
To photodeprotect or not: effect of the oxidation state of the sulfur atom of thiochromone derivatives†
Xuyang
Li‡
b,
Yan
Guo‡
a,
Tongyu
Xu
 b,
Jialin
Wang
b,
Mingdong
Dai
b,
Kiyomi
Kakiuchi
c,
Tsumoru
Morimoto
c and
Jiani
Ma
*a
b,
Jialin
Wang
b,
Mingdong
Dai
b,
Kiyomi
Kakiuchi
c,
Tsumoru
Morimoto
c and
Jiani
Ma
*a
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710119, China. E-mail: majiani@snnu.edu.cn
bKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi’an 710172, P. R. China
cDivision of Materials Science, Graduate School of Science and Technology, Nara Institute of Science and Technology (NAIST), Takayama, Ikoma, Nara, 630-0192, Japan
First published on 22nd September 2022
Abstract
Time-resolved spectroscopic experiments, assisted with DFT calculations, were employed to study the photochemical reaction mechanism of (4-oxo-3-phenyl-4H-thiochromen-2-yl) methoxycarbonyl-caged ethanol (TC) and (1,1-dioxido-4-oxo-3-phenyl-4H-thiochromen-2-yl) methoxy carbonyl-caged ethanol (TS-PPG) in different solvents. TC went through an intersystem crossing to form the triplet state with π–π* character in acetonitrile (MeCN) and protic solvents. While the n–π* triplet state was generated for TS-PPG in MeCN, which further underwent Paternò–Büchi reaction to give a biradical intermediate. Then, the C–O bond was cleaved, followed by deprotonation. Besides the similar deprotection route in MeCN, another reaction pathway existed in protic solvents, where the C–O bond heterolysis took place via the singlet excited state. The unambiguous mechanism would not only guide the efficient application of TS-PPG, but also help develop more excellent PPGs based on the thiochromone framework.
Photolabile protecting groups (PPGs) have found increasing applications due to the advantage of non-invasive spatiotemporal photodeprotection reaction through irradiation under mild conditions.1–3 Various PPGs, such as o-nitrobenzyl,4 phenacyl,5 and coumarin-4-yl-methyl,6 have been developed to be applied in organic synthesis, biochemistry, and diverse areas.7–10 Still, some shortcomings were found in these PPG applications.11–13
The chromone derivatives draw increasing attention due to their special photochemical properties arising from the expanding conjugated system over the bicyclic skeleton.14,15 Thiochromone and the oxidized counterpart thiochromone S,S-dioxide were tried as new PPG platforms to protect ethanol (such as TC and TS-PPG). After light irradiation, photodeprotection is only achieved for TS-PPG (Scheme 1).16
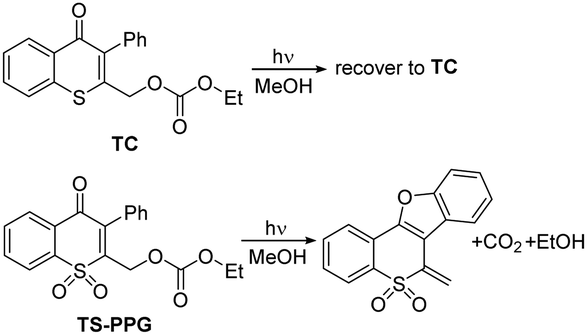 | ||
| Scheme 1 The photochemical reactions of TC and TS-PPG under irradiation. | ||
Thiochromone S,S-dioxide derivatives were selected as a new PPG platform to protect carboxylic acids, phosphates and ketones.17–21 A possible photodeprotection reaction mechanism was proposed (Scheme S1, ESI†).22 Upon excitation, TS-PPG was excited to the singlet state, followed by intersystem crossing (ISC) to the triplet state. Then, a Paternò–Büchi cyclization reaction occurred to generate an oxetane intermediate. Thereafter, the protected ethanol was released. However, due to the lack of spectra information of the transient species, some questions were still unclear. Firstly, how does the oxidation state of the sulfur atom influence the photochemical properties of thiochromone derivatives? Secondly, what is the detailed photodeprotection route for TS-PPG and the influence of the solvent effect? Previous studies unravelled that stepwise and concerted photodeprotection pathways could be possible depending on the solvent property, as well as the distance between the caged group (X) and the labile proton to be removed. For instance, in aprotic solvent MeCN, benzoin PPG, with a shorter distance between the X and the proton, underwent photodeprotection by a concerted HX elimination pathway.23 While 1,4-benzoquinone PPG, with a relatively far distance between X and the proton, underwent a stepwise photodeprotection.24 In protic aqueous solution, anthraquinone PPGs complete the deprotection by transferring the proton to X with the assistance of solvent molecules, even with the long distance between proton and X.25,26
Here, TC and TS-PPG, which recover to reactant and release the ethanol under irradiation, respectively, were selected as model compounds to investigate the photochemical reaction mechanisms of thiochromone derivatives. Femtosecond transient absorption (fs-TA), nanosecond transient absorption (ns-TA) and nanosecond transient resonance Raman (ns-TR2) spectroscopies were performed to detect the electronic and vibrational spectra of the transient and intermediates produced after photoexcitation. DFT computations were conducted to assist the identification of possible intermediates involved in the photochemical reactions and help to understand the proposed reaction mechanism.
