
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microsolvation of H2O+, H3O+, and CH3OH2+ by He in a cryogenic ion trap: structure of solvation shells†
David
Müller
and
Otto
Dopfer
 *
*
Institut für Optik und Atomare Physik, Technische Universität Berlin, Hardenbergstr. 36, 10623 Berlin, Germany. E-mail: dopfer@physik.tu-berlin.de
First published on 20th April 2022
Abstract
Due to the weak interactions of He atoms with neutral molecules and ions, the preparation of size-selected clusters for the spectroscopic characterization of their structures, energies, and large amplitude motions is a challenging task. Herein, we generate H2O+Hen (n ≤ 9) and H3O+Hen (n ≤ 5) clusters by stepwise addition of He atoms to mass-selected ions stored in a cryogenic 22-pole ion trap held at 5 K. The population of the clusters as a function of n provides insight into the structure of the first He solvation shell around these ions given by the anisotropy of the cation–He interaction potential. To rationalize the observed cluster size distributions, the structural, energetic, and vibrational properties of the clusters are characterized by ab initio calculations up to the CCSD(T)/aug-cc-pVTZ level. The cluster growth around both the open-shell H2O+ and closed-shell H3O+ ions begins by forming nearly linear and equivalent OH⋯He hydrogen bonds (H-bonds) leading to symmetric structures. The strength of these H-bonds decreases slightly with n due to noncooperative three-body induction forces and is weaker for H3O+ than for H2O+ due to both enhanced charge delocalization and reduced acidity of the OH protons. After filling all available H-bonded sites, addition of further He ligands around H2O+ (n = 3–4) occurs at the electrophilic singly occupied 2pz orbital of O leading to O⋯He p-bonds stabilized by induction and small charge transfer from H2O+ to He. As this orbital is filled for H3O+, He atoms occupy in the n = 4–6 clusters positions between the H-bonded He atoms, leading to a slightly distorted regular hexagon ring for n = 6. Comparison between H3O+Hen and CH3OH2+Hen illustrates that CH3 substitution substantially reduces the acidity of the OH protons, so that only clusters up to n = 2 can be observed. The structure of the solvation sub-shells is visible in both the binding energies and the predicted vibrational OH stretch and bend frequencies.
1. Introduction
The weak interaction of He with neutral and charged atoms and molecules is relevant for several disciplines, including molecular physics, low-temperature physics and chemistry, plasma chemistry, and astrochemistry. He atoms are quantum objects and their neutral clusters serve as models to investigate superfluidity and large-amplitude motion at the molecular level.1–8 In general, the interaction of He with cations is stronger than for neutrals due to additional electrostatic and induction forces arising from the excess positive charge, leading to deeper potential wells and larger angular anisotropy of the interaction potential.9–11 For some closed-shell and open-shell cations strong chemical bonds may be formed (e.g., H+He, He2+).9 In the case of noncovalent X+Hen clusters, their low binding energy makes their efficient production for spectroscopic characterization a challenging task.Pioneering experiments of X+Hen clusters were carried out by Kobayashi and coworkers,12–14 who injected X+ ions into drift tubes cooled by liquid He and filled with rarified He gas to generate and characterize these clusters by mass spectrometry, thereby revealing initial experimental information about their stability and structure (magic numbers). Since then, three major approaches have been applied as efficient X+Hen ion sources. In the first bottom-up approach developed around three decades ago, X+Hen clusters are generated in supersonic plasma expansions in which X+ ions are generated by electron or chemical ionization or laser desorption. The cold clusters generated can readily be characterized by infrared or optical photodissociation (IRPD, UV/visPD).10,15,16 One of the early visPD examples includes N2+ (n ≤ 3) with free internal rotation of the He atoms around the N2+ rod.17 IRPD studies of small closed-shell and open-shell protonated X+ ions include HCO+ (n = 1),18 HN2+ (n ≤ 2),19 NH2+ (n = 1),20 NH4+ (n = 1),21,22 OH+ (n = 1),23 H2O+ (n = 1),24 HO2+ (n = 1),25 HCO2+ (n = 1), CH3+ (n = 1),15,26,27 SiOH+ (n = 1),28 but also large aromatic and cycloalkane ions, such as phenol+ (n = 1),29 acetanilide+ (n = 1),30 and adamantane+ (n ≤ 3).31 However, it is often difficult to attach more than one He atom to the X+ cation as substantial energy has to be injected into the expansion to generate the ions and the expansion provides a too short time and pressure window to grow cold X+Hen clusters with larger n.
A second bottom-up approach developed later by Asmis and coworkers is growing X+Hen ions in a cryogenic trap.32–35 In this approach, ions are prepared in an arbitrary ion source, mass-selected, and trapped in a cryogenic trap by He buffer gas cooling. During the cooling process, larger X+Hen clusters can be formed and the limit of n is mostly given by the He binding energy and the cooling efficiency of the trap. As a result of the longer time for cluster growth, often larger X+Hen clusters can be formed in the trap than in the plasma expansion. This approach has been widely adopted by several groups to form X+Hen clusters around smaller and larger cations,34,36–40 such as inorganic and small hydrocarbon ions,8,35,41–46 C60+,47–49 and protonated PAH (coronene)50 and biomolecules,35,39 as well as reaction intermediates.38 However, in many of these studies, He-tagging is barely used for spectroscopy of the bare X+ ion and no or only little attention has been paid to the X+⋯He interaction potential. Because of the larger binding energies, in most cases larger rare gas atoms, H2, or N2 are used as a tag in spectroscopy experiments coupled to cryogenic traps.
A third and top-down approach to generate X+Hen clusters is ionization of doped helium nanodroplets, X@Hem. Briefly, in this technique one or more neutral atoms or molecules are picked up by very large Hem clusters (m = 103–107) and then ionized typically by electron impact. As a result of the excess ionization energy, most He atoms evaporate. The remaining population of the small X+Hen products (n ≪ m) may be analysed by mass spectrometry to reveal magic numbers and structures of solvation shells or used for photodissociation spectroscopy. This technique has been pioneered and extensively applied by Scheier and his group to a plethora of cations,51,52 including metal, PAH, and fullerene cations and their clusters.53–58 Few groups have also started recently to perform elegant ion spectroscopy in large He droplets,59–67 although this approach does not provide details about the local X+⋯He interaction potential (radial strength and angular anisotropy).
Herein, we report the generation of H2O+Hen and H3O+Hen clusters in our recently commissioned cryogenic ion trap coupled to a quadrupole/time-of-flight tandem mass spectrometer (BerlinTrap).68 As we use this bottom-up approach for X+Hen generation in this tandem mass spectrometer combination for the first time, we describe the instrument-specific data analysis in some detail and validate the approach for CH3+Hen and H3O+Hen, for which corresponding cluster growth experiments have been performed before in a quadrupole/quadrupole tandem mass spectrometer.34,42 The H2O+Hen clusters are grown for the first time and extend our previous IRPD work on H2O+He (n = 1), in which we could produce in a plasma expansion not enough larger clusters for spectroscopic interrogation.24 In that work, high-resolution rotation–vibration-tunneling spectroscopy has been performed for H2O+He and partly deuterated species, providing precise information about the 3D interaction potential by comparison to ab initio calculations at the MP2/aug-cc-pVTZ level. No spectroscopic information appears to be available for H2O+Hen≥2 and any of the H3O+Hen clusters. To this end, we employ in the present work CCSD and CCSD(T) calculations to determine the structural, energetic, and vibrational properties of H2O+Hen≤5 and H3O+Hen≤6 for investigating the solvation shell structure and for preparation of future IRPD experiments in the BerlinTrap. For comparison to H3O+Hen, we investigate also CH3OH2+Hen≤2 clusters43 using the same experimental and computational strategy to extract the effects of H → CH3 substitution on the X+⋯He interaction potential. Complexes of small interstellar hydroxy cations with He, such as OH+,69 H2O+,70 and H3O+,71 are of special interest in the context of astrophysics and considered in several models of astrochemistry.72
2. Experimental and computational techniques
