
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ-Topology and ultrafast excited-state dynamics of remarkably photochemically stabilized pentacene derivatives with radical substituents†
Nishiki
Minami
a,
Kohei
Yoshida
a,
Keijiro
Maeguchi
a,
Ken
Kato
a,
Akihiro
Shimizu
a,
Genta
Kashima
a,
Masazumi
Fujiwara
a,
Chiasa
Uragami
b,
Hideki
Hashimoto
 b and
Yoshio
Teki
*a
b and
Yoshio
Teki
*a
aDivision of Molecular Materials Science/Department of chemistry, Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: teki@osaka-cu.ac.jp
bDepartment of Applied Chemistry for Environment, Graduate School of Science and Technology, Kwansei Gakuin University, Sanda, Hyogo 669-1337, Japan
First published on 11th April 2022
Abstract
Pentacene derivatives with both π-radical- and TIPS-substituents (1m and 1p) were synthesized and their photochemical properties and excited-state dynamics were evaluated. The pentacene-radical-linked systems 1m (1p) showed a remarkable improvement in photochemical stability, which was 187 (139) times higher than that of 6,13-bis(triisopropylsilylethynyl)pentacene. Transient absorption spectroscopy showed that this remarkable photostabilization is due to the ultrafast intersystem crossing induced by effective π-conjugation between the radical substituent and pentacene moiety. The relationship between π-topology and the photochemical stability is also discussed based on the excited-state dynamics.
Introduction
Pentacene (Pn) derivatives have been intensively studied as promising candidates for organic field-effect transistors (OFETs),1 organic thin-film transistors (OTFTs),2 and organic light-emitting diodes (OLEDs)3,4 owing to their high hole mobility in the solid state.5,6 In addition, it is also a promising spin-current transport material that is useful for spintronics.7,8 Improving the photochemical stability of Pn derivatives is an important issue because the Pn framework easily decomposes by reacting with oxygen under ambient light.9,10 A typical stabilization methods involves the use of a bulky and/or electron-withdrawing substituent(s) on the Pn moiety,2,11,12 of which a representative example is 6,13-bis(triisopropylilyl-ethynyl)pentacene (TIPS-Pn) reported by J. E. Anthony et al.10,11 We reported a different strategy for the photochemical stabilization (radical photostabilization) that utilized the unique excited-state spin dynamics induced by the attachment of the π-radical substituent(s) to the Pn moiety.13–15 Thus, Pn derivatives with one13 or two14 π-radical substituent(s) were significantly more stable against photodegradation because of the enhanced/accelerated intersystem crossing (EISC) of the Pn moiety.15In this study, we report the design and synthesis of new Pn derivatives (1m and 1p in Fig. 1) with both π-radical and TIPS substituents, which have different π-orbital networks (i.e., π-topology). We evaluated their photochemical properties and their excited-state dynamics in the relation to the π-topology. The spin-state ordering and dynamics in the photoexcited states expected according to the spin-density distributions in the pohotoexcited states16,17 are also shown in Fig. 1 The π-radical-linked Pn derivatives (1m and 1p) showed remarkable improvement in photochemical stability much over the previously reported radical-linked pentacene derivatives (2, 3, and 4)13,14 or TIPS-Pn (Fig. 1). This remarkable photochemical stability has been ascribed to the ultrafast (femtosecond) EISC induced by the effective π-conjugation between the radical substituent and the pentacene moiety owing to the molecular planarity.
