
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanisms for sonochemical oxidation of nitrogen†
Thomas
Qureishy
ab,
Sverre
Løyland
 ac,
Susanne J.
Jørgensen
ad,
Eline M.
Færgestad
ad,
Truls
Norby
ab and
Einar
Uggerud
*ac
ac,
Susanne J.
Jørgensen
ad,
Eline M.
Færgestad
ad,
Truls
Norby
ab and
Einar
Uggerud
*ac
aDepartment of Chemistry, University of Oslo, Norway. E-mail: einar.uggerud@kjemi.uio.no
bCentre for Materials Science and Nanotechnology, University of Oslo, Norway
cHylleraas Centre for Quantum Molecular Sciences, University of Oslo, Norway
dCentre for Biogeochemistry in the Anthropocene, University of Oslo, Norway
First published on 9th June 2022
Abstract
N2O, and mixtures of N2 and O2, dissolved in water—both in the presence and absence of added noble gases—have been subjected to ultrasonication with quantification of nitrite and nitrate products. Significant increase in product formation upon adding noble gas for both reactant systems is observed, with the reactivity order Ne < Ar < Kr < Xe. These observations lend support to the idea that extraordinarily high electronic and vibrational temperatures arise under these conditions. This is based on recent observations of sonoluminescence in the presence of noble gases and is inconsistent with the classical picture of adiabatic bubble collapse upon acoustic cavitation. The reaction mechanisms of the first few reaction steps necessary for the critical formation of NO are discussed, illustrated by quantum chemical calculations. The role of intermediate N2O in this series of elementary steps is also discussed to better understand the difference between the two reactant sources (N2O and 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 N2:O2; same stoichiometry).
:1 N2:O2; same stoichiometry).
Introduction
Nitrate is the main ingredient in agricultural fertilizers. Today, its industrial production incorporates two coupled processes: the formation of ammonia from nitrogen and hydrogen (Haber process) followed by the oxidation of the ammonia to nitric acid (Ostwald process).1 In principle, a one-step process, avoiding the energetically unfavorable intermediate ammonia, using only nitrogen, oxygen and water as feedstock, would be more attractive. In fact, such a process, the Birkeland–Eyde process,2–5 involving nitric oxide (NO) and nitrogen dioxide (NO2) as intermediates does exist:| N2 + O2 ⇌ 2NO, ΔrxnH° = −181 kJ mol−1 | (1) |
| 2NO + O2 → 2NO2, ΔrxnH° = −100 kJ mol−1 | (2) |
| 3NO2 + H2O → 2HNO3 + NO, ΔrxnH° = −117 kJ mol−1 | (3) |
| N + O2 → NO + O | (4) |
| O + N2 → NO + N, | (5) |
NO formation (1) becomes thermodynamically more favorable the higher the temperature is. However, produced NO starts to dissociate when the temperature becomes too high, and T = 3200 K is considered an optimum.9 The second step, further oxidation to give NO2(2) is exothermic and occurs in the colder zone outside the high temperature plasma. In practice, the yield of NO + NO2 (NOx) in the Birkeland–Eyde process is only a few per cent per reaction cycle, thereby limiting the energy efficiency. For this reason, finding alternative, direct ways to nitrogen oxidation are desirable, considering that NO and NO2 production have opposite thermodynamic requirements.
Sonochemistry may provide such an alternative. It is now well established that during acoustic cavitation resulting from intense ultrasound radiation of a solution, plasma-like conditions exist transiently upon sudden bubble collapse, with local temperatures and pressure exceeding 5000 K and 100 kbar.11 More recently, systematic observations of sonoluminescence indicate even higher temperatures, up to 20000 K, with subsequent cooling rates exceeding 1012 K s−1.12 Altogether, this provides in principle an opportunity for NO production upon bubble collapse directly followed by oxidation to NO2 as the temperature lowers.
On this background it is not surprising to find that sonochemical production of nitrite and nitrate from air dissolved in water has been studied and is well documented in the literature. As early as in 1936, Schultes and Gohr determined by colorimetry and iodometry the production of H2O2, nitrous acid and nitric acid as products after applying ultrasound to air-saturated water.13 They found that NO was produced first and was further oxidized to NO2 if there was enough oxygen present. Further studies have been performed by several research groups. For instance, Virtanen and co-workers reported that hydrogen gas and carbon monoxide prevent nitrogen fixation and that the total production was pH-independent.14,15 Furthermore, it has been reported that dissolved O2 rather than H2O is the dominant source of oxygen for both NO and NO2.8,16–18
Henglein and co-workers conducted a series of experiments where Ar was added as an inert gas to gas mixtures dissolved in water for sonochemistry. The relative amount of Ar was observed to influence sonochemical nitrate and nitrite production in reactions of nitrous oxide (N2O)19,20 and N2,21 respectively, and in the formation of H2O2 upon addition of O2.22 Interestingly, in the presence of O2 the authors noticed the formation of more nitrite and nitrate indicating an “O” + N2O pathway for NO production. In neither of these papers was any active role of Ar mentioned other than it could reduce the cooling rate having a lower heat capacity ratio than the other gases.
