
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLast-step 18F-fluorination of supported 2-(aryl-di-tert-butylsilyl)-N-methyl-imidazole conjugates for applications in positron emission tomography†
Marine
Steffann
ab,
Marion
Tisseraud
a,
Guillaume
Bluet
b,
Sebastien
Roy
b,
Catherine
Aubert
b,
Eric
Fouquet
a and
Philippe
Hermange
 *a
*a
aUniv. Bordeaux, Institut des Sciences Moléculaires, UMR-CNRS 5255, 351 Cours de la Libération, 33405, Talence Cedex, France. E-mail: philippe.hermange@u-bordeaux.fr
bSanofi, Integrated Drug Discovery (IDD) Isotope Chemistry (IC), 13 Quai Jules Guesde, 94400, Vitry-sur-Seine, France
First published on 27th July 2022
Abstract
Aiming for potential applications in positron emission tomography, fully automated productions of 18F-labelled bioconjugates were achieved using heterogenous precursors obtained by anchoring imidazole-di-tert-butyl-arylsilanes to a polystyrene resin. The reactions were performed using either “batch” or “flow” procedures, avoiding both the time-consuming azeotropic drying and HPLC purifications usually required.
Positron emission tomography (PET) is a powerful imaging technique based on the injection of a radiotracer containing a β+ emitting element.1 Among the suitable isotopes, fluorine-18 exhibits convenient properties such as a 97% decay by β+ emission (635 keV positrons), a half-life of 109.8 min and the ability to form covalent bonds. For example, [18F]-2-deoxy-2-fluoro-D-glucose ([18F]FDG) has been employed routinely for decades in cancer diagnosis by PET. Looking for efficient and versatile syntheses of biologically specific [18F]tracers, numerous synthetic methods and strategies were developed,2 such as late-stage 18F-labelling,3 “click” conjugations4 or last-step fluorinations of specific prosthetic groups.5,6 However, their transfer to clinical applications remains challenging in most cases.7 Indeed, in addition to the constraints inherent to radiosyntheses, i.e. short time of synthesis (production time inferior to 3 half-lives), small scale reactions (typically 10–100 nmol) and the protection of the operator from radiation (reactions performed in closed lead-shielded fumehoods), the procedure should be fully automatable to ensure its reproducibility and operability by technicians.1a Thus, kit-like precursors that can directly deliver the isolated radio-labelled product in one step without cumbersome/time-consuming azeotropic-drying or HPLC-purification steps are greatly preferred. In particular, using precursors grafted on a solid support whose selective cleavage is triggered by the radio-labelling reaction can greatly improve the practicality of the overall process.8 Indeed, manipulation and packaging of the precursor become more convenient, and it theoretically allows a fast and easy separation of the large excess of the starting material from the radiolabelled tracer using a simple filtration method. When considering the syntheses of PET tracers, simultaneous radio-labelling and cleavage were achieved via starting materials anchored on solid supports by either electrostatic interactions, organometallic bonding or covalent linking.9 For instance, the latter one was applied to the synthesis of a protected [18F]FDG using a resin-linked sulfonate leaving group, but further deprotection was still required to obtain the final radiolabelled product (Scheme 1A(a)).9f On the other hand, the last-step 18F-labelling of supported unprotected substrates still remains extremely challenging. Indeed, heterogeneous reactions lead usually to a reactivity decrease when compared to their homogeneous versions, requiring harsher labelling conditions often non-compatible with sensitive functional groups in biomolecule-based substrates.
 | ||
| Scheme 1 Examples of simultaneous 18F-fluorination and cleavage from covalently linked supported precursors and the strategy envisaged in this work. | ||
Whereas simple [18F]fluoroarenes could be obtained in 3–14% radiochemical yields (RCY) from supported triazene9a or iodonium9i precursors (Scheme 1A(b and c)), a recent work of Kuhnast et al. demonstrated that pyridine–ammonium grafted onto a resin were radiolabelled at room temperature with RCY of 3–8% after filtration on C18 or Al2O3 cartridges (Scheme 1A(d)).9j Interestingly, when linked to a peptide, it also reacted with [18F]F− to give in 30 min the desired bioconjugate with a low RCY of 1%, but with an excellent radiochemical purity (RCP) without HPLC purification (>95%).