The fs-TA experiments of TC were performed in MeCN (Fig. 1a–c), MeOH, and MeCN–H2O (Fig. S1 and S2, ESI†), respectively, and similar results were obtained. The spectra in MeCN were selected to illustrate the photochemical reaction mechanism of TC, and were characterized with three periods. Firstly, a negative signal at 345 nm was seen immediately and two absorption bands appeared at 365 and 405 nm upon photoexcitation. The 365 nm band increased accompanying the rise of the 500–700 nm broad band, while the 405 nm signal disappeared gradually. Subsequently, the 500–700 nm signal decayed with the growth of a new band at 445 nm (2.57–11.7 ps). Finally, the absorption bands at 365 and 445 nm decayed simultaneously.
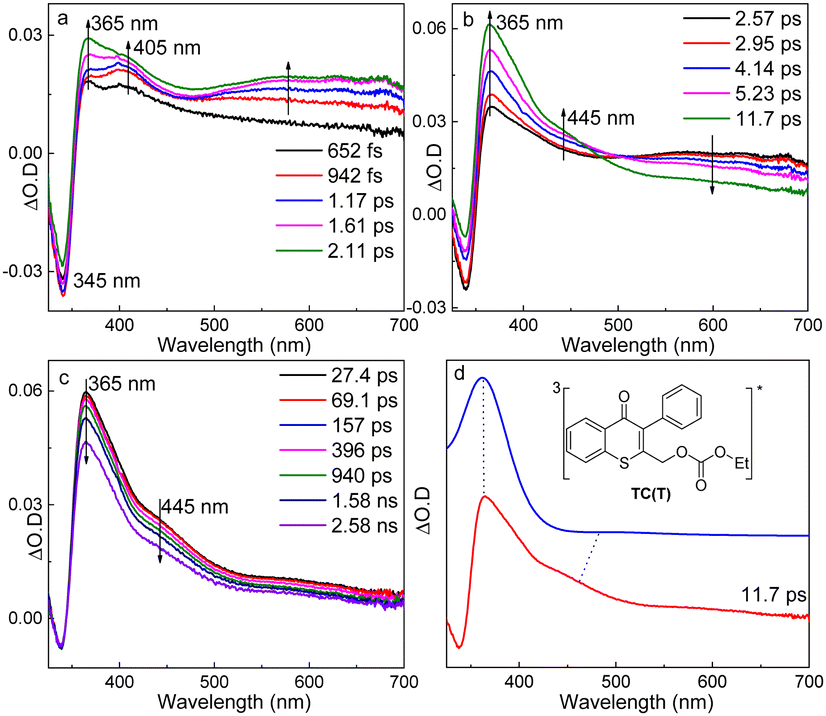 | ||
| Fig. 1 (a–c) The fs-TA results of TC in MeCN (λex = 266 nm); (d) comparison of the fs-TA spectrum of TC in MeCN recorded at 11.7 ps with the calculated UV-vis spectra of TC(T). (TD-ωB97X-D/6-311G** (MeCN) with a scale factor of 1.00 and a half-width of 2000 cm−1). | ||
The negative signal at 345 nm was due to the ground state bleaching of TC (see the UV-vis spectrum in Fig. S3, ESI†). The pump laser in fs-TA excited TC to its higher excited singlet state (405 nm, denoted as TC(Sn) hereafter), and the change in Fig. 1a was attributed to the internal conversion to its lowest excited singlet state (365 nm, denoted as TC(S1) hereafter). Then, TC(S1) converted into another species showing up at 445 nm (Fig. 1b), and a rise time of 4.1 ps was determined from the fitting at 450 nm (Fig. S4, ESI†), which was similar to the ISC rate constant of aromatic ketones.27,28 Thus, the 445 nm signal could be ascribed to the triplet TC (denoted as TC(T)). The simulated electronic spectrum of TC(T) agreed well with the fs-TA spectrum of TC recorded at 11.7 ps in MeCN, which supported the above assignment (Fig. 1d). The information of key intermediates in photochemical reaction of TC was summarized in Table S1 (ESI†).