Suslick et al. have reported optical emission in bubble cavitation experiments of concentrated sulfuric acid containing noble gases and noted the presence of both highly excited neutral noble gas atoms and ions, with intensities and spectral widths only compatible with non-thermal plasmas comprising energetic particles that give rise to electron impact dissociation and ionization, and bremsstrahlung.12,23–25 Nikitenko and co-workers compared OH emission spectra of water saturated with Ar and Xe, and concluded that the electronic and vibrational temperatures of the OH radicals in Xe are significantly higher than in Ar.26,27 They suggested that a lower ionization energy of Xe provides a plasma with higher free electron density and temperature, and that collisions of electrons with water molecules to give excited OH radicals therefore are more effective in Xe. An alternative explanation could be that Xe is more soluble, thereby creating more bubbles. In a following review paper28 the same authors write without literature citation that “it has been known for a long time that heavy noble gases like Xe and Kr result in more OH radicals than lighter noble gases such as He, Ne and Ar.”
The purpose of our work is fourfold. First, partly reproducing the previously mentioned experiments, we wanted to investigate how nitrate and nitrite production depend on the O2:N2 ratio in the sonochemical version of the Birkeland–Eyde process. Second, since it has been suggested that there is an “O” + N2O pathway towards NO, we wanted to conduct separate experiments with N2O as feedstock. Third, we considered it useful to analyze the experimental data using reaction models firmly based on quantum chemical calculations to shed light on the key reaction steps with emphasis on the role of N2O as an intermediate in the production of NO. In this, we humbly realize the inherent complexity of the plasma chemistry at work. Fourth and finally, it would be interesting to investigate how nitrate/nitrite formation is affected by the presence and nature of added noble gas, with reference to the assumption of Nikitenko that the formation of OH radicals (or similar reactive species) increases down the group of the noble gases (Ne–Xe).28
Materials and methods
Experimental
Ultrasound was applied for an hour to 150 mL liquid samples of type-II water using a Hielscher UIP1000hdT-230 ultrasonic processor, which consists of a transducer connected to a generator, and has a frequency of 20 kHz and maximum power of 1 kW. The sonotrode in contact with the liquid in the reactor had a diameter of 34 mm, and between the transducer and sonotrode was a booster horn, amplifying the signal by a factor of 2.2. The processor was set to power mode, which keeps the power stable, as opposed to amplitude mode, which keeps the amplitude stable. The power was set to 80% of the maximum value. The total power and net power dissipated into the sample were around 400 and 270 W, respectively, and were recorded continuously. The sample chamber was water-cooled to stay at temperatures within 21–26 °C. The temperature was measured by a TF100 thermocouple before and after the ultrasonication.The reactor has an arm above the level of the liquid sample. This arm was closed with a small septum. Two needles were inserted through the septum, of which one is for gas insertion. Outside the reactor it is connected with a Swagelok fitting to a 1/8 inch diameter stainless steel tube, and inside the reactor it is bent downwards and immersed deep into the liquid, so that gas is inserted near the bottom surface, providing maximum contact with and possibility to dissolve in the liquid. The other needle allows for gas to escape the reactor. It is straight and its end inside the reactor is therefore above the sample surface. In the experiments reported here, water-cooling was started one minute before turning on the ultrasound.
A stable gas flow was kept at 100 sccm (mLN min−1) for 15 minutes before applying the ultrasound and then for 1 h during ultrasonication. The gas mixtures and flow were manually controlled using calibrated variable-area rotameters with precision needle valves. Four sets of experiments were carried out (see Table S1 in ESI†). In the first, the gas mixtures consisted of nitrogen and oxygen, to check for consistency with literature data. In the second set of experiments, N2O was mixed with Ne, Ar, Kr and Xe. We used the same noble gases in the third set of experiments, but this time we mixed them with synthetic air instead of N2O. The total gas flow was fixed to 100 sccm throughout the experiments, and the flows of each individual gas are presented interchangeably as % and sccm.
It should be noted that a processor of the kind used here has several possible sources of parasitic air unintentionally entering the gas and liquid phases, such as seals and fittings in addition to the water itself and operational procedures. We took care to eliminate all of these until the yield of nitrite and nitrate was insignificant in the absence of a deliberately added source of nitrogen.
Due to degradation of the sonotrode, titanium powder residues could be found in the liquid samples. Samples were therefore filtered prior to chemical analysis in 10 mL batches using PES syringe filters with pore size 0.45 μm.
The liquid products were characterized by high-pressure ion chromatography (HPIC), using a Dionex Integrion HPIC system configured for reagent-free ion chromatography (IC). Ten standards were made using Reagecon Seven Anion Standard, which enables the quantification of fluorides, chlorides, nitrites, sulfates, borides, nitrates, and phosphates. The most and least diluted standards contained 0.002 and 10 mg L−1, respectively, of both NO2− and NO3−. Unless otherwise specified, all data points in the figures are an average of three IC measurements.
