
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanosecond solvation dynamics of the hematite/liquid water interface at hybrid DFT accuracy using committee neural network potentials†
Philipp
Schienbein
 * and
Jochen
Blumberger
*
* and
Jochen
Blumberger
*
Department of Physics and Astronomy and Thomas Young Centre, University College London, London, WC1E 6BT, UK. E-mail: p.schienbein@ucl.ac.uk; j.blumberger@ucl.ac.uk
First published on 10th June 2022
Abstract
Metal oxide/water interfaces play an important role in biology, catalysis, energy storage and photocatalytic water splitting. The atomistic structure at these interfaces is often difficult to characterize by experimental techniques, whilst results from ab initio molecular dynamics simulations tend to be uncertain due to the limited length and time scales accessible. In this work, we train a committee neural network potential to simulate the hematite/water interface at the hybrid DFT level of theory to reach the nanosecond timescale and systems containing more than 3000 atoms. The NNP enables us to converge dynamical properties, not possible with brute-force ab initio molecular dynamics. Our simulations uncover a rich solvation dynamics at the hematite/water interface spanning three different time scales: picosecond H-bond dynamics between surface hydroxyls and the first water layer, in-plane/out-of-plane tilt motion of surface hydroxyls on the 10 ps time scale, and diffusion of water molecules from the oxide surface characterized by a mean residence lifetime of about 60 ps. Calculation of vibrational spectra confirm that H-bonds between surface hydroxyls and first layer water molecules are stronger than H-bonds in bulk water. Our study showcases how state of the art machine learning approaches can routinely be utilized to explore the structural dynamics at transition metal oxide interfaces with complex electronic structure. It foreshadows that c-NNPs are a promising tool to tackle the sampling problem in ab initio electrochemistry with explicit solvent molecules.
1 Introduction
Interfaces between transition metal oxides and liquid water attracted much attention from various research disciplines ranging from heterogeneous catalysis, colloid chemistry, biology, energy storage to photocatalysis.1–8 Depending on the microscopic property of interest, experimental techniques are available which can elucidate liquid/solid interactions: structural properties, such as the surface termination and the formation of water layers at the surface can be obtained from crystal truncation rod (CTR) experiments.9 Sum frequency generation (SFG)10–13 and attenuated total reflection (ATR) IR14 spectroscopy can measure the molecular vibrations at the interface and therefore give valuable insights into the dynamics at the interface. Such experiments can also be used to monitor the H-bond composition at an oxide metal/water interface as a function of pH.15 When it comes to interpreting experiments, atomistic molecular dynamics (MD) simulations are an invaluable tool to provide a bottom-up picture. For example, it was recently unveiled that rapid O atom exchange happens across the “r-cut” (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 02) hematite/water interface16 in a joint experimental and computational study, proving that this interface is highly reactive.
02) hematite/water interface16 in a joint experimental and computational study, proving that this interface is highly reactive.
Force field MD simulations have often been conducted on oxide/water interfaces, see e.g. ref. 17. However, metal oxide interfaces are generally reactive, be it O atom exchange16 or frequent proton transfer between the metal oxide and water,18 and some materials feature a complex electronic structure.19 Such effects are generally not captured by standard non-polarizable force fields. It is therefore desirable to employ ab initio MD (AIMD, also called “DFTMD”) simulations,20 where the full electronic structure is explicitly considered using DFT. Such simulations have already been done for numerous systems,21–33 where mostly hybrid DFT functionals have been used. A large disadvantage of hybrid functionals is, that they are generally much more expensive compared to GGA functionals. MD simulations based on hybrid DFT are therefore computationally demanding and also limited to short times (about 10 picoseconds) and relatively small system sizes. Sampling dynamical properties, e.g. rate constants or residence lifetimes, require that the event of interest happens frequently along the MD trajectory. However, if the desired process is slower than the trajectory length (so-called “rare events”), the corresponding time correlation functions cannot reliably be converged. Notably, dynamical properties can also suffer from finite-size effects due to the limited size of the simulation box, which are, e.g. well known for the self-diffusion coefficient in a periodic simulation box.34–36
In the last years, Machine Learning (ML) models have been introduced to run MD simulations with the aim to replace the expensive DFT calculations by a computationally less expensive model while keeping the error reasonably small. A widely applied ML approach are Behler–Parrinello Neural Network potentials (NNPs),37 where atomic energies are trained for each atom species. The total energy of a given configuration is then obtained as the sum of all atomic energies. Here, so-called symmetry functions are used to transform the Cartesian coordinates into a representation based on intermolecular distances and angles only. Such NNPs have successfully been applied to a number of systems, including bulk liquid water,38 metal/water interfaces, e.g. Cu,39 and also metal oxide/water interfaces, e.g. ZnO18 and LixMn2O4.40 Very recently, so-called committee Neural Network potentials (c-NNP) have been introduced41 which are based on Behler–Parrinello NNPs. In a nutshell, a committee (or ensemble) of NNPs yields an average energy and a corresponding variance. The variance can be interpreted as a measure how “good” the energy prediction of a given configuration is and thereby allows active learning and automated fitting approaches which dramatically reduce the number of expensive training data needed compared to standard NNP training approaches.
In this work, we have trained a c-NNP for the hematite/liquid water interface at the hybrid DFT level of theory to uncover the structural dynamics of water at the interface over multiple time scales, from pico to nanoseconds. Hematite (Fe2O3, “rust”) is one of the most abundant metal oxide compounds on earth.42 In the field of photocatalysis, the potential use of hematite for water splitting2,7 as a source of “green” hydrogen motivated numerous studies including nanostructuring43 and doping.44 Despite all the efforts, hematite still suffers from slow reaction kinetics and a large overpotential which hinders its usage in real photoelectrochemical cells.2,45 To address these practical problems, a deep understanding of the atomistic processes (structural and dynamical) at the hematite/liquid water interface is key. Hematite is known to feature a complex electronic structure due to the antiferromagnetic spin pattern19 and the interface is a highly reactive region,16 where charge transfer and polarization effects are expected to be of major importance. We opt to describe the hematite/water system with a modified hybrid HSE06 functional, where the fraction of exact exchange is reduced from 25 to 12%. This functional was shown to give excellent agreement with experimental measurements for a wide range of different properties including crystal structure, band gap, antiferromagnetic ordering and spin density distribution.26,46 The trained c-NNP model allows us to significantly extend the simulation time (up to nanoseconds) and the size of the simulation box (up to about 3000 atoms). We show that such time and length scales are required to converge important dynamical properties such as mean residence lifetimes and diffusion coefficients of surface water molecules. This is clearly out of scope for explicit AIMD simulations, especially when using hybrid DFT, without the aid of ML. Note that even longer simulation times and larger boxes are generally possible using ML, but do not add any significant additional value to the physical properties discussed in this study. Using the c-NNP simulations, the aim of this paper is to explore the dynamics of water molecules at the hematite(001) surface. In particular, we are interested in (i) rate constants for exchange of water molecules between the surface and the bulk liquid, (ii) the intramolecular vibrational signature of surface water molecules compared to bulk water, (iii) self-diffusion of water at the interface, and finally, (iv) which time and length scales are required to converge such properties.
