
Clustering of metal dopants in defect sites of graphene-based materials†
Stephanie
Lambie
 ab,
Krista G.
Steenbergen
c,
Nicola
Gaston
*a and
Beate
Paulus
*b
ab,
Krista G.
Steenbergen
c,
Nicola
Gaston
*a and
Beate
Paulus
*b
aMacDiarmid Institute for Advanced Materials and Nanotechnology, Department of Physics, University of Auckland, Private Bag 92019, Auckland, New Zealand. E-mail: n.gaston@auckland.ac.nz
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany. E-mail: b.paulus@fu-berlin.de
cMacDiarmid Institute for Advanced Materials and Nanotechnology, School of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Wellington 6140, New Zealand
First published on 10th December 2021
Abstract
Single-atom catalysts are promising candidates for many industrial reactions. However, making true single-atom catalysts is an experimental dilemma, due to the difficulty of keeping dopant single atoms stable at temperature and under pressure. This difficulty can lead to clustering of the metal dopant atoms in defect sites. However, the electronic and geometric structure of sub-nanoscale clusters in single-atom defects has not yet been explored. Furthermore, recent studies have proven sub-nanoscale clusters of dopants in single-atom defect sites can be equally good or better catalysts than their single-atom counterparts. Here, a comprehensive DFT study is undertaken to determine the geometric and electronic structure effects that influence clustering of noble and p-block dopants in C3- and N4-defect sites in graphene-based systems. We find that the defect site is the primary driver in determining clustering dynamics in these systems.
Introduction
Atmospheric CO2 levels are currently the highest that they have been in the past 14 million years.1 One mitigation strategy is to sequester CO2 from the atmosphere and convert it into a fuel source by cleaving the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. However, CO2 is a particularly stable molecule and therefore, to have any hope of breaking this bond, a catalyst must be employed. Ideally, the catalyst would be simple to manufacture, cheap, stable and both selective and active for the CO2 reduction reaction (CO2RR). However, activity and selectivity for the CO2RR differs depending on the catalyst, pH, electrolyte and overpotential. With all of these demands, engineering a suitable catalyst is a non-trivial task.
O bond. However, CO2 is a particularly stable molecule and therefore, to have any hope of breaking this bond, a catalyst must be employed. Ideally, the catalyst would be simple to manufacture, cheap, stable and both selective and active for the CO2 reduction reaction (CO2RR). However, activity and selectivity for the CO2RR differs depending on the catalyst, pH, electrolyte and overpotential. With all of these demands, engineering a suitable catalyst is a non-trivial task.
Catalysts have been in development for the CO2RR for many years. The first catalysts to be considered were monometallic catalysts. Of the transition metals, Cu is the most active and selective for the CO2RR.2–7 However, while Cu can reduce CO2, it does so at low efficiency8 and requires a high overpotential9 rendering Cu non-viable as an industrial catalyst.
Bimetallic catalysts were then designed in an attempt to combine the activity of one metal with the selectivity of another, thus obtaining the best of both worlds. W/Au,10 Cu/In11 and Cu/Sn12 catalysts have all been shown to be active for the CO2RR. However, both mono- and bimetallic transition metal catalysts are fundamentally limited by so-called ‘scaling relations’13,14 whereby the adsorption energy of key intermediates in the reaction are related and ultimately govern the catalytic activity of the material. As such, new classes of catalysts that are not governed by scaling relations need to be explored.
In 2004, graphene was discovered.15 Graphene is a two-dimensional (2D) sheet of hexagonally bound C atoms characterised by sp2 hybridisation and resembling a honeycomb lattice. Graphene in isolation is inert and unreactive but has excellent electrical conductivity and high surface area.15 To improve graphene's catalytic activity, both metal and non-metal (e.g. N, B, S) elements can be doped on or into the sheet. However, elements cannot easily be adsorbed to a perfect graphene sheet, due to adsorbates having a high diffusion mobility on the surface under experimental conditions.16 As such, defect sites are required to limit the mobility of adsorbates on the surface. The two most commonly used sites for doping species into graphitic systems are the graphitic site (hereafter referred to as the C3-site), and the pyridinic site (hereafter referred to as the N4-site). A metal atom can then be doped into these defect sites10,17–22 giving rise to a so-called ‘single-atom’ catalyst. The interaction of the C framework, metal dopants and, in the N4-sites, the N atoms, give these single-atom catalysts unprecedented properties quite distinct from those of graphene, the defect site or, indeed, the metal dopant.23 However, how these different features interact with one another to give a material novel properties is not well understood.
Unfortunately, single-atom catalysts are extremely prone to thermal deactivation. Sintering and coalescing can occur under ambient conditions,24 at high temperature and low pressure,25 on a range of different substrate materials26 and a variety of surface coverages.27 There have been many studies that develop methods of maintaining the stability of the single-atom structures28–32 and further studies that even describe methods of producing thermally stable single-atom catalysts from nanoparticles for selected metals.33 There are advantages to maintaining the single-atom sites, such as establishing a higher activity per gram rating.34 However, it has also been found that small sub-nanoscale clusters can exhibit a higher activity than the single-atom sites with respect to time and cycles35–38 and, thus, can be equally as important, if not more so, to the overall catalytic activity of the material. For example, a Rh sub-nanoscale cluster of 4–8 Å in diameter on a TiO2 support was found to be 5–10 times more active than than its single-atom counterpart.36 Furthermore, atomic clusters of up to nine Pt atoms doped into an N4-site in graphene were shown to be more active than the single-atom sites after 4000 cycles and 16 hours for the hydrogen evolution reaction in acid.35
The relationship between structure and function in catalytic systems is well known. For example, in heterogenous catalysis by changing the surface terminations of Pt that meet to form an edge site, the catalytic activity is altered by 5 orders of magnitude for hydrogen desorption.39 A study of a Ru nanoparticle determined that available active sites have differing activities, with some sites being up to nine orders of magnitude more active than others.40,41 These studies highlight the importance of understanding the structure of the system being utilised. Very limited work has been carried out on the structure of sub-nanoscale clusters in defect sites on extended C systems – one of the primary aims of the present study.
Much work has been carried out on the incorporation of transition metals into single-atom C-based catalysts.42–46 Fe,46 Co,47 Ni,48 and Zn48 doped N4-sites have been shown to catalyse the CO2RR, producing CO as the major product. Rh-N4 was found to be most active catalyst for producing CH3OH from a range of transition metals.42 However, similar to pure transition metal catalysts, transition metal single-atom catalysts are also limited by scaling relations.42,49 One suggested way of breaking these scaling relations is to incorporate post-transition metals into graphene-based materials.42,50 To date, there is very limited research on post-transition metal doped C3- and N4-graphene based materials as single-atom catalysts for the CO2RR.
Post-transition metals have been studied in a limited number of contexts for their catalytic promise. Sn and Al doped C3-systems have been shown to be active for the CO2RR.21,51 A Bi-based metal–organic-framework, has limited propensity for the CO2RR,52 but 2D bismuthene has been reported to be a highly effective catalyst for the same reaction.53 Au is included in this study as it is a noble metal well-known for its highly size-dependent catalytic ability.54
In this study, a computational approach is used to determine the structure of noble (Au) and post-transition metal (Al, Bi, Ga and Sn) doped single-atom and sub-nanoscale cluster catalysts in C3- and N4-sites.
Computational details
All calculations were carried out in the Vienna ab initio Simulation Package (VASP)55 using plane wave density functional theory (DFT) with the PBE56 exchange-correlational functional in conjunction with the projector-augmented wave (PAW)57 method. An energy cutoff of 600 eV was used, with van der Waals (vdW) interactions included through the D3 method with Becke–Johnson damping.58,59 Calculations were electronically converged to 1 × 10−5 eV, while ionic convergence was set to 1× 10−2 eV to avoid minor optimisation fluctuations in the graphene sheets. Monkhorst–Pack k-point sampling of 3 × 3 × 1 was selected. Higher k-points were tested, but energies were altered by less than 2 × 10−4 eV atom−1 as the k-point grid was increased (Table S1, ESI†).The objective of this work was to understand how selected metal dopants localise in defect sites in graphene-based materials. The dopants considered in this study were Al, Au, Bi, Ga and Sn. Two different sites were considered; a C3-site, where a single atom has been removed from the graphene sheet (Fig. S1, ESI†) and an N4-site, where two adjacent C atoms are removed and the four under-coordinated C atoms substituted with N (Fig. S2, ESI†). Unit cell sizes of 4 × 4 hexagonal C rings were used, resulting in 50 atoms in the C3-site system and 49 atoms in the N4-site system. Size testing of the system was carried out by examining how the adsorption energy (Eads) of the metal dopant to the site varied with respect to the size of the system (Fig. S3, ESI†). Periodic systems were separated by a minimum of 15 Å of vacuum in the z-dimension. Reference single atoms were calculated as an isolated atom in a vacuum, separated from the next repeating unit by no less than 12 Å.
In the first section of this work, the structure of single metal atom doped C3- and N4-sites were examined. However, it has been experimentally proven to be extremely difficult to ensure that single-atom catalysts are truly ‘single-atom’ due to the limited control of experiments in this size regime, therefore it is likely that dopant atoms cluster in the defect sites. In the second section of this work, clustering of metal dopants in the C3- and N4-sites was considered. In this section, the stability of low energy free clusters, selected from exhaustive global optimisation studies from two to five atoms in size,60–66 were calculated. From this initial examination of low energy cluster confirmations, all 2- and 3-atom configurations were doped into the sites. For the 4- and 5-atom clusters, the three lowest energy configurations from the free cluster calculations were doped into the C3- and N4-sites.
Comparative calculations were also carried out in TURBOMOLE67 for the single-atom systems and for the free clusters. Similar trends were found between the two quantum chemical codes, providing an extra assurance that the results found in this study are reliable. Full discussion of the computational setup and results from these simulations can be found in Supplementary Note 6 (ESI†). The TURBOMOLE calculated spin states are taken as the minimum energy spin configuration of the VASP structures, due to the geometric and electronic similarity of these systems and the difficulty of extracting this information from VASP simulations.