Computational
The ORCA 5.0.1 program package29–31 was used for the quantum chemical computations. All stationary points of the potential energy hypersurface (reactants, transition states, intermediates, and products) and minimum energy crossing points (MECP) between the diabatic potential energy surfaces were characterized by complete geometry optimizations. The computations were performed using density functional theory with the hybrid functional B3LYP, full-valence CASSCF and coupled cluster with singlets, doublets and perturbational triplets (CCSD(T)) in conjunction with the augmented Dunning correlation-consistent polarized quadruple-zeta (aug-cc-pVQZ) basis set.32 The CASSCF calculations were subsequently corrected for dynamic correlation using FIC-NEVPT2.33–36 Reactants, intermediates and products were characterized by analytic or numerical frequency calculations depending on the availability for each level of theory. All reactants and intermediates have positive definite Hessian matrices, while transition state structures have a single negative eigenvalue in their diagonalized force constant matrices. The calculations were compared with existing experimental and computational data using the ANL ATcT databases.37,38Experimental results
The first series of experiments was aimed at finding the optimum ratio of O2 in O2/N2 gas mixtures. The results are presented in Fig. 1, which shows the total amount of NO2− and NO3− produced for each mixture investigated. Interestingly, a shallow maximum production is found for the gas mixture that resembles air the most.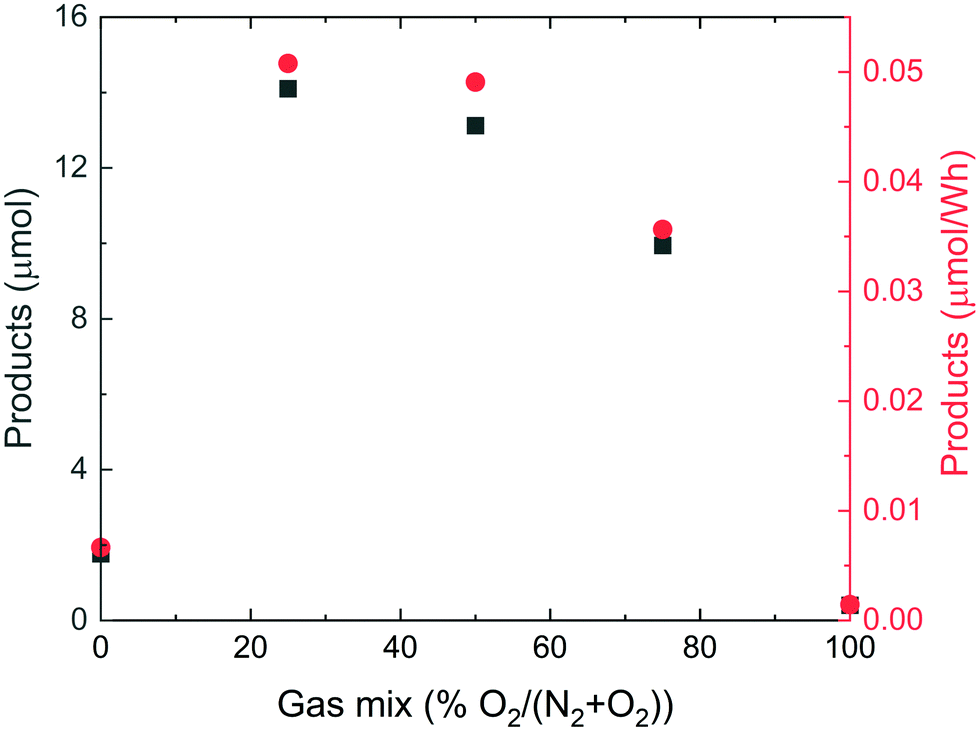 | ||
| Fig. 1 The integrated amount of nitrate and nitrite produced (black squares) and the same amount relative to the energy dissipated into the system (red circles) as a function of the relative amount of O2 in the N2 + O2 gas mixture, fed at 100 sccm. Some of the data points are overlapping. | ||
Based on these findings, we decided to apply an O2:N2 ratio of 20:80 (synthetic air) for the second series of experiments, in which equal amounts of a noble gas and synthetic air were introduced into the water. The results are presented in Fig. 2. Significantly more products (NO2− + NO3−) are formed by introducing Ar, Kr and Xe, and production increases noticeably down the group of noble gases.
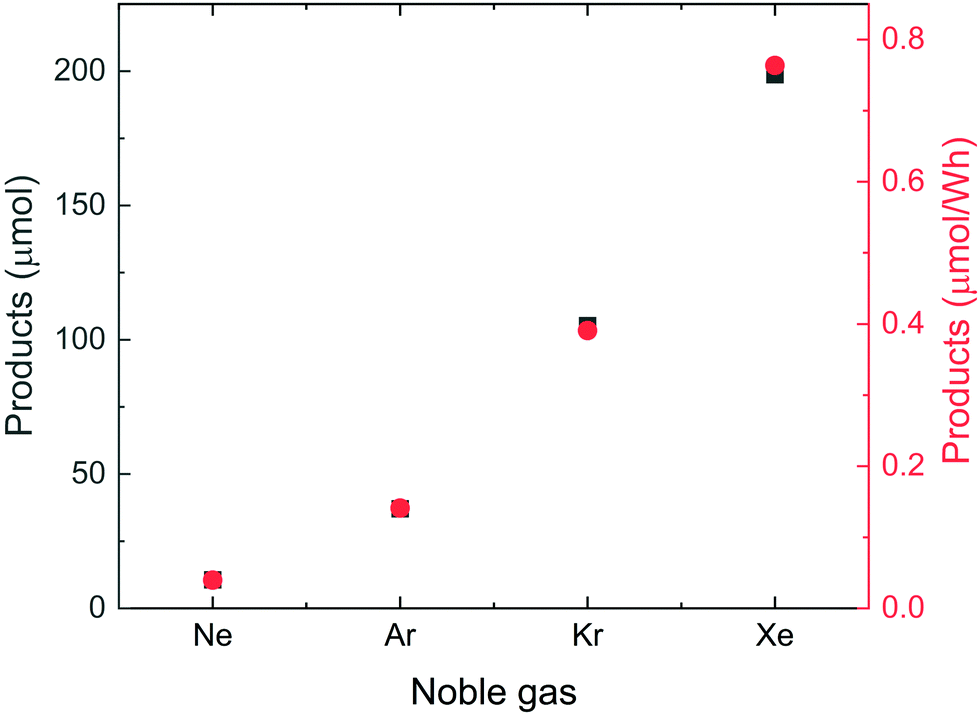 | ||
| Fig. 2 The integrated amount of nitrate and nitrite produced after flowing a mixture of 50 sccm synthetic air and 50 sccm noble gas through the liquid sample before and during ultrasonication. Some of the data points are overlapping. | ||
A third series of measurements was made for variable mixtures of N2O and Ar, Fig. 3. Maximum nitrate + nitrite production is observed around 10 sccm N2O + 90 sccm Ar. This is consistent with the results of similar measurements performed by Hart and Henglein,20 and Nikitenko and Seliverstov,39 so this mixing ratio was applied for the following experiments with the other noble gases, see below.