2 Computational details
A general issue in the context of machine learning potentials is the generation of the training set, i.e. to select the configurations used to train the model. Recently, automated, entirely data-driven approaches to select configurations into the training set have been successfully applied in many different applications, see e.g. ref. 47–49. It could be shown that such automated fitting generally improves the accuracy of the training, reduces the risk of overfitting, and also minimizes the number of configurations needed for training.41,50,51 Herein, we employ eight independent Behler–Parrinello Neural Network potentials which together form a committee, following the training protocol introduced in ref. 41 and 51. To calculate the energy of a single configuration, each of the eight NNPs is evaluated and the average of all committee members yields the total energy of the given configuration. Most importantly, also the variance of the energy can be calculated which is a measure of how well the committee members agree on that predicted energy. A large variance indicates that the predicted energies of the individual committee members are very different and therefore the committee as a whole is uncertain about the total energy of that configuration. As such, the variance can be interpreted as a measure of how good this configuration is known and it can be used to identify “most unknown” configurations which is utilized in the training protocol, see below.Here, we train our c-NNP based on a two-step procedure: In our previous work,26 about 15 ps of AIMD simulation data have been generated in total. From these available previous data, we randomly selected 20 configurations and trained a c-NNP. Thereafter, this c-NNP is used to predict the energies of all configurations of the same 15 ps AIMD trajectory. The 20 “most unknown” configurations are automatically selected, added into the training set, and a new c-NNP is trained. This cycle was repeated 10 times, such that 200 configurations are contained in our training set in total. In the second step, we further use an active learning procedure41,51 to systematically explore the phase space further and iteratively improve the quality of the c-NNP. In that protocol, we run a MD simulation of the hematite/water interface using the above trained preliminary c-NNP. While doing so, it is possible that new areas in the phase space are visited, which have not been included in the training set of the c-NNP yet. This manifests in an increase of the variance because the committee members start to extrapolate beyond the known training set and thus disagree on the energy. Importantly, the c-NNP does not constrain the MD to sample only that portion of potential energy surface which is already known, but allows to explore unknown regions. This is also desired to iteratively include more and more training data to mimic the full accessible DFT potential energy surface at a given temperature as close as possible. In full analogy to step one, we now take the 10 most unknown configurations (the ones with the largest variance) from the MD simulation. The forces for these unknown configurations are then calculated by explicit DFT calculations using the hybrid HSE06 functional52 with 12% HFX46 supplemented by D3 dispersion corrections;53 the details of the electronic structure calculations are described in Section S1 in the ESI.† We then compute the RMSE between the calculated energies/forces from the c-NNP and the explicit DFT calculation. If the RMSE is small enough, the training of the c-NNP is finished. Otherwise, the 10 new DFT energies and forces are added to the training set, a new c-NNP is trained and the whole process is repeated until the desired accuracy is met. Note that by adding only the most unknown configurations to the training set it is also ensured that only such configurations enter which are sufficiently different from the existing training set, i.e. which have not been added before. This way, 200 configurations were added to the training set such that the final training set is composed of only 400 statistically independent configurations.
All c-NNP simulations have been conducted using CP2k54 and the training of the individual Behler–Parrinello NNPs forming the committee has been done using n2p2.55 The final c-NNP used for production was validated against the previous explicit AIMD simulations26 and unknown data which was generated by the c-NNP simulations. Thereby, we ensure that the trained c-NNP mimics the explicit AIMD correctly as elaborately shown in Section S2 in the ESI.† For the final c-NNP we obtain a total force RMSE with respect to the unknown validation set of 149.8 meV Å−1. This RMSE agrees well with previous trainings of NNPs, where similar force RMSEs have been obtained, e.g. for liquid water at a Cu slab (207.5 meV Å−1),39 LixMn2O440 (240.0 meV Å−1) or liquid water at MoS2 slabs51 (190.1 meV Å−1).
It is well known in the literature that PBE functionals tend to overbind water–water interactions56–59 resulting in too rigid structures and slower dynamics than experimentally measured. This also holds true for PBE derived functionals, such as the HSE06 and PBE0 hybrid functionals.60,61 One possible way to counteract this overbinding effect is to increase the simulation temperature until the correct experimental quantities are recovered. For PBE (and derived hybrid functionals) an effective simulation temperature of 400 K has been shown to reproduce the structural dynamics, in terms of radial distribution functions and the self-diffusion coefficient, of bulk liquid water at ambient conditions satisfactorily.56,60–62 Indeed, we elaborately show in Section S4 in the ESI† that HSE06-D3 water at 400 K slightly overestimates the dynamics, but still slightly overbinds the structure and thus offers a very good compromise to describe ambient liquid water at 300 K correctly in terms of both, the structure and the dynamics. For this reason, we opt to run all simulations in this paper at that effective simulation temperature of 400 K to correctly model the structural dynamics of real liquid ambient water at 300 K.
The trained c-NNP makes possible the use of larger system sizes. We employ four different system sizes to explore the water dynamics at the hematite/water interface: the first (and smallest) box is taken from our previous simulations26 containing 93 water molecules and a height (box length perpendicular to the hematite surface) of about 47 Å and a surface area of about 10 × 10 Å2. We then expanded the dimension perpendicular to the slab to 80 Å and increased the number of water molecules to 208 while keeping the water density constant at 0.996 kg L−1. Finally, we increased the surface area of the hematite surface by a factor of two (“(1 × 2)”, 416 water molecules, 1560 atoms) and by a factor of four (“(2 × 2)”, 832 water molecules, 3120 atoms). A more detailed description and illustration of the employed boxes is given in Section S3 and Fig. S2 in the ESI.† For each box, a c-NNP simulation in the canonical (NVT) ensemble is conducted for a total trajectory length of 1 ns. From this NVT simulation we took 10 equidistant configurations as starting points for subsequent simulations in the microcanonical (NVE) ensemble. The NVE simulations were run for 400 ps each using a timestep of 0.5 fs. All the presented results in the following are averaged over 10 NVE simulations of the largest box, (2 × 2), except explicitly stated otherwise. We opted to use only the NVE simulations to obtain the presented results herein, because there are some properties (vibrational density of states, diffusion coefficient) which are calculated from atomic velocities. Within a NVT simulation, these velocities might be affected by the thermostat introducing artificial effects in those properties. Arguably, the influence of most thermostats is generally small, especially if the simulations are well equilibrated. However, if additional NVE simulations are affordable, this issue is guaranteed to be circumvented. Moreover, from a practical point of view, many independent NVE simulations can trivially be parallelized and block averaged.
Noteworthy, the hematite slab is terminated with hydroxide groups, see Fig. S2 in the ESI.† Given the length of the generated trajectories, one might expect that some of the OH groups dissociate during the simulation, but we do not find any of such dissociations. One might wonder if this could be a problem of the employed c-NNP potential instead of conducting explicit AIMD simulations. Generally, NNPs have shown to be reactive, e.g. at the ZnO/water interface,18 where proton transfer from the surface to the adjacent water molecules could be sampled using NNPs. Moreover, our employed active learning scheme described above, allows the MD to freely explore the phase space and does not put any constraint on the potential energy. Therefore it is more likely that dissociation does not occur due to physical reasons. The pKa value describing the deprotonation of these hematite(001) OH groups has been estimated before29 to be 18.5 indicating that the deprotonation is highly improbable; in fact, a terminal deprotonated O− group would even be a quite strong base. This explains why the terminal OH groups do not dissociate along the trajectories, even when going to nano second time scales.