The photochemical reaction experiments of TS-PPG were conducted in MeCN, MeOH and MeCN–H2O solutions (Fig. S5 and S6, ESI†), respectively. With the evolution of irradiation time, the absorbance at 350 nm increased gradually, which was the typical signal of the photodeprotection byproduct.16 The 350 nm absorbance was more intense and the photolysis times were shorter in MeOH and MeCN–H2O compared to that in MeCN, suggesting the more efficient photodeprotection of TS-PPG in protic solvent systems.
fs-TA of TS-PPG was tested in MeCN (Fig. 2a–c). There were three consecutive spectral changes during the timescale of fs-TA measurement. After irradiation, two bands at 360 and 575 nm rose gradually within 983 fs. Then, the features at 360 and 575 nm decayed with the generation of a new profile at 405 nm. Finally, the profile at 405 nm decayed and blue-shifted to 400 nm.
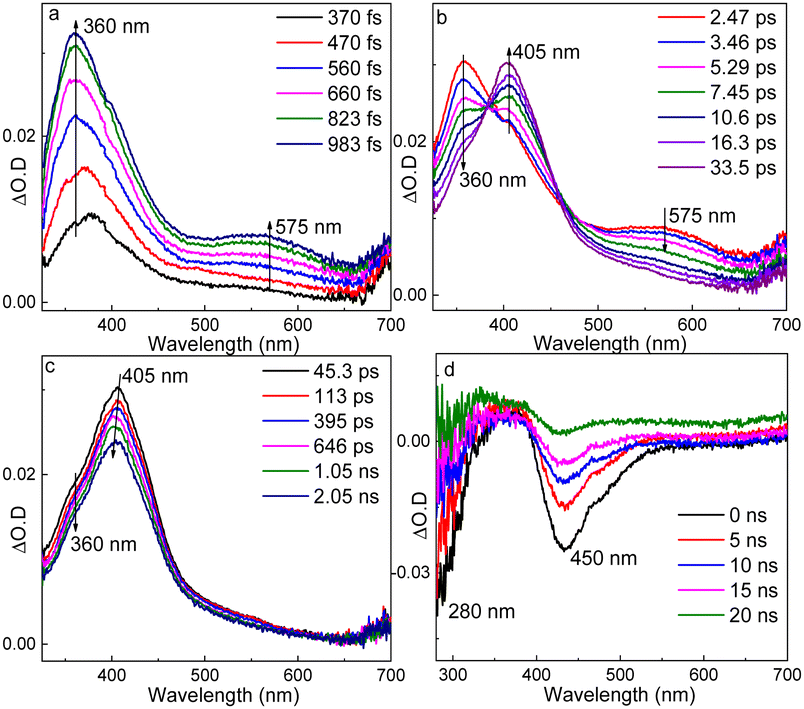 | ||
| Fig. 2 (a–c) fs-TA and (d) ns-TA spectra of TS-PPG in MeCN (λex = 266 nm). | ||
For the ns-TA spectra of TS-PPG in MeCN (Fig. 2d), the negative signals at 280 and 450 nm were attributed to the ground state bleaching of the substrate (see the UV-vis spectrum in Fig. S5, ESI†) and stimulated emission signal of the photodeprotection product, respectively.19 This result indicated that photodeprotection of TS-PPG completed within 10 ns considering the instrument response time of the ns-TA setup. Besides, ns-TR2 was performed for TS-PPG in MeOH (Fig. S7, ESI†). Dominant Raman peaks appeared at 1271, 1638, 1661 and 1710 cm−1, and resembled the calculated Raman spectrum of the photodeprotection product (Fig. S7, ESI†), which further proves the accomplishment of photodeprotection within 10 ns.