Results and discussion
Metal dopants in C3- and N4-sites
Firstly, the geometries of the single-atom doped C3- and N4-sites for all metals are considered (Fig. 1). In the C3-site, the out-of-plane height is correlated to ring strain with Al and Ga having a lower out-of-plane height of 0.81–0.83 Å and a higher ring strain of 124.3–124.6° (Fig. 1(a)(i) and (d)(i)) compared to Au, Bi and Sn which all have larger out-of-plane binding heights, between 1.38–1.49 Å, and a lower ring strain of 121.0–122.9° (Fig. 1(b)(i), (c)(i) and (eii)).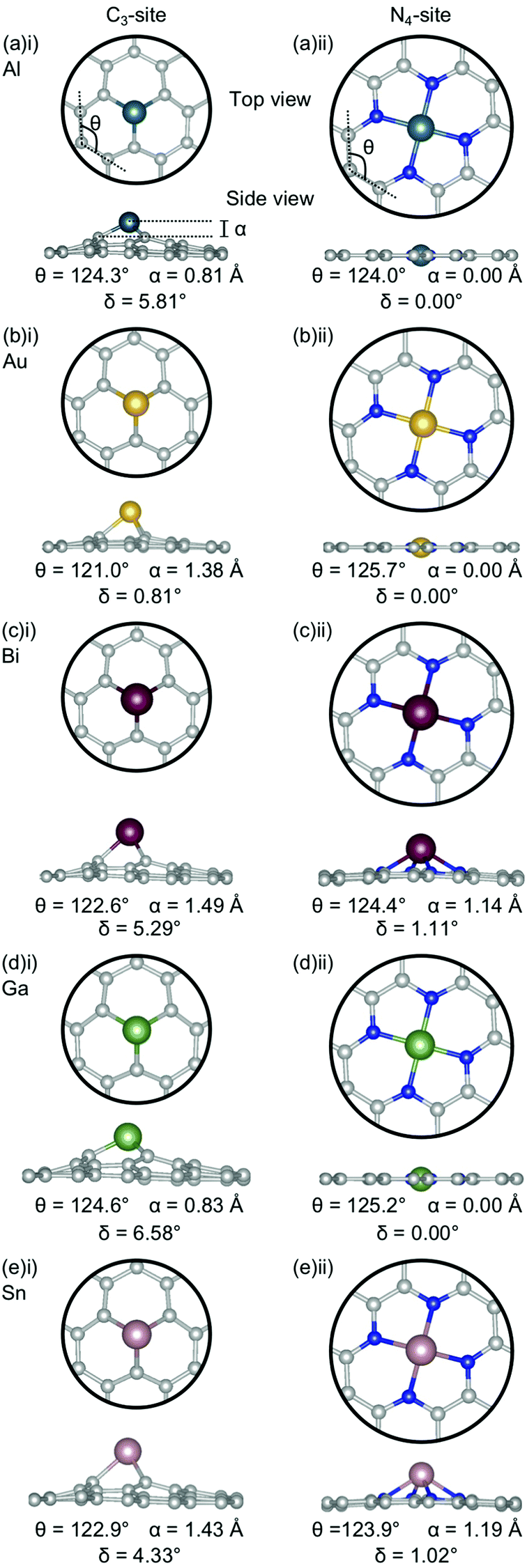 | ||
| Fig. 1 Optimised geometries for metal doped (i) C3- and (ii) N4-sites for (a) Al, (b) Au, (c) Bi, (d) Ga and (e) Sn. C atoms are grey, N atoms are blue, Al atoms are teal, Au atoms are yellow, Bi atoms are red, Ga atoms are green and Sn atoms are pink. The out-of-plane binding height is denoted by α, the average internal angle of the adjacent 6-atom ring is denoted by θ and the degree of distortion in the extended C system is given by δ. Note that the circles in the top view are to used highlight the active site and not the unit cell repetitions. | ||
For the N4-site, of those metals that bind in-plane – Al, Au and Ga – the two with larger vdW radii, Au and Ga, (2.32 Å) exhibit an increased ring strain, with θ between 125.2–125.7° (Fig. 1(b)(ii) and (d)(ii)). However, Al (2.25 Å) has a similar ring strain (θ: 124.0°; Fig. 1(a)(ii)) to the considerably larger Sn and Bi (θ: 123.9 and 124.4°, respectively; Fig. 1(c)(ii) and (e)(ii)). For Al, this reduction in ring strain is attributed to the smaller vdW radius while for Bi and Sn, the lower ring strain is attributed to the out-of-plane binding of the metal dopant as a result of their considerably larger vdW radii (2.42 and 2.52 Å, respectively).
A third parameter for describing these systems has also been defined, δ:
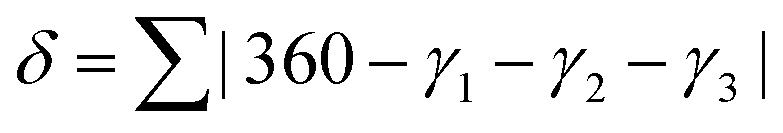 | (1) |
In the C3-site, the Au-doped system is considerably more planar (0.81°) than the other four metal-doped systems, which all have a similar degree of distortion (4.33–6.58°). In the N4-site, the three metals that bind in-plane also have a planar extended C system (Al, Au and Ga) while the Bi and Sn doped N4-systems have a small but non-zero distortion (1.02–1.11°).
The relationship between the adsorption energy, Eads, and the vdW radius68 of the metal dopant was then examined (Fig. 2a). Eads is calculated as:
| Eads = Etot − Esite − Eatom | (2) |
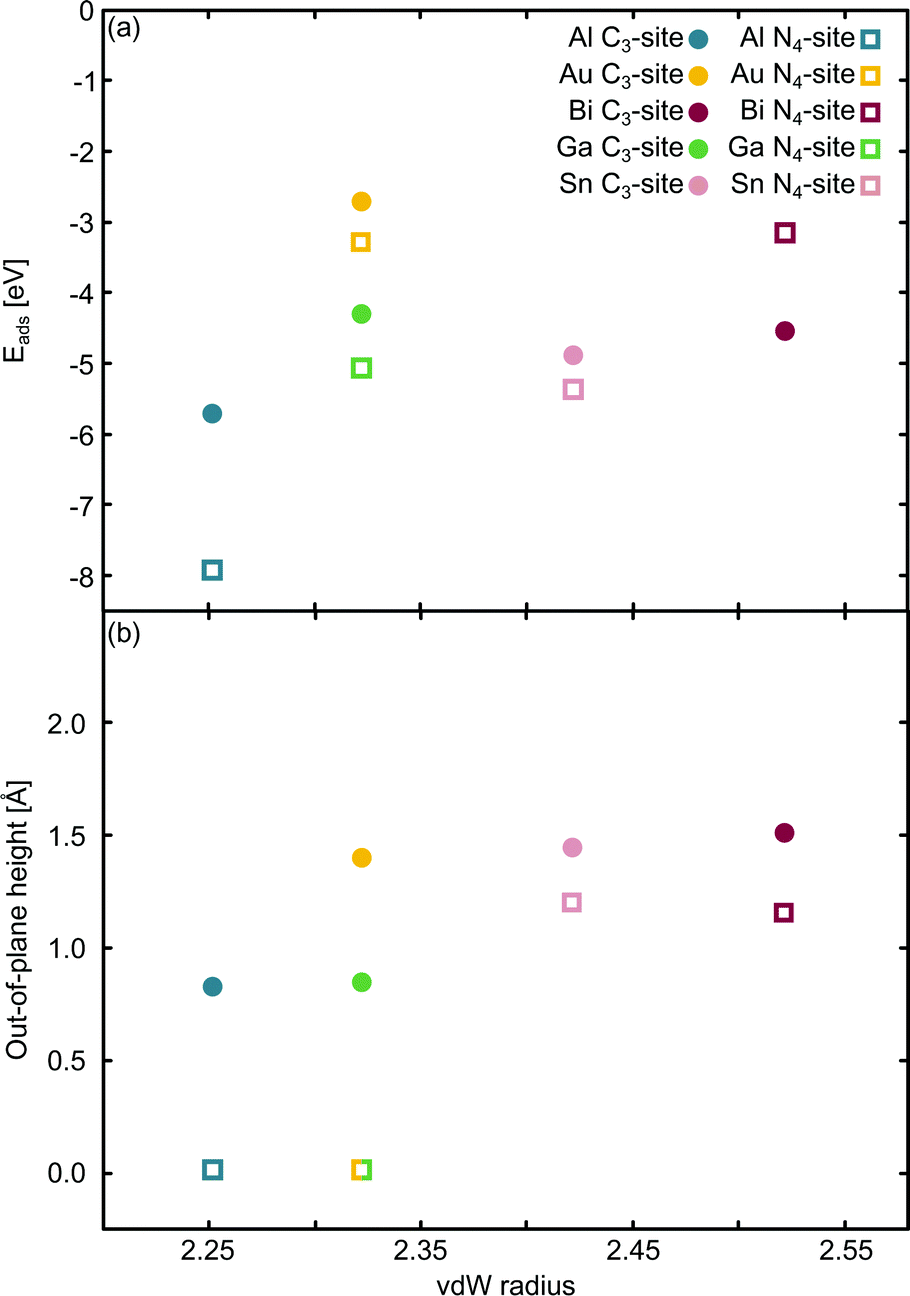 | ||
| Fig. 2 Single metal dopants in C3- and N4-sites. The relationship between the van der Waals (vdW) radius68 and (a) Eads and (b) out-of-plane binding height is shown. The green/yellow square shows the out-of-plane binding height for Ga and Au, which have the same vdW radius. | ||
E ads does not change systematically with respect to size of the dopant atom (Fig. 2a). In the C3-site, Ga, Sn and Bi all have a similar Eads, despite having considerably different vdW radii, while Al has a slightly stronger Eads. Au has a comparatively small vdW radius of 2.32 Å and a particularly weak Eads. However, size is not the only factor that dictates the interaction of the dopant with the flake; valence electrons and density of states (Fig. S5–S14, ESI†) also play a key role. Al has a valency of 3s23p1, Ga of 4p1, Sn, 5p2 and Bi, 6p3. The s2 valence electrons for Sn and Bi, are not available for bonding due to the ‘inert pair effect’ that results from the difference in energy of the s and p orbitals for elements in the lower rows of the periodic table. Ga sits on the cusp of this transition from binding to non-binding valence s2 electrons,73,74 likely explaining the similarity in Eads of these three dopants. Al, however, is able to utilise the valence 3s23p1 electrons for bonding, thus giving Al an increased flexibility in bonding configurations that it can adopt. Finally, Au, with a 6s1 valency, is simply too noble to form strong bonds with the defect site.