The cluster growth experiments are carried out in an ion-trap quadrupole/time-of-flight tandem mass spectrometer (BerlinTrap) described in detail elsewhere.68,73 Briefly, it consists of an ion source for ion generation, a quadrupole mass spectrometer (QPMS) for ion selection, an electrostatic bender for ion deflection, an octupole ion guide for transferring the ions into a cryogenic 22-pole ion trap used for trapping and cooling the ions via He buffer gas (purity 5.0, Air Liquide, impurities O2 and H2O < 3 ppm), an Einzel lens stack for focusing the ions, and finally a reflectron time-of-flight (ReTOF) mass spectrometer for detecting the product ions. For the current study, the original electrospray ionization (ESI) source is used to produce the closed-shell ions H3O+, CH3+, and CH3OH2+,68 while for the open-shell H2O+ ions the ESI source is replaced by an electron ionization (EI) source.73 H3O+ and CH3OH2+ are readily produced by ESI of solutions containing H2O or CH3OH with some addition of acetic acid. CH3+ is a major fragment of CH3OH2+ by elimination of H2O. The H2O+ cations are produced by standard electron ionization of H2O using electrons with a kinetic energy of typically 70 eV. The X+ ions generated by ESI or EI are mass-selected by the QPMS and injected in the 22-pole trap held at 5 K by a cryostat. The ions are then trapped and cooled down by an intense He pulse injected into the trap just before the ions enter the trap. Spectroscopic analysis of hot band intensities in the electronic spectra of a variety of biomolecular ions yield an effective vibrational temperature in the range of 15–30 K for trap temperatures of 4–6 K, illustrating that the ion temperature does not fully reach the trap temperature.68,74–76 During this process, X+Hen clusters grow in the trap. The resulting X+Hen cluster distribution is extracted into the extraction region of an orthogonal ReTOF (with a resolution of m/Δm > 100 in the considered size range) where they are accelerated by a high voltage pulse toward a dual stage microchannel plate detector in Chevron arrangement.One particular feature of the BerlinTrap setup is the distance between the 22-pole trap and the ReTOF (ca. 0.5 m), which acts as additional time-of-flight discriminator and leads to variable transmission efficiencies for ions with different masses via the choice of the delay between the 22-pole extraction and the ReTOF extraction. To compensate for this effect, mass spectra are recorded at varying delay times in steps of typically 2 μs and added up to cover the whole X+Hen cluster distribution. Fig. 1 illustrates this procedure for the case of H3O+Hen clusters for selected delay times and the sum spectrum.68 As can be seen, for early extraction at 60 μs delay only H3O+Hen with n = 0–2 have significant transmission. On the other hand, at late extraction at 74 μs delay only n = 2–5 clusters have measurable transmission. Thus, by varying the delay from very early to very late extraction, where at the extreme delays no ions are detected anymore, we ensure that we account reliably for the total ion population in the trap by summing up all spectra (Fig. 1). To validate this approach further, mass spectra of CH3+Hen are presented in Fig. 2 to allow for direct comparison with corresponding spectra recorded previously by Asvany and coworkers using a QPMS for ion analysis.42 Attachment of only two He atoms is observed with significant abundance in both approaches, and the n = 3 abundance in the QPMS study is lower by three orders of magnitude than those of n = 1 and 2. The spectrum in Fig. 2 is taken at relatively long delay to illustrate the sharp drop in ion population at n = 2 also using the ReTOF approach, indicating shell closure at this cluster size. This observation is in line with the previous potential energy surface calculations for CH3+Hen with n = 1–2,15,26,27 and results from rotationally-resolved IRPD and microwave spectra.26,42,77 These studies show that the first two He atoms strongly bind to the vacant and thus very electrophilic 2pz orbital of C (De = 700 cm−1 for n = 1 at MP2/aug-cc-pVTZ), while binding to the protons is much less stable (De = 100 cm−1),26 so that CH3+Hen with n ≥ 3 are very difficult to grow even in cryogenic traps.42 This view is fully supported by additional extensive IRPD studies and calculations of CH3+Rgn clusters with the larger rare gas atoms Ar (n ≤ 8) and Ne (n ≤ 2).15,78,79 While in the supersonic plasma expansion only CH3+Hen clusters with n = 1 could be produced in sufficient abundance for spectroscopy,26 in the ion trap the n = 1 and 2 clusters are readily produced with enough abundance for spectroscopic interrogation.77
 | ||
| Fig. 1 Mass spectra of the ion trap content recorded with the ReTOF, when storing mass-selected H3O+ ions in the 22-pole trap and growing H3O+Hen clusters. Individual mass spectra for variable delay times between ion extraction from the trap and ion extraction into the orthogonal ReTOF illustrate the transmission efficiency as a function of the delay. The sum spectrum is the sum of all spectra for delays between 50 and 84 μs in steps of 2 μs. In addition to the predominant H3O+Hen series, H3O+H2O (H5O2+) is observed at m/z 37. | ||
 | ||
| Fig. 2 Mass spectrum of the ion trap content recorded with the ReTOF, when storing mass-selected CH3+ ions in the 22-pole trap and growing CH3+Hen clusters. This mass spectrum is taken at a long delay to illustrate the shell closure at n = 2. | ||
Ab initio calculations at the unrestricted CCSD/aug-cc-pVTZ level with tight optimization are carried out for H2O+Hen, H3O+Hen, and CH3OH2+Hen clusters to determine their structure, binding energy, rotational constants, and vibrational frequencies.80 Single point calculations are carried out at CCSD(T) level at the minima obtained at the CCSD level. Harmonic vibrational frequencies are scaled to optimize the agreement between computed and experimental OH stretch and bend frequencies of H2O+ and H3O+. Spin contamination is negligible, with values of 〈S2〉 −0.75 < 10−2 (10−4) before (after) spin annihilation for H2O+ and its clusters in the 2A1 doublet ground state. As expected, popular DFT methods are not suitable to describe the interactions in this type of clusters. For example, our B3LYP-D3/aug-cc-pVTZ results for He2 and H2O+He show that this level severely underestimates the He⋯He interaction (De = 0.2 cm−1, Re = 2.805 Å) and strongly overestimates the H2O+He interaction (De = 708.8 cm−1, Re = 1.591 Å) compared to results at the CCSD(T)/aug-cc-pVTZ level (De = 6.9 and 487.9 cm−1, Re = 3.015 and 1.702 Å). The latter values compare favorably with the best estimate for He2 from Virial and viscosity data (De = 7.6 cm−1, Re = 2.963 Å)81 and the CCSD(T)/CBS value for H2O+He (De = 482.0 cm−1). The CCSD(T)/CBS values are obtained by extrapolation of the calculations with the aug-cc-pVnZ basis sets with n = 3–5 at the geometry of n = 3. We do not systematically include corrections for basis set superposition error (BSSE). For the global minimum of H2O+He, the CCSD(T)/aug-cc-pVTZ level yields De = 510.2 and 455.2 cm−1 without and with BSSE correction, respectively, while the CCDT(T)/CBS value amounts to 482.0 cm−1. Hence, the BSSE correction lowers De by roughly 11%, while the limited basis set overestimates it by around 6%. In addition to equilibrium geometries and dissociation energies, we also evaluate binding energies (D0) by considering harmonic zero-point vibrational corrections. We are aware that zero-point motions involve large-amplitude motions even in the ground vibrational state,23,27 and these may not be well described by the harmonic approximation. To this end, the equilibrium energies De at the CCSD(T) level are more reliable. The atomic charge distribution is evaluated using the natural bond orbital (NBO) analysis.
3. Results and discussion
3.1 H2O+Hen
Fig. 3 shows the mass spectrum obtained for the growth of H2O+Hen clusters around the mass-selected H2O+ ion. In addition to the very intense H2O+ parent ion (n = 0), H2O+Hen clusters are produced with a higher intensity for n = 1, while signals for n = 2–4 are similar and around a factor of 3–5 weaker than for n = 1. The signal for n = 5 drops again by a factor of five, and the n = 6–9 peaks are barely visible. Clusters with n ≥ 10 are below the detection limit. The relative signal intensities are 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:8:7:5:1 for n = 0–5, and <0.1 for n = 6–9. This mass spectrum suggests relatively stable and comparable binding sites for n = 1–4, while n = 5 seems less stable. After filling the solvation subshells with five He ligands, attachment of a sixth He atom seems not efficient due to even lower binding energy, and all efforts to change the experimental conditions for the He pulse (timing, intensity, duration) failed in producing H2O+Hen clusters with n ≥ 10. In addition to the main H2O+Hen series, we detect also H3O+Hen≤3 (m/z 23, 27, 31) and H3O+H2O (m/z 37), because the QPMS does not completely filter out all H3O+ ions produced in the EI source. We also detect O2+Hen≤1 clusters (m/z 32, 36), which are produced by O2 impurity in the He line and exothermic charge transfer from H2O+ to O2.
