amount of ion pairing. Instead, the method relies on the existence of some amount of ion–ion association. Since the salt dissociation in a system consisting of a salt dissolved in a polymer is driven by the coordination of ions by the polymer ligands, ΔG for the dissociation will be a measure of the ion coordination strength, i.e., the combined contribution from the electrostatic cation–ligand interactions and the CN. The extent of ion–ion association, i.e., the position of the equilibrium in eqn (2), is dependent on temperature. The ion–ion association in SPEs has been reported to increase with temperature, indicating an exothermic ion dissociation (solvation process) according to le Châtelier's principle.44,45 By determining the temperature dependence of the ion dissociation equilibrium, the thermodynamic parameters of the system can be extracted according to the van’t Hoff equation:
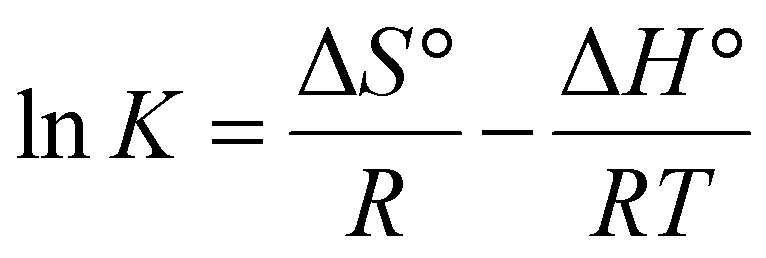 | (3) |
This equation holds under the assumption that the enthalpy and the entropy are independent of the temperature, which is reasonable at small temperature variations. By performing FTIR measurements at various temperatures, an equilibrium constant at each temperature can be determined from the integrated areas of specific TFSI− bond vibrations for the dissociation equilibrium constant in eqn (4):
 | (4) |
Since [M+] = [TFSI−] for binary salts, eqn (4) is simplified under the assumption that the number of paired anions equals the number of paired cations:
 | (5) |
For Mg2+, the equation becomes slightly different since two TFSI− are involved in the equilibrium, hence [M+] = 1/2[TFSI−] and the equilibrium constant is simplified as:
 | (6) |
For determination of the concentration of dissociated anions, the S–N band vibration in TFSI− with the main peak of non-associated anions at 740 cm−1, ion pairs and aggregates (associated TFSI−) at slightly higher wavenumbers and a shoulder of additional non-associating TFSI− conformers at slightly lower wavenumbers is suitable, since it shows little or no overlap with the absorption bands in PEO, PCL and PTMC (Fig. S6, ESI†).27,28,46–49 However, at temperatures below the melting point of PCL, C–H band vibrations in crystalline PCL appear at 732 cm−1 and 711 cm−1 (Fig. S6, ESI†), making it difficult to deconvolve the absorption band with certainty.50 These measurements have therefore been disregarded in the analysis in order not to misinterpret the results. The concentrations of the associated and non-associated TFSI−, respectively, are calculated by multiplying the ratio of the area fraction of associated/non-associated anions from the deconvolution with the total salt concentration, derived by measuring the density of the polymer electrolytes (Table S1, ESI†). In Fig. S7 (ESI†), an example of the deconvolution at different temperatures is shown.
By performing FTIR measurements on solvent-free samples, only containing polymers and salts at various temperatures, the coordination strength in systems containing Li+, Na+ and Mg2+ was resolved by determining the dissociation energies of their TFSI salts in PEO, PCL and PTMC. In Fig. 4, van’t Hoff plots of the dissociation of LiTFSI, NaTFSI and Mg(TFSI)2 in PEO, PCL and PTMC are displayed, showing a linear dependence of the natural logarithmic equilibrium constant of the inverse temperature. The plot confirms that the enthalpy and entropy are independent of the temperature (in the measured interval) and can be extracted from the van’t Hoff plot, from the y-axis intercept and the slope of the linear fit of eqn (3). The extracted values of the enthalpy, entropy and derived Gibbs free energy at 25 °C are presented in Table 1 for all investigated systems.