In the N4-sites, Al has a particularly strong Eads. Bi and Au have the weakest Eads of the metals considered here and Ga and Sn exhibit a similar Eads, between that of Al, and Au and Bi. The strong interaction of Al with the N4-site is not surprising as it has 3 valence electrons to form both σ and π bonds with the site and sits in-plane. Furthermore, because Al and N are close on the periodic table, their orbitals have similar energetics and thus, good overlap, ultimately leading to a strong interaction. Au is a noble metal and only has a 6s1 electron available for binding, with the addition of a small capability for dispersive binding due to polarisation of the d-shell. When compared to, for example, Ga, with the same vdW radius, Ga has a valence configuration of 4p1. Ga having a p valency allows for better orbital overlap with the valence N p states thus enabling a stronger adsorption interaction with the defect site.
When comparing the C3-site to the N4-site, the Eads varies across a smaller range for the former. Eads is consistently less in the C3-site than in the N4-site, with the exception of Bi, which, counter-intuitively, binds more strongly to the smaller C3-site than the bigger N4-site. Au, Ga and Sn all display a similar difference in Eads between the C3- and N4-sites, while Al has a considerably stronger Eads in the N4-site compared to the C3-site. The increased stability for N4-binding over C3-binding is likely due to the increased versatility of the N atoms compared to the C atoms. That is, N atoms provide opportunity for both σ and π bonding, while sp2 hybridised C only offers σ bonding.
Subsequently, the relationship between the vdW radius of the metal dopant and the out-of-plane binding height was investigated (Fig. 2b). Two possible binding heights for the metal dopants in each of the C3- and N4-sites were observed; in the C3-site, if the metal dopant has a vdW radius ≤2.32 Å (Al and Ga), it sits ∼0.8 Å out-of-plane, while if the vdW radius is ≥2.42 Å (Bi and Sn), the metal dopant sits ∼1.5 Å out-of-plane. The out-of-plane binding height of Au in the C3-site is an anomaly as it has the same vdW radius as Ga, but an out-of-plane binding height comparable to Bi and Sn (Fig. 2b). This is likely due to Au having a full valence d-shell, making it ‘hard sphere’ compared to Ga, which has a more diffuse valence shell. In the N4-site, however, Al, Au and Ga all fit within the cavity and bind in-plane with the extended C system, while Sn and Bi are larger and, therefore, bind at a height of ∼1.2 Å (Fig. 2b). We also highlight that the difference in the binding height between the dopants in the C3-site is considerably less (∼0.7 Å) compared to the N4-site (∼1.2 Å). Due to the more similar binding height of the dopants in the C3-site, it is plausible that the Eads is less variable for these sites. In contrast, the marked difference in binding height in the N4-site, in conjunction with the variance in valency, is a probable explanation for the increased variance in Eads of this site. Finally, we suggest that the particularly weak binding of Bi in the N4-site is the result of both the large out-of-plane binding height and the doublet spin state of Bi giving rise to a comparatively destabilised structure. This is in contrast to the Sn doped N4-site, which has a similar out-of-plane binding height to Bi but adopts a triplet spin state, thus Sn is likely able to better interact with the N p orbitals than Bi (Fig. S10 and S14, ESI†).
Chan et al. found that in the C3-site, Al, Ga, Sn and Bi were covalently bound to the neighbouring C atoms.75 Electron localisation function (ELF) analysis carried out in this study shows that there is a degree of covalency to the metal dopant-C bond, but that this bond is not truly covalent, when compared to, for example, the sp2 hybridised C–C bond (Table S2, ESI†). It has previously been found that a covalent bonding interaction between the C and the dopant atom results in large in-plane distortions.76 Here, we find that both the size of the metal dopant and Eads factor into the distortion in the graphitic flake. In the C3-site, because all of the metal dopants considered in this study are too large to site within the defect, Eads appears to be the determining factor in the degree of distortion in the graphene flake. Au doped C3-systems have the weakest Eads of any metal considered here, thus, we attribute the planarity of the extended C system to the comparatively weak interaction between Au and the C system. In contrast, Al, Bi, Ga and Sn all display a similar degree of distortion within the graphene flake (δ: 4.33–6.48°). In the N4-sites, however, the size of the metal dopant is the determining factor for the degree of planarity in the graphitic flake, with Al, Au and Ga all binding in-plane and maintaining the planarity of the graphene flake (δ: 0.00°). In contrast to this, Bi and Sn bind out-of-plane (α: 1.14–1.19 Å) which results in the graphene flake no longer being truly planar (δ: 1.02–1.11°).
In order to further probe the single-atom dopant systems, the charge density distribution was examined by means of the charge density (Table S3, ESI†) and Bader charge analyses (Tables S4 and S5, ESI†). These show that the Bader charge on all the metal dopants is lower than the formal charge, which is unsurprising due to the delocalisation of electrons. It is interesting that Ga, in the same group as Al, has a Bader charge similar to that of Bi and Sn but a very similar geometric structure to Al. However, this is likely due to the inert pair effect that needs to be taken into account for the Ga, Bi and Sn. Al, Au, Bi and Ga doped C3- and N4-sites all resulted in the doublet being the lowest energy spin state, while the lowest energy spin states for Sn doped systems were found to be a singlet and triplet state in the C3- and N4-site, respectively (Table S10, ESI†).
Free clusters (N = 2–5)
Making true ‘single-atom’ catalysts is the goal of experiments, however, in reality it is extremely difficult to control how many dopant atoms locate in the cavities under experimental conditions.24–27 Therefore, in this study, the structure of these sub-nanoscale metal clusters doped into C3- and N4-sites are explicitly explored. To do this, low energy configurations of free metal clusters 2–5 atoms in size are considered (Table S6, ESI†) and the cohesive energy (Ecoh) was calculated (Fig. 3). Ecoh is defined as:77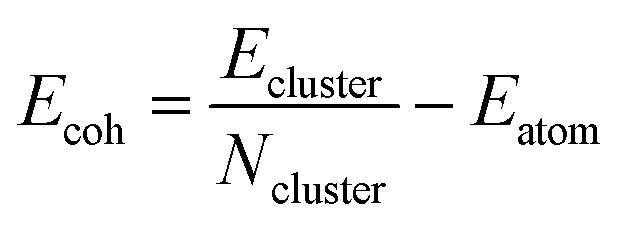 | (3) |
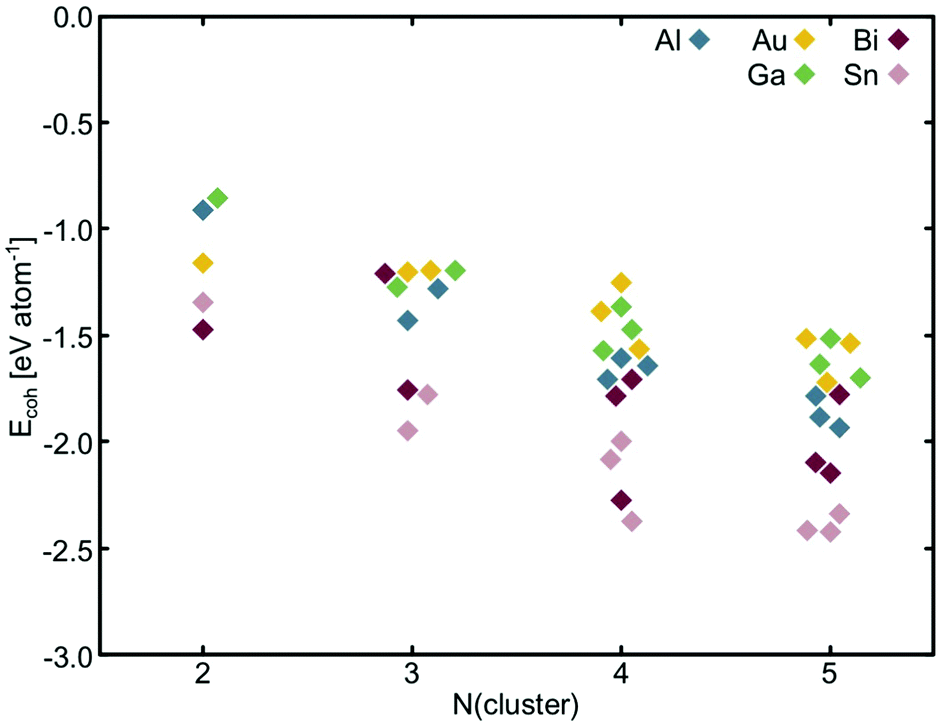 | ||
| Fig. 3 Cohesive energy per metal atom for selected free clusters. Points are offset on the x-axis only to make them visible and are associated with the nearest integer value. | ||
Of the free metal clusters, Sn generally displays the strongest Ecoh. Bi exhibits an intermediate Ecoh between Sn, and Al, Au and Ga. It is interesting to note, that Bi has the largest range in Ecoh of all of the metals considered here. Al, Au and Ga all display similar Ecoh however, the range in Ecoh for Au is lower across the N = 2–5 atom size range than for Al and Ga. The trends between the metal Ecoh values calculated here are consistent with those calculated by various methods in the literature (Fig. S15, ESI†).