In this context, we developed recently a new 2-(aryl-di-tert-butylsilyl)-N-methyl-imidazole tag (Imidazole-Silicon Fluoride Acceptors = ImidSiFA)10 that could be fluorinated efficiently by aqueous [18F]F−.11 Thus, anchoring the imidazole leaving group on a solid support was envisaged as the next step for the full automation of the radiolabelling process. In particular, the possibility to achieve the conjugation after grafting on the solid support was explored using copper-catalysed Huisgen cycloaddition (CuAAC).12 Indeed, it would enhance the versatility of this method by allowing the preparation of various conjugated precursors from a unique starting resin (Scheme 1B).
Firstly, various pathways were screened to introduce a linking moiety on the imidazole leaving group of the ImidSiFA tag, but post-functionalization of the previously described 2-(4-(methoxymethoxy)phenyl-di-tert-butylsilyl)-N-methyl-imidazole 110 proved to be the most efficient and reliable one (Scheme 2). Indeed, iodination,13 Sonogashira coupling hydrogenation led to product 5, exhibiting unprotected alcohol and phenol moieties (45% yield from 1). Interestingly, 5 was crystallised and analysed by X-ray diffraction,‡ which confirmed the highly hindered structure of this silane (Scheme 2). Finally, the phenol was alkylated with a PEG chain functionalized by an alkyne moiety, and product 6 was consecutively anchored by esterification onto a polystyrene resin with benzoyl chloride residues to give the supported precursor PS-7. The efficiency of the grafting was confirmed by fluorination of a portion of the resin beads in model conditions with aqueous [19F]fluoride (0.1 M, 1.3 equiv., in THF at 70 °C for 2 h), which yielded quantitatively the corresponding fluorosilane 8 based on the theoretical resin loading of 6. This highly encouraging result led us to explore the conjugation of the PS-7 resin by CuAAC with azides present on biomolecule derivatives (Scheme 3). Model molecules containing thymidine, glucose, biotine, and estradiol cores (i.e.9a, 9b, 9c and 9d, respectively) were selected and engaged in the heterogeneous “click” reaction. After optimisation, a Cu(II) complex with tris(benzyltriazolylmethyl)amine) as ligand (TBTA)14 was identified as the best catalyst with either classical heating or micro-wave activation depending on the substrate (Scheme 3).
 | ||
| Scheme 2 Synthesis of the resin supported ImidSiFA bearing a pendant alkyne moiety and 19F-labelling in model conditions. aMercury drawing of the crystalline structure of 5 obtained by X-ray diffraction. | ||
 | ||
| Scheme 3 Conjugation of the resin supported ImidSiFA PS-7 and 19F-labelling in model conditions. aYields over two steps. For the exact experimental conditions, see the ESI.† | ||
Then, the resulting beads were simply washed and dried, and a fraction of each was subjected to 19F-fluorination using the model conditions previously described (aqueous HF at 0.1 M in THF for 2 h at 70 °C). In this case, the desired fluorinated bioconjugates were obtained in 54% to 100% yields over the conjugation/fluorination steps after simple filtration.§ Moreover, no fluorinated alkyne 8 was detected by 1H NMR and HPLC analyses in the isolated products, which confirmed full conversions for the CuAAC reactions after optimisation.