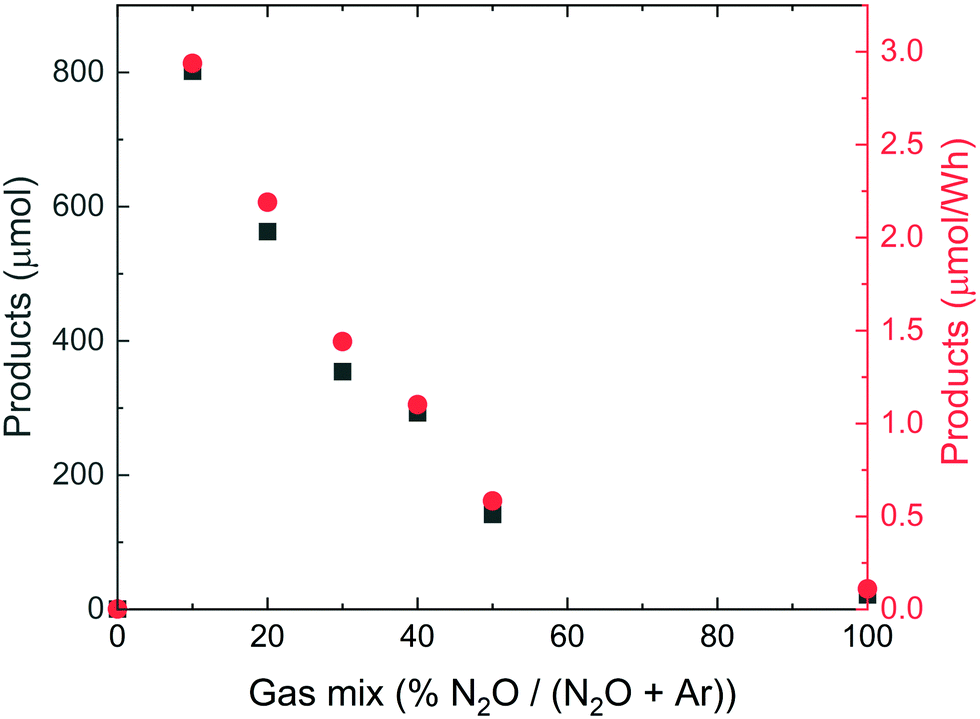 | ||
| Fig. 3 The integrated amount of nitrate and nitrite produced (black squares) and the same amount relative to the energy dissipated into the system (red circles) as a function of the relative amount of N2O in the N2O + Ar gas mixture, fed at 100 sccm. Some of the data points are overlapping. | ||
Fig. 4 shows the results of the fourth and final series of measurements that were conducted for mixtures of N2O/E (E = Ne, Ar, Kr, Xe), with 10% N2O and 90% of the noble gas. Also in this case, a significant periodic trend Ne < Ar < Kr < Xe is observed.