We take the spin states calculated in TURBOMOLE to be the lowest energy electronic configurations (Tables S11 and S12, ESI†). Al and Ga both have one p valence electron and when there are an uneven number of atoms in the cluster prefer a doublet spin state. However, when there are an even number of atoms in the cluster, the high spin triplet state is preferred over the singlet state, in keeping with prior studies on the spin states of Ga clusters.61,78 Bi has three valence p electrons and Au has one valence s electron but, in contrast to Al and Ga, these metals flip between the singlet state, when the number of the atoms in the cluster is even, to the low spin doublet state, when the number of atoms in the cluster is uneven. Sn has two p valence electrons and is more stable in the high spin triplet configuration when Ncluster ≤ 3 atoms and the low spin singlet state when Ncluster ≥ 4 atoms.
Clustering of metal dopants in C3- and N4-sites
The clusters for which the Ecoh was calculated were then doped into C3- and N4-sites to simulate dopant clustering in the cavity. What follows is a detailed analysis of the lowest energy configurations of clustered metal dopants in C3- and N4-sites and their relation to the lowest energy free cluster configurations. All cluster geometries and relative electronic energies can be found in the ESI† (Tables S7 and S8).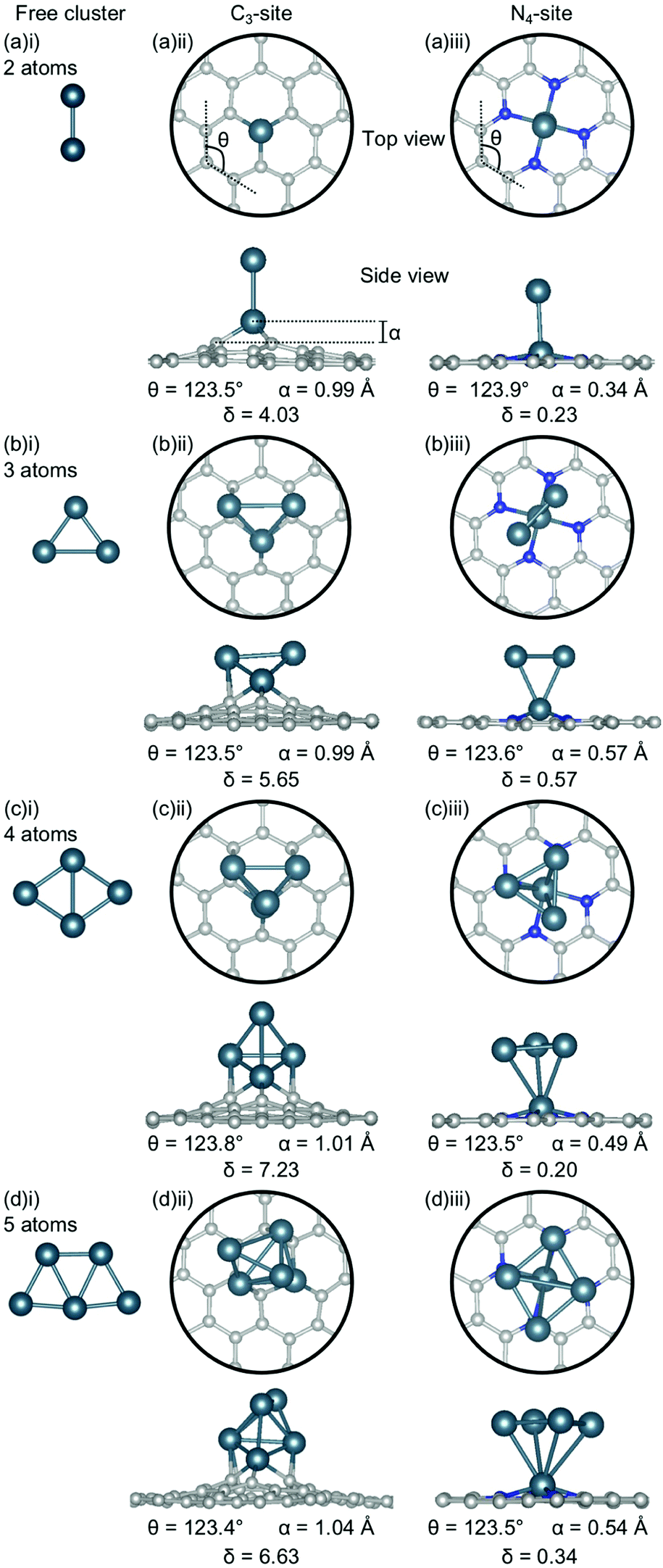 | ||
| Fig. 4 Lowest energy structures for Al systems of (a) 2-atoms, (b) 3-atoms, (c) 4-atoms and (d) 5-atoms in size as (i) a free cluster and doped in the (ii) C3- and (iii) N4-site. Al is shown in teal, C in grey and N in blue. The average out-of-plane binding height is denoted by α, the average internal angle of the adjacent 6-atom ring is denoted by θ and the degree of distortion in the extended C system is given by δ. Note that the circles in the top view highlight the active site and not the unit cell repetitions. | ||
 | ||
| Fig. 5 Lowest energy structures for Ga systems of (a) 2-atoms, (b) 3-atoms, (c) 4-atoms and (d) 5-atoms in size as (i) a free cluster and doped in the (ii) C3- and (iii) N4-site. Ga is shown in green, C in grey and N in blue. The out-of-plane binding height is denoted by α, the average internal angle of the adjacent 6-atom ring is denoted by θ and the degree of distortion in the extended C system is given by δ. Note that the circles in the top view highlight the active site and not the unit cell repetitions. | ||
In the C3-sites, Al doped systems are considerably different to the free clusters of the same size. In particular, the preference for planarity is lifted. At cluster sizes of 3–5 atoms, Al is in close proximity to more than one C atom (Fig. 4(b)(ii), (c)(ii) and (d)(ii)), possibly due to the small size of Al allowing an increased proximity. The interaction of the metal cluster to the graphene flake through more than one C atom is also observed for Ga at N = 4 atoms (Fig. 5(c)(ii)). For Al, the ring strain is very consistent between all clustered sites, ranging from 123.4–123.8°, and is slightly lower than the single-atom doped system (124.3°). The out-of-plane binding height of the Al also remains very similar between the lowest energy clustered configurations (0.99–1.04 Å) but is slightly higher in all clustered cases than the single-atom doped system (α: 0.88 Å). In the Ga doped systems, a slightly higher out-of-plane binding height (0.98–1.11 Å) than the single-atom doped system (0.83 Å), and an associated lower ring strain (123.1–124.0° compared to 124.6° for the single-atom doped system) is observed. The Al doped C3-sites have a considerably distorted extended C system with δ values ranging from 4.03–7.23°, however, this is in range of the single-atom doped system (δ: 5.81°). In the Ga-systems, high δ values are also observed (Fig. 5(a)(ii), (b)(ii), (c)(ii), (d)(ii)) in the range of 4.16–6.48°. In both the Al and Ga C3-systems, this high degree of distortion in the extended C system could be the result of the clustered metal atoms interacting with the graphene flake through multiple atoms, and therefore, these C atoms being pulled out of plane. As a final note, the Ga systems also appear to have a preference for the clustered Ga atoms orienting over C atoms associated with the C3-site.
There are two points of interest for clustering in the N4-site. Firstly, in all cases, the doped metal atom is pulled out of the graphitic plane (for Al, α: 0.34–0.57 Å and, for Ga, α: ∼0.78 Å; Fig. 4 and 5(a)(iii), (b)(iii), (c)(iii), (d)(iii)), in contrast to the optimised single-atom configuration where the dopant atom sat perfectly in-plane with the extended C system for both Al and Ga. While the δ values are low for all of the clustered systems (Al; 0.20–0.57°, Ga; 0.50–0.56°), these values are, notably, non-zero. Secondly, when considering a N atom cluster, the lowest energy configuration is that of the optimised single-atom doped N4-site with the N − 1 free cluster above the metal-doped site. For example, the lowest energy structure for the N4-site doped with a 4-atom (N) Al cluster (Fig. 4(c)(iii)) is the metal doped N4-site (Fig. 1(a)ii) beneath the 3-atom (N − 1) subcluster (Fig. 4(b)(i)). When clusters of 4- and 5-atoms are doped into the N4-site, there appears to be a preference for the Al atoms locating directly over the N atoms. The ring strain in the Al-doped N4-systems is very similar across all cluster sizes (123.5–123.9°) and also to the singly doped system (124.0°). For the clustered Ga systems, associated with an increased out-of-plane binding height is a lower ring strain (∼123.9°) compared to the single-atom doped system (125.2°).