With these precursors in hands, 18F-fluorination was then examined using an AllinOne automated PET tracer synthesizer (see the ESI† for details). The [18F]fluoride obtained from the cyclotron (in solution in H2[18O]O) was firstly purified and concentrated by trapping onto a quaternary ammonium cartridge (QMA) and further eluted by an aqueous solution of sodium chloride (Step (1), Table 1). Then, in a first set-up (“batch procedure”, Table 1), the resulting [18F]F− saline solution was transferred to the reaction vial previously loaded with a supported precursor PS-10, the organic solvent and a small quantity of acetic acid to activate the imidazole leaving group (Step (2), Table 1). After heating the suspension, the corresponding labelled compound [18F]11 was purified by adding water in the reaction vial (Step (3), Table 1) and filtering the resulting mixture through a Sep-Pak C18 cartridge in order to remove the unreacted [18F]fluoride (Step (4), Table 1). Finally, the radiotracer was isolated by elution with acetonitrile in a collecting vial (Step (5), Table 1). The activity was measured to calculate the RCY, and the radiochemical purity (RCP) was determined by radio HPLC. It should be noted that the optimisation of the conditions was performed with a batch of PS-10a with a conversion of ≈80% for the CuAAC step.¶ In a first experiment (entry 1, Table 1), the conditions developed for the homogenous 18F-labelling of ImidSiFAs were employed (i.e. THF, 10 equiv. of AcOH and heating at 100 °C for 15 min).10
|
|
|||
|---|---|---|---|
| Entry (number of runs) | Substrate (mass) | Conditionsa | RCY of [18F]11b (%) (RCP in %)c |
| a Using the batch procedure otherwise indicated. b Calculated as the activity in the collected vial (corrected by the decay) divided by the activity of the reaction vial before heating, and multiplied by the RCP, mean ± standard deviation of the n experiments, see the ESI for details. c Determined by radio-HPLC. d Experiments performed from a batch of PS-10-a with only 80% conversion for the CuAAC step. e Using the “flow procedure”. | |||
| 1d (n = 1) | PS-10a (5 mg) | THF (1.7 mL), AcOH (10 equiv.), 100 °C, 15 min | 8 (81) |
| 2d (n = 1) | PS-10a (5 mg) | CH3CN (1.7 mL), AcOH (10 equiv.), 100 °C, 15 min | 16 (67) |
| 3d (n = 1) | PS-10a (5 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 15 min | 17 (65) |
| 4d (n = 1) | PS-10a (10 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 15 min | 14 (71) |
| 5d (n = 1) | PS-10a (5 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 30 min | 19 (76) |
| 6 (n = 1) | PS-10a (5 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 15 min | 12 (97) |
| 7 (n = 3) | PS-10a (5 mg) | CH3CN (1.7 mL), AcOH (10 equiv.), 100 °C, 15 min | 13 ± 3 (97 ± 3) |
| 8 (n = 3) | PS-10b (5 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 15 min | 17 ± 2 (>98) |
| 9 (n = 3) | PS-10c (5 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 15 min | 13 ± 1 (86 ± 7) |
| 10 (n = 3) | PS-10d (5 mg) | CH3CN (1.7 mL), AcOH (60 equiv.), 100 °C, 15 min | 9 ± 4 (71 ± 11) |