 | ||
| Fig. 4 Van’t Hoff plots of the equilibrium constant K as a function of the inverse temperature for the dissociation equilibrium of (a) LiTFSI, (b) NaTFSI and (c) Mg(TFSI)2 in PCL, PTMC and PEO. Dashed lines represent the linear fit to the data. For PCL, only values at higher temperatures are presented due to overlap with peaks for crystalline PCL in the FTIR spectra below its melting point.50 | ||
| Salt | Polymer | ΔHo/kJ mol−1 | ΔSo/J mol−1 K−1 | ΔGo/kJ mol−1 |
|---|---|---|---|---|
| LiTFSI | PEO | −4.6 | 18.7 | −10.2 |
| PCL | −6.8 | 5.1 | −8.4 | |
| PTMC | −4.6 | 5.9 | −6.3 | |
| NaTFSI | PEO | −6.5 | 10.0 | −9.5 |
| PCL | −10.2 | −3.4 | −9.2 | |
| PTMC | −9.6 | −6.8 | −7.6 | |
| Mg(TFSI)2 | PEO | −8.8 | −4.0 | −7.6 |
| PCL | −12.6 | −23.8 | −5.5 | |
| PTMC | −9.4 | −15.9 | −4.6 |
In agreement with the observations with the previous approaches, the dissociation energy of the salt is most negative for PEO in all systems, indicating that the strongest coordination is attained for PEO regardless of the cation. This effect originates, as mentioned, from the chelating nature of PEO when it wraps itself around the cations.29 In a similar manner, PTMC consistently shows the most positive dissociation energy regardless of the cation, indicating the weakest coordination strength. Overall, the trend of the dissociation energy is the same regardless of cation, and follows from positive to negative dissociation energy: PTMC > PCL > PEO. This trend is identical to the T+ trend observed for Li+ in the same polymer systems in earlier studies, where a higher T+ was observed for the weaker-coordinating polymer.5,24 This correlation further suggests a higher T+ for Na+ and Mg2+ in PCL and PTMC than PEO, which indeed was observed for Na+ in a study by Sångeland et al.51 For Mg2+, however, these findings are particularly compelling, since the T+ for Mg2+ is difficult to determine with more available methods like the Bruce–Vincent method because of the difficulties associated with Mg stripping/plating.52 Therefore, by assuming that the same correlation between the coordination strength and T+ observed for Li+ and Na+ also applies for Mg2+, it can be anticipated that a higher T+ would be observed in PCL and PTMC than in PEO also for Mg2+. This also accords with observations by Eriksson et al., seeing a weaker electrostatic force and binding energy between the cation and coordinating ligand in polycarbonates than in polyesters.53 Considering the dissociation enthalpy and entropy of the salts, the observed trends deviate from the dissociation energy trend. Herein, the following order is observed for ΔH0 for all cations PEO > PTMC > PCL, while ΔS0 is most positive for PEO and varies between PCL and PTMC. This implies that the dissociation of the salts in PEO is more entropy-driven, whilst being more enthalpy-driven in PCL and PTMC. The large ΔS0 variations between the polymers (which in many cases is negative), is not uncommon for polymer electrolytes and imply that the loss of entropy of the chains upon salt dissolution exceeds the gain due to ion disordering. Compared to liquid electrolytes, negative entropies upon dissolution of salt are less common due to greater entropy losses for solvents.54
Comparing the salts, the dissolution energy order varies between the polymers and the order for PEO is as follows from weakly to strongly coordinating Mg(TFSI)2 > NaTFSI > LiTFSI, while for PCL and PTMC the order is Mg(TFSI)2 > LiTFSI > NaTFSI. It implies that the weakest coordination strength is observed for Mg2+ in all investigated polymers, which is opposite to the previous methods, where the equilibrium in eqn (1) was studied. However, considering that the reference point for the ion dissociation is the associated ion pair – which is different for the different cations – and that the high charge density of the Mg2+ ion is likely to lead to strong ion–ion interactions, this is not unreasonable. Nevertheless, the more positive dissociation energy observed for NaTFSI compared to LiTFSI in PEO, which implies weaker coordination strength for Na+, has previously been explained by the optimal coordination structure obtained for Li+ in PEO.22 In addition, the smaller charge/radius ratio for Na+ leads to increased ion–ion association, ultimately reducing the dissociation energy.13 In PCL and PTMC, however, the dissociation energy is more positive for LiTFSI than for NaTFSI. A more positive dissociation energy for Li-based salts was also observed by Nguyen et al. by DFT calculations in tetraglyme solvents, who discovered that smaller cations like Li+ and Mg2+ tend to adopt a CN of 5–6, while larger cations like Na+ prefer a CN of 8, interpreted to originate from the size difference.15 The higher CN for Na+ should have a direct impact on the dissociation energy of the salt, leading to a more negative dissociation energy for Na+ as observed in PCL and PTMC.