:25:8:7:5:1 for n = 0–5, and <0.1 for n = 6–9. This mass spectrum suggests relatively stable and comparable binding sites for n = 1–4, while n = 5 seems less stable. After filling the solvation subshells with five He ligands, attachment of a sixth He atom seems not efficient due to even lower binding energy, and all efforts to change the experimental conditions for the He pulse (timing, intensity, duration) failed in producing H2O+Hen clusters with n ≥ 10. In addition to the main H2O+Hen series, we detect also H3O+Hen≤3 (m/z 23, 27, 31) and H3O+H2O (m/z 37), because the QPMS does not completely filter out all H3O+ ions produced in the EI source. We also detect O2+Hen≤1 clusters (m/z 32, 36), which are produced by O2 impurity in the He line and exothermic charge transfer from H2O+ to O2.
 | ||
| Fig. 3 Sum mass spectrum of the ion trap content recorded with the ReTOF, when storing mass-selected H2O+ ions in the 22-pole trap and growing H2O+Hen clusters. The filled circles and the asterisk indicate the formation of H3O+Hen (m/z 23, 27, 31) and H3O+H2O (H5O2+, m/z 37), while the open circles are due O2+Hen≤1 (m/z 32, 36), which are produced by O2 impurity in the He line and exothermic charge transfer from H2O to O2. | ||
In an effort to assign the observed H2O+Hen cluster structures, we consider again the CCSD calculations, and the most stable isomers are shown in Fig. 4. Ionization of H2O (1A1, C2v) into its cation ground state (2B1, C2v) occurs by removal of an electron from the nonbonding b1 orbital, which is essentially the 2pz orbital of O and thus perpendicular to the molecular plane. The structural data for H2O+ (re = 1.0001 Å, θe = 109.3°) and rotational constants (Ae = 28.119, Be = 12.572, Ce = 8.688 cm−1) are in excellent agreement with the experimental values (re = 0.9992(6) Å, θe = 109.3(1)°, Ae = 27.789, Be = 12.588, Ce = 8.700 cm−1).82 The scaled harmonic vibrational frequencies of the symmetric and antisymmetric OH stretch modes of ν1 = 3211 and ν3 = 3261 cm−1 are also close to the measured fundamentals, ν1 = 3213 and ν3 = 3259 cm−1.82
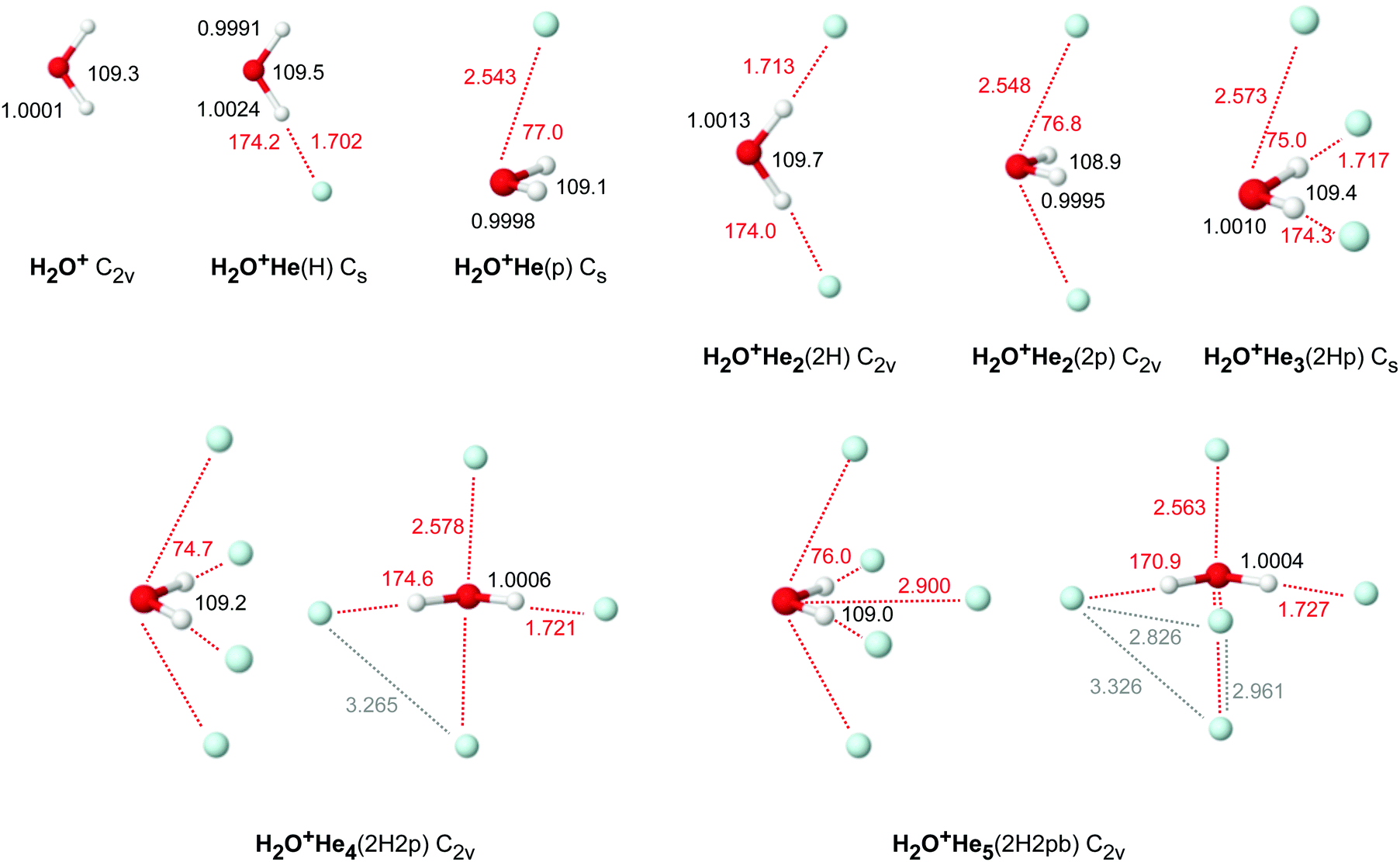 | ||
| Fig. 4 Minimum structures (in Å and degree) of H2O+Hen with n = 0–6 obtained at the CCSD/aug-cc-pVTZ level. | ||
The potential energy surface of H2O+Rg dimers with Rg = He–Ar has been characterized in detail before by calculations at the MP2 and CCSD(T) levels.24,83–89 It contains two nonequivalent minima, in which the Rg atom binds either to one of the protons forming a nearly linear OH⋯Rg ionic H-bond (two equivalent minima) or to the 2pz orbital of O (also two equivalent minima), while other parts of the potential are less attractive. The H-bonded minima are stabilized by dispersion and induction forces (charge-induced dipole), while the p-bonded minima are stabilized by charge transfer from Rg into the 2pz singly occupied orbital (SOMO). The latter is weak for small Rg atoms but becomes more pronounced for larger Rg atoms, because the ionization potential of Rg gets closer to that of H2O (IP = 24.6, 21.6, 15.8, 14.0, 12.1 eV for He–Xe and 12.6 eV for H2O),90 Thus, for Rg = He–Ar the H-bonded isomer is the global minimum on the H2O+Rg potential and the p-bonded structure is clearly a higher lying local minimum, in agreement with the experimental IRPD spectra.24,83,84,87 On the other hand, for Rg = Kr the hemi-bonded p-isomer is slightly more stable than the H-bonded isomer.91,92
For H2O+He, the CCSD calculations predict for H2O+He(H) a planar structure (Cs) with a nearly linear OH⋯He bond, with intermolecular bond parameters (Re = 1.702 Å, βe = 174.2°, Ae = 21.539 cm−1, Be = 0.68900 cm−1, Ce = 0.66765 cm−1, De = 488 cm−1, D0 = 207 cm−1), which are in good agreement with previous computational data24,86 and experimental values from IRPD spectroscopy (R0 = 1.656(4) Å, β0 = 175(5)°, (B0 + C0)/2 = 0.6535 cm−1).24 The proton donor O–H bond considerably elongates (by 2.3 mÅ) upon H-bonding, while the free O–H bond slightly contracts (by 1.0 mÅ). As a result, the ν1 and ν3 frequencies display both red shifts of −27 and −8 cm−1, in qualitative agreement with the observed values (−15 and −5 cm−1).24 The weak interaction of H2O+ with He is insufficient to decouple ν1 and ν3 into the two equivalent OH stretch local modes, leading to a red shift in ν3 (mostly free OH stretch) although the free O–H bond contracts upon forming the OH⋯He H-bond. In fact, the barrier between the two equivalent H-bonded minima at a planar transition state is rather low so that tunneling splittings for hindered internal rotation could be resolved in the IRPD spectra of H2O+He, which are absent in the HDO+He spectra. This tunneling motion, along with the involved large amplitude motion, also explains the overestimation of the computed Δν1/3 shifts calculated for the rigid H2O+He(H) equilibrium structure because the He moves away from the linear OH⋯He configuration even for the zero-point level. H-bond formation slightly increases the H2O bend angle by 0.2°. It also increases the H2O+ bend frequency by Δν2 = +4 cm−1, due to the additional retarding force of the OH⋯He bond.