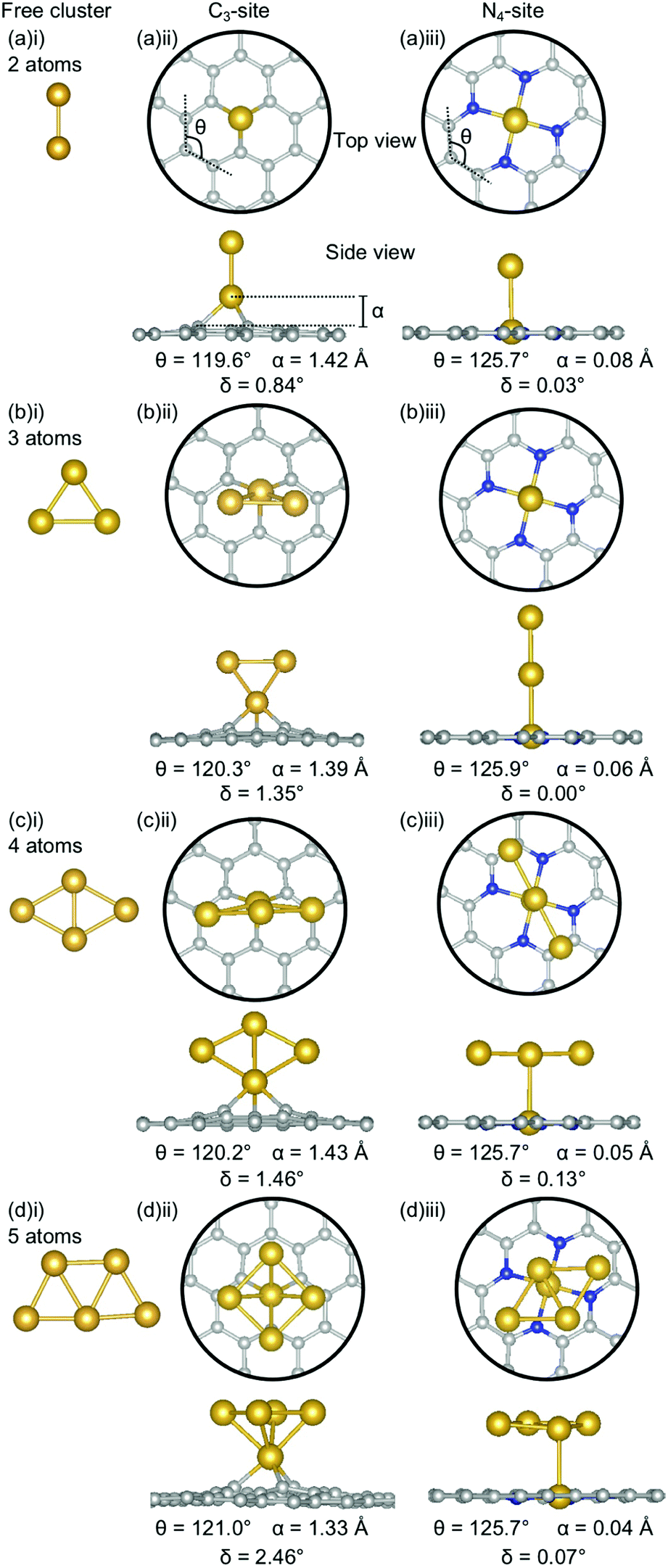 | ||
| Fig. 6 Lowest energy structures for Au systems of (a) 2-atoms, (b) 3-atoms, (c) 4-atoms and (d) 5-atoms in size as (i) a free cluster and doped in the (ii) C3- and (iii) N4-site. Au is shown in yellow, C in grey and N in blue. The out-of-plane binding height is denoted by α, the average internal angle of the adjacent 6-atom ring is denoted by θ and the degree of distortion in the extended C system is given by δ. Note that the circles in the top view highlight the active site and not the unit cell repetitions. | ||
The clustering of Au in the C3-sites is interesting, because, compared to all of the other metals, the graphitic flake is more planar (Fig. 6(a)(ii), (b)(ii), (c)(ii), and (d)(ii)) with δ values between 0.84–2.46°. It is also interesting to note, that the δ values increase monotonically as the cluster size increases. The 2-atom Au doped system has a similar δ value to that of the single-atom doped system (0.83° and 0.81°, respectively). For N = 2–4 atoms, the lowest energy cluster simply inserts into the C3-site, and the cluster geometry of the lowest energy free cluster is retained. This is not the case for the 5-atom system, where a square planar N − 1 cluster sits above the Au doped C3-site. The ring strain for the clustered Au systems is fairly consistent (119.6–121.0°) as is the out-of-plane binding height of the binding Au atom (1.33–1.43 Å). Both of these values are comparable to the single-atom doped system (θ: 121.0° and α: 1.38 Å).
In the N4-site, Au generally exhibits the same trends as Al and Ga, with the lowest energy structure being the N − 1 free cluster above the metal-doped site (Fig. 6(a)(iii), (b)(iii), (c)(iii), (d)(iii)). However, for Au, the cluster binds directly through the Au atom in the N4-site and perpendicular to the sheet. There is no preference for keeping the doped metal atoms close in proximity to the C-based system. In the N = 3 atom case, the competitive energetics of the triangle and linear trimer cluster are highlighted, with the insertion of the N − 1 subcluster adopting a linear structure rather than the lower surface area triangle geometry. Again, the Au doped N4-systems are highly planar, with very low δ values of 0.00–0.13° and α values of 0.04–0.08 Å. The planarity of the Au-doped systems likely arises from the weak Eads of Au with the defect sites resulting from the 6s1 valence electron.
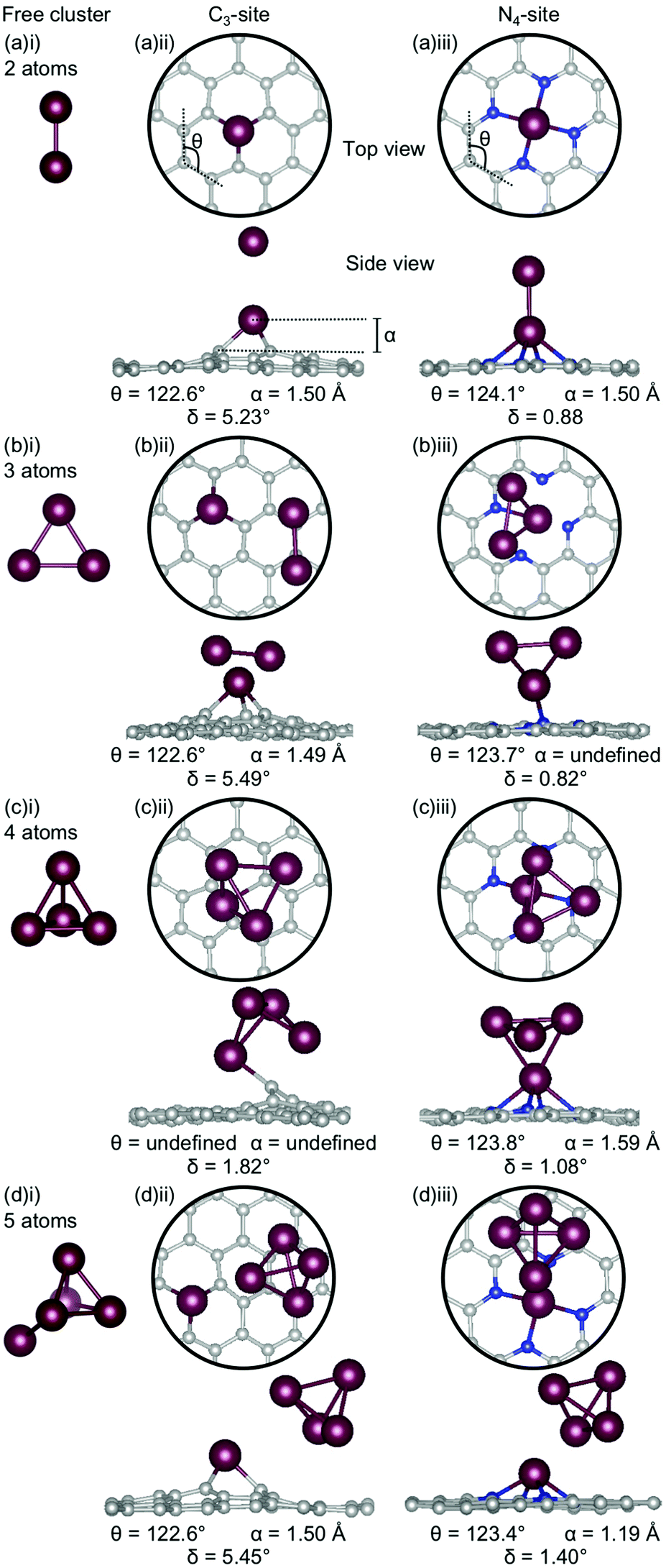 | ||
| Fig. 7 Lowest energy structures for Bi systems of (a) 2-atoms, (b) 3-atoms, (c) 4-atoms and (d) 5-atoms in size as (i) a free cluster and doped in the (ii) C3- and (iii) N4-site. Bi is shown in red, C in grey and N in blue. The out-of-plane binding height is denoted by α, the average internal angle of the adjacent 6-atom ring is denoted by θ and the degree of distortion in the extended C system is given by δ. Note that the circles in the top view highlight the active site and not the unit cell repetitions. | ||
When Bi becomes clustered in the C3-site, a number of the Bi-Bi bond distances become anomalously long (Fig. 7(a)(ii), (b)(ii), (c)(ii), (d)(ii)). For example, the Bi–Bi dimer has a bond length of 2.65 Å (Fig. 7(a)(i)), in good agreement with previous experimental and calculated values,80,81 however when this dimer is doped into the C3-site, the Bi-Bi bond distance is increased to 3.74 Å – longer than the reported non-covalent Bi-Bi bond of 3.53 Å.82 Furthermore, in the N = 3 atom cluster (Fig. 7(b)(ii)), the Bi-Bi bond distance between the Bi doped in the C3-site and the 2-atom, N − 1, subcluster is 4.20 Å – 1.5 times that of the calculated Bi-Bi dimer. The N = 4 atom cluster is unique because the Bi atom is bound to the extended C system through a single C atom, rather than through the three available in the C3-site, resulting in one of the hexagonal C rings associated with the C3-site being altered such that a 5-membered ring is formed. In the N = 5 atom cluster, the minimum bond distance between the single-atom doped site and the N − 1 subcluster is 3.96 Å making the subcluster effectively unbound. However, covalent bonding within the subclusters is expected, due to the proximity of the Bi atoms.82
The distortion (δ) in the flake is fairly consistent for the Bi doped C3-systems (5.23–5.49°), and very similar to the singly doped Bi C3-system, which has a δ of 5.29°. The similarity in the geometric parameters between the single-atom doped system and the clustered systems is rationalised by the clustered systems essentially being single-atom doped systems with loosely bound Bi clusters. The exception to this is N = 4 atoms, which has a much lower distortion of 1.82°. Again, the 4-atom Bi cluster is a particularly stable structure as a unit82 and, therefore, interacts weakly with graphene flake, thereby not distorting the flake and explaining this exception. Finally, we note that a larger out-of-plane binding height is not necessarily associated with reduced ring strain for these systems.