 | ||
| Fig. 1 (a) Chemical structures for π-radical-linked pentacene derivatives (1m, 1p, 2, 3, and 4), and TIPS-Pn. (b) Expected excited-state dynamics for 1m and 1p. | ||
Results and discussion
1m was synthesized from 6,13-pentacenedione as the starting material, according to the procedures shown in Scheme 1. 5 was synthesized from 6,13-pentacenedione according to the similar procedures described in the literature.186m was synthesized from 5 by a lithiation reaction with 40% yield and, then, reduced in the dark to 7m under acidic condition with 74% yield.19 A condensation reaction of 7m with 1,3-diamino-1,3-dimethylurea in a (CH2Cl)2/methanol mixture afforded 1mpre. Finally, oxidation of 1mpre by Ag2CO3 on Celite (48 wt%) produced 1m as a greenish-blue powder after recrystallization (19% yield). 1p was synthesized using similar procedures. The detailed synthetic procedures for 1m and 1p are described in the ESI.†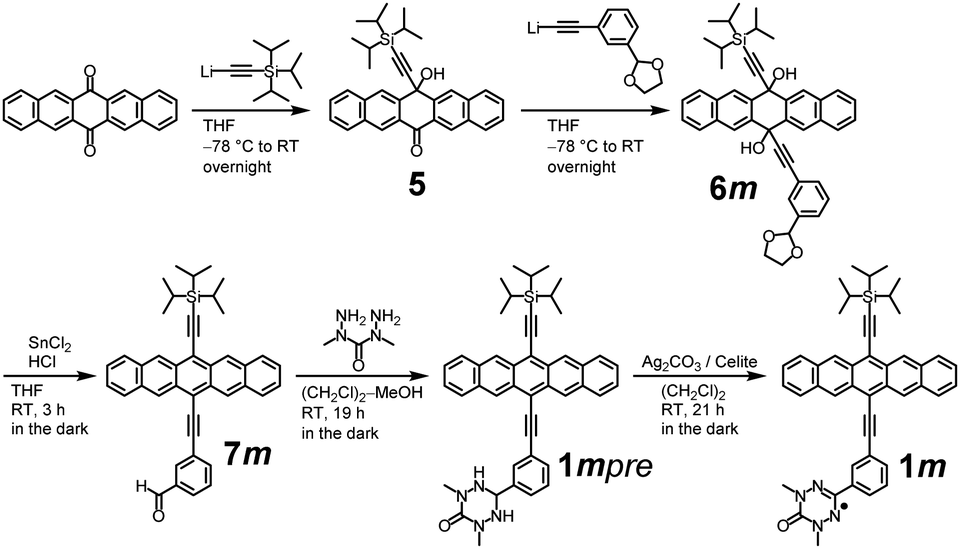 | ||
| Scheme 1 Synthetic route for 1m. | ||
To characterize the electronic structures of 1m and 1p, electron spin resonance (ESR) and cyclic voltammetry (CV) measurements were performed. The obtained and simulated ESR spectra of 1m and 1p are shown in Fig. S13 (ESI†), which was adequately characterized as a dimethyloxoverdazyl radical according to their g values and the hyperfine splitting, which are similar to those of the phenyl-dimethyl-substituted oxoverdazyl radical.20 The cyclic voltammograms (CV) of 1m, 1p, TIPS-Pn and 4 are shown in Fig. S14 (ESI†). The CV curves of 2 and 313 are not shown here, but their redox potentials are listed in Table 1, and their photochemical stabilities are discussed later. The first oxidation potential of 1m (1p) is 0.332 V (0.322 V), which is close to the middle between TIPS-Pn (0.402 V) and 2 (0.294 V) or 3 (0.298 V). Notably, the oxidation potential of the verdazyl radical overlaps with that of the pentacene moiety.21 These findings obtained from the CV data show that the electronic states of the pentacene moiety in their ground state are almost unchanged by the attachment of the radical substituent similar to shown already in our previous work.13 Although the luminescence of 1m and 1p were completely eliminated by the ultrafast EISC shown later, their ground-state properties of the pentacene moiety are not affected by the radical substituent, leading to the application to OFETs, OTFTs, spintronics devices and so forth. It has been actually shown in our preliminary experiment that 1p acts as the efficient hole transport material in the OFETs (not shown). In addition, the luminescent property of the pentacene is expected to recover by a weak acid treatment after making devices, leading to the possible application for OLEDs.