The CCSD calculations predict for the local H2O+He(p) minimum (Cs) a structure, in which the He is attached to H2O in the plane perpendicular to the molecular plane. The He ligand is slightly tilted away from the 2pz orbital toward the OH protons. The predicted intermolecular bond parameters for the O⋯He bond are Re = 2.543 Å, βe = 77.0°, Ae = 9.0835 cm−1, Be = 0.78853 cm−1, Ce = 0.75945 cm−1, De = 270 cm−1, and D0 = 99 cm−1. The O–H bonds slightly contract upon He attachment at the 2pz orbital (by 0.3 mÅ) and, as a result, the computed ν1 and ν3 frequencies display both minor blue shifts of 4 cm−1. This effect is mostly caused by the small electron transfer from He into the electrophilic 2pz SOMO orbital of H2O+, which makes the cation slightly less positively charged and thus shifts the low-frequency ν1/3 modes of H2O+ toward the higher-frequency modes of neutral H2O. Such blue-shifted vibrational transitions are absent in the IRPD spectrum, clearly confirming that the H-bonded isomer is the global minimum on the H2O+He potential.24 In contrast to the H-bonded isomer, p-bonding decreases the H2O bond angle by 0.2° but also increases ν2 by 3 cm−1. Overall, the CCSD results for H2O+He(p) are close to those reported earlier at the MP2 level.24,86
In the planar global minimum structure of H2O+He2(2H) with C2v symmetry, two equivalent OH⋯He H-bonds are formed, which are only slightly weaker than that in the H2O+He(H) dimer, with Re = 1.713 Å, βe = 174.0°, De = 472 cm−1, and D0 = 228 cm−1. In principle, the two He atoms do not interact much with each other and one expects near additivity in the properties and effects upon sequential He solvation. The observation of slightly less than additivity is consistent with the noncooperative threebody forces of interior ion solvation. This effect arises from nonadditive induction forces and charge delocalization, which are both small due to the weak interaction. Both proton donor O–H bonds considerably elongate (by 1.2 mÅ) upon H-bonding, causing total red shifts of −21 and −41 cm−1 in ν1 and ν3. H-bond formation slightly increases the H2O bend angle by 0.4° and ν2 by +7 cm−1 compared to bare H2O+, again nearly twice the changes caused by the first OH⋯He bond.
For comparison, we have also computed the H2O+He2(2p) local minimum with C2v symmetry. As expected, it has two equivalent O⋯He p-bonds, which are only slightly weaker than that in the H2O+He(p) dimer due to slightly noncooperative threebody forces (Re = 2.548 Å, βe = 76.8°, De = 266 cm−1, D0 = 139 cm−1). The free O–H bonds contract by 0.6 mÅ, resulting in total blue shifts of 9 cm−1 in both ν1 and ν3 from the values of bare H2O+. The H2O bond angle increases by 0.4° and ν2 increases by 5 cm−1.
The most stable n = 3 and n = 4 clusters are obtained by adding two p-bonded He ligands to H2O+He2(2H), resulting in H2O+He3(2Hp) and H2O+He4(2H2p) with Cs and C2v symmetry. The p-bonds in n = 3 and 4 are slightly longer (Re = 2.573 and 2.578 Å) and weaker (De = 264 and 261 cm−1, D0 = 160 and 158 cm−1) than in the p-bonded isomers of n = 1 and 2. Again, p-bonding slightly contracts the O–H bonds compared to H2O+He2(2H) by 0.3 and 0.6 mÅ, causing blue shifts in both ν1 and ν3 of 5/10 cm−1 for n = 3/4. The H2O bond angle decreases by 0.2° and ν2 increases by 3 cm−1 for each p-bonded ligand. Moreover, p-bonding destabilizes the OH⋯He bonds, which elongates by 4 mÅ for each p-bonded ligand. This trend is in line with strengthening the intramolecular O–H bonds, which reduces their acidity and thus their propensity to form H-bonds.
After all four minima on the H2O+He dimer potential are filled with two H-bonded and two p-bonded He ligands, the fifth ligand occupies a less stable binding site, which is not a minimum on the dimer potential. Inspection of the in-plane and out-of-plane potential characterized by MP2 suggests that the O end is not very favorable because of the repulsive in-plane lone pair of O.24 On the other hand, the proton side is rather attractive. Hence, a natural attractive binding site is in the H2O+ plane on the C2 axis between the two H-bonded He ligands, leading to a H2O+He5 structure with C2v symmetry and binding energies (De = 251 and D0 = 161 cm−1) not much lower than those of the p-bonded ligands. Indeed, the p-bonded minimum on the MP2 dimer potential is very shallow and has a low barrier for migration toward the position of the fifth He ligand.24 At this bridged position, it has a distance of 2.900 Å from O, 2.458 Å from the two protons, and 2.827 and 2.961 Å from the H-bonded and p-bonded He atoms. The latter distances are close to the He–He distance in He2 of 2.805 Å, consistent with close packing. Attachment of the fifth ligand weakens and thus lengthens the OH⋯He H-bonds (by 6 mÅ), while the O⋯He p-bonds become slightly shorter (by 16 mÅ). As a result of the weaker H-bonds and the stronger p-bonds, the O–H bonds become shorter (by 0.2 mÅ), resulting in blue shifts of ν1 and ν3 by +4 and +3 cm−1. The H2O bend angle becomes smaller (by 0.2°) and ν2 decreases by 3 cm−1.
The H2O+Hen cluster growth deduced from the CCSD calculations suggests the initial formation of the in-plane H-bonded minima with binding energies of De ∼ 480 cm−1, followed by out-of-plane p-bonded He-ligands with much weaker binding of De ∼ 260 cm−1 and the bridged ligand with similar interaction (De ∼ 250 cm−1) (Fig. 5 and Table 1). At n = 5, all favorable binding sites are occupied by He atoms, as suggested by the H2O+He dimer potential, which suggests for the O binding site only weak He binding (De ∼ 60 cm−1) due to repulsion from the lone pair.24 Apparently, the mass spectrum in Fig. 3 is fully consistent with this view, showing only minor abundances for H2O+Hen clusters with n ≥ 6. Interestingly, the intensity of n = 5 is substantially reduced compared to n = 3–4, although these cluster sizes have similar He binding energies. This result may indicate the quantum nature of the He motion. While in H2O+Hen with n = 1–4 the He ligands can readily undergo large amplitude motions, the structure of the n = 5 cluster is rather compact and more rigid due to close-packing of all five ligands, thereby strongly reducing the possibility for large amplitude motions even at 0 K, whereby this effect is larger for the angular than the stretching motions. As a result, even though the interaction energy for the fifth He atom is quite favorable, the large zero-point motions may make it difficult for the He ligand to squeeze into this binding site, thereby reducing its effective binding energy and population. The cluster growth deduced herein from mass spectrometry and CCSD calculations is fully consistent with the one derived for H2O+Arn clusters derived from IRPD spectroscopy (n = 1–14) and MP2 calculations (n = 1–4).84,88,93 As expected, the charge transfer from H2O+ to the individual He ligands is small, and the NBO charges per He atom are 9, 1, and 0.3 me for the H-bonded, p-bonded, and bridged ligands, and decrease slightly as the cluster grows (Table S1 in ESI†).
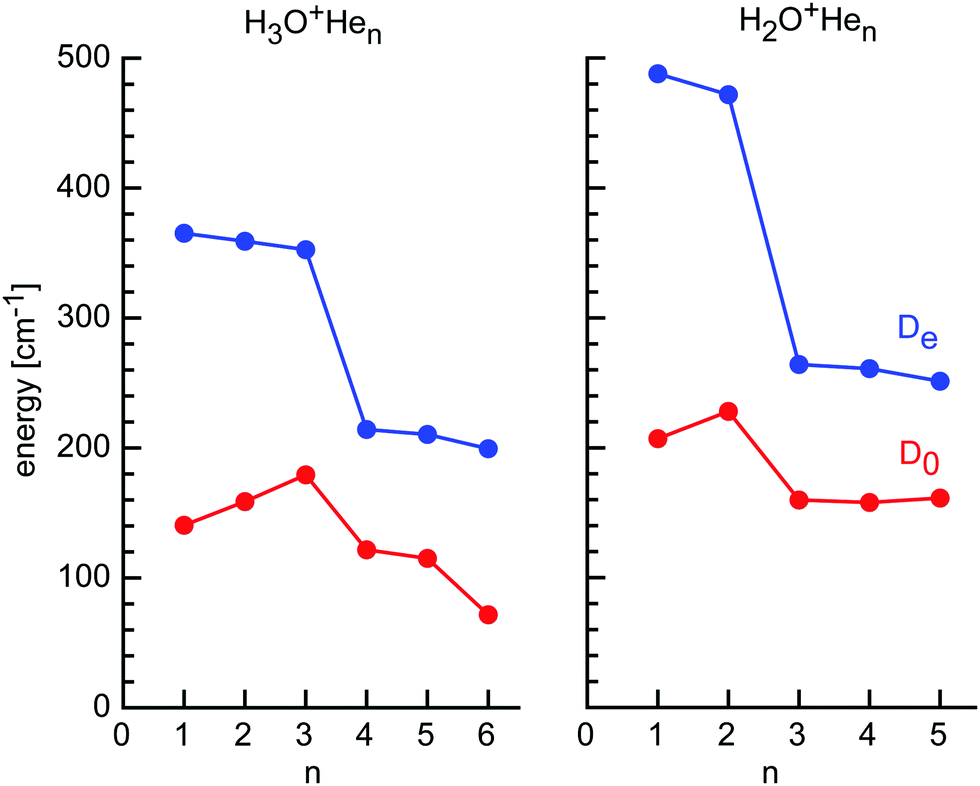 | ||
| Fig. 5 Incremental He binding energies (De and D0) of H3O+Hen and H2O+Hen clusters calculated at the CCSD/aug-cc-pVTZ level. | ||
| n | H2O+ | H3O+ | CH3OH2+ | |||
|---|---|---|---|---|---|---|
| D e | D 0 | D e | D 0 | D e | D 0 | |
| a Numbers in parentheses are CCSD(T) energies (single-point calculations at the CCSD geometry). | ||||||
| 1 (H) | 487.9 (510.2) | 207.0 | 365.2 (384.9) | 139.9 | 283.6 (301.7) | 140.0 |
| 1 (p) | 269.7 (282.1) | 99.4 | ||||
| 2 (2H) | 471.8 (492.5) | 228.0 | 359.1 (378.1) | 158.7 | 281.8 (299.4) | 140.1 |