To summarize the findings, one quantitative method has been presented alongside two qualitative methods to determine the ion coordination strength of Li+, Na+ and Mg2+ in PEO, PCL and PTMC systems. Comparing the polymers, the coordination strength order is from strong to weak PEO > PCL > PTMC, in all cases but for Na+ determined with the NMR titration method (where the order PEO > PTMC > PCL was observed). The strong coordination strength for PEO can be ascribed to the chelating effect observed upon coordination to the cation, while the weak coordination of PTMC originates from the weak ion–dipole interaction with the cation. Considering the coordination strength order for the cations, the order in the quantitative analysis in PEO is from strong to weak Li+ > Na+ > Mg2+, and for the carbonyls Na+ > Li+ > Mg2+. The observations for Mg2+ are contradictive to the results from the qualitative methods, where Mg2+ clearly displayed the strongest coordination. However, due to the higher charge density of Mg2+, it is likely that a higher degree of ion–ion association exists. This revelation along with the diverging coordination strength order for the cations between PEO and the polycarbonyls (observed with the quantitative method) indicate that it is an interplay between the cation charge and size, and the polymer structure in the system that defines the dissociation energy and ultimately the coordination strength for the salts in the polymer. Nonetheless, the attained results considering the order of the polymer neatly confirm earlier observations outlined by Rosenwinkel et al. and Eriksson et al., simultaneously as they quantitatively explain the resulting mobilities and transference numbers for PEO and PCL–PTMC systems.5,24
Conclusion
The ion coordination strength is one out of three important parameters that control ion transport in polymer electrolytes, yet it is still relatively unexplored and its influence neglected. In this study, the ion coordination strength was investigated through the use of qualitative and quantitative methods based on NMR and FTIR spectroscopy. For the qualitative methods, the competitive equilibria of cations coordinating to polymer ligands vs. solvent molecules were investigated by NMR and FTIR measurements. Although different physical properties are observed for the techniques, similar trends of declining chemical shifts are obtained (and expected) since the methods basically measure the same thing, i.e. the chemical environmental and how it changes upon addition of polymer. Comparing the methods, the FTIR method has two advantages over the NMR method. Firstly, it is independent on what cation is observed, since it probes the vibrational bonds in the coordinating ligand rather than the magnetic spin of the nucleus of the ion, permitting any cation to be probed. Secondly, the CN for the cation can be determined if the extinction coefficient is known for the system. Considering the quantitative method, which studies the dissociation equilibria of a salt in a solvent-free polymer with FTIR, it has the major advantage of being able to determine quantitative values relating to the coordination strength in the system. Furthermore, the quantitative method excludes any assumptions about a negligible amount of ion–ion association for the determination of the coordination strength and can therefore be applied on systems with relevant SPE salt concentrations. However, since it is defined by the dissociation of a salt in a polymer, comparisons between salts should be performed with caution.Considering the observed systems, the strongest ion coordination strength was clearly observed for PEO with all approaches in all cation systems, stemming from its strong chelation of cations. In contrast, PTMC exhibited the lowest coordination strength for all cations in the investigated polymers, originating from weaker electrostatic interactions with coordinating cations. Interestingly, Mg2+ displayed strong coordination strength with the qualitative methods, while for the quantitative method, the weakest coordination strength was observed for Mg2+. Considering that the reference point for the dissociation studies is the association ions, the weaker measured coordination strength likely originates in the strong ion–ion interactions. For Li+ and Na+, the relative order changes between PEO and the carbonyl-coordinating polymers, where Li+ coordination is stronger in PEO and Na+ coordination is stronger in PCL and PTMC. These findings seem to be more dependent on the interplay of charge/radius ratios of the cation in combination with polymer structures and steric hindrance.