The increase of the 360 and 575 nm signals may be attributed to the ISC process from the excited singlet state of TS-PPG (denoted as TS-PPG(S1) hereafter) to the triplet state (denoted as TS-PPG(T) hereafter) due to the similar profile with the reported triplet state spectra of isoflavone derivatives, having a similar molecular chemical skeleton.29 Besides, the calculated UV-vis spectrum of TS-PPG(T) was similar to the fs-TA spectrum of TS-PPG obtained at 983 fs in MeCN (Fig. S8, ESI†). The ISC rate of TS-PPG (<1 ps) was obviously faster than that of TC (4.1 ps). According to the previous reports, when the sulfur atom was oxidized to sulfone, the singlet–triplet energy difference decreases and the spin–orbit coupling value increases, which resulted in the augmentation of the ISC rate.30
The growth of the 405 nm signal could be ascribed to the generation of the reactive intermediate relating with the photodeprotection process. We tried to optimize the oxetane intermediate by DFT computation, as proposed in Scheme S1 (ESI†), while the minimum point was not located. Based on the reported work on Paternò–Büchi reaction, a biradical intermediate (IM1) was possibly produced from TS-PPG(T).31 IM1 may be depopulated via two different reaction routes, namely a concerted elimination of the protective group and the proton to be leaving, or a stepwise manner by cleavage of the protective group generating the cation intermediate, followed by deprotonation. As discussed earlier, due to the aprotic solvent and the long distance between the protective group and the proton, IM1 was inclined to undergo a stepwise deprotection reaction in MeCN. That is, IM1 underwent C–O bond heterolysis to produce the cation species (IM2), and then the deprotonation reaction took place. TD-DFT calculations were performed to simulate the spectra of IM1 and IM2, and the results were in good agreement with the fs-TA recorded at 7.45 ps and 2.05 ns of TS-PPG, respectively (Fig. S9, ESI†).
The photodeprotection reaction route of TS-PPG in MeCN was simulated (Fig. S10, ESI†). TS-PPG(T) overcame a 1.9 kcal mol−1 reaction energy barrier to generate the triplet IM1, which converted to its open shell singlet state. Then open shell singlet biradical underwent C–O bond cleavage to produce IM2 with an 8.3 kcal mol−1 barrier. Finally, the photodeprotection product was yielded.
fs-TA data of TS-PPG in MeCN were globally analyzed and give the decay-associated difference spectra (DADS) (Fig. S11, ESI†). The 8 ps DADS showed the positive band at 365 and 575 nm with the negative band at 405 nm, which indicated that the TS-PPG(T) converted into IM1. The DADS of the 799 ps component displayed positive features at 365 and 405 nm corresponding to the process of the C–O bond cleavage reaction of IM1. The infinite (out of the instrument detection) DADS exhibited the decay of IM2. The absorption and kinetics of key intermediates in the photochemical reaction of TS-PPG are given in Table S2 (ESI†). With the combination of experimental and calculated results, the photodeprotection reaction mechanism of TS-PPG in MeCN is proposed in Scheme 2.