| 2 (2p) | 266.0 (278.1) | 138.7 | ||||
| 3 | 264.1 (276.2) | 159.8 | 352.6 (371.1) | 179.3 | ||
| 4 | 260.9 (259.0) | 157.9 | 214.2 (225.9) | 121.6 | ||
| 5 | 251.2 (277.5) | 161.3 | 210.4 (221.4) | 115.0 | ||
| 6 | 199.4 (212.9) | 71.6 | ||||
Significantly, while the H2O+He dimer has been produced in a supersonic plasma expansion and spectroscopically characterized by IRPD previously, the larger H2O+Hen clusters with n = 2–9 are generated herein for the first time. Similar to the CH3+Hen case, this result demonstrates the superior performance of ion traps over supersonic expansions in growing larger X+Hen clusters. This is in contrast to H2O+Rgn with the larger Rg atoms Ar and Kr, which have been grown and studied up to n = 14 and 3, respectively.93,94Fig. 6 summarizes the predicted evolution of the OH stretch and H2O bend frequencies ν1–3 as a function of cluster size for the size range n = 0–5 as basis for future IRPD spectroscopy and includes the available experimental data for n = 0 and 1 (Table S2 in ESI†). As can be seen, the H-bonded ligands cause large incremental red shifts in ν1 and ν3 due to destabilization of the O–H bonds by the formation of OH⋯He H-bonds (∼15 cm−1 on average) and smaller blue shifts of 4 cm−1 in ν2 due to the additional retarding force of the H-bonds. On the other hand, p-bonded ligands cause only minor blue shifts in ν1 and ν3 (∼5 cm−1) due to modest charge transfer into the 2pz orbital of O which strengthens the O–H bonds and similarly small blue shifts in ν2 (∼3 cm−1). Interestingly, the bridged He ligand (n = 5) causes a similarly small blue shift in ν1 and ν3 as the p-bonded ligand but a red shift in ν2. Hence, the pattern in the ν1–3 frequencies will readily allow for distinguishing the various isomeric structures. In addition to the frequency pattern, the computed IR intensities are reported in Table S2 in ESI.† All fundamentals of H2O+ are strongly IR active. While H-bonded He ligands strongly increase the IR activity in ν1 and ν3 and slightly reduce that of ν2, p-bonded ligands have little impact on the intensity of all three modes. Overall, the spectral pattern predicted for H2O+Hen is similar to that predicted and observed for H2O+Nen and H2O+Arn, whereby the effects scale with the polarizability of the Rg atom. A detailed comparison of the H2O+Rg dimer potentials, structures, energies, and spectral shifts has been presented elsewhere.24
 | ||
| Fig. 6 OH stretch (ν1 and ν3, scaled by 0.9442) and O–H bend (ν2, scaled by 0.9492) frequencies calculated for H2O+Hen clusters (CCSD/aug-cc-pVTZ) compared to available experimental values for n = 0 and 1 (crosses). | ||
3.2 H3O+Hen
Fig. 1 shows the mass spectrum obtained for the growth of H3O+Hen clusters around the mass-selected H3O+ ion. In addition to the very intense H3O+ parent ion (n = 0), H3O+Hen clusters are produced with a higher intensity for n = 1–3, while signals drop for n = 4–5 and disappear for n = 6. The relative signal intensities are roughly 125:50:30:30:10:1 for n = 0–5. This mass spectrum suggests relatively stable and comparable binding sites for n = 1–3 by forming OH⋯He ionic H-bonds to the three available OH protons. After filling this solvation subshell with three He ligands, further attachment occurs at less stable binding sites. In addition to the H3O+Hen series, we detect again H3O+H2O (m/z 37) from H2O impurity in the He line or residual gas pressure in the trap. The observed mass spectrum is similar to that obtained recently in a cryogenic trap coupled to a QPMS, again validating our approach.34 Actually, the latter study observed the n = 6 cluster about a factor 10 weaker than the n = 5 cluster.
To rationalize the observed H3O+Hen cluster growth, we consider again the CCSD calculations and the most stable isomers are shown in Fig. 7. Protonation of H2O produces the pyramidal H3O+ cation with C3v symmetry and a low barrier for inversion through the planar D3h transition state. The equilibrium structural data for H3O+ (re = 0.9769 Å, θe = 111.7°) and rotational constants (Be = 11.028, Ce = 6.4006 cm−1) are in excellent agreement with the experimental values (re = 0.974(1) Å, θe = 113.6(1)°, Be = 11.2329(26), Ce = 6.2913(66) cm−1).95 The scaled harmonic vibrational frequencies of the symmetric and degenerate antisymmetric OH stretch modes of ν1(a) = 3439 and ν3(e) = 3529 cm−1 are also close to the measured fundamentals, ν1 = 3440 and ν3 = 3528 cm−1 (average of tunneling components).95,96
 | ||
| Fig. 7 Minimum structures (in Å and degree) of H3O+Hen with n = 0–6 obtained at the CCSD/aug-cc-pVTZ level. | ||
The global minima for the H3O+Hen clusters with n = 1–3 are characterized by n equivalent and nearly linear OH⋯He ionic H-bonds, leading to structures with Cs, Cs, and C3v symmetry. Due to small noncooperative threebody forces, the strength of the H-bonds decreases roughly linearly with n, as indicated by the intermolecular bond lengths of Re = 1.800, 1.808, and 1.816 Å, and the equilibrium binding energies, De = 365, 359, and 353 cm−1 (Table 1), respectively. This trend is also visible in the charge transfer from H3O+ per He atom of 8.5, 8.1, and 7.9 me (Table S1 in ESI†). The zero-point corrected energy shows the opposite trend, D0 = 140, 159, and 179 cm−1, illustrating the subtle effect of the vibrations on the dissociation energy, although the harmonic correction may not be reliable for these floppy clusters. The OH⋯He bond angle changes from 175.0 to 174.8° for n = 1 and 3, respectively, while the H3O+ ion gets more planar (θe = 111.7 and 111.9° for n = 0 and 3). The bound O–H bonds elongate upon H-bonding (by 1.4, 0.8, and 0.3 mÅ for n = 1–3), while the free O–H bonds contract (by 0.4 and 0.9 mÅ for n = 1–2). Overall, the properties derived here for H3O+He are consistent with previous CCSD calculations of the dimer potential.97,98
In contrast to the open-shell H2O+ cation with its partly occupied and electrophilic 2pz orbital, the closed-shell H3O+ cation has a filled and thus for He repulsive 2pz orbital of the central O atom. As a result, this region of p-bonding to H3O+ is not favorable for He ligands. For example, the O-bound structure of H3O+He with C3v symmetry is a transition state between the H-bound minima with a very low binding energy (De = 95 cm−1, D0 = 39 cm−1, Re = 3.049 Å). Hence, they prefer to occupy positions between the H-bonded He ligands, leading to structures with Cs, Cs, and C3v symmetry for n = 4–6. The plane of the n = 4–6 ligands is only slightly above the plane of the n = 1–3 ligands, so that the He ligands in H3O+He6 form a slightly distorted regular hexagon with He–He distances of 2.8 Å in close agreement with the van der Waals distance in He2 (2.805 Å). The equilibrium binding energies in this second shell are much lower than in the first H-bonded shell, with De = 214, 210, and 199 cm−1 for n = 4–6, respectively, and this trend is also visible in the reduced charge transfer from H3O+ per He atom of 1.9 me. The slight decrease in De with size is again due to small noncooperativity and results in an increase in the O–He distance from 2.797 to 2.809 Å for n = 4–6. Interestingly, the dissociation energies show a significant drop for n = 4–6 (D0 = 122, 115, and 72 cm−1), and the drop is particularly large for the last ligand. Due to enhanced charge delocalization, the formation of the second He ring also weakens the bonds to the first H-bonded He ring, whose bond lengths elongate from 1.816 Å in n = 3 to 1.834 Å in n = 6. Formation of the second He shell makes the H3O+ ion again slightly less planar and the pyramidal angle in n = 6 is again the same as in n = 0 (111.7°).