3 Results
The layering of the water molecules at the hematite(001) surface has already been discussed in our previous publication26 in detail, however, understanding the structure is key for the following analyses, therefore the main conclusions of our previous, short AIMD simulations are briefly summarized here. In Fig. 1 we show the atom density of water molecules as a function of distance to the hematite slab which is located at 0 Å by convention. To further illustrate the interface we also present a snapshot from our c-NNP simulation. In a nutshell, we find that the terminating oxide hydroxyl groups are either oriented parallel to the slab (maximum at 0 Å, “in-plane” orientation) or point towards the liquid phase (maximum at around 1 Å, “out-of-plane” orientation). We find that a first water layer is clearly formed as deduced from the prominent peak of the O density around 2.5 Å. The second peak of the O density is located around 5.5 Å and both maxima are separated by a broad minimum indicating that the first layer is well distinguishable from the water molecules beyond. The OH bonds of the water molecules can point towards the oxide slab which is indicated by a prominent maximum of the H atom density around 1.7 Å. If the oxide hydroxyl group points towards the liquid phase or if a water OH group points towards the oxide, H-bonds are formed in between which is illustrated by the simulation snapshot.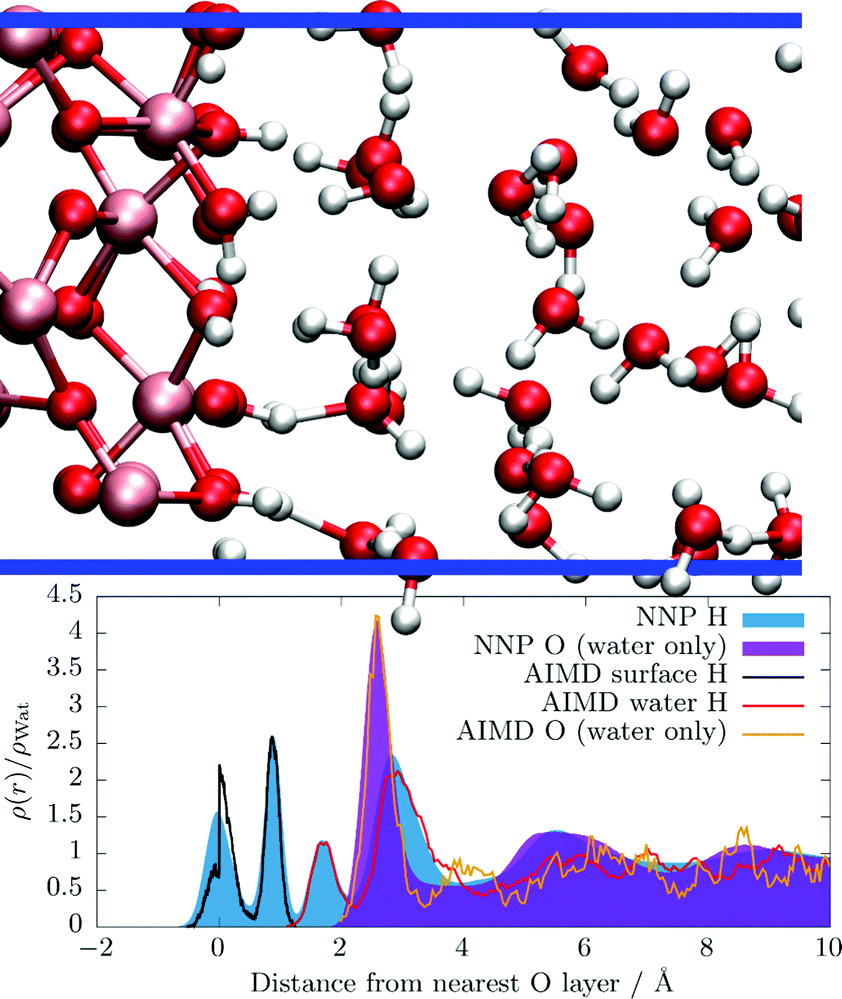 | ||
| Fig. 1 Side view of the water layering at the hematite slab (top), in the previously26 used 10 × 10 × 47 Å box, see Section S3 in the ESI† for details, where Fe, O, and H atoms are painted ochre, red, and white, respectively. The blue horizontal lines illustrate the periodic box. Atom densities (bottom) obtained from the c-NNP simulations trained to mimic the hybrid HSE06 functional in this work of H atoms (filled blue curve) and O atoms (filled purple curve). For reference, the previously published atom densities obtained from explicit AIMD simulations of the terminal hydroxyl H atoms (black solid line), of the water H atoms (red solid line) and of the water O atoms (orange solid line) are shown. All atom densities are normalized by their respective value in bulk ambient water. The average location of the interfacial oxide O layer is set to a distance of 0 Å by convention. | ||
In Fig. 1 we also compare the atom densities as obtained by the previously published explicit AIMD simulations (pico-second timescale) with the ones obtained by the new c-NNP simulations (nano-second timescale). First, we deduce that the trained c-NNP simulations qualitatively recover the very same layering as the explicit AIMD simulations as expected. Quantitatively, we observe that the immediate layering at the slab up to about 3 to 4 Å is described well with the explicit AIMD simulations. At distances further away, the structure becomes much less clear because the density simply is not fully converged statistically. The c-NNP simulations adding about three orders of magnitude more data are fully converged also at large distances. In terms of the structure, explicit AIMD simulations are therefore sufficient to properly describe the immediate surrounding of the slab (in terms of the statistical quality of the results), however, did not provide enough data to converge the structure at larger distances.
The kinetics of water exchange between the first and second or higher water layers can be obtained using the stable-state-picture (SSP) introduced by Laage and Hynes.63 Within this technique, a time correlation function
 | (1) |
In Fig. 2C(t) is shown as a function of time. We observe that the time correlation function slowly but systematically converges from a total trajectory length of 200 to 400 ps. Note that the shortest length considered here is already about ten times longer than the original AIMD simulations26 and is thus clearly out of scope for present-day AIMD simulations at hybrid DFT level. The inset shows the resulting residence lifetimes τ determined from the respective correlation functions as a function of the total trajectory length. At 200 ps we find a residence lifetime of about 32.5 ps, whereas at 400 ps we find one of about 49 ps. The residence lifetimes fit remarkably well using a standard exponential “limited growth” function (f(t) = S − (S − A)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−kt], where S, A, and k are fitting constants and S marks the upper limit) which yields a residence lifetime of about 58.3 ps for an infinitely long simulation. The error bars are obtained by block averaging over the N water molecules in eqn (1). Here, we have averaged over 485 water molecules being in the first solvation layer at t = 0; recall that the data is collected from 10 independent NVE simulations of 400 ps each (4 ns in total), where each trajectory contained 3120 atoms (largest box investigated). To decrease the statistical error further, even larger simulation boxes or even more trajectories would therefore be required. Given the size of the error bars for the obtained residence lifetime, we estimate the error of our extrapolated result to be about 10 ps.
exp[−kt], where S, A, and k are fitting constants and S marks the upper limit) which yields a residence lifetime of about 58.3 ps for an infinitely long simulation. The error bars are obtained by block averaging over the N water molecules in eqn (1). Here, we have averaged over 485 water molecules being in the first solvation layer at t = 0; recall that the data is collected from 10 independent NVE simulations of 400 ps each (4 ns in total), where each trajectory contained 3120 atoms (largest box investigated). To decrease the statistical error further, even larger simulation boxes or even more trajectories would therefore be required. Given the size of the error bars for the obtained residence lifetime, we estimate the error of our extrapolated result to be about 10 ps.