In the N4-site, where N = 3 atoms, the Bi-Bi bond distance between the Bi single-atom doped flake and the 2-atom (N − 1) subcluster is ∼3.0 Å. In the 4- and 5-atom clusters the same geometry is seen for both the C3- and N4-sites; a point of difference between Bi, and Al, Ga and Au. All of the Bi doped N4-systems have a similar degree of planarity, with δ values between 0.82–1.40°, similar to the singly doped Bi N4-system, which has a δ value of 1.11°.
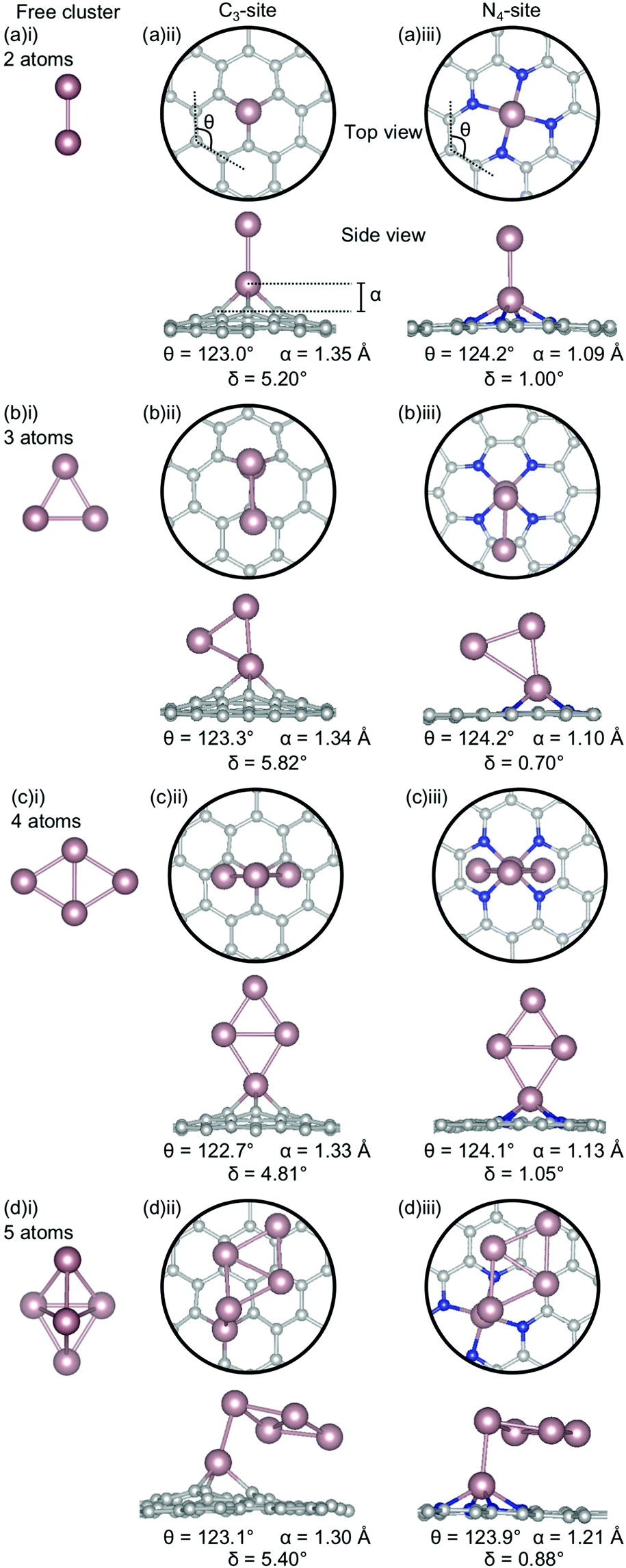 | ||
| Fig. 8 Lowest energy structures for Sn systems of (a) 2-atoms, (b) 3-atoms, (c) 4-atoms and (d) 5-atoms in size as (i) a free cluster and doped in the (ii) C3- and (iii) N4-site. Sn is shown in pink, C in grey and N in blue. The out-of-plane binding height is denoted by α, the average internal angle of the adjacent 6-atom ring is denoted by θ and the degree of distortion in the extended C system is given by δ. Note that the circles in the top view highlight the active site and not the unit cell repetitions. | ||
Sn has a unique behaviour amongst the metals considered here. In both the C3- and N4-sites, the lowest energy cluster configurations are identical (Fig. 8(ii), (iii)), making it the only dopant considered in this study to be completely agnostic to site. The out-of-plane binding height for the Sn clustered C3-sites (1.30–1.35 Å) are consistently less than for the single-atom system (1.43 Å). Interestingly, despite, the lower out-of-plane binding height of the clustered systems, the ring strain is comparable between the clustered and single-atom systems (122.70–123.3° and 122.9° respectively). However, the distortion in the extended C system is slightly higher in the clustered systems (4.81–5.82°) than for the single-atom system (4.33°).
In the N4-sites, the out-of-plane binding between the clustered and single-atom systems is comparable (1.09–1.21 Å for the clustered sites and 1.19 Å for the singly doped site), as is the ring strain (123.9–124.2° compared to 123.9°) and the distortion in the extended C system (0.70–1.05° compared to 1.02°). There appears to be a preference for Sn–Sn interaction being through a single Sn atom and the Sn–Sn interaction being oriented perpendicular to the extended C system, excepting N = 4 atoms. In the N = 5 atom case, the Sn-Sn interaction bond length is 2.96 Å; comparable to the Sn-Sn metallic bond length in β-Sn, 3.02 Å.83 It is possible that the particularly strong Ecoh of Sn free clusters (Fig. 3) makes the lowest energy Sn clusters more difficult to alter thus providing a hypothesis for the consistency of Sn cluster configurations, irrespective of site.
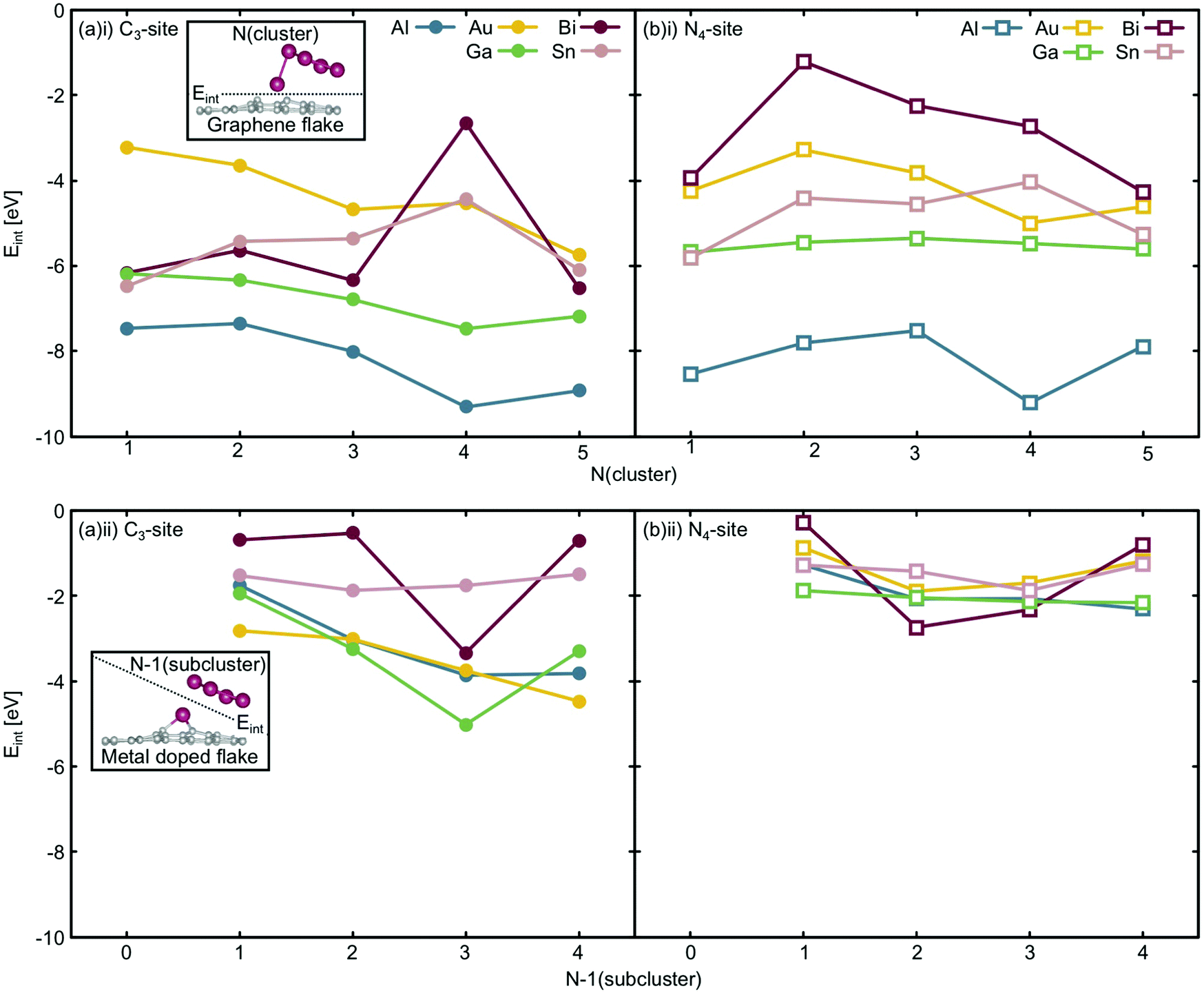 | ||
| Fig. 9 Energy of interaction (Eint) between (i) the N-atom cluster and the graphene flake and (ii) the N − 1 atom subcluster and the metal doped graphene flake in the (a) C3-sites (filled circles) and (b) N4-sites (unfilled squares). Al is shown in teal, Au in gold, Bi in red, Ga in green and Sn in pink. Insets depict the definition of Eint for a generic system with the dashed line showing where the systems were differentiated. | ||
Considering the interaction energy of a single metal atom with the flake i.e. Eint at N = 1 atom, all are stabilised by more than 3.0 eV (Fig. 9(a)(i) and (b)(i)). From the free cluster energies of the metal dimer (N = 2 atoms), it is clear that Ecoh is no more than 1.5 eV atom−1 (Fig. 3). This highlights that if the graphitic flakes were defect rich and metal dopant poor, the expected thermodynamic result would be single atom catalysts rather than clustered sites because it is more energetically favourable for the second metal dopant atom to locate in a defect site than it is for this atom to cluster at the site. The only exception to this is the 4-atom Bi cluster in the C3-site, where Ecoh and the Eint are competitive as this Bi cluster has a closed electronic shell and therefore is a particularly stable configuration,65 as has been previously discussed.