The H3O+Hen cluster growth deduced from the CCSD calculations is fully consistent with the mass spectrum shown in Fig. 1. The first solvation shell has three H-bonded H-ligands with similarly high binding energies, giving rise to similar intensities in the mass spectrum. The signals drop for the three bridged He atoms, which have substantially lower binding energies. Similar to the H2O+Hen case, it appears not easy to squeeze in the last two ligands (n = 5 and n = 6), probably again by quenching the possibility for large-amplitude motion of the first ligands. This causes higher intermolecular frequencies and thus larger zero-point energy, which reduces the binding energies, as seen for the low D0 value computed for n = 6. The deduced cluster growth is fully consistent with the H3O+He dimer potential,97,98 and the computational and spectroscopic studies of H3O+Ln clusters with larger Rg atoms and small inert ligands (L = N2, CO2).99–104
While the vibrational spectroscopy of H3O+ is known for long time by high-resolution spectroscopy, no spectral data appear to be available for its H3O+Hen clusters. In the C3v symmetric equilibrium structure, H3O+ has two OH stretch normal modes, namely the symmetric ν1(a) and antisymmetric degenerate ν3(e) mode, which are split by inversion tunneling. Herein, we do not consider this tunneling motion, which will be strongly affected by He solvation, and thus focus on the average frequencies of the two tunneling components, measured as ν1 = 3440 and ν3 = 3528 cm−1.95,96 Complexation with n = 1, 2, 4, and 5 He atoms reduces the three-fold symmetry and thus removes the degeneracy of ν3. The predicted evolution of the ν1 and ν3 components of H3O+Hen as a function of n is illustrated in Fig. 8 (Table S3 in ESI†), along with the average OH stretch frequency (νav). The spectral shifts and splittings are larger for the H-bonded He ligands due to the stronger interaction, and an overall red shift of 13 cm−1 per atom is obtained. On the other hand, the bridged He atoms cause incremental blue shifts of 3 cm−1 for n = 4 and 5, while shell closure at n = 6 causes a small red shift (4 cm−1). H3O+Ln clusters with larger Rg ligands and small ligands show a similar pattern as a function of cluster size but with larger shifts and splittings due to the stronger H3O+⋯L interaction.99–104
 | ||
| Fig. 8 OH stretch (ν1 and ν3) frequencies calculated for H3O+Hen clusters (CCSD/aug-cc-pVTZ, scaling factor 0.9505, filled circles) compared to available experimental values for n = 0 (crosses). | ||
3.3 CH3OH2+Hen
Fig. 9 shows the mass spectrum obtained for the growth of CH3OH2+Hen clusters around the mass-selected CH3OH2+ ion, which is in good agreement with a recent report.43 In addition to the prominent CH3OH2+ parent peak (n = 0), only CH3OH2+Hen clusters with n = 1–2 are produced with significant abundance (relative ratio of roughly 10:3:1 for n = 0–2). This mass spectrum suggests relatively stable and comparable binding sites for n = 1–2 by forming OH⋯He ionic H-bonds to the two available OH protons. In contrast to the related H3O+ ion, no further He atoms can be attached under our experimental conditions, because the CH3 group is not attractive for He. In addition, the overall ROH2+⋯He interaction becomes weaker upon H → CH3 substitution due to enhanced charge delocalization. As a result, the OH protons in CH3OH2+ are less acidic than in H3O+ leading to weaker OH⋯He bonds. Moreover, the binding energy for the bridged binding site becomes too weak in CH3OH2+He3 and thus is not observed. To rationalize the observed cluster growth and the qualitative conclusions, we consider again the CCSD calculations.
 | ||
| Fig. 9 Sum mass spectrum of the ion trap content recorded with the ReTOF, when storing mass-selected CH3OH2+ ions in the 22-pole trap and growing CH3OH2+Hen clusters. | ||
Our CCSD optimization for CH3OH2+ yields a pyramidal structure, which agrees well with previous calculations at a similar level (Fig. 10).43 The O–H bonds are slightly shorter than in H3O+ (by 4 mÅ), leading to symmetric and antisymmetric OH stretch frequencies of νs/aOH = 3510 and 3585 cm−1, which are substantially higher than for H3O+ using the same scaling factor (3439 and 3529 cm−1, Table S4 in ESI†). The oxonium ion is less pyramidal in CH3OH2+, although the HOH bond angle is substantially smaller, βe = 109.3 vs. 111.7°. So far, no IR spectrum of cold and isolated CH3OH2+ has been reported, preventing any direct comparison to experiment. An IRPD spectrum of warm CH3OH2+ produced by CO2 elimination of protonated methyl formate shows only a single broad band centered at 3450 cm−1 (width 300 cm−1), which is substantially red shifted from the spectrum predicted for cold CH3OH2+ due to high internal temperature.105
 | ||
| Fig. 10 Minimum structures (in Å and degree) of CH3OH2+Hen with n = 0–2 obtained at the CCSD/aug-cc-pVTZ level. | ||
The only previous experimental and computational study for CH3OH2+Hen reports only B3LYP calculations for n = 1, which strongly overestimates the interaction energy. Our CCSD data yield nearly linear intermolecular OH⋯He bonds with Re = 1.905 and 1.910 Å, βe = 109.5 and 109.6°, and De = 284 and 282 cm−1 for n = 1 and 2, respectively. The weaker OH⋯He bonds go along with smaller charge transfer from the cation to the He ligands (3 me per He, Table S1 in ESI†). As the O–H bonds in CH3OH2+ are stronger and less acidic than in H3O+, the intermolecular OH⋯He bonds are somewhat weaker in CH3OH2+Hen. Consequently, the corresponding O–H bond elongations and red shifts in the OH stretch frequencies are smaller (ΔravOH = 1.5 and 0.5 mÅ, Δνs/aOH = −1.7/−3.7 and −1.6/−7.8 cm−1 for n = 1 and 2). The computed frequencies for n = 1, νs/aOH = 3508/3581 cm−1, agree well with the measured ones, 3504/3571 cm−1.43 No spectral OH stretch data are available for n = 2. Interestingly, the C–O bond of CH3OH2+ is also substantially affected by He complexation, with bond contractions of 1.5 mÅ per He atom and blue shifts in the CO stretch mode near 800 cm−1 of 5 cm−1. In the limit of considering CH3OH2+ as dative bond between CH3+ and OH2, the strengthening of the C–O bond may be rationalized by a lowering of the ionization energy of the H2O moiety, leading to a slightly stronger bond. Finally, we note that our CCSD structure of CH3OH2+He (Re = 1.905 Å) is rather different from the B3LYP and B3LYP-D3 structures reported recently (Re = 1.833 and 1.809 Å),43 again due to the failure of these DFT methods to properly describe the X+⋯He interaction (severe overestimation).
4. Conclusions
In summary, He cluster growth around the small cations X = H2O+, H3O+, and CH3OH2+ in a cryogenic ion trap has been characterized by mass spectrometry and high level CCSD(T) calculations to determine the structural, vibrational, and energetic properties of X+Hen clusters. The abundances of the cluster ions provide valuable experimental insight about the stepwise solvation process and the structure of the first solvation shell. Clearly, the cluster growth is fully controlled by the radial and angular properties of X+⋯He dimer potential, because the He⋯He interaction is much weaker. In all cases, the cluster growth begins with the formation of equivalent OH⋯He ionic H-bonds leading to highly symmetric structures (first coordination), before less stable binding sites are occupied. In the case of the open-shell H2O+Hen clusters, the latter are p-bonds to the partly filled electrophilic 2pz orbital of O (second coordination). These binding sites are not favorable for H3O+Hen, because the filled 2pz orbital is not attractive for the electron-avoiding He atoms. As a result, the second coordination shell has He atoms between the H-bonded He atoms, leading to a nearly planar and almost regular hexagon for n = 6. Overall, the strengths of the OH⋯He bonds scale with the proton affinity of the proton donor, which varies as PA = 593.2 < 691 < 754.3 kJ mol−1 for OH, H2O, and CH3OH.106 The higher the PA, the stronger and less acidic the intermolecular O–H bonds (re = 0.9991, 0.9769, 0.9721 Å), the weaker the OH⋯He bonds (Re = 1.702, 1.800, 1.905 Å; De = 488, 365, 284 cm−1), and the smaller the charge transfer to He (q = 9, 5, 3 me). As a consequence of the weaker OH⋯He bonds, their impact on the O–H bond elongation and reduction in the OH stretch frequencies are smaller, too. Overall, the binding energies for equivalent binding sites are almost the same, apart from a very minor noncooperative three-body effect, resulting mostly from nonadditive induction. Indeed, the dipole moments induced in the He atoms are not aligned favorably in the structures given by the strong X+⋯He dimer two-body potential. Overall, the He atoms are arranged such that they have roughly the He⋯He van der Waals distance of ∼3 Å. The more He atoms are added to the solvation shell, the more rigid the cluster structure. As a result of restricted possibility for large-amplitude motions (in particular in the angular direction), the zero-point energy increases, which reduces the binding energy (D0), and this leads to reduced population of equivalent binding sites as n increases. This effect may be probed in the future by advanced quantum calculations, including nuclear quantum effects, dynamics, and effects of temperature and the bosonic character of He. In general, the structured growth of the X+Hen clusters is not only visible in their geometric and energetic properties but nicely reflected also in the evolution of the vibrational properties (both in frequency and IR intensity).This work paves the way for several future directions. So far, from the clusters considered herein, only for the H2O+He dimer potential has been characterized in detail by high-resolution IRPD spectroscopy at the rotation–vibration-tunneling level. To this end, the larger H2O+Hen clusters produced herein for the first time are further attractive targets for future IRPD experiments using the reliable CCSD predictions for these clusters. Similarly, no spectroscopic information is available yet for any of the H3O+Hen clusters, and our presented CCSD predictions provide again a useful guide for such experiments. The open-shell H2O+Hen clusters offer also the potential for optical spectroscopy as a probe for cluster structure.107
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by Deutsche Forschungsgemeinschaft (DFG, grant DO 729/8). We thank A. Günther, P. Nieto and D. Hauer-Vidal for support in the initial stage of this study.References
- S. Grebenev, J. P. Toennies and F. Vilesov Andrei, Science, 1998, 279, 2083–2086 CrossRef CAS PubMed.