 | ||
| Fig. 4 The integrated amount of nitrate and nitrite produced for each noble gas after flowing mixtures of 10 sccm N2O and 90 sccm noble gas mixtures through the liquid. Some of the data points are overlapping. | ||
Discussion
From Fig. 1 we estimate that the sum of nitrite and nitrate products in a 2:1 mixture of N2 and O2, in the absence of a noble gas, amounts to 13 μmol, which is 95% of that measured at the maximum of the curve, for synthetic air (4:1). In the presence of Ar (Fig. 2), after normalizing to the same total gas flow as in Fig. 2, the product yield increases to 2 × 35 μmol = 70 μmol. However, when we instead use N2O (also having N:O = 2:1) as reactant, and also with Ar present, the yield increases to 10 × 800 μmol = 8000 μmol (Fig. 4), when normalized to the same reactant gas flow as used in the Fig. 1 experiments. For these considerations, we need to realize that N2O is 20 times more soluble in water than O2 and 40 times more soluble than N2 (see ESI†). Therefore, if we assume that N2 solubility is the limiting factor in the N2 + O2 case, this shows the amount of produced nitrite and nitrate should be 8000 μmol/40 = 200 μmol, approximately three times higher. In other words, when N2O is used as reactant, the amount of products formed is three times higher than when an N2/O2 mixture with the same stoichiometric ratio is used. This rules out thermodynamic equilibrium in the two experiments. Although our simple analysis is not based on any systematic kinetic study, it is tempting based on the available data to hypothesize that nitrous oxide may be an intermediate during nitric oxide formation in the reaction between N2 and O2, i.e.,| N2 + O2 → N2O → NO. | (6) |
The fact that reactivity increases down the group Ne < Ar < Kr < Xe is consistent with the results of the elegant sonoluminescence experiments reported from the laboratories of Suslick and Nikitenko.12,23–27 We note that the observed trends in reactivity show good correlation with noble gas solubility. In the case of N2 + O2, the correlation is linear, while for N2O it is slightly curved, see Fig. S1 and S2 in ESI.† Whether the correlations between solubility and reactivity relate to the number of bubbles created, the electron temperature of the transient plasma due to the ease of ionization or is coincidental—in the sense that most physical properties change monotonically down the group of noble gases can, of course, not be straightforwardly determined based on the present data. We will, despite this critical comment, relate the rest of our discussion to the electron temperature scenario substantiated by the two named laboratories.
As already explained, a short-lived plasma is formed during bubble collapse upon sonication of a liquid. In general, a manifold of chemical reactions is involved in this high-energy event and in the following period as the liquid quickly approaches room temperature. The extremely fast temperature and pressure changes make a complete, quantitative description of the chemical system very demanding, if not essentially impossible. However, a great deal is known about the general features of plasma chemistry, irrespective of the plasma is formed in electric discharges, in flames, in shock waves from detonations, upon electromagnetic radiation, or by sonication. In the present context, a comprehensive and recent account of the chemistry in N2 + O2 plasmas by Guerra et al.40 provides a good guide to this fascinating landscape. Just to describe the reactions between the numerous neutral and charged species formed from these two diatomic molecules alone (in the absence of water) the authors involve 198 different elementary reactions, each with a given rate coefficient. With humble recognition of the overall complexity, we will extend the simplified Zel’dovich model (eqn (4) and (5)) by including N2O explicitly as an intermediate and relate this to the non-adiabatic high electron temperature regime advocated by Suslick and Nikitenko.
In doing so, we will first examine how energy rich electrons interact with O2 and N2, respectively, to provide atomization. From a thermodynamic perspective, since the bond dissociation energy of O2 is half that of N2 (498 vs. 945 kJ mol−1), one could expect that this gives rise to more O atoms than N atoms. But in a plasma with a myriad of fast electrons the dominating process for atomization will be by electron-impact dissociation:
| e− + N2 → e− + 2N | (7) |
| e− + O2 → e− + 2O. | (8) |
 | ||
| Fig. 5 Potential energy diagram for lowest energy reactions (all electronic doublet states) between N and O2 obtained by B3LYP calculations, see text for description. All energies in kJ mol−1. | ||
It should be noted that none of the computational methods employed are able to fully describe all features of the systems studied. While CCSD(T) works very well for the minima and give values close to the experimental values with low T1 diagnostics, it does not handle the multireference character during bond-breaking in transition states as well and breaks down completely for the minimum energy crossing points (MECPs). On the other hand, CASSCF presumably handles the MECPs better, but it is less suited for e.g. N2O as has been previously demonstrated.43,44 B3LYP seems to handle both the stationary points and the MECPs somewhat well and is thus used throughout the discussion. Because of these discrepancies, we advise that the computed values are taken merely as qualitative. We also note that some of the experimental values used for comparison are not entirely trustworthy and some of the listed intermediates are debated whether they even exist as stable minima.45–48 Comparisons of the different computational methods and experimental values are listed in the ESI.†
The left hand side of Fig. 5 shows two reactant states with the lowest quartet state of nitrogen atom, 4S, with the doublet 2D lying 264 kJ mol−1 higher in energy, compared to the spectroscopic value of 230 kJ mol−1.49 The right hand side displays two product states, with the lowest triplet state of oxygen atom, 3P, with the singlet 1D being 266 kJ mol−1 higher in energy, compared to the spectroscopic value of 190 kJ mol−1.49 Regarding the reaction mechanism, the potential energy surfaces of N + O2 reactions have been studied in greater detail by others,50–53 and here we limit ourselves to display only a few characteristic features relevant to the present context. Starting from the ground doublet reactant state (4N + 3O2), after passing a modest barrier of 25 kJ mol−1, a local minimum corresponding to the peroxy radical NOO is reached. The existence of this species has been debated in the literature,50 but both the aforementioned quantum chemical studies and a recent velocity-map imaging study54 indicate that NOO corresponds to a local minimum albeit with a limited lifetime. Ultimately, the products 2NO + 3O are formed after passing the submerged barrier at −88 kJ mol−1. From Fig. 5 we also see that the same products are formed in a barrierless reaction from the electronically excited state 2N + O2, while there is only a small barrier for 2N + O2 → NO + 1O. All the three essentially direct exothermic reactions therefore constitute efficient channels for forming NO, in accordance with the first Zel’dovich reaction (eqn (4)). In Fig. 5 we have also included nitrogen dioxide (ONO), which is the lowest energy point of the potential energy surface. There seems to be no other route from NOO to ONO other than via the 2NO + 3O asymptote as indicated in the diagram. On the other hand, this shows that NO is easily oxidized to NO2 in direct reaction with 3O once the temperature and the pressure allows for trapping this product by third body collisions.