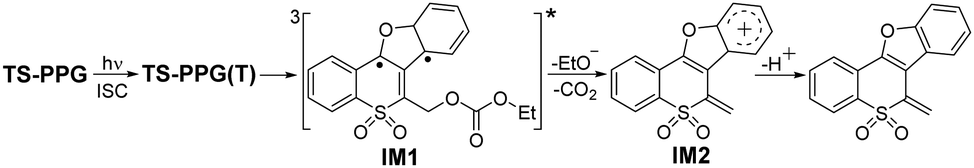 | ||
| Scheme 2 Proposed photochemical reaction mechanism of TS-PPG in MeCN. | ||
To explain the absent detection of photodeprotection for TC, the spin distributions of TC(T) and TS-PPG(T) were calculated (Fig. S12, ESI†). It is suggested that the electronic state of TS-PPG(T) exhibited n–π* character with the spin distribution mainly locating on the carbonyl group, while the electronic state of TC(T) was the π–π* state. When the π-electron density increased, the triplet state became more favorable to the π–π* character. Therefore, TC(T) turned into π–π* electronic character, because the lone pair electrons on the sulfur atom could conjugate with the bicyclic skeleton.32 Based on the previous study, the excited carbonyl compounds with n–π* character were required to undergo Paternò–Büchi with an alkene. And the low Paternò–Büchi efficiency of TC caused the failure of photodeprotection.33 The cyclization reaction of TC(T) was also simulated, and the remarkably higher barrier (15.8 kcal mol−1) supported the above suggestion (Fig. S13, ESI†).
The fs-TA of TS-PPG in MeOH (Fig. S14, ESI†) was similar to that in MeCN-H2O (Fig. S15, ESI†). Along with the detection of TS-PPG(T), a new band at 510 nm was seen within 1 ps (Fig. S15a, ESI†) and blue shifted to 505 nm rapidly (Fig. S15b, ESI†). Except for this signal, the position and evolution of other bands in MeCN–H2O resembled that in MeCN. Combining with the photochemical reaction results, the profile absorbed at 510 nm may be related with the higher photodeprotection efficiency of TS-PPG in protic solvents.
With the increase of the solvent polarity, the efficiency of heterolytic cleavage reactions enhanced with the lower reaction barrier.34 For instance, the process of reaction changes from the inefficient, solvent-independent homolysis of haloanilines to the efficient heterolysis observed in polar protic solvents.35 And the benzoin type PPGs exhibit C–O bond heterolysis directly from its excited state in aqueous MeCN solution, which can compete with the cyclization reaction.23 It is therefore reasonable to propose that along with the occurrence of the ISC process, TS-PPG(S1) might face competition from C–O bond heterolysis in MeOH and MeCN–H2O.
The C–O bond distances for TS-PPG(S0) and TS-PPG(S1) in MeCN, MeOH, and H2O are listed in Fig. S16 and S17 (ESI†). The C–O bond of TS-PPG(S0) had no obvious change in different solvents, except for a slight elongation when a H2O molecule was added in the system (Fig. S16, ESI†), while for the TS-PPG(S1), the C–O bond distances were elongated from 1.437 (MeCN) to 1.442 (MeOH) to 1.443 Å (H2O), and when a H2O molecule was added in the system, the C–O bond was further elongated (1.449 Å) (Fig. S17, ESI†). That is, the C–O bond cleavage of TS-PPG(S1) would be favored in protic solvent systems. The same tendency was observed for the signal of 510 nm in MeOH and MeCN–H2O, suggesting that the 510 nm band could be attributed to the cation intermediate (denoted as IM3(S1)) formed by C–O bond heterolysis from TS-PPG(S1). Subsequently, IM3(S1) converted to its ground state (denoted as IM3) through internal conversion. The UV-vis spectra of IM3(S1) and IM3 were compared with fs-TA of TS-PPG recorded at 979 fs and 3.61 ps in MeCN–H2O, respectively (Fig. S18, ESI†). The reasonable resemblance further verified the above assignments. On the other hand, as suggested by DFT calculations, for the cation intermediate produced by heterolytic cleavage of TC(S1), the conjugation degree between the carbon cation and the side-chain benzene ring is lower than that in IM3 generated from TS-PPG(S1) (Fig. S19, ESI†). This suggested that the heterolytic cleavage was not preferred for TC(S1) as the instability of the cation intermediate, in line with the absence of the cation signal in the transient spectra of TC (Fig. 1 and Fig. S1 and S2, ESI†).