The change in Eint from N = 1 atom to N = 2 atoms shows, similarly, only weak changes in comparison to the single atom interaction energies, reinforcing the potential for these systems to produce single-atom catalysts. In the case of the C3-site, for Al, Sn and Bi there is an initial destabilisation of Eint as the number of dopant atoms is increased. In contrast, for Ga and Au, there is a slight stabilisation of Eint upon adding a second dopant atom to the defect site. By comparison, in the N4-site, Eint is destabilised for all the dopants upon the addition of a second dopant atom to the defect site. While the changes in Eint in the C3-site are small, they highlight that the metallic interaction energies vary more in this site, than in the N4-site.
Finally, Eint between the doped metal flake and the remaining cluster atoms in their adsorbed configuration (N − 1) (Fig. 9(a)(ii) and (b)(ii)) was considered. Generally, the changes in Eint of the N − 1 subcluster in the C3-site are much greater than in the N4-site. In the N4-site, the single metal dopant atom is able to sit in the defect site and the N − 1 subcluster is comparatively free to adopt a preferred minimum energy configuration within the N − 1 subcluster. The ability of the metal dopants to form the minimum energy N − 1 subcluster, and for all the metal dopants to behave in this way, reduces both the magnitude of Eint and the variation in this value, as is reflected in Fig. 9(b)(ii). In contrast, the C3-site tends to encourage 3D cluster geometries that are less favoured for free clusters, as seen from the lowest energy free cluster calculations (Fig. 4–8 and Table S6, ESI†). Due to the fact that the metal dopants adopt a different cluster geometry than the minimum energy cluster, Eint of the N − 1 cluster with the metal doped flake is highly variable and metal dependent, resulting in larger magnitude and variation in Eint in the C3-site, compared to the N4-site.
While no explicit simulations at finite temperatures are carried out in the present research, we expect that the SACs studied here are likely to remain SACs at room temperature, and indeed, well above. Using Boltzmann's kinetic theory as an approximate gauge of average kinetic energy, kinetic effects would destabilise Eint by ∼0.04 eV at room temperature. Given that our interaction energies are between 13–230 times stronger than the destabilising kinetic effect, we conclude that typical experimental temperatures will minimally alter the behaviour described in this work.
Conclusions
In conclusion, the interaction of the metal dopant with the defect site is dictated by the valency and size of the dopant and the size and bonding flexibility of the defect site. Clustering of the dopants at the site is highly likely, experimentally. However, the considerable additional stabilisation that is gained by locating metal dopant atoms in unoccupied defect sites over clustering in an occupied site suggests that both C3- and N4-sites may be selective for true single-atom catalysts. Under conditions that support clustering, such as high dopant concentration, the defect site exerts considerable control over the clustering geometry; the smaller C3-site with reduced bonding options drives dopant clusters to adopt 3D structures. In contrast, the N4-site is larger with more bonding possibilities, thus a single metal dopant atom sits in the defect site and the N − 1 subcluster is able to sit, loosely bound, above it. As such, we conclude that if clustering at a site does occur, the C3-site is more likely to maintain this clustering than the N4-site.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors wish to acknowledge the use of New Zealand eScience Infrastructure (NeSI) high performance computing facilities and consulting support as part of this research. New Zealand's national facilities are provided by NeSI and funded jointly by NeSI's collaborator institutions and through the Ministry of Business, Innovation & Employment's Research Infrastructure programme. URL https://www.nesi.org.nz. S. L. acknowledges funding from the Deutscher Akademischer Austauschdienst to support this collaboration and Jiang Liang Low for useful discussions. The Zentraleinrichtung für Datenverarbeitung (ZEDAT) of the Freie Universität Berlin is also acknowledged for supplying computational resources.References
- Y. Zhang, M. Pagani, Z. Liu, S. Bohaty and R. DeConto, A 40-million-year history of atmospheric CO2, Philos. Trans. R. Soc. A, 2013, 371, 20130096 CrossRef.
- K. Schouten, Z. Qin, E. Pérez Gallent and M. Koper, Two pathways for the formation of ethylene in CO reduction on single-crystal copper electrodes, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS.
- W. Durand, A. Peterson, F. Studt, F. Abild-Pederson and J. Nørskov, Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- M. Gattrell, N. Gupta and A. Co, A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- A. Peterson, F. Abild-Pederson, F. Studt, J. Rossmeisl and J. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- D. DeWulf, T. Jin and A. Bard, Electrochemical and surface studies of carbon dioxide reduction to methane and ethylene at copper electrodes in aqueous solutions, J. Electrochem. Soc., 1989, 136, 1686–1691 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, Electrochemical reduction of CO at a copper electrode, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- L. Zhang, Z. Zhao and J. Gong, Nanostructured materials for heterogeneous electrocatalytic CO2 reduction and their related reaction mechanisms, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS.
- Y. Hori, Electrochemical CO2 reduction on metal electrodes, 2008 Search PubMed.
- S. Back, H. Kim and Y. Jung, Selective heterogeneous CO2 electroreduction to methanol, ACS Catal, 2015, 5, 965–971 CrossRef CAS.
- G. Larrazábal, A. Martín, S. Mitchell, R. Hauert and J. Pérez-Remírez, Enhanced reduction of CO2 to CO over Cu–In electrocatalysts: catalyst evolution is the key, ACS Catal., 2016, 6, 6265–6274 CrossRef.
- S. Sarfraz, A. Garcia-Esparza, A. Jedidi, L. Cavallo and K. Takanabe, Cu–Sn bimetallic catalyst for selective aqueous electroreduction of CO2 to CO, ACS Catal., 2016, 6, 2842–2851 CrossRef CAS.
- J. Nørskov, T. Bligaard, J. Rossmeisl and C. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. Munter, P. Moses, E. Skúlason, T. Bligaard and J. Nørskov, Scaling properties of adsorption energies for hydrogencontaining molecules on transition-metal surfaces, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS.
- K. Novoselov, A. Geim, S. Morozov, D. Jiang, Y. Zhang, S. Dubonos, I. Grigorieva and A. Firsov, Electric Field effect in atomically thin carbon Films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. Nakada and A. Ishii, Migration of adatom adsorption on graphene using DFT calculation, Solid State Commun., 2011, 151, 13–16 CrossRef CAS.
- S. Kattel, P. Atanassov and B. Keifer, Stability, electronic and magnetic properties of inplane defects in graphene: a First principles study, J. Phys. Chem. C, 2012, 116, 8161–8166 CrossRef CAS.
- A. Titov, P. Zapol, P. Král, D. Liu, H. Iddir, K. Baishya and L. Curtiss, Catalytic Fe-xN sites in carbon nanotubes, J. Phys. Chem. C, 2009, 113, 21629–21634 CrossRef CAS.
- D. Lee, W. Lee, W. Lee, S. Kim and Y. Kim, Theory, synthesis, and oxygen reduction catalysis of Fe-porphyrin-like carbon nanotube, Phys. Rev. Lett., 2012,(106), 175502 Search PubMed.
- M. Kaukonen, A. Krasheninnikov, E. Kauppinen and R. Nieminen, Doped graphene as a material for oxygen reduction reaction in hydrogen fuel cells: a computational study, ACS Catal., 2012, 3, 159–165 CrossRef.
- M. Esrafili and N. Saeidi, Sn-embedded graphene: an active catalyst for CO oxidation to CO2?, Physica E, 2015, 74, 382–387 CrossRef CAS.
- Y.-H. Lu, M. Zhou, C. Zhang and Y.-P. Feng, Metal-embedded graphene: a possible catalyst with a high activity, J. Phys. Chem. C, 2009, 113, 20156–20160 CrossRef CAS.
- T. Ma, Q. Fan, X. Li, J. Qiu, T. Wu and Z. Sun, Graphene-based materials for electrochemical CO2 reduction, J. CO2 Util., 2019, 30, 168–182 CrossRef CAS.
- M. Asoro, D. Kovar, Y. Shao-Horn, L. Allard and P. Ferreira, Coalescence and sintering of Pt nanoparticles: in situ observation by aberration-corrected HAADF-STEM, Nanotechnology, 2009, 21, 025701 CrossRef PubMed.
- S. Simonsen, I. Chorkendorff, S. Dahl, M. Skoglundh, J. Sehested and S. Helveg, Direct observations of oxygen-induced platinum nanoparticle ripening studied in situ TEM, J. Am. Chem. Soc., 2010, 132, 7968–7975 CrossRef CAS PubMed.
- T. Risse, S. Shaikhutdinov, N. Nilius, M. Sterrer and H.-J. Freund, Gold supported on thin oxide Films: from single atoms to nanoparticles, Acc. Chem. Res., 2008, 41, 949–956 CrossRef CAS PubMed.
- C. Campbell, S. Parker and D. Starr, The effects of size-dependent nanoparticle energetics on catalyst sintering, Science, 2002, 298, 811–814 CrossRef CAS.
- R. Qin, P. Liu, G. Fu and N. Zheng, Strategies for stabilizing atomically dispersed metal catalysts, Small Methods, 2018, 2, 1700286 CrossRef.
- H. Tang, F. Liu, J. Wei, B. Qiao, H. Zhao, Y. Su, C. Jin, L. Li, J. Liu, J. Wang and T. Zhang, Ultrastable hydroxyapatite/titanium-dioxide-supported gold nanocatalyst with strong metal-support interaction for carbon monoxide oxidation, Angew. Chem., Int. Ed., 2016, 128, 10764–10769 CrossRef.