Through the presented approaches to determine the coordination strength in polymer electrolytes, the importance and its connection to the transport properties of the cations can be verified. With this, we move one step closer to attaining a complete comprehension of the ion transport properties of polymer electrolytes.
Experimental
Materials
PEO (Polymer Source, Canada, Mn = 4000 g mol−1) and PCL (Perstorp, Sweden, Capa 2402, Mn = 4000 g mol−1) were dried overnight at 50 °C in a vacuum oven. PTMC (Mn = 4000 g mol−1) were synthesized by ring-opening polymerization of the monomer trimethylene carbonate (TMC, Richman Chemical) with the initiator 1,3-propandiol (98%, Sigma Aldrich) and the catalyst stannous 2-ethylhexanoate (95%, Sigma-Aldrich) as described elsewhere.5,55 Briefly, in an argon-filled glovebox, the materials were added to a reactor and sealed before they were transferred to an oven outside the glovebox for polymerization for 72 h at 130 °C. During the initial few hours, the reactor was shaken regularly to ensure properly mixing of the materials. After the polymerization, the reactor was transferred back into an argon-filled glovebox where the polymer was recovered.Lithium bis(trifluoromethylsulphonyl)imide (LiTFSI, Sigma Aldrich) and sodium bis(trifluoromethylsulphonyl)imide (NaTFSI, Solvionic) were dried at 120 °C for 48 h in a vacuum oven and magnesium bis(trifluoromethylsulphonyl)imide (Mg(TFSI)2, Solvionic) was dried at 200 °C for 24 h in a vacuum oven before use. Acetonitrile (ACN) and propionitrile (PPN) were used as received.
NMR titrations
In an argon-filled glovebox, solutions of LiTFSI and polymers in ACN were prepared. The concentration of LiTFSI was kept constant at 50 mM in all solutions, while the polymer concentrations ranged between 0 and 2 M. The polymers studied were PEO, PCL and PTMC with Mn = 4000 g mol−1. The polymers were left overnight to dissolve in ACN under heating at 40 °C and stirring.The NMR-titrations of Li+ and Na+ were conducted at 25 °C on a 400 MHz NMR spectrometer (JEOL ECZ 400S). The NMR titration experiment were performed by first measuring the 7Li and 23Na chemical shift of solutions only containing dissolved LiTFSI or NaTFSI in ACN and then repeatedly measuring the chemical shifts after adding an increasing amount of polymer solution until the ratio of coordinating ligands to cations (n = [O]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[M+]) was 30. The concentration of LiTFSI/NaTFSI was kept constant at 50 mM in the measured solutions throughout the experiments.
:[M+]) was 30. The concentration of LiTFSI/NaTFSI was kept constant at 50 mM in the measured solutions throughout the experiments.
FTIR titrations
The FTIR measurements were conducted on a Bruker Vertex 70v FT-IR spectrometer with a liquid nitrogen-cooled MCT detector and RT-DLaTGs detector. The measurements were performed in transmission mode with an Omni transmission cell from Specac with either CaF2 or KBr glass windows and a 0.012 mm thick Mylar spacer in between the windows. The scanning velocity and aperture settings were adjusted to achieve an appropriate transmittance of 0.5–0.7 and an amplitude above 15000. 256 scans were performed for the experiments between the wavenumbers 8000–500 cm−1 with a resolution of 1–4 cm−1.