| 11e (n = 2) | PS-10a (5 mg) | CH3CN (4.0 mL) +0.4% AcOH over 15 min, 100 °C | 7 ± 1 (87 ± 3) |
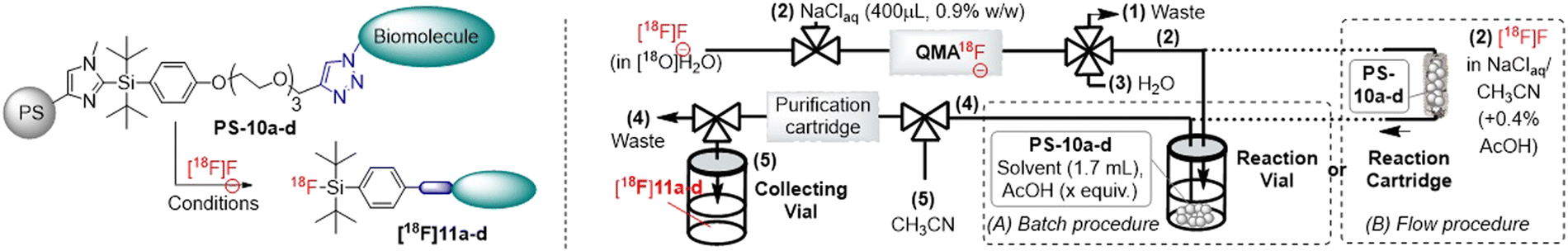
Interestingly, the desired [18F]11a was obtained with promising 8% RCY and 81% RCP. Moreover, switching the solvent to acetonitrile doubled the radiochemical yield to 16% (entry 2, Table 1), while only slightly lowering the radiochemical purity to 67%. On the contrary, increasing the quantities of AcOH or PS-10a gave no significant improvement (RCY of 17% and 14%, respectively, entries 3 and 4, Table 1). Furthermore, as heating the reaction mixture for 30 min only improved the radiochemical yield by 3% (entry 5 compared to entry 2, Table 1), a reaction time of 15 min was preferred for the consecutive experiments in order to shorten as much as possible the overall procedure. With these optimised conditions in hand, the last-step radio-labelling was performed using a fully conjugated batch of supported precursors PS-10a, yielding [18F]11a in satisfactory 12% RCY using 60 equivalents of AcOH (entry 6) or 13 ± 3% RCY (n = 3) with 10 equivalents of AcOH (entry 7).|| Moreover, in both cases, an excellent radiochemical purity of 97% was determined without the need for an HPLC purification, which fully demonstrated the advantage of supported ImidSiFA. Then, 18F-fluorination was performed for supported precursors PS-10b, PS-10c and PS-10d using similar conditions to evaluate the versatility of this supported tag. Interestingly, the glucose-based bioconjugate [18F]11b was produced smoothly with a radiochemical yield of 17 ± 2% (n = 3) and an excellent radiochemical purity (>98%) for each experiment (entry 8). A similar result was observed for the synthesis of the biotin-based tracer from resin beads PS-10c, [18F]11c being obtained in 13 ± 1% RCY (n = 3) with a good radiochemical purity of 86 ± 7% (entry 9, Table 1). Finally, the radiosynthesis employing the estradiol-based precursor PS-10d yielded compound [18F]11d in 9 ± 4% RCY (n = 3), while the radiochemical purity was slightly lower and less reproducible in this case (71 ± 11%, entry 10, Table 1). However, taken together, these results confirmed the versatility of the alkyne-functionalized PS-7 by easily producing various [18F]bioconjugates from the same starting resin.
Then, the set-up was modified to perform the 18F-fluorination in flow through a pre-packed “reaction cartridge” in stainless steel filled with the supported precursor PS-10a and a packing resin (see the ESI† for details). In this case, the [18F]F− saline solution was firstly diluted with CH3CN (+0.4% AcOH) and consecutively injected through the column over 15 min while heating it at 100 °C. The downstream solvent was diluted with H2O, and filtered through the Sep-Pack C18 cartridge, and the desired radiolabelled compound was recovered by elution with acetonitrile. This fully automated procedure provided [18F]11a in a slightly lower RCY of 7 ± 1% (n = 2), but with a good radiochemical purity (87 ± 3, entry 11, Table 1), which established the proof-of-concept for potentially developing kit set-ups using this technology.