- H. Jeong, J. Bae, J. Woo Han and H. Lee, Promoting effects of hydrothermal treatment on the activity and durability of Pd/CeO2 catalysts for CO oxidation, ACS Catal., 2017, 7, 7097–7105 CrossRef CAS.
- S. Schmidt, C. Greczynski, C. Goyenola, C. K. Gueorguiev, X. S. Czigány, J. Jensen, I. G. Ivanov and L. Hultman, CFx thin solid Films deposited by high power impulse magnetron sputtering: synthesis and characterization, Surf. Coat. Technol., 2011, 206, 646–653 CrossRef CAS.
- G. K. Gueorguiev, C. Goyenola, S. Schmidt and L. Hultman, CFx: a First-principles study of structural patterns arising during synthetic growth, Chem. Phys. Lett., 2011, 516, 62–67 CrossRef CAS.
- S. Wei, A. Li, J.-C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W.-C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Direct observation of noble metal nanoparticles transforming to thermally stable single atoms, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed.
- S. Yang, J. Kim, Y. Joo Tak, A. Soon and H. Lee, Single-atom catalyst of platinum supported on titanium nitride for selective electrochemical reactions, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS.
- M. Sun, J. Ji, M. Hu, M. Weng, Y. Zhang, H. Yu, J. Tang, J. Zheng, Z. Jiang, F. Pan, C. Liang and Z. Lin, Overwhelming the performance of single atoms with atomic clusters for platinum-catalyzed hydrogen evolution, ACS Catal., 2019, 9, 8213–8223 CrossRef CAS.
- H. Guan, J. Lin, B. Qiao, X. Yang, L. Li, S. Miao, J. Liu, A. Wang, X. Wang and T. Zhang, Catalytically active Rb sub-nanoclusters on TiO2 for CO oxidation at cryogenic temperatures, Angew. Chem., Int. Ed., 2016, 55, 2820–2824 CrossRef CAS.
- X. Cheng, Y. Li, L. Zheng, Y. Yan, Y. Zhang, G. Chen, S. Sun and J. Zhang, Highly active, stable oxidized platinum clusters as electrocatalysts for the hydrogen evolution reaction, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W.-C. Cheong, R. Yu, L. Gu, J. Gond, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Carbon nitride support Fe2 cluster catalysts with superior performance for alkene epoxidation, Nat. Commun., 2018, 9, 2353 CrossRef.
- E. Skúlason, A. Faraj, L. Kristinsdóttir, J. Hussain, A. Garden and H. Jónsson, Catalytic activity of Pt nano-particles in H2 formation, Top. Catal., 2014, 57, 273–281 CrossRef.
- J. Gavnholt and J. Schiøtz, Structure and reactivity of ruthenium nanoparticles, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035404 CrossRef.
- S. Dahl, A. Logadottir, R. Egeberg, J. Larsen, I. Chorkendorff, E. Törnqvist and J. Nørskov, Role of steps in N2 activation on Ru(0001), Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- V. Tripkovich, M. Vanin, M. Karamad, M. Björketun, K. Jacobsen, K. Thygesen and J. Rossmeisl, Electrochemical CO2 and CO reduction on metal-functionalized porphyrinlike graphene, J. Phys. Chem. C, 2013, 117, 9187–9195 CrossRef.
- H. Zhou, X. Zou, X. Wu, X. Yang and J. Li, Coordination engineering in cobalt–nitrogenfunctionalised materials for CO2 reduction, J. Phys. Chem. Lett., 2019, 10, 6551–6557 CrossRef CAS.
- K. Ogura and I. Yoshida, Electrocatalytic reduction of CO2 to methanol. 9. Mediation with metal porphyrins, J. Mol. Catal., 1988, 47, 51–57 CrossRef CAS.
- J. Shen, M. Kolb, A. Göttle and M. Koper, DFT study on the mechanism of the electrochemical reduction of CO2 catalysed by cobalt porphyrins, J. Phys. Chem. C, 2016, 120, 15714–15721 CrossRef CAS.
- T. Huan, N. Ranjbar, G. Rousse, M. Sougrati, A. Zitolo, V. Mougel, F. Jaouen and M. Fontecave, Electrochemical reduction of CO2 catalysed by Fe–N–C materials: a structureselectivity study, ACS Catal., 2017, 7, 1520–1525 CrossRef CAS.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, Unveiling active sites of CO2 reduction on nitrogen-coordinated and atomically dispersed iron and cobalt catalysts, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- H. Yang, S. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. Chen, C. Li, T. Zhang and B. Liu, Atomically dispersed Ni(I) as the active site for electrochemical CO2 reduction, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- J. Li, P. Pršlja, T. Shinagawa, A. Fernández, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou, R. Garía-Muelas, N. López, J. Pérez-Ramírez and F. Jaouen, Volcano trend in electrocatalytic CO2 reduction activity over atomically dispersed metal sites on nitrogen-doped carbon, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS.
- J. Wu, T. Sharifi, Y. Gao, T. Zhang and P. Ajayan, Emerging carbon-based heterogeneous catalysts for electrochemical reduction of carbon dioxide into value-added chemicals, Adv. Mater., 2019, 31, 1804257 CrossRef.
- Y. Tang, X. Dai, Z. Yang, Z. Liu, L. Pan, D. Ma and Z. Lu, Tuning the catalytic property of non-noble metallic impurities in graphene, Carbon, 2014, 71, 139–149 CrossRef CAS.
- E. Zhang, T. Wang, K. Yu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, Bismuth single atoms resulting from transformation of metal-organic frameworks and their use as electrocatalysts for CO2 reduction, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- F. Yang, A. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Bismuthene for highly efficient carbon dioxide electroreduction reaction, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed.
- M. Haruta, When gold is not noble: catalysis by nanoparticles, Chem. Record, 2003, 3, 75–87 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- N. Drebov and R. Ahlrichs, Small clusters of aluminium and tin: highly correlated calculations and validation of density functional procedures, J. Chem. Phys., 2011, 134, 124308 CrossRef.
- N. Drebov, F. Weigend and R. Ahlrichs, Structures and properties of neutral gallium clusters: a theoretical investigation, J. Chem. Phys., 2011, 135, 044314 CrossRef.
- X. Gong and E. Tosatti, Structure of small gallium clusters, Phys. Lett. A, 1992, 166, 369–372 CrossRef CAS.
- B. Assadollahzadeh, S. Schäfer and P. Schwerdtfeger, Electronic properties for small tin clusters Snn (n < 20) from density functional theory and convergence toward the solid state, J. Comput. Chem., 2009, 31, 929–937 Search PubMed.
- B. Assadollahzadeh and P. Schwerdtfeger, A systematic search for minimum structures of small gold clusters Aun (n = 2–20) and their electronic properties, J. Chem. Phys., 2009, 131, 064306 CrossRef PubMed.
- H. Yuan, H. Chen, A. Kuang, Y. Miao and Z. Xiong, Density-functional study of small neutral and cationic bismuth clusters Bin and Bin+ (n = 2–24), J. Chem. Phys., 2008, 128, 094305 CrossRef CAS PubMed.
- P. Jain, A DFT-based study of the low energy electronic structures and properties of small gold clusters, Struct. Chem., 2005, 16, 421–426 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Turbomole, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- S. Alvarez, A cartography of the van der Waals territories, Dalton Trans., 2013, 42, 8617–8636 RSC.
- S. S. Batsanov, Calculation of van der Waals radii of atoms from bond distances, J. Mol. Struct., 1999, 468, 151–159 CrossRef CAS.
- S. S. Batsanov, van der Waals radii of elements, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- M. Mantina, A. Chamberlin, R. Valero, C. Cramer and D. Truhlar, Consistent van der Waals radii for the whole main group, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- A. Bondi, van der Waals volume and radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- P. Power, π-bonding and the lone pair effect in multiple bonds between heavier group elements, Chem. Rev., 1999, 99, 3463–3503 CrossRef CAS.
- W. Kutzelnigg, Chemical bonding in higher main group elements, Angew. Chem., 1984, 23, 272–295 CrossRef.
- K. Chan, J. Neaton and M. Cohen, First-principles study of metal adatom adsorption on graphene, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235430 CrossRef.
- X. Liu, C. Wang, Y. Yao, W. Lu, M. Hupalo, M. Tringides and K. Ho, Bonding and charge transfer by metal adatom adsorption on graphene, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 235411 CrossRef.
- T. Pawluk, Y. Hirata and L. Wang, Studies of iridium nanoparticles using density functional theory calculations, J. Phys. Chem. B, 2005, 109, 20817–20823 CrossRef CAS PubMed.
- N. Gaston and A. Parker, On the bonding of Ga2, structures of Gan clusters and the relation to the bulk structure of gallium, Chem. Phys. Lett., 2011, 501, 375–378 CrossRef CAS.
- R. Ahlrichs and S. Elliott, Clusters of aluminium, a density functional theory, Phys. Chem. Chem. Phys., 1998, 1, 13–21 Search PubMed.
- L. Gao, P. Li, H. Lu, S. Li and Z. Guo, Size- and charge-dependent geometric and electronic structures of Bin (Bin−) clusters (n = 2–13) by First principles simulations, J. Chem. Phys., 2008, 128, 194304 CrossRef PubMed.
- C. Effantin, A. Topouzkhanian, J. Figuet, J. d'Incan, R. F. Barrow and J. Verges, Electronic states of Bi2 studied by laser-excited uorescence, J. Phys. B: Atom. Mol. Phys., 1982, 15, 3829 CrossRef CAS.
- H. Matsushima, S.-W. Lin, S. Morin and O. M. Magnussen, In situ video-STM studies of the mechanisms and dynamics of electrochemical bismuth nanostructure formation on Au, Faraday Discuss., 2016, 193, 171–185 RSC.
- M. Musgrave, On the relation between grey and white tin (α-Sn and β-Sn), Proc. R. Soc. A, 1963, 272, 503–528 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp05008g |
| This journal is © the Owner Societies 2022 |