 | ||
| Fig. 2 Time correlation functions according to eqn (1) describing the residence lifetime of water molecules in the first solvation layer at the hematite surface as a function of total simulation time, where the brown, light brown, light green, and green curves correspond to total simulation times of 200, 300, 350, and 400 ps, respectively. The inset shows the corresponding residence lifetimes τ which have been determined by fitting an exponential function (see text) to the respective correlation function as a function of total simulation length, using the same color code. The black solid line shows an exponential growth function which fitted to all τ values to extrapolate towards an infinite simulation length. The black horizontal dotted line indicates the limit of that growth function of 59.3 ps corresponding to an infinite total simulation length. | ||
Having determined the residence lifetime, we now turn to H-bond dynamics at the interface. H-Bond lifetimes have been computed from MD simulations for a long time, however, they are no physical observables and depend on the H-bond definition used. Moreover, lifetimes itself can be defined in many different ways, e.g. “continuous” and “intermittent” lifetimes64 or within the more sophisticated reactive-flux methodology,65 and their interpretation is slightly different. Here, we employ the continuous H-bond lifetime definition64
 | (2) |
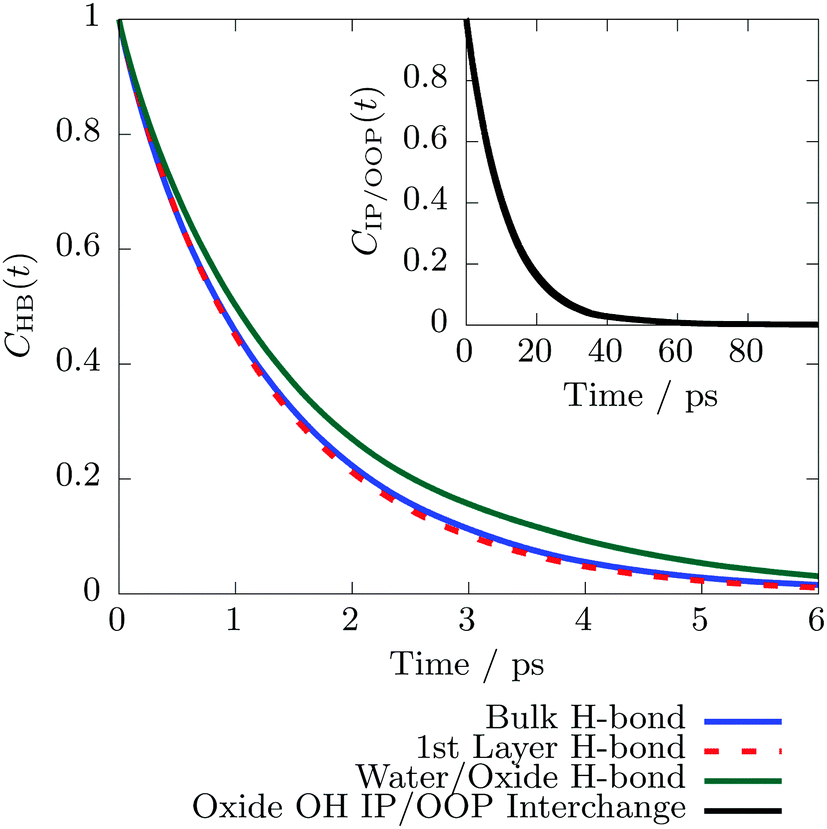 | ||
| Fig. 3 Continuous H-bond lifetime correlation functions64CHB(t), see eqn (2), for H-bonds between two water molecules in the bulk phase (solid blue), between water molecules where at least one is located in the first solvation layer (dashed red), and between water and the hematite surface (green). The inset shows the residence lifetime of a terminal hematite O–H group in the IP or OOP state, respectively, see Fig. 1 which was calculated using the steady-state picture63 and eqn (1). | ||
Previously we have reported that the exposed terminal OH groups at the hematite surface can interchange between an “in-plane” (IP) and “out-of-plane” (OOP) configuration,26 where the OH bond vector is parallel or perpendicular to the surface plane, respectively (see Fig. 1). As shown in Section S6 in the ESI,† these two states are clearly separated by a transition state. We can therefore again apply the SSP in eqn (1) as employed above for the residence time of water molecules in the first solvation layer. The corresponding time correlation function is shown in the inset of Fig. 3 and yields an average lifetime of 11.26 ± 3.28 ps. The latter quantity describes how long the OH group resides in the IP or OOP configuration until it interchanges and is settled in the respective other state. We find that the correlation function is converged after roughly 300 ps total simulation time. Therefore an extrapolation to an infinite simulation duration, as it was done above for the residence time in the first solvation layer, see Fig. 2, is not necessary. Consequently, the IP/OOP interchange lifetime is significantly shorter compared to the average residence time of water molecules in the first layer lifetime, but substantially longer than the interfacial H-bond lifetimes.
A common (indirect) measure of the H-bond strength in liquid water is the spectral signature of the intramolecular OH stretch vibration.70 If a strong H-bond is formed between two water molecules, the intramolecular OH bond is weakened, causing a red-shift of the frequency and vice versa. Note that the intermolecular H-bond stretch can also directly be measured in bulk water using IR spectroscopy in the THz frequency range, also at elevated temperatures and pressures.68,71 Still, the intramolecular OH stretch has been used more frequently in the past, because it is readily available from the vibrational density of states (VDOS) of the H atoms,24,68,72 while the intermolecular H-bond mode does not appear in the H or O VDOS at all since charge transfer and polarization effects play a crucial role.71 The H VDOS is computed as usual from the Fourier transform of the velocity time auto correlation function of H atoms
 | (3) |
![[v with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0076_20d1.gif) (i)H(t) is the velocity of the i-th H atom at time t and the sum runs over all selected H atoms NH. We again distinguish between water molecules being in the first solvation layer at the hematite surface and bulk water molecules. Note that as soon as a water molecule leaves the first solvation layer according to the SSP, see above, we stop evaluating its corresponding H VDOS. Given that 〈(i)H(0)(i)H(t)〉 converges within a couple of pico seconds and this is well within the mean residence time of a water molecule in the first solvation layer (see Fig. 2), water molecules stay sufficiently long to obtain reasonable time correlation functions. Note that the calculated VDOS can be used to qualitatively interpret measured vibrational spectra, such as IR, Raman, ATR, or vibrational SFG spectra. However, the VDOS does not exactly correspond to these techniques since it only captures the motion of masses, while all named spectroscopies obey different selection rules, e.g. dipole moment changes which can also modulate the spectrum.
(i)H(t) is the velocity of the i-th H atom at time t and the sum runs over all selected H atoms NH. We again distinguish between water molecules being in the first solvation layer at the hematite surface and bulk water molecules. Note that as soon as a water molecule leaves the first solvation layer according to the SSP, see above, we stop evaluating its corresponding H VDOS. Given that 〈(i)H(0)(i)H(t)〉 converges within a couple of pico seconds and this is well within the mean residence time of a water molecule in the first solvation layer (see Fig. 2), water molecules stay sufficiently long to obtain reasonable time correlation functions. Note that the calculated VDOS can be used to qualitatively interpret measured vibrational spectra, such as IR, Raman, ATR, or vibrational SFG spectra. However, the VDOS does not exactly correspond to these techniques since it only captures the motion of masses, while all named spectroscopies obey different selection rules, e.g. dipole moment changes which can also modulate the spectrum.
In Fig. 4 we show ΛH(ω) considering only H atoms belonging to water molecules in the first layer compared to H atoms belonging to water molecules in the bulk liquid phase. We find that the OH stretch peak of bulk water molecules is centered around 3500 cm−1 (black line). In case of the first layer water molecules we further differentiate between OH vibrations which are perpendicular to the surface and vibrations which are parallel to the surface (blue line). This can trivially be achieved by computing the time correlation functions in eqn (3) for each Cartesian component individually; note that the hematite surface is constructed such that it is parallel to the xy plane. We find that water OH bonds which are oriented perpendicular to the hematite surface are significantly red-shifted compared to those which are oriented mostly parallel to the slab. This observation can be understood in terms of the structure (see Fig. 1), where we showed that water actively donates H-bonds to the hematite slab. In such a case, the water OH bond points towards the surface and the bond vector is consequently also oriented perpendicular to the surface plane. By dissecting the spectrum into parallel and perpendicular contributions, we can thus deduce that H-bonds donated to the hematite slab are stronger compared to H-bonds between water molecules. Recall that the intramolecular OH stretch vibration is an indirect measure of the H-bond strengths.70 A red-shift of that band indicates that the intramolecular vibration is weakened due to a stronger intermolecular H-bond. Remarkably, we cannot find any difference in H-bonding for water/water H-bonds, even if at least one of the two molecules is in the first solvation layer. This implies that the water/water H-bond network at the surface is not changed compared to bulk, but only the hematite/water H-bonds differ from H-bonds in bulk liquid water.