The potential energy surfaces of N2O have been surveyed in photodissociation55–57 and electron-impact dissociation experiments,58,59 and by quantum chemical calculations.55,60,61 These studies provided the background for our own quantum chemical model displayed in Fig. 6.
 | ||
| Fig. 6 Potential energy diagram for lowest energy reactions (blue: electronic singlet state, red: electronic triplet state) between O and N2 obtained by B3LYP calculations, see text for description. For linear geometries intersystem (singlet/triplet) crossing may occur at the points marked with green circles. As only lighter period 2 elements contribute to this reaction path, the spin–orbit coupling effect is small compared to the MECP barriers. All energies in kJ mol−1. | ||
From the energy diagram of Fig. 6 we see that the second Zel’dovich reaction (O + N2 → NO + N, eqn (5)) is endothermic, irrespective of whether the reactant oxygen atom is the singlet or the triplet or whether the product nitrogen atom is the doublet or the quartet. Only at very high vibrational temperatures of N2, any of the indicated channels will therefore be open. In addition, N atoms formed by electron-impact dissociation are more prevalent in a hot electron plasma than O atoms, see above, so we must conclude that the O + N2 pathway to NO is clearly less important than the corresponding N + O2 pathway discussed above. All this is consistent with the kinetic data collected and presented by Guerra et al.40
The global minimum of Fig. 6 corresponds to nitrous oxide, which exists en route to NO on the singlet surface. If the temperature is not too high or the pressure too low, this species may be trapped by third body collisions, which explains why it has been observed as an end product in sonochemical reactions of air, albeit in small quantities.21 Alternatively, the metal residue produced by sonotrode disintegration may catalyze the spin inversion from the triplet to the singlet state to form and stabilize N2O. Both the ease of electron-impact dissociation of N2O58,59 and its high reactivity with other species present (see below) make this species fragile in a plasma. Despite this, N2O appears to play an important supporting role in the reactions studied here. In our sonochemical experiments using pure N2O as reactant, we observed that this gives rise to more products when we react an N2:O2 mixture of the same stoichiometry. Second, due to its vulnerability for electron-impact dissociation, it will be the main source for O atoms that in a next step may react with intact N2O molecules to give NO. This is illustrated in Fig. 7, which we have obtained by quantum chemical calculation, capitalizing on the results from earlier studies.58,62,63
 | ||
| Fig. 7 Potential energy diagram for lowest energy reactions (blue: electronic singlet state, red electronic triplet state) between O and N2O obtained by B3LYP calculations, see text for description. All energies in kJ mol−1. | ||
In agreement with the references cited immediately above we find that the reaction between triplet oxygen atom and N2O is highly favorable, since this leads directly to two NO molecules in an exothermic, barrierless reaction via the elusive ON⋯NO complex. To which degree this species actually corresponds to a shallow potential energy minimum is questionable, since the computational methods give different answers to this. In any case this provides a rationale for the aforementioned “O” + N2O pathway17–22,39 now identifying the reactive oxygen species “O” to be 3O. Whether the oxygen source is N2O or O2 seems to be of less importance than the observation that 3O is the reactive species rather than OH radical formed by sonolysis of water. Fig. 7 also shows that 1O also will react with N2O, but with a more demanding mechanism, via the covalently bonded O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NO intermediate that requires the passage of a transition state.
N–NO intermediate that requires the passage of a transition state.
Conclusion
The energy diagrams in Fig. 5–7 help us suggest the major mechanism for formation of NO from N2 and O2 in the transient plasma formed upon bubble collapse, namely:| N2 → 24N (upon electron-impact dissociation) | (9a) |
| O2 → 23O (upon electron-impact dissociation) | (9b) |
| 4N + O2 → NO + 3O | (10) |
| 3O + N2 → NO + 4N | (11a) |
| → N2O | (11b) |
| 3O + N2O → 2NO | (12) |
The fact that the addition of noble gases increases product formation with the observed reactivity order Ne < Ar < Kr < Xe is best understood from the hot-electron model of Suslick and Nikitenko. Among the four noble gases, Xe is the most soluble and has the lowest ionization energy, giving rise to the highest flux of free electrons and most likely the most energetic ones. This in turn provides more atomization and thereby higher product yield.
A spectroscopic study of the sonoluminescence during NOx formation would be valuable in elucidating the suggested mechanisms. However, our current experimental setup does not permit such experiments.
As a final comment, our nitrate + nitrite product yield is unimpressive from an industrial perspective, to state it mildly. Even in the presence of Xe the synthetic air mixture gives rise to modest 0.8 μmol W−1 h−1 (from Fig. 2) = 0.06 mg W−1 h−1 = 0.06 g kW−1 h−1 (based on the molecular mass of nitric acid). In comparison, the electric-arc Birkeland–Eyde process gives 60 g kW−1 h−1.5 A first step for improvement of sonochemical nitrogen oxidation would be to eliminate water using a non-volatile solvent, and consequently separate NO production from the following reaction steps (oxidation to nitrogen dioxide and hydrolysis). Virtanen noticed already seven decades ago that hydrogen is unavoidably present in water upon sonolysis, and therefore has an inhibitory effect on nitrate formation since it is a reductant.14,15 More recent studies have shown that the low viscosity and considerable vapor pressure of water, even at room temperature, counteract fast energy release during bubble collapse and specifically introduce unproductive OH radicals.17,18 The sonochemical process could also be improved by increasing the reactor pressure64–66 and/or identifying more effective promotors than the noble gases that may facilitate nitrogen activation.
Author contributions
Thomas Qureishy: investigation, methodology, writing – review & editing. Sverre Løyland: investigation, writing – review & editing. Susanne J. Jørgensen: resources. Eline M. Færgestad: resources. Truls Norby: conceptualization, supervision, writing – review & editing, funding acquisition. Einar Uggerud: conceptualization, supervision, funding acquisition, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding and support from the Norwegian Research Council through Grant No. 280495 (Novel concepts for cold activation of energy-intensive chemical processes (COLD)), the Hylleraas Centre for Quantum Molecular Sciences No. 262695/F50 435 through their Centre of Excellence program, and the Norwegian Supercomputing Program (NOTUR) through a grant of computer time (Grant NN4654K). The authors are grateful to the advisory board members and Dr Quanbao Ma of the COLD project for inspiring discussions.References
- V. Smil, Enriching the Earth, Fritz Haber, Carl Bosch, and the Transformation of World Food Production, The MIT Press, Boston, 2001 Search PubMed.
- K. Birkeland, Trans. Faraday Soc., 1906, 2, 98–116 RSC.
- S. Eyde, J. R. Soc. Arts, 1909, 57, 568–576 CAS.
- N. Cherkasov, A. O. Ibhadon and P. Fitzpatrick, Chem. Eng. Process., 2015, 90, 24–33 CrossRef CAS.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2021, 14, 2520–2534 RSC.
- H. Cavendish, Philos. Trans. R. Soc. London, 1785, 75, 372–384 CrossRef.
- R. D. Hill, R. G. Rinker and H. D. Wilson, J. Atmos. Sci., 1980, 37, 179–192 CrossRef CAS.
- J. Yao, L. Chen, X. Chen, L. Zhou, W. Liu and Z. Zhang, Ultrason. Sonochem., 2018, 42, 42–47 CrossRef CAS PubMed.
- J. Lee, H. Sun, S.-k Im and M. Soo Bak, J. Appl. Phys., 2017, 122, 083303 CrossRef.
- Y. B. Zel'dovich and Y. P. Raizer, in Physics of Shock Waves and High-Temperature Hydrodynamic Phenomena, ed. W. D. Hayes and R. F. Probstein, Dover Publications, Mineola, New York, 1967, ch. 8, vol. 2, pp. 564–573 Search PubMed.
- E. A. Neppiras, Phys. Rep., 1980, 61, 159–251 CrossRef.
- K. S. Suslick and D. J. Flannigan, Annu. Rev. Phys. Chem., 2008, 59, 659–683 CrossRef CAS PubMed.
- H. Schultes and H. Gohr, Angew. Chem., 1936, 49, 420–423 CrossRef CAS.
- A. I. Virtanen and N. Ellfolk, Acta Chem. Scand., 1950, 4, 93–102 CrossRef CAS.
- A. I. Virtanen and N. Ellfolk, J. Am. Chem. Soc., 1950, 72, 1046–1047 CrossRef CAS.
- P. K. Supeno, Ultrason. Sonochem., 2000, 7, 109–113 CrossRef CAS PubMed.
- D. Lohse, M. P. Brenner, T. F. Dupont, S. Hilgenfeldt and B. Johnston, Phys. Rev. Lett., 1997, 78, 1359–1362 CrossRef CAS.
- Y. T. Didenko and K. S. Suslick, Nature, 2002, 418, 394–397 CrossRef CAS PubMed.
- A. Henglein, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40b, 100–107 CrossRef CAS.
- E. J. Hart and A. Henglein, J. Phys. Chem., 1986, 90, 5992–5995 CrossRef CAS.
- E. J. Hart, C. H. Fischer and A. Henglein, J. Phys. Chem., 1986, 90, 5989–5991 CrossRef CAS.
- E. J. Hart and A. Henglein, J. Phys. Chem., 1985, 89, 4342–4347 CrossRef CAS.
- D. J. Flannigan and K. S. Suslick, Phys. Rev. Lett., 2005, 95, 044301 CrossRef PubMed.
- N. C. Eddingsaas and K. S. Suslick, J. Am. Chem. Soc., 2007, 129, 3838–3839 CrossRef CAS PubMed.
- K. S. Suslick, N. C. Eddingsaas, D. J. Flannigan, S. D. Hopkins and H. Xu, Acc. Chem. Res., 2018, 51, 2169–2178 CrossRef CAS PubMed.
- R. Pflieger, H.-P. Brau and S. I. Nikitenko, Chem. – Eur. J., 2010, 16, 11801–11803 CrossRef CAS PubMed.