For each measurement, a background spectrum was recorded, before the sample was injected into the cell and a second spectrum of the sample was recorded. Measurements were conducted on the salts LiTFSI, NaTFSI and Mg(TFSI)2 and the polymers PEO, PCL and PTMC (Mn = 4000 g mol−1) dissolved in ACN or PPN. The concentration of the salt was kept at a constant concentration of 100 mM for all measurements, while the polymer concentrations were varied between 0–2 M with respect to the coordinating oxygens.
FTIR temperature sweeps
The experiments were performed with a PerkinElmer Spectrum 100 FT-IR spectrometer with a MIR TGS detector in ATR mode with a Golden Gate ATR module (Specac). The measurements were conducted with a temperature interval of 15 °C in the range 25–85 °C with a Specac West 6100+ heater. 32 scans were completed in the wavenumber range 4000–600 cm−1 with a resolution of 4 cm−1.A background measurement was recorded for each sample and temperature. Measurements were performed on the dissolved LiTFSI, NaTFSI and Mg(TFSI)2 salts in PEO, PCL and PTMC with the concentration n = [O]/[M+] = 10, along with the pure polymers. The samples containing salts were solution-cast as described elsewhere.55 For the determination of the non-associated TFSI concentration in the FTIR spectra, the sum of the two TFSI− conformers were used for the calculations.56 The concentrations of the salts in the polymers were calculated from the determined densities of the polymer electrolytes.
Density measurements
The density of the prepared NaTFSI and Mg(TFSI)2-based polymer electrolytes were determined with a 5 ml glass pycnometer. About 0.1–0.5 g sample were used for the density measurements and cyclohexanone was used as the working liquid (in which the polymer electrolytes were insoluble in). The density of the LiTFSI-based polymer electrolytes was determined by Rosenwinkel et al.5,23Author contributions
RA performed all the experiments. RA, GH and JM all contributed to the planning, analyzing and writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
STandUP for Energy is acknowledged for financial support. The Swedish NMR centre at Umeå University is acknowledged for support with 25Mg NMR measurements. Samuel Emilsson and the Division of Coating Technology at KTH are acknowledged for their support with the FTIR temperature sweep measurements.References
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- Q. Zhang, K. Liu, F. Ding and X. Liu, Nano Res., 2017, 10, 4139–4174 CrossRef.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- D. Brandell, J. Mindemark and G. Hernández, Polymer-based Solid State Batteries, De Gruyter, Berlin, Boston, 2021 Search PubMed.
- M. P. Rosenwinkel, R. Andersson, J. Mindemark and M. Schönhoff, J. Phys. Chem. C, 2020, 124, 23588–23596 CrossRef CAS.
- M. Ebadi, T. Eriksson, P. Mandal, L. T. Costa, C. M. Araujo, J. Mindemark and D. Brandell, Macromolecules, 2020, 53, 764–774 CrossRef CAS PubMed.
- K. Timachova, H. Watanabe and N. P. Balsara, Macromolecules, 2015, 48, 7882–7888 CrossRef CAS.
- Q. Zheng, D. M. Pesko, B. M. Savoie, K. Timachova, A. L. Hasan, M. C. Smith, T. F. Miller, G. W. Coates and N. P. Balsara, Macromolecules, 2018, 51, 2847–2858 CrossRef CAS.
- S. D. Jones, N. S. Schauser, G. H. Fredrickson and R. A. Segalman, Macromolecules, 2020, 53, 10574–10581 CrossRef CAS.
- N. S. Schauser, A. Nikolaev, P. M. Richardson, S. Xie, K. Johnson, E. M. Susca, H. Wang, R. Seshadri, R. J. Clément, J. Read de Alaniz and R. A. Segalman, ACS Macro Lett., 2020, 10, 104–109 CrossRef PubMed.