- M. Hartmann, R. E. Miller, J. P. Toennies and A. Vilesov, Phys. Rev. Lett., 1995, 75, 1566–1569 CrossRef CAS PubMed.
- J. Tang, Y. Xu, A. R. W. McKellar and W. Jäger, Science, 2002, 297, 2030–2033 CrossRef CAS PubMed.
- L. A. Surin, A. V. Potapov, B. S. Dumesh, S. Schlemmer, Y. Xu, P. L. Raston and W. Jäger, Phys. Rev. Lett., 2008, 101, 233401 CrossRef CAS PubMed.
- M. J. Weida, J. M. Sperhac, D. J. Nesbitt and J. M. Hutson, J. Chem. Phys., 1994, 101, 8351–8363 CrossRef CAS.
- J. M. Hutson, Annu. Rev. Phys. Chem., 1990, 41, 123–154 CrossRef CAS.
- K. B. Whaley, Int. Rev. Phys. Chem., 1994, 13, 41–84 Search PubMed.
- A. G. Császár, T. Szidarovszky, O. Asvany and S. Schlemmer, Mol. Phys., 2019, 117, 1559–1583 CrossRef.
- F. Grandinetti, Int. J. Mass Spectrom., 2004, 237, 243–267 CrossRef CAS.
- E. J. Bieske and O. Dopfer, Chem. Rev., 2000, 100, 3963–3998 CrossRef CAS.
- F. Marinetti, E. Bodo and F. A. Gianturco, ChemPhysChem, 2007, 8, 93–100 CrossRef CAS PubMed.
- N. Kobayashi, T. Kojima and Y. Kaneko, J. Phys. Soc. Jpn., 1988, 57, 1528–1531 CrossRef CAS.
- T. M. Kojima, N. Kobayashi and Y. Kaneko, Z. Phys. D, 1992, 23, 181–185 CrossRef CAS.
- H. Tanuma, J. Sanderson and N. Kobayashi, J. Phys. Soc. Jpn., 1999, 68, 2570–2575 CrossRef CAS.
- O. Dopfer, Int. Rev. Phys. Chem., 2003, 22, 437–495 Search PubMed.
- O. Dopfer, Z. Phys. Chem., 2005, 219, 125–168 CrossRef CAS.
- E. J. Bieske, A. S. Soliva, A. Friedmann and J. P. Maier, J. Chem. Phys., 1992, 96, 28–34 CrossRef CAS.
- S. A. Nizkorodov, J. P. Maier and E. J. Bieske, J. Chem. Phys., 1995, 103, 1297–1302 CrossRef CAS.
- M. Meuwly, S. A. Nizkorodov, J. P. Maier and E. J. Bieske, J. Chem. Phys., 1996, 104, 3876–3885 CrossRef CAS.
- O. Dopfer, D. Roth and J. P. Maier, Chem. Phys. Lett., 1999, 310, 201 CrossRef CAS.
- O. Dopfer, S. A. Nizkorodov, M. Meuwly, E. J. Bieske and J. P. Maier, Chem. Phys. Lett., 1996, 260, 545–550 CrossRef CAS.
- N. M. Lakin, R. V. Olkhov and O. Dopfer, Faraday Discuss., 2001, 118, 455 RSC.
- D. Roth, S. A. Nizkorodov, J. P. Maier and O. Dopfer, J. Chem. Phys., 1998, 109, 3841–3849 CrossRef CAS.
- D. Roth, O. Dopfer and J. P. Maier, Phys. Chem. Chem. Phys., 2001, 3, 2400 RSC.
- S. A. Nizkorodov, D. Roth, R. V. Olkhov, J. P. Maier and O. Dopfer, Chem. Phys. Lett., 1997, 278, 26–30 CrossRef CAS.
- R. V. Olkhov, S. A. Nizkorodov and O. Dopfer, J. Chem. Phys., 1999, 110, 9527 CrossRef CAS.
- O. Dopfer and D. Luckhaus, J. Chem. Phys., 2002, 116, 1012 CrossRef CAS.
- R. V. Olkhov and O. Dopfer, Chem. Phys. Lett., 1999, 314, 215–222 CrossRef CAS.
- N. Solcà and O. Dopfer, J. Phys. Chem. A, 2001, 105, 5637–5645 CrossRef.
- M. Schmies, A. Patzer, M. Schütz, M. Miyazaki, M. Fujii and O. Dopfer, Phys. Chem. Chem. Phys., 2014, 16, 7980–7995 RSC.
- A. Patzer, M. Schütz, T. Möller and O. Dopfer, Angew. Chem., Int. Ed., 2012, 51, 4925–4929 CrossRef CAS PubMed.
- M. Brümmer, C. Kaposta, G. Santambrogio and K. R. Asmis, J. Chem. Phys., 2003, 119, 12700–12703 CrossRef.
- K. R. Asmis and J. Sauer, Mass Spectrom. Rev., 2007, 26, 542–562 CrossRef CAS PubMed.
- O. Asvany, S. Brünken, L. Kluge and S. Schlemmer, Appl. Phys. B, 2014, 114, 203–211 CrossRef CAS.
- C. J. Johnson, A. B. Wolk, J. A. Fournier, E. N. Sullivan, G. H. Weddle and M. A. Johnson, J. Chem. Phys., 2014, 140, 221101 CrossRef PubMed.
- D. Gerlich, J. Chin. Chem. Soc., 2018, 65, 637–653 CrossRef CAS.
- I. Savić, D. Gerlich, O. Asvany, P. Jusko and S. Schlemmer, Mol. Phys., 2015, 113, 2320–2332 CrossRef.
- J. Roithova, A. Gray, E. Andris, J. Jasik and D. Gerlich, Acc. Chem. Res., 2016, 49, 223–230 CrossRef CAS.
- O. Boyarkin, Int. Rev. Phys. Chem., 2018, 37, 559–606 Search PubMed.
- P. J. Kelleher, C. J. Johnson, J. A. Fournier, M. A. Johnson and A. B. McCoy, J. Phys. Chem. A, 2015, 119, 4170–4176 CrossRef CAS PubMed.
- T. Salomon, M. Töpfer, P. Schreier, S. Schlemmer, H. Kohguchi, L. Surin and O. Asvany, Phys. Chem. Chem. Phys., 2019, 21, 3440–3445 RSC.
- M. Töpfer, T. Salomon, H. Kohguchi, O. Dopfer, K. M. T. Yamada, S. Schlemmer and O. Asvany, Phys. Rev. Lett., 2018, 121, 143001 CrossRef PubMed.
- P. Jusko, S. Brünken, O. Asvany, S. Thorwirth, A. Stoffels, L. van der Meer, G. Berden, B. Redlich, J. Oomens and S. Schlemmer, Faraday Discuss., 2019, 217, 172–202 RSC.
- O. Asvany and S. Schlemmer, Phys. Chem. Chem. Phys., 2021, 23, 26602–26622 RSC.
- M. Töpfer, A. Jensen, K. Nagamori, H. Kohguchi, T. Szidarovszky, A. G. Császár, S. Schlemmer and O. Asvany, Phys. Chem. Chem. Phys., 2020, 22, 22885–22888 RSC.
- J. A. Davies, N. A. Besley, S. Yang and A. M. Ellis, J. Chem. Phys., 2019, 151, 194307 CrossRef PubMed.
- E. K. Campbell, M. Holz, D. Gerlich and J. P. Maier, Nature, 2015, 523, 322–323 CrossRef CAS PubMed.
- D. Gerlich, J. Jašík and J. Roithová, Int. J. Mass Spectrom., 2019, 438, 78–86 CrossRef CAS.
- D. Gerlich, J. Jasik, V. D. Strelnikov and J. Roithova, Astrophys. J., 2018, 864, 62 CrossRef.
- C. A. Rice, F. X. Hardy, O. Gause and J. P. Maier, J. Phys. Chem. Lett., 2014, 5, 942–945 CrossRef CAS PubMed.
- T. Gonzalez-Lezana, O. Echt, M. Gatchell, M. Bartolomei, J. Campos-Martinez and P. Scheier, Int. Rev. Phys. Chem., 2020, 39, 465–516 Search PubMed.
- A. Mauracher, O. Echt, A. M. Ellis, S. Yang, D. K. Bohme, J. Postler, A. Kaiser, S. Denifl and P. Scheier, Phys. Rep., 2018, 751, 1–90 CrossRef CAS.