 | ||
| Fig. 4 Vibrational density of states (VDOS) of H atoms (ΛH(ω), eqn (3)) showing the intramolecular OH stretch of water molecules located in the pure bulk phase (black) and in the first solvation layer at the hematite surface. In the latter case, the VDOS is split into its xy components (blue) and z component (red) which are parallel and perpendicular to the surface, respectively. | ||
The dynamics of water molecules in a simulation box are commonly quantified in terms of the self-diffusion coefficient. To compute the diffusion coefficient we use the Green–Kubo formulation74
 | (4) |
i(t) is the center of mass velocity of the i-th water molecule, d is the dimensionality, and the sum runs over all NW water molecules of interest. Using the Green–Kubo (or equivalently the Einstein relation), the self diffusion coefficient perpendicular to the surface is not reasonably defined. The reason is, that the particle is confined between the two limiting hematite slabs in that dimension. Therefore, we restrict ourselves to discuss the self-diffusion coefficient only in the two dimensions parallel to the surface, D‖PBC, which can again trivially be achieved by evaluating eqn (4) only along the xy components. Moreover we again distinguish between water molecules in the first layer at the hematite surface and bulk water molecules. Technically, this distinction is made by checking if a molecule is in the first layer at time zero or not; recall that the velocity time correlation function converges quickly, see above. Therefore, water molecules stay long enough in the first solvation layer to yield a converged velocity auto correlation function.
In Fig. 5 we show D‖PBC as a function of the simulation box size. Remarkably, it significantly depends on the size of the simulation box: upon increasing the dimension perpendicular to the surface from 47 to 80 Å the diffusion coefficient is greatly enhanced and after increasing the surface area from 100 to 400 Å2 the diffusion coefficient decreases again. Surprisingly, in case of the 10 × 10 × 80 box, we find a diffusion coefficient of the bulk molecules around 7 which is more than twice as large as the diffusion coefficient calculated for a cubic bulk water box using the very same c-NNP as for the hematite/water interface (dashed horizontal line, see ESI† for details). Simultaneously, the error bars behave the same way. We further observe, that the diffusion coefficient of water molecules being in the first layer is generally smaller compared to the one of water molecules being in the bulk phase, irrespective of the simulation box size.
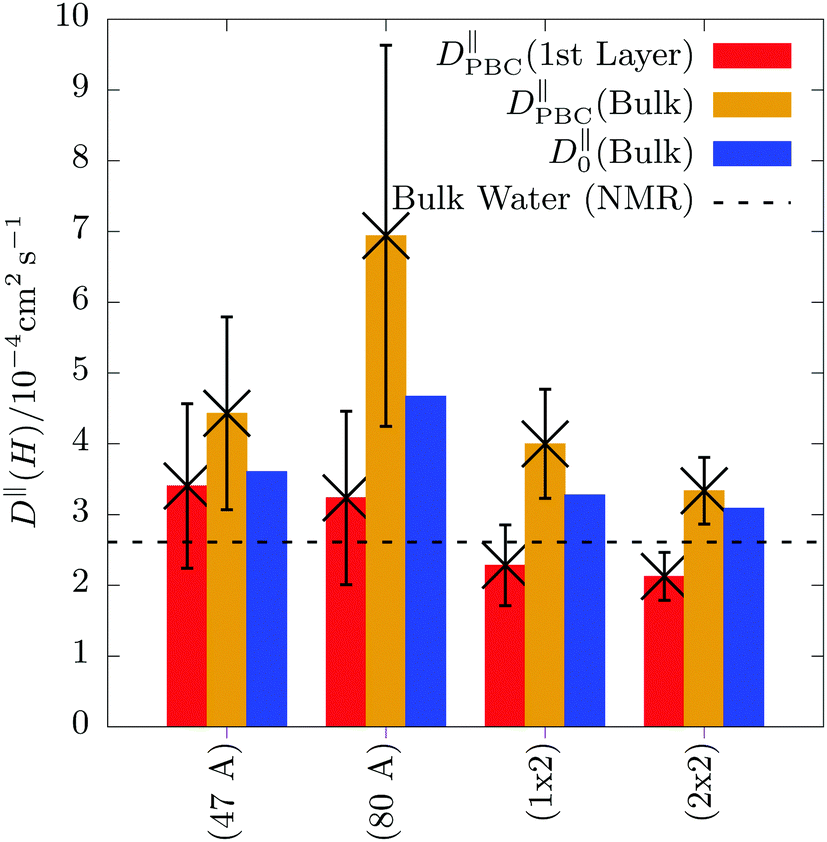 | ||
| Fig. 5 Self-diffusion coefficient D‖PBC computed by eqn (4), but only considering the x and y Cartesian components which are parallel to the hematite surface as a function of system size. The simulation boxes and simulation sizes are given in Section S3 in the ESI.† The red and orange bars show D‖PBC of water molecules located in the first solvation layer at the surface and in the bulk liquid phase, respectively. The error bars are obtained by block averaging. The horizontal black dashed line represents the experimental self-diffusion coefficient of pure bulk liquid water (2.61 × 10−5 cm2 s−1) obtained by NMR measurements.73 | ||
The behavior of DPBCxy as a function of the simulation box size implies the presence of significant finite-size effects. It is well-known for cubic simulation boxes that the diffusion coefficient depends on the simulation box size and there are formulae available to quantify this finite-size effect as well as to extrapolate the value to its value at infinity.34,35 Finite-size effects for confined systems are less well-known, however, they have been reported.36 The authors find that for wide, thin pores, i.e. when the simulation box size in the confined direction is large, but the corresponding surface area is small, DPBCxy “can be several times larger than Dbulk”.36 The opposite effect, i.e. that DPBCxy converges to its bulk value, is observed when the surface area is large compared to the length of the confined dimension. Qualitatively, we exactly find this behavior and thus ascribe it to a finite-size effect arising due to the confinement.
Moreover, we applied the given finite-size correction equation36 (see eqn (S2) in the ESI†) to estimate the error of the self-diffusion coefficient as a function of system size. The correction term requires knowledge of the viscosity, η, which we estimated from pure bulk liquid water simulations, see Section S5 in the ESI† for details. Due to the strong interactions between water and the hematite slab, we expect that the viscosity of bulk water molecules and molecules in the first solvation layer are different. Therefore, we only applied the correction to bulk water molecules in the hematite/water box and not to those water molecules being in the first solvation layer. Due to these two approximations (η estimated for pure bulk water and the probable dependence of η on the distance from the slab), we interpret the finite-size correction only as an estimate of the error of D as a function of system size. Inspecting Fig. 5 again, we indeed find that the finite-size correction is largest for the 80 Å box, where the surface area (about 10 × 10 Å) is comparably small with respect to the 80 Å in the perpendicular dimension. In contrast, for the largest box (20 × 20 × 80 Å) the finite-size correction is quite small. Therefore, we conclude that the calculated self-diffusion coefficients with the latter box should be a reliable estimate. This conclusion is further supported by available experimental self-diffusion coefficient of pure bulk liquid water which closely agrees with the calculated one in the largest box. Importantly, when using explicit AIMD simulations, usually only a single simulation box is affordable. Fig. 5 shows that if a unsuitable simulation box is chosen accidentally, the calculated self-diffusion coefficients can potentially be far off due to substantial finite-size effects.