To conclude, anchoring the imidazole-di-tert-butyl-silane 6 onto a polystyrene resin provided the versatile platform PS-7, while its pendant alkyne moiety was directly functionalized by CuAAC. Using aqueous [18F]F−, the various heterogeneous precursors were simultaneously radio-labelled and cleaved from the resin as the last-step of the radiosynthesis. Thymidine-, glucose-, biotin- and estradiol-based conjugates [18F]11a–d were produced with automated “batch” or “flow” set-ups in RCY up to 19%. Good to excellent RCP were obtained in most cases after a simple filtration through the Sep-Pack C18 cartridge and consecutive elution. Removing the need for HPLCs, this “supported-ImidSiFA” method is highly promising for the development of 18F-fluorination kits for automated syntheses of PET tracers. Other types of “click” bioconjugations are currently explored to widen synthetic and biological application scopes.
This study was supported by a public grant from the French ANR within the context of the Investments for the Future Program, referred to as ANR-10-LABX-57 and named TRAIL (SUPSIFLU project, PhD grant for M. T.) and by an ANRT grant with Sanofi (CIFRE 2019/0768, PhD grant for M. S.). NMR and mass analyses were performed at the CESAMO (ISM UMR 5255).
Conflicts of interest
M. S., G. B., S. R. and C. A. are Sanofi employees and may hold shares and/or stock options in the company.Notes and references
- (a) N. J. Long, R. Vilar, P. W. Miller and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef PubMed; (b) S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed; (c) Z. Li and P. S. Conti, Adv. Drug Delivery Rev., 2010, 62, 1031–1051 CrossRef CAS PubMed; (d) X. Deng, J. Rong, L. Wang, N. Vasdev, L. Zhang, L. Josephon and S. H. Liang, Angew. Chem., Int. Ed., 2019, 58, 2580–2605 CrossRef CAS PubMed.
- For selected recent reviews, see: (a) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed; (b) H. S. Krishnan, L. Ma, N. Vasdev and S. H. Liang, Chem. – Eur. J., 2017, 23, 15553–15577 CrossRef CAS PubMed; (c) S. Specklin, F. Caillé, M. Roche and B. Kuhnast, in Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals, ed. G. Haufe and F. R. Leroux, Academic Press, 2019, pp. 425–458 Search PubMed.
- For reviews, see: (a) A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. H. Scott, Chem. Sci., 2014, 5, 4545–4553 RSC; (b) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef CAS PubMed; (c) R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS PubMed ; for selected recent examples, see: ; (d) J. Rickmeier and T. Ritter, Angew. Chem., Int. Ed., 2018, 57, 14207–14211 CrossRef CAS PubMed; (e) Z. Yuan, M. B. Nodwell, H. Yang, N. Malik, H. Merkens, F. Bénard, R. E. Martin, P. Schaffer and R. Britton, Angew. Chem., Int. Ed., 2018, 57, 12733–12736 CrossRef CAS PubMed.
- For reviews, see: (a) M. Glaser and E. G. Robins, J. Labelled Compd. Radiopharm., 2009, 52, 407–414 CrossRef CAS; (b) J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef CAS PubMed; (c) C. Mamat, M. Gott and J. Steinbach, J. Labelled Compd. Radiopharm., 2018, 61, 165–178 CrossRef CAS PubMed.
- For selected reviews on Si-, B- or Al-based prosthetic groups, see: (a) O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed; (b) J.-L. Zeng, J. Wang and J.-A. Ma, Bioconjugate Chem., 2015, 26, 1000–1003 CrossRef CAS PubMed; (c) V. Bernard-Gauthier, J. J. Bailey, Z. Liu, B. Wängler, C. Wängler, K. Jurkschat, D. M. Perrin and R. Schirrmacher, Bioconjugate Chem., 2016, 27, 267–279 CrossRef CAS PubMed.
- For other examples of prosthetic groups, see: (a) D. O’Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed; (b) Q. Zheng, H. Xu, H. Wang, W.-G. Han Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, J. Am. Chem. Soc., 2021, 143, 3753–3763 CrossRef CAS PubMed; (c) C. Wang, L. Zhang, Z. Mou, W. Feng, Z. Li, H. Yang, X. Chen, S. Lv and Z. Li, Org. Lett., 2021, 23, 4261–4266 CrossRef CAS PubMed.