fs-TA data of TS-PPG in MeCN–H2O were globally analyzed (Fig. S20, ESI†). The 7 ps DADS showed positive bands at 365, 510 and 575 nm with the negative ones at 405 and 505 nm, which indicated that TS-PPG(T) converted into IM1 and the transformation from IM3(S1) to IM3, while the rate constant of the later process was not obtained due to the influence of the former conversion. The 451 ps DADS component corresponded to the C–O bond cleavage reaction of IM1, and the shorter lifetime of the conversion, compared with the situation in MeCN (799 ps), was due to the solvent effect. The infinite DADS was the decay of IM2 and IM3.
The photodeprotection mechanism of TS-PPG in protic solvents was concluded (Scheme 3). Upon excitation, the generated TS-PPG(S1) underwent competing reactions. One was the C–O heterolysis reaction to produce excited state ion pair intermediates, which decayed into the ground state and transformed into IM2 via cyclization reaction. The other is that TS-PPG(S1) transformed into TS-PPG(T), which produced IM1 via a Paternò–Büchi reaction, and then IM1 underwent C−O bond cleavage to obtain IM2, followed by deprotonation.
 | ||
| Scheme 3 Proposed photodeprotection mechanism of TS-PPG. | ||
TC could not undergo photodeprotection in MeCN due to the less efficient Paternò–Büchi reaction of the π–π* TC(T), while TS-PPG(T) exhibited n–π* character. In polar protic solvents, the occurrence of the photodeprotection for TS-PPG via a heterolysis pathway is due to the stabilized cation species for the more conjugated degree between the carbon cation and the side-chain benzene ring, compared to the situation for TC. The different photodeprotection mechanisms of TS-PPG in MeCN and protic solvents would guide its efficient applications, and the development of novel PPGs.
This research was sponsored by grants from the National Natural Science Foundation of China (21973075), the Shaanxi Science Fund for Distinguished Young Scholars (2021JC-38), the Shaanxi Science and Technology New Star Project (2020KJXX-010), and the China Postdoctoral Science Foundation (2018T111089) to J.M.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Vidal, Angew. Chem., Int. Ed., 2019, 58, 13628 CrossRef PubMed.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- S. B. Salunke and J. A. Malla, Angew. Chem., Int. Ed., 2019, 58, 5354–5358 CrossRef CAS PubMed.
- F. Salahi, V. Purohit, G. Ferraudi, C. Stauffacher, O. Wiest and P. Helquist, Org. Lett., 2018, 20, 2547–2550 CrossRef CAS PubMed.
- Q. Lin, L. Yang, Z. Wang, Y. Hua, D. Zhang, B. Bao, C. Bao, X. Gong and L. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3722–3726 CrossRef CAS PubMed.
- X. J. Tang, Y. Wu, R. Zhao, X. Kou, Z. Dong, W. Zhou, Z. Zhang, W. Tann and X. Fang, Angew. Chem., Int. Ed., 2020, 59, 18386–18389 CrossRef CAS PubMed.
- M. Lee, R. Rizzo, F. Surman and M. Zenobi-Wong, Chem. Rev., 2020, 120, 10950–11027 CrossRef CAS PubMed.
- K. Morihiro, T. Ishinabe, M. Takatsu, H. Osumi, T. Osawa and A. Okamoto, J. Am. Chem. Soc., 2021, 143, 3340–3347 CrossRef CAS.
- I. Elamri, C. Abdellaoui, J. K. Bains, K. F. Hohmann, S. L. Gande, E. Stirnal, J. Wachtveitl and H. Schwalbe, J. Am. Chem. Soc., 2021, 143, 10596–10603 CrossRef CAS PubMed.
- T. Schmierer, S. Laimgruber, K. Haiser, K. Kiewisch, J. Neugebauer and P. Gilch, Phys. Chem. Chem. Phys., 2010, 12, 15653–15664 RSC.
- L. Zayat, C. Calero, P. Alborés, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 125, 882–883 CrossRef CAS PubMed.