- L. Kranabetter, N. K. Bersenkowitsch, P. Martini, M. Gatchell, M. Kuhn, F. Laimer, A. Schiller, M. K. Beyer, M. Ončák and P. Scheier, Phys. Chem. Chem. Phys., 2019, 21, 25362–25368 RSC.
- C. Leidlmair, Y. Wang, P. Bartl, H. Schöbel, S. Denifl, M. Probst, M. Alcamí, F. Martín, H. Zettergren, K. Hansen, O. Echt and P. Scheier, Phys. Rev. Lett., 2012, 108, 076101 CrossRef PubMed.
- A. Schiller, M. Meyer, P. Martini, F. Zappa, S. A. Krasnokutski, F. Calvo and P. Scheier, J. Phys. Chem. A, 2021, 125, 7813–7824 CrossRef CAS PubMed.
- M. Meyer, P. Martini, A. Schiller, F. Zappa, S. A. Krasnokutski and P. Scheier, Astrophys. J., 2021, 913, 136 CrossRef CAS.
- M. Gatchell, P. Martini, F. Laimer, M. Goulart, F. Calvo and P. Scheier, Faraday Discuss., 2019, 217, 276–289 RSC.
- S. Spieler, M. Kuhn, J. Postler, M. Simpson, R. Wester, P. Scheier, W. Ubachs, X. Bacalla, J. Bouwman and H. Linnartz, Astrophys. J., 2017, 846, 6 CrossRef.
- N. B. Brauer, S. Smolarek, X. Zhang, W. J. Buma and M. Drabbels, J. Phys. Chem. Lett., 2011, 2, 1563–1566 CrossRef CAS.
- D. A. Thomas, E. Mucha, S. Gewinner, W. Schöllkopf, G. Meijer and G. von Helden, J. Phys. Chem. Lett., 2018, 9, 2305–2310 CrossRef CAS PubMed.
- X. Zhang, N. B. Brauer, G. Berden, A. M. Rijs and M. Drabbels, J. Chem. Phys., 2012, 136, 044305 CrossRef PubMed.
- F. Bierau, P. Kupser, G. Meijer and G. von Helden, Phys. Rev. Lett., 2010, 105, 133402 CrossRef PubMed.
- E. Mucha, A. I. González Flórez, M. Marianski, D. A. Thomas, W. Hoffmann, W. B. Struwe, H. S. Hahm, S. Gewinner, W. Schöllkopf, P. H. Seeberger, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 11248–11251 CrossRef CAS PubMed.
- S. Erukala, A. Feinberg, A. Singh and A. F. Vilesov, J. Chem. Phys., 2021, 155, 084306 CrossRef CAS.
- D. Verma, S. Erukala and A. F. Vilesov, J. Phys. Chem. A, 2020, 124, 6207–6213 CrossRef CAS.
- S. Erukala, D. Verma and A. Vilesov, J. Phys. Chem. Lett., 2021, 12, 5105–5109 CrossRef CAS.
- J. A. Davies, S. Yang and A. M. Ellis, Phys. Chem. Chem. Phys., 2021, 23, 27449–27459 RSC.
- A. Günther, P. Nieto, D. Müller, A. Sheldrick, D. Gerlich and O. Dopfer, J. Mol. Spectrosc., 2017, 332, 8–15 CrossRef.
- F. Wyrowski, K. M. Menten, R. Güsten and A. Belloche, Astron. Astrophys., 2010, 518, A26 CrossRef.
- V. Ossenkopf, et al. , Astron. Astrophys., 2010, 518, L111 CrossRef.
- J. M. Hollis, E. B. Churchwell, E. Herbst and F. C. De Lucia, Nature, 1986, 322, 524–526 CrossRef CAS.
- A. Das, M. Sil, B. Bhat, P. Gorai, S. K. Chakrabarti and P. Caselli, Astrophys. J., 2020, 902, 131 CrossRef CAS.
- P. B. Crandall, D. Müller, J. Leroux, M. Förstel and O. Dopfer, Astrophys. J., 2020, 900, L20 CrossRef CAS.
- P. Nieto, D. Müller, A. Sheldrick, A. Günther, M. Miyazaki and O. Dopfer, Phys. Chem. Chem. Phys., 2018, 20, 22148–22158 RSC.
- D. Müller and O. Dopfer, J. Photochem. Photobiol., 2020, 3–4, 100009 CrossRef.
- D. Müller and O. Dopfer, J. Phys. Chem. A, 2021, 125, 3146–3158 CrossRef.
- M. Töpfer, P. C. Schmid, H. Kohguchi, K. M. T. Yamada, S. Schlemmer and O. Asvany, Mol. Phys., 2019, 117, 1481–1485 CrossRef.
- R. V. Olkhov, S. A. Nizkorodov and O. Dopfer, J. Chem. Phys., 1998, 108, 10046–10060 CrossRef CAS.
- O. Dopfer, R. V. Olkhov and J. P. Maier, J. Chem. Phys., 2000, 112, 2176 CrossRef CAS.
- M. J. Frisch, et al., Gaussian16, Rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- R. A. Aziz, F. R. W. McCourt and C. C. K. Wong, Mol. Phys., 1987, 61, 1487–1511 CrossRef CAS.
- T. R. Huet, C. J. Pursell, W. C. Ho, B. M. Dinelli and T. Oka, J. Chem. Phys., 1992, 97, 5977 CrossRef CAS.
- O. Dopfer, D. Roth and J. P. Maier, J. Chem. Phys., 2001, 114, 7081 CrossRef CAS.
- O. Dopfer, J. Phys. Chem. A, 2000, 104, 11693–11701 CrossRef CAS.
- O. Dopfer and V. Engel, J. Chem. Phys., 2004, 121, 12345 CrossRef CAS.
- X. Chen and M. Thachuk, Int. J. Quant. Chem., 2005, 101, 1–7 CrossRef CAS.
- J. P. Wagner, D. C. McDonald and M. A. Duncan, J. Chem. Phys., 2017, 147, 7 Search PubMed.
- H. Zhou, R. Yang, X. Jin and M. Zhou, J. Phys. Chem. A, 2005, 109, 6003–6007 CrossRef CAS PubMed.
- G. E. Lopez, J. Comput. Chem., 1995, 16, 768–776 CrossRef CAS.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standards and Technology, Gaithersburg MD, 20899, 2001, https://webbook.nist.gov/ Search PubMed.
- D. F. Liu, T. Wyttenbach and M. T. Bowers, Int. J. Mass Spectrom., 2004, 236, 81–90 CrossRef CAS.
- H. S. Kim, C. H. Kuo and M. T. Bowers, J. Chem. Phys., 1990, 93, 5594–5604 CrossRef CAS.
- O. Dopfer, D. Roth and J. P. Maier, J. Phys. Chem. A, 2000, 104, 11702–11713 CrossRef CAS.
- J.-M. Liu, T. Nishigori, T. Maeyama, Q.-R. Huang, M. Katada, J.-L. Kuo and A. Fujii, J. Phys. Chem. Lett., 2021, 12, 7997–8002 CrossRef CAS PubMed.
- J. Tang and T. Oka, J. Mol. Spectrosc., 1999, 196, 120–130 CrossRef CAS PubMed.
- M. H. Begemann and R. J. Saykally, J. Chem. Phys., 1985, 82, 3570–3579 CrossRef CAS.
- S. Borocci, P. Cecchi, M. Giordani and F. Grandinetti, Eur. J. Mass Spectrom., 2015, 21, 171–181 CrossRef CAS PubMed.
- H. El Hanini, F. Najar, M. Naouai and N.-E. Jaidane, Phys. Chem. Chem. Phys., 2019, 21, 11705–11713 RSC.
- J.-W. Li, M. Morita, K. Takahashi and J.-L. Kuo, J. Phys. Chem. A, 2015, 119, 10887–10892 CrossRef CAS PubMed.
- A. B. McCoy, T. L. Guasco, C. M. Leavitt, S. G. Olesen and M. A. Johnson, Phys. Chem. Chem. Phys., 2012, 14, 7205–7214 RSC.
- J. A. Tan, J.-W. Li, C.-C. Chiu, H.-Y. Liao, H. T. Huynh and J.-L. Kuo, Phys. Chem. Chem. Phys., 2016, 18, 30721–30732 RSC.
- Q.-R. Huang, T. Nishigori, M. Katada, A. Fujii and J.-L. Kuo, Phys. Chem. Chem. Phys., 2018, 20, 13836–13844 RSC.
- B. Bandyopadhyay, T. C. Cheng and M. A. Duncan, Int. J. Mass Spectrom., 2010, 297, 124–130 CrossRef CAS.
- J. Yang, X. Kong and L. Jiang, Chem. Phys., 2018, 501, 1–7 CrossRef CAS.
- K. Chatterjee and O. Dopfer, Astrophys. J., 2020, 898, 92 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- H. Lew, Can. J. Phys., 1976, 54, 2028–2049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01192a |
| This journal is © the Owner Societies 2022 |