4 Discussion
Merging all presented results together yields a rather complete picture of liquid water dynamics at the hematite(001) surface. First of all, we find that water molecules reside about 55–65 ps (see Fig. 2) at the hematite surface on average, i.e. substantially longer than the average H-bond lifetime. This implies that the H-bond network between first layer waters and surface hydroxyls breaks and reforms many times until a molecule leaves the surface for bulk. Generally, it seems that the first layer is therefore tightly bound to the hematite surface. The bond strengths of H-bonds can be assessed using H-bond lifetimes. Here, we indeed find that the H-bond lifetime between water and hematite (1.58 ps, see Fig. 3) is slightly larger than the H-bond lifetime between two water molecules (1.28 ps). Moreover, strong H-bonds are further supported by the VDOS, where we found a substantial red-shift (see Fig. 4) of the intramolecular OH bond of water molecules in the first solvation layer pointing towards the hematite slab. Generally, such red-shift of the intramolecular OH bond is assigned to a stronger intermolecular H-bond.70A similar red-shift of the intramolecular OH stretch of water was also measured previously at the Al2O3(11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0)/water interface by vibrational SFG spectroscopy12 and assigned to strongly H-bonded species. By computing the VDOS of interfacial water molecules at Al2O3(0001) and SiO2(0001) interfaces, it could also be shown that H-bonds donated to and accepted from the oxide material can show different spectral signals.24 Two different bands could indeed also be measured by vibrational SFG spectra at the Al2O3/water interface and their respective band strengths change as function of pH value.15
0)/water interface by vibrational SFG spectroscopy12 and assigned to strongly H-bonded species. By computing the VDOS of interfacial water molecules at Al2O3(0001) and SiO2(0001) interfaces, it could also be shown that H-bonds donated to and accepted from the oxide material can show different spectral signals.24 Two different bands could indeed also be measured by vibrational SFG spectra at the Al2O3/water interface and their respective band strengths change as function of pH value.15
Besides the H-bond kinetics at the interface, also the in-plane/out-of-plane kinetics of the terminal OH groups play an important role. We find that the IP/OOP interchange occurs every 11 ps on average (see Fig. 3). Note that this value, obtained from extensive sampling, is about one order of magnitude slower than previously reported by short explicit AIMD simulations.26 In contrast, the average H-bond lifetime between a terminal OH group and a water molecule is 1.58 ps. The difference between these two values (about one order of magnitude) indicate that the IP/OOP interchange and H-bond dynamics between the hematite slab and adjacent water molecules are two different dynamical processes which reside on different time scales, but which must obviously be coupled. Previously, we could show26 that there is a concerted IP/OOP interchange involving many different surface hydroxyls. It is suggestive, that this requires the first layer water molecules to form some pre-organization H-bond network state, which is capable to accept (or even to facilitate) such a concerted IP/OOP interchange of multiple surface hydroxyl groups. This might explain why the H-bond dynamics at the interface is faster than the concerted interchange of multiple surface hydroxyls.
In our previous publication, H-bond lifetimes on the order of 150 fs have been reported. We also came to the conclusion, that H-bond lifetimes between the hematite and adjacent water molecules are shorter compared to water/water H-bonds.26 This seeming discrepancy between the previous and current H-bond lifetimes can be understood in terms of the definition of the employed H-bond criteria: previously,26 the renowned Luzar–Chandler criterion has been used which had also been employed before in conjunction with the reactive flux technique to determine H-bond lifetimes.65 However, this criterion does not cut through the saddle point between H-bonded and non H-bonded states, see Section S7 in the ESI.† Therefore, it is known to include transient ruptures of the H-bond due to fast librational motion of the water molecules.75 This means that as soon as a participating water molecule slightly rotates out of the H-bond criterion (which is guaranteed to happen on the librational motion time scale), the H-bond is counted as broken. Note that the reactive flux technique explicitly accounts for such short time bond ruptures65,75 such that the ultra-short discontinuities of the H-bond are not counted as its rupture. Using the continuous H-bond definition64 together with the Luzar–Chandler geometrical H-bond criterion therefore maps the librational motion which resides in between 300 and 1000 cm−1 in case of liquid ambient water,76 corresponding to vibration periods in between about 34 and 111 fs. Clearly, the H-bond lifetime on the order of 150 fs reported previously falls into that time regime. It is known that the libration frequency of a water molecule blue-shifts, i.e. the vibration becomes faster, if the H-bond strength increases.68 In this work, we clearly show that the hematite/water H-bond is stronger than a water/water H-bond and thus the librational motion of first layer water molecules becomes faster than in bulk water. Since the continuous H-bond definition employing the Luzar–Chandler criterion exactly recovers that vibrational period, it is clear that the previously calculated lifetime26 was found to be shorter than the water/water H-bond lifetime.
Finally, the self-diffusion coefficient of first layer water molecules is significantly smaller compared to water molecules in the bulk phase (see Fig. 5) implying that water molecules in the first layer are systematically hindered or slowed down. Clearly, in case of hematite, the slowdown is mainly caused because of the strong H-bonds across the hematite/liquid water interface, see above. From a macroscopic perspective, a slowdown of the water molecules at the interface comes at no surprise, because friction is generally expected,36 also for solids which do not form any H-bonds with water, such as graphene and boron nitride77 or metals, such as Cu.39 However, the quantitative strength of this slowdown clearly depends on the atomistic structure of the solid and the strength of the interfacial interactions. It can also depend on the exposed facet as shown in case of the Cu/water interface39 as well as on the exposed microstructure, e.g. if solid defects are present at the interface.78
5 Conclusion
In summary, we have assessed the dynamics of water molecules at the hematite interface in terms of residence and H-bond lifetimes as well as vibrational spectra and self-diffusion coefficients. All data has been gathered from a total trajectory length of 4 ns and a simulation box containing 3120 atoms using a trained committee Neural Network potential emulating hybrid DFT. We find that water molecules are tightly bound to the hematite surface due to strong H-bonds between hematite and water. We unveil three different time scales: water/water H-bond network dynamics on the order of 1 ps at the hematite slab which is not different from H-bond dynamics in pure bulk liquid water, in-plane/out-of-plane interchange kinetics of surface hydroxyl groups on the 10 ps time scale, and residence lifetimes of water molecules in the first solvation layer of 55–65 ps time scales. Importantly, on the explicit AIMD time scale the latter two events must clearly be regarded as “rare events” and converging the associated time correlation functions is impossible for explicit AIMD. Utilizing machine learning and active learning approaches (in this case c-NNPs) is therefore mandatory to obtain converged, quantitative results. This work offers a blueprint, how state of the art c-NNPs can straightforwardly and routinely be applied for transition metal oxide/liquid water interfaces featuring a complex electronic structure. This approach will therefore be most helpful in computationally demanding realms of e.g. ab initio electrochemistry with explicit water at the DFT level, where free energy differences need to be reliably converged by thermodynamic integration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Christoph Schran (Cambridge) and Guido Falk von Rudorff (Wien) for helpful discussions. This work was supported by an individual postdoc grant to P. S. funded by the German National Academy of Sciences Leopoldina under grant number LPDS 2020-05. Computational resources have been provided by PRACE through an individual grant to P. S. giving access to HAWK at GCS@HLRS, Germany, and by ARCHER2, the UK National Supercomputing Service (https://www.archer2.ac.uk) through an EPSRC Pioneer Project grant to P. S. and J. B.References
- S. V. Yanina and K. M. Rosso, Linked reactivity at mineral–water interfaces through bulk crystal conduction, Science, 2008, 320, 218–222 CrossRef CAS PubMed
.
- K. Sivula, F. Le Formal and M. Grätzel, Solar water splitting: Progress using hematite (α-Fe2O3) photoelectrodes, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed
- H. Kuhlenbeck, S. Shaikhutdinov and H.-J. Freund, Well-ordered transition metal oxide layers in model catalysis – A series of case studies, Chem. Rev., 2013, 113, 3986–4034 CrossRef CAS PubMed
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Metal oxides and oxysalts as anode materials for Li ion batteries, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS
- A. R. Ravishankara, Y. Rudich and D. J. Wuebbles, Physical chemistry of climate metrics, Chem. Rev., 2015, 115, 3682–3703 CrossRef CAS PubMed
- J. Blumberger, Recent advances in the theory and molecular simulation of biological electron transfer reactions, Chem. Rev., 2015, 115, 11191–11238 CrossRef CAS PubMed
- Q. Wang and K. Domen, Particulate photocatalysts for light-driven water splitting: Mechanisms, challenges, and design strategies, Chem. Rev., 2019, 120, 919–985 CrossRef PubMed
- S. Corby, R. R. Rao, L. Steier and J. R. Durrant, The kinetics of metal oxide photoanodes from charge generation to catalysis, Nat. Rev. Mater., 2021, 6, 1136–1155 CrossRef CAS
- T. P. Trainor, A. M. Chaka, P. J. Eng, M. Newville, G. A. Waychunas, J. G. Catalano and G. E. Brown, Structure and reactivity of the hydrated hematite(0001) surface, Surf. Sci., 2004, 573, 204–224 CrossRef CAS
- J. A. McGuire and Y. R. Shen, Ultrafast vibrational dynamics at water interfaces, Science, 2006, 313, 1945–1948 CrossRef CAS PubMed
- Y. R. Shen and V. Ostroverkhov, Sum-frequency vibrational spectroscopy on water interfaces: Polar orientation of water molecules at interfaces, Chem. Rev., 2006, 106, 1140–1154 CrossRef CAS
- A. Tuladhar, S. Dewan, J. D. Kubicki and E. Borguet, Spectroscopy and ultrafast vibrational dynamics of strongly hydrogen bonded OH Species at the α-Al2O3(1120)/H2O interface, J. Phys. Chem. C, 2016, 120, 16153–16161 CrossRef CAS
- A. M. Gardner, K. H. Saeed and A. J. Cowan, Vibrational sum-frequency generation spectroscopy of electrode surfaces: Studying the mechanisms of sustainable fuel generation and utilisation, Phys. Chem. Chem. Phys., 2019, 21, 12067–12086 RSC
- J.-M. Andanson and A. Baiker, Exploring catalytic solid/liquid interfaces by in situ attenuated total reection infrared spectroscopy, Chem. Soc. Rev., 2010, 39, 4571–4584 RSC
- L. Zhang, C. Tian, G. A. Waychunas and Y. R. Shen, Structures and charging of α-alumina(0001)/water interfaces studied by sum-frequency vibrational spectroscopy, J. Am. Chem. Soc., 2008, 130, 7686–7694 CrossRef CAS PubMed
- Z. Jakub, M. Meier, F. Kraushofer, J. Balajka, J. Pavelec, M. Schmid, C. Franchini, U. Diebold and G. S. Parkinson, Rapid oxygen exchange between hematite and water vapor, Nat. Commun., 2021, 12, 6488 CrossRef CAS PubMed
- S. Kerisit, Water structure at hematite–water interfaces, Geochim. Cosmochim. Acta, 2011, 75, 2043–2061 CrossRef CAS
- V. Quaranta, M. Hellström and J. Behler, Proton-transfer mechanisms at the water–ZnO interface: The role of presolvation, J. Phys. Chem. Lett., 2017, 8, 1476–1483 CrossRef CAS PubMed
- K. M. Rosso, D. M. A. Smith and M. Dupuis, An ab initio model of electron transport in hematite (α-Fe2O3) basal planes, J. Chem. Phys., 2003, 118, 6455–6466 CrossRef CAS
-
D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, Cambridge, 2009 Search PubMed
- J. Cheng and M. Sprik, Acidity of the aqueous rutile TiO2(110) surface from density functional theory based molecular dynamics, J. Chem. Theory Comput., 2010, 6, 880–889 CrossRef CAS PubMed
- L.-M. Liu, C. Zhang, G. Thornton and A. Michaelides, Structure and dynamics of liquid water on rutile TiO2(110), Phys. Rev. B, 2010, 82, 161415 CrossRef
- J. Cheng, M. Sulpizi, J. VandeVondele and M. Sprik, Hole localization and thermochemistry of oxidative dehydrogenation of aqueous rutile TiO2(110), ChemCatChem, 2012, 4, 636–640 CrossRef CAS
- M.-P. Gaigeot, M. Sprik and M. Sulpizi, Oxide/water interfaces: How the surface chemistry modifies interfacial water properties, J. Phys.: Condens. Matter, 2012, 24, 124106 CrossRef PubMed
- J. Cheng, J. VandeVondele and M. Sprik, Identifying trapped electronic holes at the aqueous TiO2 interface, J. Phys. Chem. C, 2014, 118, 5437–5444 CrossRef CAS
- G. F. von Rudorff, R. Jakobsen, K. M. Rosso and J. Blumberger, Fast interconversion of hydrogen bonding at the hematite(001)–liquid water interface, J. Phys. Chem. Lett., 2016, 7, 1155–1160 CrossRef CAS PubMed
- G. F. von Rudorff, R. Jakobsen, K. M. Rosso and J. Blumberger, Hematite(001)–liquid water interface from hybrid density functional-based molecular dynamics, J. Phys.: Condens. Matter, 2016, 28, 394001 CrossRef PubMed
- M. E. McBriarty, G. F. von Rudorff, J. E. Stubbs, P. J. Eng, J. Blumberger and K. M. Rosso, Dynamic stabilization of metal oxide–water interfaces, J. Am. Chem. Soc., 2017, 139, 2581–2584 CrossRef CAS PubMed
- O. R. Gittus, G. F. von Rudorff, K. M. Rosso and J. Blumberger, Acidity constants of the hematite–liquid water interface from ab initio molecular dynamics, J. Phys. Chem. Lett., 2018, 9, 5574–5582 CrossRef CAS PubMed
- P. Gono, J. Wiktor, F. Ambrosio and A. Pasquarello, Surface polarons reducing overpotentials in the oxygen evolution reaction, ACS Catal., 2018, 8, 5847–5851 CrossRef CAS
- K. Ulman, E. Poli, N. Seriani, S. Piccinin and R. Gebauer, Understanding the electrochemical double layer at the hematite/water interface: A first principles molecular dynamics study, J. Chem. Phys., 2019, 150, 041707 CrossRef PubMed
- J. Wiktor and A. Pasquarello, Electron and hole polarons at the BiVO4–water interface, ACS Appl. Mater. Interfaces, 2019, 11, 18423–18426 CrossRef CAS PubMed
- Z. Futera and N. J. English, Water breakup at Fe2O3–hematite/water interfaces: Influence of external electric fields from nonequilibrium ab initio molecular dynamics, J. Phys. Chem. Lett., 2021, 12, 6818–6826 CrossRef CAS PubMed
- B. Dünweg and K. Kremer, Molecular dynamics simulation of a polymer chain in solution, J. Chem. Phys., 1993, 99, 6983–6997 CrossRef
- I.-C. Yeh and G. Hummer, System-size dependence of diffusion coeffcients and viscosities from molecular dynamics simulations with periodic boundary conditions, J. Phys. Chem. B, 2004, 108, 15873–15879 CrossRef CAS
- P. Simonnin, B. Noetinger, C. Nieto-Draghi, V. Marry and B. Rotenberg, Diffusion under confinement: Hydrodynamic finite-size effects in simulation, J. Chem. Theory Comput., 2017, 13, 2881–2889 CrossRef CAS PubMed
- J. Behler and M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy surfaces, Phys. Rev. Lett., 2007, 98, 146401 CrossRef
- T. Morawietz, A. Singraber, C. Dellago and J. Behler, How van der Waals interactions determine the unique properties of water, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8368–8373 CrossRef CAS PubMed
- S. K. Natarajan and J. Behler, Neural network molecular dynamics simulations of solid–liquid interfaces: Water at low-index copper surfaces, Phys. Chem. Chem. Phys., 2016, 18, 28704–28725 RSC
- M. Eckhoff and J. Behler, Insights into lithium manganese oxide–water interfaces using machine learning potentials, J. Chem. Phys., 2021, 155, 244703 CrossRef CAS PubMed