- L. A. P. Antony, T. Slanina, P. Šebej, T. Šolomek and P. Klán, Org. Lett., 2013, 15, 4552–4555 CrossRef CAS.
- K. R. Huffman, M. Loy and E. F. Ullman, J. Am. Chem. Soc., 1965, 87, 5417–5423 CrossRef CAS.
- V. Rossollin, V. Lokshin, A. Samat and R. Guglielmetti, Tetrahedron, 2003, 59, 7725–7731 CrossRef CAS.
- S. Kitani, K. Sugawara, K. Tsutsumi, T. Morimoto and K. Kakiuchi, Chem. Commun., 2008, 2103–2105 RSC.
- Y. Zhang, H. Tanimoto, Y. Nishiyama, T. Morimoto and K. Kakiuchi, Synlett, 2012, 367–370 Search PubMed.
- R. Sugiura, R. Kozaki, S. Kitani, Y. Gosho, H. Tanimoto, Y. Nishiyama and T. Morimoto, Tetrahedron, 2013, 69, 3984–3990 CrossRef CAS.
- S. Hikage, Y. Sasaki, T. Hisai, H. Tanimoto, T. Morimoto, Y. Nishiyama and K. Kakiuchi, J. Photochem. Photobiol., A, 2016, 331, 175–183 CrossRef CAS.
- S. Hikage, Y. Nishiyama, Y. Sasaki, H. Tanimoto, T. Morimoto and K. Kakiuchi, ACS Omega, 2017, 2, 2300–2307 CrossRef CAS PubMed.
- C. Ma, Y. Zhang, H. Zhang, J. Li, Y. Nishiyama, H. Tanimoto, T. Morimoto and K. Kakiuchi, Synlett, 2017, 560–564 CAS.
- Y. Zhang, H. Zhang, C. Ma, J. Li, Y. Nishiyama, H. Tanimoto, T. Morimoto and K. Kakiuchi, Tetrahedron Lett., 2016, 57, 5179–5184 CrossRef CAS.
- C. Ma, Y. Du, W. M. Kwok and D. L. Phillips, Chem. – Eur. J., 2007, 13, 2290–2305 CrossRef CAS PubMed.
- Y. Chen and M. G. Steinmetz, J. Org. Chem., 2006, 71, 6053–6060 CrossRef CAS PubMed.
- Y. Guo, Q. Song, J. Wang, J. Ma, X. Zhang and D. L. Phillips, J. Org. Chem., 2018, 83, 13454–13462 CrossRef CAS PubMed.
- Y. Guo, Q. Song, T. Xu, J. Ma and D. L. Phillips, Phys. Chem. Chem. Phys., 2019, 21, 14598–14604 RSC.
- D. R. Kearns and W. A. Case, J. Am. Chem. Soc., 1966, 88, 5087–5097 CrossRef CAS.
- P. J. Wagner, A. E. Kemppainen and H. N. Schott, J. Am. Chem. Soc., 1973, 95, 5604–5614 CrossRef CAS.
- Y. X. Tian, R. M. Han, L. M. Fu, J. P. Zhang and L. H. Skibsted, J. Phys. Chem. B, 2008, 112, 2273–2280 CrossRef CAS.
- E. Varathan and A. Patnaik, J. Phys. Chem. A, 2019, 123, 8755–8765 CrossRef CAS PubMed.
- M. D’Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 CrossRef PubMed.
- T. Shimokage, T. Ikoma, K. Akiyama, S. Tero-Kubota, M. Yamaji and H. Shizuka, J. Phys. Chem. A, 1997, 101, 9253–9256 CrossRef CAS.
- D. R. Arnold, R. L. Hinman and A. H. Glick, Tetrahedron Lett., 1964, 5, 1425–1430 CrossRef.
- J. C. Walton, J. Org. Chem., 2019, 84, 12606–12616 CrossRef CAS PubMed.
- M. Freccero, M. Fagnoni and A. Albini, J. Am. Chem. Soc., 2003, 125, 13182–13190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data. See DOI: https://doi.org/10.1039/d2cc04650d |
| ‡ Xuyang Li and Yan Guo contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |