
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into the role of reduced graphene oxide in enhancing photocatalytic hydrogen evolution in disordered carbon nitride†
Mohammad Z.
Rahman
*,
Partha
Maity
 *,
Omar F.
Mohammed
and
Jorge
Gascon
*
*,
Omar F.
Mohammed
and
Jorge
Gascon
*
KAUST Catalysis Center (KCC), Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Kingdom of Saudi Arabia. E-mail: jorge.gascon@kaust.edu.sa; mohammadziaur.rahman@kaust.edu.sa; partha.maity@kaust.edu.sa
First published on 12th April 2022
Abstract
Compared to crystalline carbon nitride, the performance of disordered carbon nitride (d-CN) as a hydrogen production photocatalyst is extremely poor. Owing to its disordered atomic orientation, it is prone to numerous defect states. These energy states are potential sites for trapping and recombination of photogenerated charge carriers. As a result, rapid recombination of photogenerated charge carriers places a fundamental photophysical challenge in charge separation and transport, which inhibits its photocatalytic activity. In the presence of reduced graphene oxide (rGO), d-CN shows enhanced photocatalytic production of hydrogen. However, photophysical insight into the tacit role of rGO is not well understood which limits the rational design of d-CN as a photocatalyst. Particularly, understanding of the early time-scale (in fs to ps) recombination mechanism and the charge transport kinetics has not yet been achieved. With the help of ultrafast transient absorption spectroscopy, femtosecond time-resolved photoluminescence spectroscopy and transient photocurrent measurements, this article deciphers the ultrafast dynamics of the separation and transport of photogenerated charge carriers in d-CN facilitated by rGO. It is found that rGO substantially suppresses the bimolecular and trap-assisted recombination and enables a faster separation of charge carriers. As a result, it increases the lifetime of the charge carriers to be transported to the surface catalytic sites, and therefore, augments the rate of hydrogen production almost by an order of magnitude. Our findings therefore offer a proof-of-concept for overcoming the trap-mediated recombination problems in disordered carbon nitride.
1. Introduction
Carbon nitride, arguably, is the most popular metal-free polymeric semiconductor for various energy related conversion and storage applications, particularly for photocatalytic hydrogen production from water.1,2 Broadly, carbon nitride as a photocatalyst can be categorized into two types, namely, crystalline and amorphous carbon nitride. Due to its disordered atomic orientation and resultant defect states, amorphous carbon nitride is also termed as disordered carbon nitride (d-CN). Crystalline carbon nitride is an intensively studied photocatalyst evident from hundreds of archival publications, while there are only a few publications (<10 articles) on amorphous carbon nitride or d-CN.1,2 Considering the apparent quantum efficiency (AQE) of hydrogen production, d-CN is extremely poor (AQE < 6%) compared with crystalline carbon nitride (AQE > 60%).3,4The poor photocatalytic performance of d-CN is mainly attributed to its poor transport of charge carriers.5–7 This is largely due to the non-aromatic connection between the polymer strands of CN which causes photogenerated singlet excitons to be typically confined to the tri-s-triazine (or heptazine) unit of CN.8 The excitons in CN therefore experience a larger binding energy (≫kBT, where kB is the Boltzmann constant and T is the temperature in Kelvin; at room temperature, kBT is ∼0.26 mV) to be split into free electrons and holes due to electron–lattice and electron–electron Coulomb interactions.8,9 To achieve a charge separated state, the excitons should be dissociated within their lifetime, which is in the picosecond time scale.10,11 Additionally, the carriers must survive recombination before reaching the catalytic sites.7
The charge transport becomes further exacerbated by the residual chemical defects (e.g., dangling bonds, interstitials, vacancies, etc.) in the crystal structure due to the incomplete polycondensation of precursor monomers or doping/adsorption of heteroatoms in C–N networks.4,12 Pendant amine groups are the root cause of surface dangling bonds that create shallow trap states in CN and act as potential sites for charge carrier trapping.3,13 Being in this regime, charge trapping detrimentally reduces the charge carrier mobility and makes the interfacial charge transfer difficult. It causes a kinetic competition between charge separation and non-radiative recombination at the semiconductor–water interfaces, and therefore, presents a significant photophysical challenge.14
The efficiency of charge extraction depends on the ratio of the rate constants for charge recombination and charge separation.5 Generally, a high rate of hydrogen production requires charge recombination processes to occur on a longer time scale than the charge separation and extraction processes.15,16 It is therefore important to determine the time scale for charge recombination processes. Transient absorption spectroscopy (TAS) and time-resolved photoluminescence (TRPL) spectroscopy are the widely accepted techniques used to assess the individual dynamics of interfacial charge transfer processes.5,17
Because pristine d-CN is a poor photocatalyst,18 most often a noble metal co-catalyst (e.g. Pt) is employed to enhance the rate of hydrogen production on the surface of d-CN.19 Given the scarcity and price of noble metals, metal-free co-catalysts are highly desirable. In lieu of Pt, incorporation of reduced graphene oxide (rGO) has been shown to enhance the performance of the catalyst.20–24 However, little is known about how rGO contributes to the overall charge transport process. It is therefore at the research forefront to understand what limits the performance of d-CN and why the incorporation of rGO enhances its performance.
This situation demands a systematic research endeavor to probe into the kinetics of photoexcited charge carriers and to delineate the directional transfer of the photoexcited electrons between the photocatalyst and co-catalyst or vice versa. In this regard, here, we employed femtosecond TAS and TRPL spectroscopy complemented by transient photocurrent measurements to provide insight into the charge separation and transport of d-CN facilitated by rGO for photocatalytic hydrogen production.
2. Results and discussion
2.1 Synergistic physicochemical effects is induced by the overlapped rGO nanolayers on d-CN
We synthesized d-CN via polycondensation of thiourea and dicyandiamide at 620 °C using slightly modified synthesis procedures reported previously.3 The d-CN/rGO hybrid was synthesized by mixing rGO powder (Sigma-Aldrich) with the CN precursors following the same procedures of temperature treatment (see the Experimental section in the ESI† for details). The as-synthesized d-CN shows a morphology of disorderly stacked nanosheets under a transmission electron microscope (TEM; Fig. 1A). We did not see any noticeable changes in the morphology under the TEM after incorporation of rGO into the d-CN skeleton (Fig. 1B). This is also confirmed from the identical X-ray diffractogram (Fig. S1, ESI†). However, when compared with d-CN, the XRD pattern of d-CN/rGO shows a relatively flat peak at ∼13° which describes the in-plane structural packing motif and a broader inter-planar stacking peak at ∼27.0°. These reflections indicate that rGO is not connected to the lattice of d-CN but overlapped (also confirmed from the TEM images, Fig. 1A and B) and is connected to the surface terminal groups.25,26 In support of this observation, in the FTIR spectra, the C–O stretching vibrations of epoxy groups at 1096 cm−1 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of COOH groups at 1725 cm−1 for rGO are absent in d-CN/rGO, but the vibrations of typical C–N heterocycles (C–N, CN) at 1000 to 1500 cm−1 are present (see Fig. S2, ESI†).27 This is further supported by the XPS deconvoluted C1s spectrum (Fig. S3, ESI†) which consists only of CC–C and NC–N groups.28
O stretching vibrations of COOH groups at 1725 cm−1 for rGO are absent in d-CN/rGO, but the vibrations of typical C–N heterocycles (C–N, CN) at 1000 to 1500 cm−1 are present (see Fig. S2, ESI†).27 This is further supported by the XPS deconvoluted C1s spectrum (Fig. S3, ESI†) which consists only of CC–C and NC–N groups.28
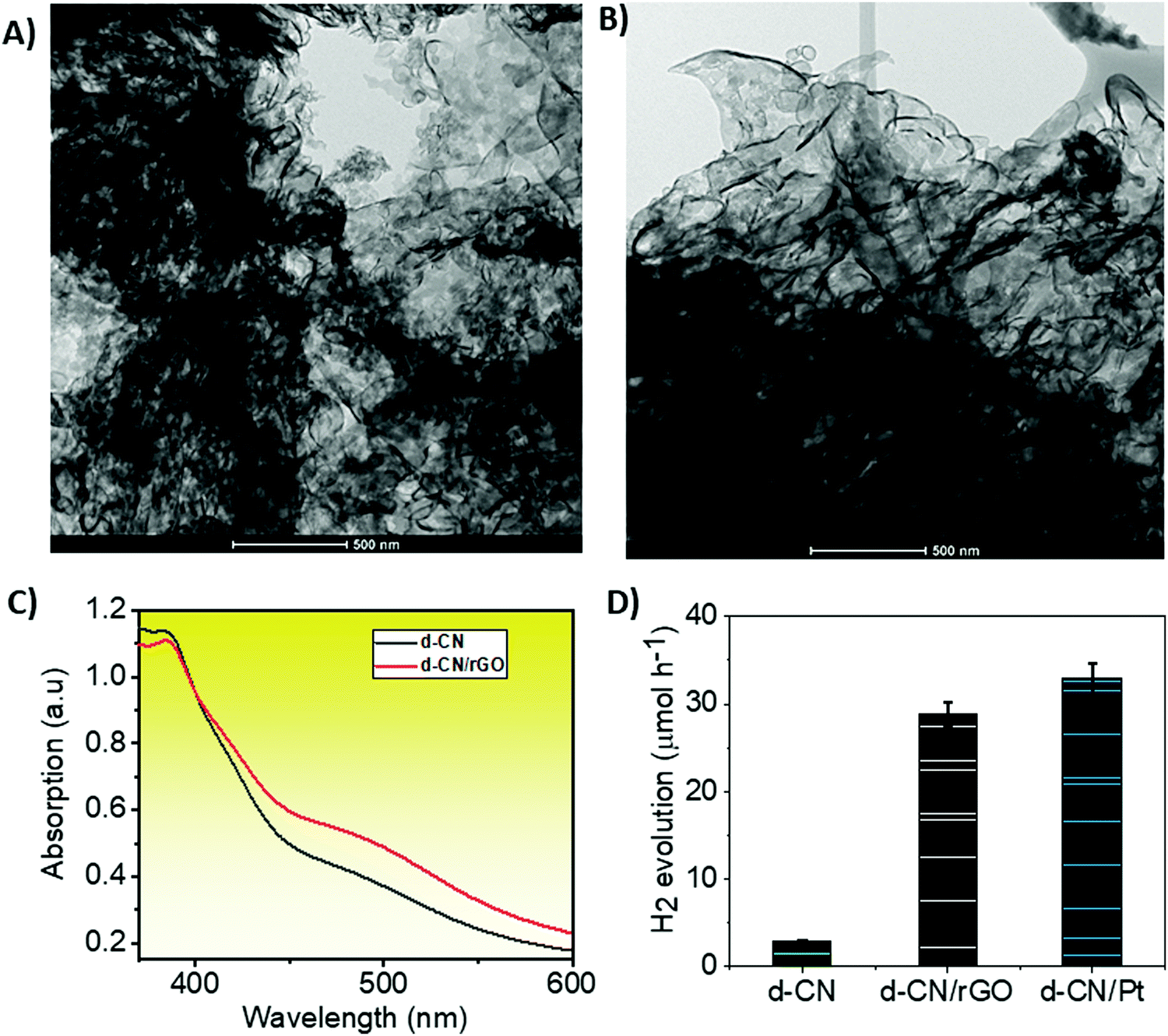 | ||
| Fig. 1 Morphology under an electron microscope, and optical absorption and photocatalytic performance. TEM images of (A) d-CN and (B) d-CN/rGO. (C) Combined UV-vis absorption spectra. (D) Rate of hydrogen evolution by d-CN, d-CN/rGO, and d-CN/Pt from 10 vol% triethanolamine solution when irradiated by a 300 W Xe lamp mounted with a 420 nm cut-off filter at STP. | ||
The absence of epoxy, carboxyl and carboxylic groups is indicative of the functionalization of the rGO periphery with pyridinic and pyrrolic nitrogen when N-rich precursors (thiourea and dicyandiamide) and rGO were copolymerized onto the d-CN/rGO hybrid.29 We previously reported3 that, during the synthesis of amorphous carbon nitride from polycondensation of N-rich precursors, significant amounts of ammonia were eliminated which could possibly destroy the regular stacking of rGO. A decrease of d-spacing (∼0.05 nm) in the case of d-CN/rGO is observed, which infers the compact overlapped layers between rGO and d-CN (Fig. S1, ESI†. A synergistic physicochemical effect can be expected due to this strong coupling between d-CN and rGO.
2.2 Incorporation of rGO red-shifts the visible light absorption
UV-Vis absorption spectroscopy gives further details about the strength of the electronic transition between the valence band (VB) and the conduction band (CB). A smooth and monotonically increasing absorption with decreasing wavelength marks the indirect bandgap transitions for both d-CN and d-CN/rGO (Fig. 1C). Unlike direct bandgap transitions, the indirect bandgap transitions require phonon assistance or changes in the crystal momentum (k-vector) which invariably influence the relaxation of excited states due to an electron–phonon coupling effect.30Relative to d-CN, a slightly red-shifted absorption peak in the case of d-CN/rGO infers that the electronic transitions from the VB to the excitonic states in the CB or the states located just below the bottom of the CB will require a lower exciton binding energy to be overcome.31 We have extrapolated the bandgap of d-CN (2.76 eV) and d-CN/rGO (2.66 eV) from the Kubelka–Munk (K–M) plot (Fig. S4, ESI†). We and other groups have also shown that the decomposition of N-rich precursors to CN incorporates nitrogen atoms into the edges and induces defect sites in rGO.27 Particularly, the graphitic-N links CN and rGO which reduces the electrical resistivity of the resulting hybrid.32 This would positively affect the charge carrier photogeneration and transfer. We therefore expect that d-CN/rGO would be a functional photocatalyst.
2.3 rGO enhances the rate of visible light (λ ≥ 420 nm) H2 production by d-CN
We measured the visible light (from a 300 W Xe lamp mounted with a 420 nm cut-off filter) photocatalytic H2 production activities of the d-CN/rGO system in a batch reactor where 50 mg of the catalyst was dispersed in 80 mL of an aqueous solution containing 10 vol% triethanolamine as a sacrificial reagent. The rate of H2 production was quantified using gas chromatography. Bare d-CN showed a H2 production rate of 2.9 μmol h−1, while d-CN/rGO showed 28.8 μmol h−1 (Fig. 1B). We observed a gradual increase of activity with increasing the ratio of d-CN to rGO, which reaches a peak at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. We observed a decreasing trend of activity for any further increase in the rGO content. d-CN also shows a comparable hydrogen production rate to d-CN/Pt (32.9 μmol h−1), and there was no noticeable degradation in cyclic hydrogen production for 24 h (data are not shown here, but available on request). As can be seen, there was almost an order of magnitude increase in the H2 production rate in d-CN/rGO compared to bare d-CN. Intuitively, this may be due to the favorable charge transport dynamics that stems from the synergistic effect between rGO and d-CN. We therefore examined the d-CN/rGO system with steady-state and femtosecond TRPL spectroscopy, followed by TAS and transient photocurrent measurements, to understand the charge transport mechanisms.