| Derivatives | First redox potentials vs. Fc/Fc+E[Ox]/V E[Red]/V | τ dec/hour | |
|---|---|---|---|
| 1m | 0.332 | −1.32, −1.47 | 178 ± 6 (97%, dominant) |
| 3.03 ± 0.43 (3%, minor) | |||
| 1p | 0.322 | −1.32, −1.46 | 132 ± 4 (96%, dominant) |
| 0.90 ± 0.16 (4%, minor) | |||
| TIPS-Pn | 0.402 | −1.45 | 0.950 ± 0.015 |
| 2 | 0.2947,13 | −1.3813 | 0.737 ± 0.014 |
| 3 | 0.29813 | −1.3713 | 2.64 ± 0.05 |
| 4 | 0.405 | −1.24 | 15.4 ± 0.1 |
The photochemical stabilities of 1m and 1p were evaluated by comparing them with TIPS-Pn, 2, 3, and 4 in air-saturated CH2Cl2 solution at room temperature (ca. 20 °C) under irradiation with visible light (light power 70 mW; λex = 650 ± 25 nm for 1m, 1p and TIPS-Pn; λex = 600 ± 25 nm for 2, 3 and 4). The setup of the irradiation system is shown in Fig. S18 (ESI†). Fig. 2a–c show the photoirradiation-induced time-variations in the steady-state absorption spectra of 1m, 1p, and TIPS-Pn, respectively, in CH2Cl2. Only a 13(17)% decrease in the absorbance was observed for 1m (1p) for 20 h, showing their remarkable photochemical stabilities. In contrast, TIPS-Pn, a well-known commercially available photochemically stable pentacene derivative, showed a significant decay of the 1π–π* absorption band of the Pn moiety within 2 h. The time variations in the absorption spectra of the other compounds are provided in the ESI.† The 1π–π* transition of the Pn moiety in 1m (1p) was observed at the peak wavelength of 653.5 nm (658 nm), which was ca. 10.5 (15) nm red-shifted than that for TIPS-Pn. The molar absorption coefficient (ε) of 1m (1p) in CH2Cl2 was determined to be 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 cm−1 at 653.5 nm (38000 M−1 cm−1 at 658 nm), which is almost identical to that of TIPS-Pn (ε = 28900 (27100) M−1 cm−1 at 640 nm (643 nm) in THF (CH2Cl2)).14 The n–π transition of the verdazyl radical was masked by the overlap with the 1π–π* transition. These characteristics were well reproduced by the TD-DFT calculations (Fig. S19–S21 in ESI†). Fig. 2d shows the decay profiles of the low-energy 1π–π* absorption peaks of all compounds studied in this work (1m: λ = 653.5 nm, 1p: λ = 658 nm, TIPS-Pn: λ = 643 nm, 2: λ = 589.5 nm, 3: λ = 589.5 nm, and 4: λ = 602 nm). The decay profiles of 2, 3, 4 and TIPS-Pn were analyzed by using the following eqn;14
600 M−1 cm−1 at 653.5 nm (38000 M−1 cm−1 at 658 nm), which is almost identical to that of TIPS-Pn (ε = 28900 (27100) M−1 cm−1 at 640 nm (643 nm) in THF (CH2Cl2)).14 The n–π transition of the verdazyl radical was masked by the overlap with the 1π–π* transition. These characteristics were well reproduced by the TD-DFT calculations (Fig. S19–S21 in ESI†). Fig. 2d shows the decay profiles of the low-energy 1π–π* absorption peaks of all compounds studied in this work (1m: λ = 653.5 nm, 1p: λ = 658 nm, TIPS-Pn: λ = 643 nm, 2: λ = 589.5 nm, 3: λ = 589.5 nm, and 4: λ = 602 nm). The decay profiles of 2, 3, 4 and TIPS-Pn were analyzed by using the following eqn;14
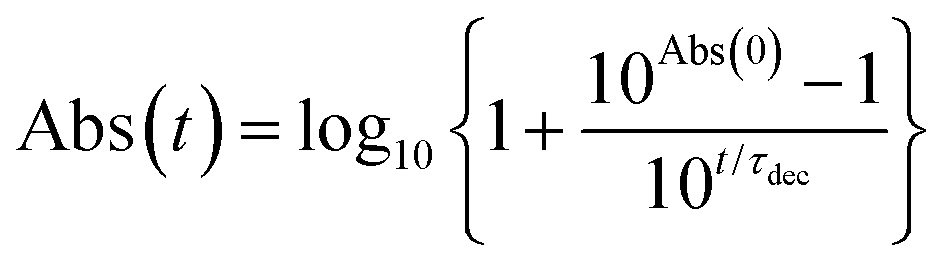 | (1) |
 | (2) |
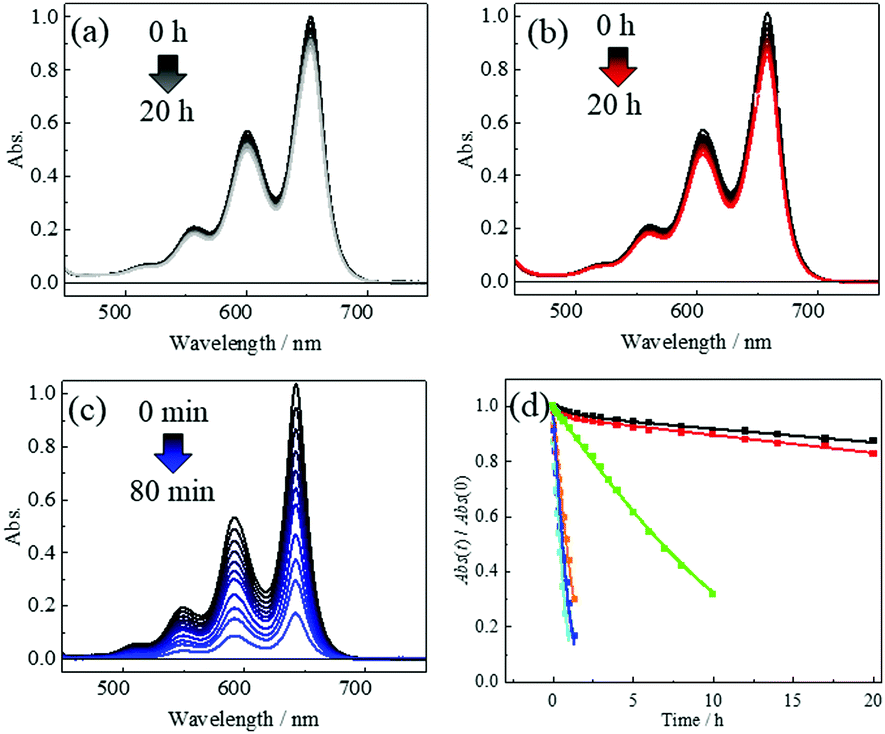 | ||
| Fig. 2 Photochemical stability of 1m (black), 1p (red), TIPS-Pn (blue), 2 (light blue), 3 (yellowish brown) and 4 (yellow green) in air-saturated CH2Cl2 at room temperature. Time variations in the absorption spectra of (a) 1m, (b) 1p, and (c) TIPS-Pn. (d) Time variations in absorbance of the peaks in 0 ← 0 band for 1m, 1p, TIPS-Pn, 2, 3, and 4. Their curve fittings were obtained using eqn (1) for TIPS-Pn, 2, 3, and 4 or eqn (2) for 1m and 1p. Abs(0) values of 1m, 1p, TIPS-Pn, 2, 3, and 4 were 1.001, 1.013,1.035, 1.036, 1.037, and 1.014, respectively. | ||
We reported that intersystem crossing (ISC) in Pn and anthracene derivatives was enhanced/accelerated by introducing π-radical substituent(s),15,16,22 and a similar phenomenon was observed for ultrafast ISC for perylene-3,4,9,10-bis-(dicarboximide) derivatives with radical substituent(s).23,24 The excited singlet states (1Pn*) in 1m and 1p were expected to be efficiently converted to the excited triplet state (3Pn*) by the EISC. Thus, the enhanced/accelerated ISC mechanism in the Pn moiety from 1Pn* to 3Pn* works in 1m and 1p, leading to the observed remarkable photochemical stability, because the photochemical reaction of the Pn derivative with oxygen occurs predominantly in 1Pn* states.25 The enhancement in the photostability was much larger than that of 2, 3, 4, and TIPS-Pn. It should be noted that the increase in the number of radical or TIPS substituents leads to an additional effect on the photostability, which is “linearly” (or close to linearly) proportional to the number of substituents.13,14,19 Therefore, the significant increase in the photochemical stability of 1m and 1p is due to their molecular planarity, which leads to the ultrafast (enhanced) ISC by the effective π-conjugation between the radical substituent and pentacene moiety. The synergetic effect of the radical photostabilization and the electronic effect of the TIPS substituent may also contribute to this enormous photostability.
To confirm the above speculation and gain further insight into the excited-state dynamics, we performed transient absorption (trA) measurements. The experimental details are described in the ESI.†Fig. 3 shows the color maps of the trA spectra and time courses at the characteristic wavelengths. Typical time courses of the spectra are provided in the ESI.† The transitions at 446 nm and 574 nm in Fig. 3a correspond to the wavelength of the trA peaks of 1m at an early stage, which was assigned to the singlet excited state of the Pn moiety (1Pn*). The peak wavelength of the triplet excited state of the Pn moiety (3Pn*) corresponds to 497 nm, while that of the bleach corresponds to 644 nm (see ESI†). The time-dependent decays of the trA intensities (ΔAbs) at 446 nm and 574 nm were analyzed using a double exponential function (ΔAbs(t) = CSexp(−t/τS) + CTexp(−t/τT), where, τS and τT refer to the singlet state and triplet state lifetimes of Pn* moiety, respectively), showing that the trA spectra of 1Pn* and 3Pn* overlapped. It should be noted that the total spin states of the whole molecule are sing-doublet, 2(1Pn*–R) for 1Pn*, and trip-doublet 2(3Pn*–R) and trip-quartet 4(3Pn*–R) for 3Pn*, respectively. Therefore, τS and τT denote the lifetimes of 1Pn*–R and 3Pn*–R, respectively. The τS value was estimated from the fit of the trA decay at 574 nm, where the trA of 1Pn* was the most dominant (the τS estimated from the fit of the trA decay at 446 nm was 560 ± 43 fs which was within the experimental error). The recovery of the bleach at 644 nm was used to estimate the average decay time (〈τT〉) of 3Pn* because the process related to 3Pn* is slightly complicated, as follows. The trA intensity at 497 nm first decreased, second increased and then decreased, which may suggest the existence of an indirect pathway from 1Pn* to 3Pn* (see ESI†). This behavior is difficult to fit using a simple double exponential function. The triplet lifetime (τT) was roughly estimated from the least-squares fit using a triple exponential function (see ESI†). The τS and 〈τT〉 values of 1p were estimated by the double exponential fit of the decay of the TrA peak at 584 nm and by the single exponential fit of the recovery of the bleach at 659 nm in Fig. 3b, respectively. The peak at 584 nm corresponds to the trA peak of 1p (see ESI†) at an early stage. The τT value was roughly estimated from the fit using a triple exponential function (see ESI†), because the indirect pathway from 1Pn* to 3Pn* is also indicated from the time course of the trA intensity at 501 nm. The obtained lifetimes (τS, τT, and 〈τT〉) are listed in Table 2 along with the lifetimes (τS and τT) of 2, 3 and TIPS-Pn. The lifetimes of the precursors (1mpre and 1ppre) were similar to those of the TIPS-Pn (see ESI†).
 | ||
| Fig. 3 Typical transient absorption data. (a) 1m (Left: Color maps of the transient absorption spectra. Right: Time decays at the characteristic wavelengths.) (b) 1p (Left: Color maps of the transient absorption spectra. Right: Time decays at the characteristic wavelengths.). | ||
The τS of 1m (1p) was ca. 1.8 × 104 (7.0 × 104) times shorter than that of TIPS-Pn because of the EISC and 35 (140) times shorter than that of 2 due to the effective π-conjugation, as a result of the molecular planarity (the dependency on the radical species was less than 4 times judging from the τS of 2 and 3). The 〈τT〉 of 1m (1p) was ca. 8.8 × 104 (5.8 × 104) times shorter than τT of TIPS-Pn and 17 (11) times shorter than that of 2 (On the discussion related to the photochemical stability, 〈τT〉 is better than τT.). It is known that the difference in the exchange-couplings between the unpaired electron on the radical and the electrons in half-filled orbitals on the excited chromophore is important in the EISC rather than the magnitude of the exchange coupling,26,27 although the effective π-conjugation is expected to be one of the critical factors in EISC of 1m and 1p. The detailed discussions of the expected mechanism for the ultrafast quenching of the photoexcited states is described in the ESI.† Because the photochemical stability of 1m (1p) is 242(179) times higher than that of 2, a synergetic effect between the radical photo-stabilization and TIPS electronic stabilization seems to exist, but the dominant effect is due to the ultrafast EISC owing to the effective π-conjugation. It should be noted that τT and 〈τT〉 of 1m are shorter than those of 1p, which is consistent with the spin-state ordering of 2(3Pn*–R) and 4(3Pn*–R). Thus, as shown in Fig. 1b, the expected lowest excited state of 1m is 2(3Pn*–R) with a low-lying higher energy 4(3Pn*–R) state. Therefore, the spin-allowed transition to the ground-state is expected to occur efficiently from 2(3Pn*–R). In contrast, the expected lowest excited state of 1p is 4(3Pn–R), from which the transitions to the ground state can occur indirectly via the low-lying higher-energy 2(3Pn*–R) state. This indicates that the excited-state dynamics can be controlled by π-topology. Notably, the photochemical stability of 1m was higher than that of 1p, although the τS of 1p was shorter than that of 1m. This is understandable as the complete suppression of the reaction pathway via1Pn* is due to the extremely short τS value for 1m and 1p, although the dominant decomposition pathway of pentacene is known to occur via1Pn*.28 Thus, the remaining effective decomposition pathways occur in 3Pn*.
Conclusions
A remarkable improvement in photochemical stability of 1m (1p) was achieved, which was 187 (139), 242(179), and 11.6 (8.6) times higher than that of TIPS-Pn, 2, and 4, respectively. It is also worthy of notice that the intersystem crossing of the purely organic compounds without heavy atoms is realized in the sub-femtosecond region. The trA spectroscopy clarified that the ultrafast enhanced/accelerated ISC was realized owing to the effective π-conjugation between the radical and the pentacene moieties. In contrast, no enhanced/accelerated ISC and no photochemical stability improvement were reported for the Pn derivative attached to the TEMPO radical.19,29 The strong conjugation of the π-radical with the Pn moiety is essential for the enormous photochemical stabilization of 1m and 1p. In addition, π-topological control of the excited-state dynamics was realized and applied to the enhancement of photochemical stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Grant-in-Aid for Scientific Research (B) (No. 16H04136, No. 20H02715) and Grant-in-Aid for Challenging Exploratory Research (No. 18K19062) from the Japan Society for the Promotion of Science (JSPS). The authors acknowledge to Ms Yumi Umehara (Research Assistant at the Osaka City University) for the help with the sample purification.Notes and references
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed.
- S. K. Park, T. N. Jackson, J. E. Anthony and D. A. Mourey, Appl. Phys. Lett., 2007, 91 Search PubMed.
- M. A. Wolak, B. B. Jang, L. C. Palilis and Z. H. Kafafi, J. Phys. Chem. B, 2004, 108, 5492 CrossRef CAS.
- M. A. Wolak, J. Delcamp, C. A. Landis, P. A. Lane, J. Anthony and Z. Kafafi, Adv. Funct. Mater., 2006, 16, 1943–1949 CrossRef CAS.
- O. D. Jurchescu, J. Baas and T. M. Palstra, Appl. Phys. Lett., 2004, 84, 3061 CrossRef CAS.