- A. A. Ndiaye, R. Pflieger, B. Siboulet and S. I. Nikitenko, Angew. Chem., Int. Ed., 2013, 52, 2478–2481 CrossRef CAS PubMed.
- S. I. Nikitenko and R. Pflieger, Ultrason. Sonochem., 2017, 35, 623–630 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- Y. Guo, K. Sivalingam, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 147, 064110 CrossRef PubMed.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J. P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- B. Ruscic, R. E. Pinzon, M. L. Morton, G. von Laszevski, S. J. Bittner, S. G. Nijsure, K. A. Amin, M. Minkoff and A. F. Wagner, J. Phys. Chem. A, 2004, 108, 9979–9997 CrossRef CAS.
- B. Ruscic, R. E. Pinzon, G. v Laszewski, D. Kodeboyina, A. Burcat, D. Leahy, D. Montoy and A. F. Wagner, J. Phys.: Conf. Ser., 2005, 16, 561–570 CrossRef CAS.
- S. I. Nikitenko and A. F. Seliverstov, Russ. Chem. Bull., 2000, 49, 2077–2079 CrossRef CAS.
- V. Guerra, A. Tejero-del-Caz, C. D. Pintassilgo and L. L. Alves, Plasma Sources Sci. Technol., 2019, 28, 073001 CrossRef CAS.
- P. C. Cosby, J. Chem. Phys., 1993, 98, 9544–9553 CrossRef CAS.
- P. C. Cosby, J. Chem. Phys., 1993, 98, 9560–9569 CrossRef CAS.
- D.-Y. Hwang and A. M. Mebel, Chem. Phys., 2000, 259, 89–97 CrossRef CAS.
- J. M. L. Martin and T. J. Lee, J. Chem. Phys., 1993, 98, 7951–7957 CrossRef CAS.
- H. A. Duarte, E. Proynov and D. R. Salahub, J. Chem. Phys., 1998, 109, 26–35 CrossRef CAS.
- E. A. Wade, J. I. Cline, K. T. Lorenz, C. Hayden and D. W. Chandler, J. Chem. Phys., 2002, 116, 4755–4757 CrossRef CAS.
- K. A. Nguyen, M. S. Gordon, J. A. Montgomery, Jr. and H. H. Michels, J. Phys. Chem., 1994, 98, 10072–10078 CrossRef CAS.
- M. A. Vincent, I. H. Hillier and L. Salsi, Phys. Chem. Chem. Phys., 2000, 2, 707–714 RSC.
- C. E. Moore and J. W. Gallagher, Tables of spectra of hydrogen, carbon, nitrogen, and oxygen atoms and ions, 1993 Search PubMed.
- S. P. Walch, J. Chem. Phys., 1995, 102, 4189–4192 CrossRef CAS.
- M. Braunstein and J. W. Duff, J. Chem. Phys., 2000, 113, 7406–7413 CrossRef CAS.
- V. Kurkal, P. Fleurat-Lessard and R. Schinke, J. Chem. Phys., 2003, 119, 1489–1501 CrossRef CAS.
- M. González, I. Miquel and R. Sayós, Chem. Phys. Lett., 2001, 335, 339–347 CrossRef.
- B. A. Laws, S. J. Cavanagh, B. R. Lewis and S. T. Gibson, J. Phys. Chem. Lett., 2017, 8, 4397–4401 CrossRef CAS PubMed.
- P. Felder, B. M. Haas and J. Robert Huber, Chem. Phys. Lett., 1991, 186, 177–182 CrossRef CAS.
- T. F. Hanisco and A. C. Kummel, J. Phys. Chem., 1993, 97, 7242–7246 CrossRef CAS.
- D. Yuan, S. Yu, T. Xie, W. Chen, S. Wang, Y. Tan, T. Wang, K. Yuan, X. Yang and X. Wang, J. Phys. Chem. A, 2018, 122, 2663–2669 CrossRef CAS PubMed.
- D. A. Shutov, S.-Y. Kang, K.-H. Baek, K.-S. Suh and K.-H. Kwon, J. Korean Phys. Soc., 2009, 55, 1836–1840 CrossRef CAS.
- K. Katsonis and C. Berenguer, Int. J. Aerosp. Eng., 2013, 2013, 737463 Search PubMed.
- J. A. Schmidt, M. S. Johnson, U. Lorenz, G. C. McBane and R. Schinke, J. Chem. Phys., 2011, 135, 024311 CrossRef CAS PubMed.
- R. Schinke, J. Chem. Phys., 2011, 134, 064313 CrossRef CAS PubMed.
- T. Takayanagi and H. Akagi, Chem. Phys. Lett., 2002, 363, 298–306 CrossRef CAS.
- R. Li and R. E. Continetti, J. Phys. Chem. A, 2002, 106, 1183–1189 CrossRef CAS.
- I. G. Polotskii, Zh. Obshch. Khim., 1947, 6, 1048–1054 Search PubMed.
- K. Gaddam and H. M. Cheung, Ultrason. Sonochem., 2001, 8, 103–109 CrossRef CAS PubMed.
- M. Haïssinsky, R. Klein and P. Rivayrand, J. Chim. Phys. Phys.-Chim. Biol., 1962, 59, 611–622 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01995g |
| This journal is © the Owner Societies 2022 |