:1. We observed a decreasing trend of activity for any further increase in the rGO content. d-CN also shows a comparable hydrogen production rate to d-CN/Pt (32.9 μmol h−1), and there was no noticeable degradation in cyclic hydrogen production for 24 h (data are not shown here, but available on request). As can be seen, there was almost an order of magnitude increase in the H2 production rate in d-CN/rGO compared to bare d-CN. Intuitively, this may be due to the favorable charge transport dynamics that stems from the synergistic effect between rGO and d-CN. We therefore examined the d-CN/rGO system with steady-state and femtosecond TRPL spectroscopy, followed by TAS and transient photocurrent measurements, to understand the charge transport mechanisms.
2.4 Photophysical insight into the role of rGO in enhancing the photocatalytic performance of d-CN
 | ||
| Fig. 2 Steady state and time-resolved photoluminescence (PL) measurements. (A) Combined steady state PL spectra of d-CN and d-CN/rGO. Deconvoluted steady state PL spectra of (B) d-CN and (C) d-CN/rGO. (D) TRPL decay traces for d-CN and d-CN/rGO measured in TCSPC and fluorescence upconversion (inset) in response to 370 nm optical excitation. Solid black lines represent the exponential fitting of the experimental data. | ||
Although the PL intensity is dominated by the band-edge emission for typical semiconductor nanoparticles, the PL spectrum can also be used as a sensitive probe to extract useful information about bandgap states. Trap states are typically located within the bandgap, and consequently photons emitted from trapped electrons should exhibit a red-shifted emission relative to band-edge emission. We deconvoluted the steady state PL spectra using a Gaussian fit in order to qualitatively understand the contribution of band-edge emission and trap state emission. As can be seen in Fig. 2B, in line with previous studies,34,35 the band centered near 2.83 eV is attributed to the bandgap emission, and those at 2.54 and 2.26 eV are attributed to intra-bandgap state (shallow and deep traps, respectively) emission for d-CN. From the K–M plot, the bandgap for d-CN was extrapolated to be 2.76 eV (Fig. S4, ESI†). Therefore, the intra-bandgap emissive states are ∼0.22 eV below the absorption band edges for shallow traps and ∼0.5 eV for deep traps.36 The deconvoluted PL spectrum for d-CN/rGO is shown in Fig. 2C. For d-CN/rGO, the peak centered at 2.7 eV is assigned to bandgap emission and the peak centered at 2.4 eV is assigned to the emission from shallow trap states. Both of these emission peaks are red-shifted when compared with that of d-CN, as shown in Fig. 2B. This is also in good agreement with the corresponding absorption spectra shown in Fig. 1C. Given the K–M extrapolated bandgap of 2.66 eV for d-CN/rGO, intra-bandgap emissive states are ∼0.26 eV below the absorption band edges which may be assigned to shallow traps or emissive states associated with rGO. The absence of deep trap state emission in the case of d-CN/rGO implies that incorporation of rGO is likely to passivate the defects in d-CN (see also ESI† note S1). We have estimated the highest amount of energy contained in the excited state to be 2.92 eV and 2.87 eV from the cross-over of the normalized UV-vis and PL spectra that cross at 424 nm and 432 nm for d-CN and d-CN/rGO, respectively (see Fig. S5, ESI†).13 Therefore, excited state electrons in d-CN are seemingly more prone to undergo electron–electron and electron–lattice interactions before relaxation to the conduction band minimum.
Comparing the steady-state PL spectra of d-CN with and without rGO, we can see the drastic reduction in the intensity of the respective PL spectra. As the PL intensity is proportional to the emitted photons due to electron–hole recombination, we can say that radiative recombination of photogenerated electron–hole pairs in d-CN/rGO is significantly reduced relative to bare d-CN (see also ESI† note S1). However, steady-state PL does not provide information on the relaxation and trapping of excited state electrons before falling back to the ground state, and therefore, cannot provide decisive information on whether the trap assisted recombination dominates radiative recombination or/and charge carrier separation happens before recombination. Therefore, we used time resolved photoluminescence (TRPL) and absorption measurements in the femtosecond to nanosecond time regime to understand the trapping and trap-assisted recombination of charge carriers. Details of femtosecond transient absorption spectroscopy (TAS) and fluorescence upconversion can be found in our previous reports37–39 as well as in the ESI.†
TAS helps us to resolve the temporal transfer of electrons on an ultrafast time-scale from excited states to trap or interfacial states (see also ESI† note S2). The fs-TA measurements were performed in the presence of 10% TEOA in water. In our TAS measurements, we have chosen 370 nm as the excitation wavelength of the femtosecond pump pulses to promote the electrons effectively from the valence band to the conduction band of d-CN. Fig. 3A and B present the fs-TA spectra at different time delays of d-CN and d-CN/rGO in response to 370 nm optical excitation. In both cases, persistent negative (known as ground state bleach, GSB) and positive (known as photoinduced absorption, PIA) absorption changes can be discerned. The TA signals in the NIR region and visible region are due to photoexcited electrons and a combination of electrons and holes, respectively. The GSB signal (in the visible region) is contributed by electrons and holes. In the presence of TEOA the hole concentration is decreased; however, the electron concentration remains unchanged. That is why the signal intensity in the low wavelength region is reduced (and appears as a low S/N) as compared to the electron signal in the NIR region. There are two main reasons for the observed differences in the S/N ratio between the visible and NIR regions: (i) the probe light intensity after absorption of the sample is lower in the visible as compared to the NIR region, which is due to d-CN dominating absorption in the visible region (Fig. 1C); (ii) the absorption cross-section of a conduction band electron is greater than that of a valence band hole. Moreover, d-CN absorbs light in the visible region, which affects unequally the probe light intensity in both regions.
 | ||
| Fig. 3 Transient absorption spectroscopy. Femtosecond transient absorption spectra at different time delays for (A) d-CN and (B) d-CN/rGO in response to 370 nm excitation. (C) Normalized bleach recovery kinetics of d-CN and d-CN/rGO at 435 nm in response to 370 nm excitation. Solid black lines represent the exponential fitting of the experimental data. | ||
The GSB spectral position consistent with steady-state absorption of the S1 excitonic band which manifests the bleaching of the ground state is due to depletion of valence band (VB) electrons and appears from state filling at the bandgap.37 The broad PIA is attributed to the absorption of excited state electrons generated in the CB.40,41 The excited electrons are not different in the IR region. However, in the TA spectra it appears as a positive band (known as photoinduced absorption) in the IR region and a negative band in the visible region (GSB) because of different directions of transition. The NIR spectrum in Fig. 3B shows the excited electron in the CB of d-CN not rGO. The excited state dynamics of rGO is much faster (hundreds of fs to a few ps) than what we presented in Fig. 3. The rGO concentration is 10 wt% of d-CN, and therefore upon excitation, mostly d-CN is excited, not rGO. In the case of d-CN, as can be seen in Fig. 3A, the GSB signal gradually recovers and in 3 ns it reaches zero. On the other hand, the GSB for d-CN/rGO (Fig. 3B) shows also fast recovery at an early time scale; however, it remains unchanged for a longer time, which could be attributed to efficient charge separation in the d-CN/rGO composite. In contrast, the PIA of d-CN/rGO decays at a faster rate than d-CN. In the present study, we are interested in the charge separation dynamics in d-CN/rGO. That is why our TA measurements were focused on in the sub-ns time scale. As can be seen the excited state lifetime of d-CN is >5 ns.
To explore the charge transfer behavior, we compared the GSB and PIA kinetics in Fig. 3C and Fig. S6 (ESI†), respectively. With an exponential fitting of the kinetic traces acquired at 435 nm, we have retrieved the characteristic relaxation time constants. The exponential fitting results are τ1 = 0.33 ± 0.01 ps (65%) and τ2 = 900 ± 40 ps (35%) for bare d-CN, which gives a mean relaxation lifetime of ∼315 ± 15 ps. The two decay times are indicative of exciton relaxation pathways to two trap states with different trap depths, namely shallow traps and deep traps.31,42 Considering the proton reduction potential of −410 mV vs. SHE at pH = 7 (pure water) or −590 mV vs. SHE at pH = 10 (for the triethanolamine–water mixture), a loss of 0.6 eV by deeply trapped electrons would therefore result in a substantial loss of driving force for proton reduction regardless of the type of solution. It is therefore not surprising that d-CN shows a low performance in the production of hydrogen.
In contrast, the GSB kinetics can fit with two time constants τ1 = 0.30 ± 0.01 ps (47%) and τ2 = 14 ± 0.6 ps (31%) for d-CN/rGO. Similar to bare d-CN the very fast component (0.30 ps) remains unchanged, which indicates de-excitation in d-CN/rGO associated through the trapping mediated process. However, the weight percentage is reduced as compared to the bare d-CN, confirming that trap assisted processes are reduced as electrons are transferred from d-CN to the conduction band of rGO. In addition, the time constant of 14 ps represents a quick charge recombination between separated charge carriers generated after electron transfer. The remaining charge recombination (∼22%) in d-CN/rGO occurs at a >5 ns time scale, clearly representing a longer charge carrier lifetime.
Normalized TA kinetics at 870 nm of d-CN and d-CN/rGO is presented in Fig. S6 (ESI†). The kinetics of bare d-CN in the NIR region (e.g., 870 nm) is due to excited electrons, which undergo trapping during the deexcitation process. The intermediate trap state lifetime is higher as compared to the initial excited state. That is why the rise is observed in PIA decay in the NIR region after a 1 ns time delay. Fig. 3C highlights the kinetics comparison of d-CN and d-CN/rGO at the GSB position (435 nm). It can be seen that the GSB kinetics of bare d-CN recovers completely and appears as a positive signal at a >5 ns time delay. In addition to Fig. S6, Fig. 3C confirms the presence of a mid-band longer lived trap state in bare d-CN.
It can be seen that the excited state of d-CN/rGO decays faster than that of bare d-CN, clearly suggesting photoexcited electron transfer, which is consistent with the decay measured from TRPL (Fig. 2D). The slower charge recombination and faster excited state decay in d-CN/rGO are attributed to the fast interfacial transfer and efficient charge separation between d-CN and rGO. The very fast electron transfer (within the temporal resolution, <100 fs) from d-CN to rGO and slow charge recombination (>5 ns) are the key factors for the enhancement of the water reduction reaction in d-CN/rGO. It can be safely inferred according to the data extracted from TA spectroscopy that incorporation of rGO into d-CN induces a channel for excited electron transfer from d-CN to rGO, and therefore, provides a means for enhancement of hydrogen production.
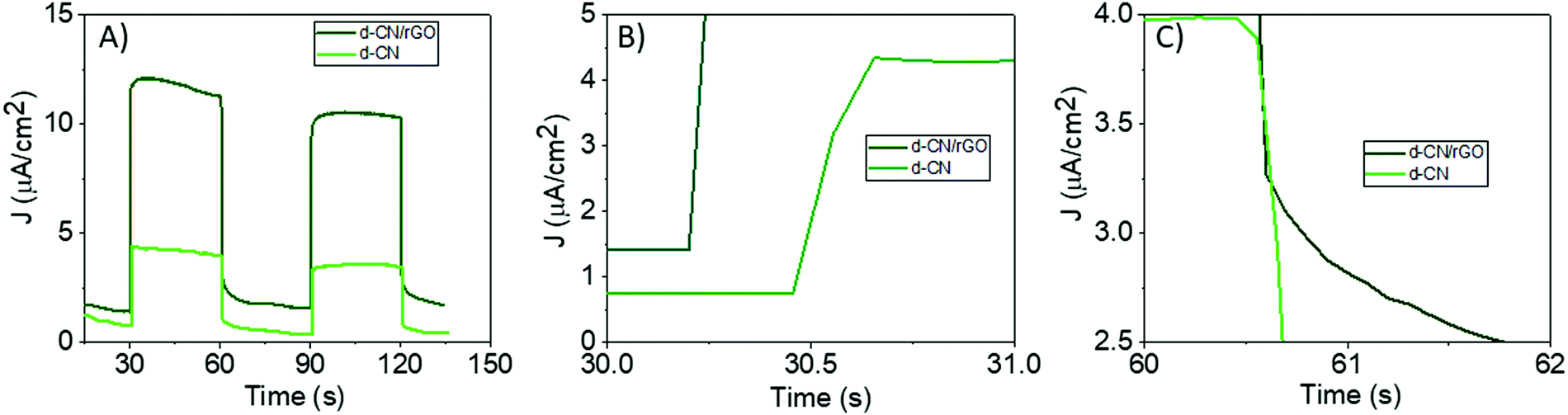 | ||
| Fig. 4 Transient photocurrent response showing (A) typical on–off cycles of intermittent visible-light irradiation. (B) Rise-time dynamics and (C) decay-time dynamics of the transient photocurrent. The measurement was carried out in three-electrode configurations (working electrode, reference electrode and counter electrode) using a 0.2 M Na2S + 0.05 M Na2SO3 mixed aqueous solution. | ||
Before reaching the steady state, the rise–decay dynamics of transient photocurrent provides direct information on trapping and detrapping of charge carriers.45 As can be seen in Fig. 4B, d-CN/rGO exhibits a faster increase in the steady state current than d-CN. This refers to the fact of faster detrapping of trapped-charge population in d-CN/rGO before reaching equilibrium, while the delayed rise to steady-state current observed in bare d-CN indicates the increased trap population17,45,46 which induces a strong trap-mediated recombination in d-CN that results in a decreased current density (Fig. 4A). At the turn-off of the illumination, d-CN/rGO shows a long photocurrent tail for more than 750 μs (Fig. 4C). This infers the continuous extraction of charge carriers because of suppressed recombination evident from the strong quenching of the PL intensity (Fig. 2A).
Due to the lattice mismatch between rGO and d-CN, layers of rGO impart considerable strain onto the surface of d-CN with an increased likelihood of defect site formation (which is also confirmed by Raman spectroscopy measurements, Fig. S7, ESI†). Because of weak electron–acoustic phonon coupling in graphene,48 lattice distortion couples excitons with higher-energy optical phonons through deformation potential interactions known as Fröhlich coupling (in which the phonons generated oscillating polarization which modulates the periodic potential that determines the electronic band energies).49–51 The red-shift in the UV absorbance of d-CN/rGO is also a reflection of the scattering of incident photonic radiation through rGO.52 Correspondingly, we also observed a red-shift in the PL emission spectra of d-CN/rGO. We have hypothesized that exciton–phonon coupling would dephase the electronic transition to produce phonon sidebands that would displace emission energetically from the zero-phonon transition.53,54 As a result, we observed spectral shifts that corroborate the photoinduced fluctuations in local electric fields.53–55 However, in the UV-vis spectra, there was only a smooth and monotonic increase of absorption with decreasing wavelength, but there were no added red-shifted excitonic peaks. Therefore, quantum confinement effects are less favorable due to the lack of sharp or well-defined spectral peaks.31
Previously it has been reported that the ground-state excitons in 2D nanosheets of rGO consist of multiple optically active electronic fine-structure states.56,57 Emissions can originate from multiple electronic states each with its own phonon coupling properties. It was shown that coupling to longitudinal optical (LO) phonons had a major contribution to the spectral broadening because of an increased Fröhlich interaction due to a decreased wave function overlap between the electrons and holes (i.e. surface charge localization).47,58 d-CN/rGO forms a quasi-type II heterostructure where the electron wave function of d-CN leaks into rGO (rGO acts as an electron sink), while the hole remains confined in d-CN. Therefore, a decrease in the overlap between the electron and hole wave functions is expected. This will correspondingly increase the polarization of charge carriers that would enhance the coupling to LO phonons (see also ESI† note S3).48,51,59
Based on our TAS and TRPL results, here we have provided a mechanistic picture to illustrate the direction of the transport and separation of charge carriers in the d-CN/rGO system (see Scheme 1). The above TAS and TRPL results have clearly demonstrated that the addition of rGO positively influences the charge transport kinetics by suppressing the band-edge recombination, and interfacial transfer and temporal separation of electrons which lead to enhanced hydrogen production in d-CN.