- H. Gudla, Y. Shao, S. Phunnarungsi, D. Brandell and C. Zhang, J. Phys. Chem. Lett., 2021, 12, 8460–8464 CrossRef CAS PubMed.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, J. Electrochem. Soc., 2013, 160, A2160–A2165 CrossRef CAS.
- E. Jonsson and P. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 10774–10779 RSC.
- T. A. Pham, K. E. Kweon, A. Samanta, V. Lordi and J. E. Pask, J. Phys. Chem. C, 2017, 121, 21913–21920 CrossRef CAS.
- L. H. B. Nguyen, T. Picard, N. Sergent, C. Raynaud, J. S. Filhol and M. L. Doublet, Phys. Chem. Chem. Phys., 2021, 23, 26120–26129 RSC.
- G. W. Neilson, M. C. R. Symons, P. E. Mason, S. Ramos and D. Sullivan, Philos. Trans. R. Soc., A, 2001, 359, 1575–1591 CrossRef CAS.
- Y. Kameda, S. Saito, A. Saji, Y. Amo, T. Usuki, H. Watanabe, N. Arai, Y. Umebayashi, K. Fujii, K. Ueno, K. Ikeda and T. Otomo, J. Phys. Chem. B, 2020, 124, 10456–10464 CrossRef CAS PubMed.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- K. Yuan, H. Bian, Y. Shen, B. Jiang, J. Li, Y. Zhang, H. Chen and J. Zheng, J. Phys. Chem. B, 2014, 118, 3689–3695 CrossRef CAS PubMed.
- R. Matsuoka, M. Shibata, K. Matsuo, R. Sai, H. Tsutsumi, K. Fujii and Y. Katayama, Macromolecules, 2020, 53, 9480–9490 CrossRef CAS.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- F. M. Gray, Polymer Electrolytes, Royal Society of Chemistry, Cambridge, UK, 1997 Search PubMed.
- M. P. Rosenwinkel and M. Schönhoff, J. Electrochem. Soc., 2019, 166, A1977–A1983 CrossRef CAS.
- T. Eriksson, A. Mace, J. Mindemark and D. Brandell, Phys. Chem. Chem. Phys., 2021, 23, 25550–25557 RSC.
- I. Gerz, E. M. Lindh, P. Thordarson, L. Edman, J. Kullgren and J. Mindemark, ACS Appl. Mater. Interfaces, 2019, 11, 40372–40381 CrossRef CAS PubMed.
- E. G. Bloor and R. G. Kidd, Can. J. Chem., 1968, 46, 3425–3430 CrossRef CAS.
- S. Abbrent, J. Lindgren, J. Tegenfeldt and Å. Wendsjö, Electrochim. Acta, 1998, 43, 1185–1191 CrossRef CAS.
- A. Bakker, S. Gejji, J. Lindgren, K. Hermansson and M. M. Probst, Polymer, 1995, 36, 4371–4378 CrossRef CAS.
- C.-S. Chung, Inorg. Chem., 1979, 18, 1321–1324 CrossRef CAS.
- R. Hoffman, J. Magn. Reson., 2022, 335, 107105 CrossRef CAS PubMed.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacín, J. Mater. Chem. A, 2015, 3, 22–42 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- E. N. Howe, M. Bhadbhade and P. Thordarson, J. Am. Chem. Soc., 2014, 136, 7505–7516 CrossRef CAS PubMed.
- S. Ramesh, T. F. Yuen and C. J. Shen, Spectrochim. Acta, Part A, 2008, 69, 670–675 CrossRef CAS PubMed.
- M. Jaipal Reddy and P. P. Chu, J. Power Sources, 2002, 109, 340–346 CrossRef CAS.