- C. Schran, K. Brezina and O. Marsalek, Committee neural network potentials control generalization errors and enable active learning, J. Chem. Phys., 2020, 153, 104105 CrossRef CAS PubMed
- T. W. Hamann, Splitting water with rust: Hematite photoelectrochemistry, Dalton Trans., 2012, 41, 7830–7834 RSC
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Grätzel, Influence of feature size, film thickness, and silicon doping on the performance of nanostructured hematite photoanodes for solar water splitting, J. Phys. Chem. C, 2009, 113, 772–782 CrossRef CAS
- Z. Zhou, P. Huo, L. Guo and O. V. Prezhdo, Understanding hematite doping with group IV elements: A DFT+U study, J. Phys. Chem. C, 2015, 119, 26303–26310 CrossRef CAS
- J. Brillet, M. Cornuz, F. L. Formal, J.-H. Yum, M. Grätzel and K. Sivula, Examining architectures of photoanode–photovoltaic tandem cells for solar water splitting, J. Mater. Res., 2010, 25, 17–24 CrossRef CAS
- Z. D. Pozun and G. Henkelman, Hybrid density functional theory band structure engineering in hematite, J. Chem. Phys., 2011, 134, 224706 CrossRef PubMed
- J. S. Smith, B. Nebgen, N. Lubbers, O. Isayev and A. E. Roitberg, Less is more: Sampling chemical space with active learning, J. Chem. Phys., 2018, 148, 241733 CrossRef PubMed
- Y. Zhai, A. Caruso, S. Gao and F. Paesani, Active learning of many-body configuration space: Application to the Cs+–water MB-nrg potential energy function as a case study, J. Chem. Phys., 2020, 152, 144103 CrossRef CAS PubMed
- C. Schran, J. Behler and D. Marx, Automated fitting of neural network potentials at coupled cluster accuracy: Protonated water clusters as testing ground, J. Chem. Theory Comput., 2020, 16, 88–99 CrossRef PubMed
-
P. Sollich and A. Krogh, Learning with ensembles: How overfitting can be useful, in Advances in Neural Information Processing Systems, ed. D. Touretzky, M. Mozer and M. Hasselmo, MIT Press, 1995, vol. 8 Search PubMed
- C. Schran, F. L. Thiemann, P. Rowe, E. A. Muller, O. Marsalek and A. Michaelides, Machine learning potentials for complex aqueous systems made simple, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110077118 CrossRef CAS PubMed
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Inuence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schimann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borstnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package – Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed
- A. Singraber, T. Morawietz, J. Behler and C. Dellago, Parallel multistream training of high-dimensional neural network potentials, J. Chem. Theory Comput., 2019, 15, 3075–3092 CrossRef CAS PubMed
- E. Schwegler, J. C. Grossman, F. Gygi and G. Galli, Towards an assessment of the accuracy of density functional theory for first principles simulations of water. II, J. Chem. Phys., 2004, 121, 5400–5409 CrossRef CAS PubMed
- K. Tonigold and A. Groß, Dispersive interactions in water bilayers at metallic surfaces: A comparison of the PBE and RPBE functional including semiempirical dispersion corrections, J. Comput. Chem., 2012, 33, 695–701 CrossRef CAS PubMed
- T. Morawietz and J. Behler, A density-functional theory-based neural network potential forwater clusters including van der Waals corrections, J. Phys. Chem. A, 2013, 117, 7356–7366 CrossRef CAS PubMed
- M. J. Gillan, D. Alfè and A. Michaelides, Perspective: How good is DFT for water?, J. Chem. Phys., 2016, 144, 130901 CrossRef PubMed
- C. Zhang, D. Donadio, F. Gygi and G. Galli, First principles simulations of the infrared spectrum of liquid water using hybrid density functionals, J. Chem. Theory Comput., 2011, 7, 1443–1449 CrossRef CAS PubMed
- A. P. Gaiduk, C. Zhang, F. Gygi and G. Galli, Structural and electronic properties of aqueous NaCl solutions from ab initio molecular dynamics simulations with hybrid density functionals, Chem. Phys. Lett., 2014, 604, 89–96 CrossRef CAS
- P. Schienbein, G. Schwaab, H. Forbert, M. Havenith and D. Marx, Correlations in the solute–solvent dynamics reach beyond the first hydration shell of ions, J. Phys. Chem. Lett., 2017, 8, 2373–2380 CrossRef CAS PubMed
- D. Laage and J. T. Hynes, On the residence time for water in a solute hydration shell: Application to aqueous halide solutions, J. Phys. Chem. B, 2008, 112, 7697–7701 CrossRef CAS PubMed
- D. C. Rapaport, Hydrogen bonds in water, Mol. Phys., 1983, 50, 1151–1162 CrossRef CAS
- A. Luzar and D. Chandler, Hydrogen-bond kinetics in liquid water, Nature, 1996, 379, 55–57 CrossRef CAS
- R. Kumar, J. R. Schmidt and J. L. Skinner, Hydrogen bonding definitions and dynamics in liquid water, J. Chem. Phys., 2007, 126, 204107 CrossRef CAS PubMed
- P. Schienbein and D. Marx, Assessing the properties of supercritical water in terms of structural dynamics and electronic polarization effects, Phys. Chem. Chem. Phys., 2020, 22, 10462–10479 RSC
- P. Schienbein and D. Marx, Supercritical water is not hydrogen bonded, Angew. Chem., Int. Ed., 2020, 59, 18578–18585 CrossRef CAS PubMed
- D. Ojha, K. Karhan and T. D. Kühne, On the hydrogen bond strength and vibrational spectroscopy of liquid water, Sci. Rep., 2018, 8, 16888 CrossRef PubMed
- H. J. Bakker and J. L. Skinner, Vibrational spectroscopy as a probe of structure and dynamics in liquid water, Chem. Rev., 2010, 110, 1498–1517 CrossRef CAS PubMed
- M. Heyden, J. Sun, S. Funkner, G. Mathias, H. Forbert, M. Havenith and D. Marx, Dissecting the THz spectrum of liquid water from first principles via correlations in time and space, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12068–12073 CrossRef CAS PubMed
- J. Marti, J. Padro and E. Guardia, Molecular dynamics calculation of the infrared spectra in liquid H2O–D2O mixtures, J. Mol. Liq., 1994, 62, 17–31 CrossRef CAS
- K. Yoshida, C. Wakai, N. Matubayasi and M. Nakahara, A new high-temperature multinuclearmagnetic-resonance probe and the self-diffusion of light and heavy water in sub- and supercritical conditions, J. Chem. Phys., 2005, 123, 164506 CrossRef PubMed
-
J. P. Hansen and I. R. McDonald, Cheory of Simple Liquids, Academic Press, Amsterdam, Boston, London, New York, Oxford, Paris, San Diego, San Francisco, Singapore, Sydney, Tokyo, 1986 Search PubMed
- A. Luzar, Resolving the hydrogen bond dynamics conundrum, J. Chem. Phys., 2000, 113, 10663–10675 CrossRef CAS
- G. E. Walrafen, Raman spectrum of water: transverse and longitudinal acoustic modes below 300 cm−1 and optic modes above 300 cm−1, J. Phys. Chem., 1990, 94, 2237–2239 CrossRef CAS
- G. Tocci, L. Joly and A. Michaelides, Friction of water on graphene and hexagonal boron nitride from ab initio methods: Very different slippage despite very similar interface structures, Nano Lett., 2014, 14, 6872–6877 CrossRef CAS PubMed
- L. Joly, G. Tocci, S. Merabia and A. Michaelides, Strong coupling between nanouidic transport and interfacial chemistry: How defect reactivity controls liquid-solid friction through hydrogen bonding, J. Phys. Chem. Lett., 2016, 7, 1381–1386 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01708c |
| This journal is © the Owner Societies 2022 |