 | ||
| Scheme 1 Hypothetical illustration of charge transport kinetics. CBM: conduction band maximum, VBM: valence band maximum, h+: hole, e−: electron. Lines are not drawn to scale. Incorporation of rGO into the d-CN skeleton enhances the charge transfer 2.5-fold and slows down the recombination ∼5-fold compared to bare d-CN. | ||
3. Conclusion
In conclusion, this study has explored the role of rGO in enhancing the photocatalytic activity of d-CN toward hydrogen production by delving into the kinetic competition of charge separation and transport using femtosecond TAS and TRPL complemented by transient photocurrent measurements. Our findings reveal that rGO, when incorporated into d-CN, passivates traps that not only help in suppression of the band edge bimolecular recombination as well as the trap-assisted recombination, but also enhance the spatial separation of charge carriers in d-CN. As a result, d-CN/rGO increased the rate of hydrogen production by almost an order of magnitude compared to bare d-CN. By providing an insight into the kinetic competition of different photophysical processes, our study may therefore guide the pathway to ‘metal-free photocatalysts by design’.Experimental section
See the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the funding from King Abdullah University of Science and Technology (KAUST).References
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- M. Z. Rahman, P. C. Tapping, T. W. Kee, R. Smernik, N. Spooner, J. Moffatt, Y. Tang, K. Davey and S.-Z. Qiao, Adv. Funct. Mater., 2017, 27, 1702384 CrossRef.
- M. Z. Rahman, K. Davey and C. B. Mullins, Adv. Sci., 2018, 5, 1800820 CrossRef PubMed.
- M. Z. Rahman and C. B. Mullins, Acc. Chem. Res., 2019, 52, 248–257 CrossRef CAS PubMed.
- L. Kong, P. Song, F. Ma and M. Sun, Mater. Today Energy, 2020, 17, 100488 CrossRef.
- M. Z. Rahman, T. Edvinsson and J. Gascon, Nat. Rev. Chem., 2022, 6, 243–258 CrossRef.
- C. Merschjann, T. Tyborski, S. Orthmann, F. Yang, K. Schwarzburg, M. Lublow, M. C. Lux-Steiner and T. Schedel-Niedrig, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 205204 CrossRef.
- M. Z. Rahman, Y. Tang and P. Kwong, Appl. Phys. Lett., 2018, 112, 253902 CrossRef.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- M. Rahman, G. Boschloo, A. Hagfeldt and T. Edvinsson, Adv. Mater., 2020, 32, e1905653 CrossRef PubMed.
- M. Rahman and K. Davey, Phys. Rev. Mater., 2018, 2, 125402 CrossRef CAS.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 139, 5216–5224 CrossRef CAS PubMed.
- K. Appavoo, M. Liu, C. T. Black and M. Y. Sfeir, Nano Lett., 2015, 15, 1076–1082 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- M. Z. Rahman, M. Batmunkh, M. Bat-Erdene, J. G. Shapter and C. B. Mullins, J. Mater. Chem. A, 2018, 6, 18403–18408 RSC.
- M. Z. Rahman, J. Moffatt and N. Spooner, Mater. Horiz., 2018, 5, 553–559 RSC.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- M. Rahman, H. Tian and T. Edvinsson, Angew. Chem., Int. Ed., 2020, 59, 16278–16293 CrossRef CAS PubMed.
- M. Z. Rahman, J. Zhang, Y. Tang, K. Davey and S.-Z. Qiao, Mater. Chem. Front., 2017, 1, 562–571 RSC.
- Q. Xiang and J. Yu, J. Phys. Chem. Lett., 2013, 4, 753–759 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- X. Li, R. Shen, S. Ma, X. Chen and J. Xie, Appl. Surface Sci., 2018, 430, 53–107 CrossRef CAS.
- W. Lin, E. Cao, L. Zhang, X. Xu, Y. Song, W. Liang and M. Sun, Nanoscale, 2018, 10, 5482–5488 RSC.