- B. Sun, J. Mindemark, E. V. Morozov, L. T. Costa, M. Bergman, P. Johansson, Y. Fang, I. Furo and D. Brandell, Phys. Chem. Chem. Phys., 2016, 18, 9504–9513 RSC.
- Y. Shintani and H. Tsutsumi, J. Power Sources, 2010, 195, 2863–2869 CrossRef CAS.
- W.-H. Hou and C.-Y. Chen, Electrochim. Acta, 2004, 49, 2105–2112 CrossRef CAS.
- J. Mindemark, S. Tang, H. Li and L. Edman, Adv. Funct. Mater., 2018, 28, 1801295 CrossRef.
- S. Varma and S. B. Rempe, Biophys. Chem., 2006, 124, 192–199 CrossRef CAS PubMed.
- E. Flores, G. Åvall, S. Jeschke and P. Johansson, Electrochim. Acta, 2017, 233, 134–141 CrossRef CAS.
- A. V. Cresce, S. M. Russell, O. Borodin, J. A. Allen, M. A. Schroeder, M. Dai, J. Peng, M. P. Gobet, S. G. Greenbaum, R. E. Rogers and K. Xu, Phys. Chem. Chem. Phys., 2016, 19, 574–586 RSC.
- F. C. Jentoft, J. Kröhnert, I. R. Subbotina and V. B. Kazansky, J. Phys. Chem. C, 2013, 117, 5873–5881 CrossRef CAS.
- A. J. Ringsby, K. D. Fong, J. Self, H. K. Bergstrom, B. D. McCloskey and K. A. Persson, J. Electrochem. Soc., 2021, 168, 080501 CrossRef CAS.
- G. Åvall, J. Mindemark, D. Brandell and P. Johansson, Adv. Energy Mater., 2018, 8, 1703036 CrossRef.
- J. C. Lassègues, J. Grondin, R. Holomb and P. Johansson, J. Raman Spectrosc., 2007, 38, 551–558 CrossRef.
- J.-K. Kim, L. Niedzicki, J. Scheers, C.-R. Shin, D.-H. Lim, W. Wieczorek, P. Johansson, J.-H. Ahn, A. Matic and P. Jacobsson, J. Power Sources, 2013, 224, 93–98 CrossRef CAS.
- D. M. Seo, P. D. Boyle, R. D. Sommer, J. S. Daubert, O. Borodin and W. A. Henderson, J. Phys. Chem. B, 2014, 118, 13601–13608 CrossRef CAS PubMed.
- L. Suo, F. Zheng, Y.-S. Hu and L. Chen, Chin. Phys. B, 2016, 25, 016101 CrossRef.
- N. B. Erdal, G. A. Lando, A. Yadav, R. K. Srivastava and M. Hakkarainen, Polymers, 2020, 12, 1849 CrossRef CAS PubMed.
- C. Sångeland, R. Younesi, J. Mindemark and D. Brandell, Energy Storage Mater., 2019, 19, 31–38 CrossRef.
- B. Park, R. Andersson, S. G. Pate, J. Liu, C. P. O’Brien, G. Hernández, J. Mindemark and J. L. Schaefer, Energy Mater. Adv., 2021, 2021, 1–14 CrossRef.
- T. Eriksson, Cation Conduction and Coordination in Carbonyl-Containing Compounds: Li+ Transport in Alternative Polymer Electrolyte Host Materials, PhD thesis, Uppsala University (Uppsala: Acta Universitatis Upsaliensis), 2021 Search PubMed.
- P. G. Bruce, Solid State Electrochemistry, Cambridge University Press, Cambridge, 1994 Search PubMed.
- B. Sun, J. Mindemark, K. Edström and D. Brandell, Solid State Ionics, 2014, 262, 738–742 CrossRef CAS.
- M. Herstedt, M. Smirnov, P. Johansson, M. Chami, J. Grondin, L. Servant and J. C. Lassègues, J. Raman Spectrosc., 2005, 36, 762–770 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01904c |
| This journal is © the Owner Societies 2022 |