- J. L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef CAS PubMed.
- Y. Tani, Y. Teki and E. Shikoh, Appl. Phys. Lett., 2015, 107 Search PubMed.
- Y. Tani, T. Kondo, Y. Teki and E. Shikoh, Appl. Phys. Lett., 2017, 110, 032403 CrossRef.
- I. Lewis and L. Singer, J. Phys. Chem., 1981, 85, 352 Search PubMed.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980 CrossRef CAS.
- J. E. Anthony, D. L. Eaton and S. R. Parkin, Org. Lett., 2002, 4, 15–18 CrossRef CAS PubMed.
- S. S. Palayangoda, R. Mondal, B. K. Shah and D. C. Neckers, J. Org. Chem., 2007, 72, 6584 CrossRef CAS PubMed.
- Y. Kawanaka, A. Shimizu, T. Shinada, R. Tanaka and Y. Teki, Angew. Chem., Int. Ed., 2013, 52, 6643 CrossRef CAS PubMed.
- A. Shimizu, A. Ito and Y. Teki, Chem. Commun., 2016, 52, 2889–2892 RSC.
- A. Ito, A. Shimizu, N. Kishida, Y. Kawanaka, D. Kosumi, H. Hashimoto and Y. Teki, Angew. Chem., Int. Ed., 2014, 53, 6715 CrossRef CAS PubMed.
- Y. Teki, S. Miyamoto, M. Nakatsuji and Y. Miura, J. Am. Chem. Soc., 2001, 123, 294 CrossRef CAS PubMed.
- Y. Teki, T. Toichi and S. Nakajima, Chem. – Eur. J., 2006, 12, 2329 CrossRef CAS PubMed.
- D. Lehnherr, R. McDonald and R. R. Tykwinski, Org. Lett., 2008, 10, 4163 CrossRef CAS PubMed.
- E. T. Chernick, R. Casillas, J. Zirzlmeier, D. M. Gardner, M. Gruber, H. Kropp, K. Meyer, M. R. Wasielewski, D. M. Guldi and R. R. Tykwinski, J. Am. Chem. Soc., 2015, 137, 857 CrossRef CAS PubMed.
- F. A. Neugebauer and H. Fischer, Angew. Chem., Int. Ed. Engl., 1980, 19, 724 CrossRef.
- J. B. Gilroy, S. D. McKinnon, B. D. Koivisto and R. G. Hicks, Org. Lett., 2007, 9, 4837 CrossRef CAS PubMed.
- K. Kanemoto, A. Fukunaga, M. Yasui, D. Kosumi, H. Hashimoto, H. Tamekuni, Y. Kawahara, Y. Takemoto, J. Takeuchi, Y. Miura and Y. Teki, RSC Adv., 2012, 2, 5150 RSC.
- E. M. Giacobbe, Q. Mi, M. T. Colvin, B. Cohen, C. Ramanan, A. M. Scott, S. Yeganeh, T. J. Marks, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 3700 CrossRef CAS PubMed.
- S. Yeganeh, M. R. Wasielewski and M. A. Ratner, J. Am. Chem. Soc., 2009, 131, 2268 CrossRef CAS PubMed.
- W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071 CrossRef CAS PubMed.
- R. L. Ake and M. Gouterman, Theor. Chim. Acta, 1969, 15, 20 CrossRef CAS.
- B. W. Stein, C. R. Tichnell, J. Chen, D. A. Shultz and M. L. Kirk, J. Am. Chem. Soc., 2018, 140, 2221 CrossRef CAS PubMed.
- A. R. Reddy and M. Bendikov, Chem. Commun., 2006, 1179 RSC.
- C. E. Avalos, S. Richert, E. Socie, G. Karthikeyan, G. Casano, G. Stevanato, D. J. Kubicki, J. E. Moser, C. R. Timmel, M. Lelli, A. J. Rossini, O. Ouari and L. Emsley, J. Phys. Chem. A, 2020, 124, 6068 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: https://doi.org/10.1039/d2cp00683a |
| This journal is © the Owner Societies 2022 |