- P. Niu, L. C. Yin, Y. Q. Yang, G. Liu and H. M. Cheng, Adv. Mater., 2014, 26, 8046–8052 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- L.-C. Chen, C.-Y. Teng, C.-Y. Lin, H.-Y. Chang, S.-J. Chen and H. Teng, Adv. Energy Mater., 2016, 6, 1600719 CrossRef.
- P. Wurfel, Physics of Solar Cells, Wiley-VCH, 2nd edn, 2010 Search PubMed.
- D. A. Wheeler and J. Z. Zhang, Adv. Mater., 2013, 25, 2878–2896 CrossRef CAS PubMed.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- J. B. Baxter, C. Richter and C. A. Schmuttenmaer, Annu. Rev. Phys. Chem., 2014, 65, 423–447 CrossRef CAS PubMed.
- A. L. Stroyuk, Y. V. Panasiuk, A. E. Raevskaya and S. Y. Kuchmy, Theor. Exp. Chem., 2015, 51, 243–251 CrossRef CAS.
- Z. Gan, Y. Shan, J. Chen, Q. Gui, Q. Zhang, S. Nie and X. Wu, Nano Res., 2016, 9, 1801–1812 CrossRef CAS.
- J. Xu, M. Shalom, F. Piersimoni, M. Antonietti, D. Neher and T. J. K. Brenner, Adv. Opt. Mater., 2015, 3, 913–917 CrossRef CAS.
- P. Maity, N. A. Merdad, J. Yin, K. J. Lee, L. Sinatra, O. M. Bakr and O. F. Mohammed, ACS Energy Lett., 2021, 6, 2602–2609 CrossRef CAS.
- I. Dursun, P. Maity, J. Yin, B. Turedi, A. A. Zhumekenov, K. J. Lee, O. F. Mohammed and O. M. Bakr, Adv. Energy Mater., 2019, 9, 1900084 CrossRef.
- J. Wang, et al. , ACS Energy Lett., 2022, 7, 10–16 CrossRef CAS.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francas, S. Corby, C. L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, J. Am. Chem. Soc., 2019, 141, 11219–11229 CrossRef CAS PubMed.
- W. Bi, L. Zhang, Z. Sun, X. Li, T. Jin, X. Wu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, ACS Catal., 2016, 6, 4253–4257 CrossRef CAS.
- L. M. Peter and K. G. Upul Wijayantha, Chem. Phys. Chem., 2014, 15, 1983–1995 CrossRef CAS PubMed.
- Y. H. Ng, I. V. Lightcap, K. Goodwin, M. Matsumura and P. V. Kamat, J. Phys. Chem. Lett., 2010, 1, 2222–2227 CrossRef CAS.
- C. R. McNeill, I. Hwang and N. C. Greenham, J. Appl. Phys., 2009, 106, 024507 CrossRef.
- Z. Li and C. R. McNeill, J. Appl. Phys., 2011, 109, 074513 CrossRef.
- J. Cui, A. P. Beyler, I. Coropceanu, L. Cleary, T. R. Avila, Y. Chen, J. M. Cordero, S. L. Heathcote, D. K. Harris, O. Chen, J. Cao and M. G. Bawendi, Nano Lett., 2016, 16, 289–296 CrossRef CAS PubMed.
- A. Laitinen, M. Oksanen, A. Fay, D. Cox, M. Tomi, P. Virtanen and P. J. Hakonen, Nano Lett., 2014, 14, 3009–3013 CrossRef CAS PubMed.
- A. M. Kelley, ACS Nano, 2011, 5, 5254–5262 CrossRef CAS PubMed.
- A. M. Kelley, J. Phys. Chem. Lett., 2010, 1, 1296–1300 CrossRef CAS.
- C.-H. Park, F. Giustino, M. L. Cohen and S. G. Louie, Nano Lett., 2008, 8, 4229–4233 CrossRef CAS PubMed.
- D. P. Singh, V. Kumar, A. Kumar, R. Manohar, R. Pasricha, B. Duponchel, Y. Boussoualem, A. H. Sahraoui and A. Daoudi, RSC Adv., 2017, 7, 12479–12485 RSC.
- S. A. Empedocles, R. Neuhauser, K. Shimizu and M. G. Bawendi, Adv. Mater., 1999, 11, 1243–1256 CrossRef CAS.
- D. T. Nguyen, C. Voisin, P. Roussignol, C. Roquelet, J. S. Lauret and G. Cassabois, Chem. Phys., 2013, 413, 102–111 CrossRef CAS.
- S. A. Empedocles and M. G. Bawendi, Science, 1997, 278, 2114–2117 CrossRef CAS PubMed.
- W. E. Moerner and M. Orrit, Science, 1999, 283, 1670–1676 CrossRef CAS PubMed.
- A. L. Efros and M. Rosen, Annu. Rev. Mater. Sci., 2000, 30, 475–521 CrossRef CAS.
- T. D. Krauss and F. W. Wise, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 9860–9865 CrossRef CAS.
- E. Groeneveld and C. de Mello Donegá, J. Phys. Chem. C, 2012, 116, 16240–16250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp00200k |
| This journal is © the Owner Societies 2022 |