- (a) R. H. Mach and S. W. Schwarz, PET Clin., 2010, 5, 131–153 CrossRef PubMed; (b) M. G. Campbell, J. Mercier, C. Genicot, V. Gouverneur, J. M. Hooker and T. Ritter, Nat. Chem., 2017, 9, 1–3 CrossRef CAS PubMed.
- For reviews, see: (a) S. Boldon, I. S. R. Stenhagen, J. E. Moore, S. K. Luthra and V. Gouverneur, Synthesis, 2011, 3929–3953 CAS; (b) R. Hoareau and P. J. H. Scott, in Solid-Phase Organic Synthesis: Concepts, Strategies, and Applications, ed. P. H. Toy and Y. Lam, John Wiley & Sons, 2012, pp. 415–426 Search PubMed.
- For an example of electrostatic interactions, see: (a) P. J. Riss, S. Kuschel and F. I. Aigbirhio, Tetrahedron Lett., 2012, 53, 1717–1719 CrossRef CAS ; for representative examples of organometallic linking, see: ; (b) L. P. Szajek, M. A. Channing and W. C. Eckelman, Appl. Radiat. Isot., 1998, 49, 795–804 CrossRef CAS; (c) A. Tabey, H. Audrain, E. Fouquet and P. Hermange, Chem. Commun., 2019, 55, 7587–7590 RSC; (d) A. Tabey, T. Christine, E. Fouquet and P. Hermange, Appl. Organometal. Chem., 2020, 34, e5779 CrossRef CAS; (e) M. Cormier, A. Tabey, T. Christine, H. Audrain, E. Fouquet and P. Hermange, Dalton Trans., 2021, 50, 10608–10614 RSC ; for representatives examples of covalent linking, see: ; (f) L. J. Brown, D. R. Bouvet, S. Champion, A. M. Gibson, Y. Hu, A. Jackson, I. Khan, N. Ma, N. Millot, H. Wadsworth and R. C. D. Brown, Angew. Chem., Int. Ed., 2007, 46, 941–944 CrossRef CAS PubMed; (g) A. C. Topley, V. Isoni, T. A. Logothetis, D. Wynn, H. Wadsworth, A. M. R. Gibson, I. Khan, N. J. Wells, C. Perrio and R. C. D. Brown, Chem. – Eur. J., 2013, 19, 1720–1725 CrossRef CAS PubMed; (h) R. Takeuchi, Y. Sakai, H. Tanaka and T. Takahashi, Bioorg. Med. Chem. Lett., 2015, 25, 5500–5503 CrossRef CAS PubMed; (i) R. Edwards, W. de Vries, A. D. Westwell, S. Daniels and T. Wirth, Eur. J. Org. Chem., 2015, 6909–6916 CrossRef CAS; (j) M. Richard, S. Specklin, M. Roche, F. Hinnen and B. Kuhnast, Chem. Commun., 2020, 56, 2507–2510 RSC.
- M. Tisseraud, J. Schulz, D. Vimont, M. Berlande, P. Fernandez, P. Hermange and E. Fouquet, Chem. Commun., 2018, 54, 5098–5101 RSC.
- For key articles on SiFA, see: (a) R. Ting, M. J. Adam, T. J. Ruth and D. M. Perrin, J. Am. Chem. Soc., 2005, 127, 13094–13095 CrossRef CAS PubMed; (b) R. Schirrmacher, G. Bradtmöller, E. Schirrmacher, O. Thews, J. Tillmanns, T. Siessmeier, H. G. Buchholz, P. Bartenstein, B. Wängler, C. M. Niemeyer and K. Jurkschat, Angew. Chem., Int. Ed., 2006, 45, 6047–6050 CrossRef CAS PubMed; (c) L. Mu, A. Hhne, P. A. Schubiger, S. M. Ametamey, K. Graham, J. E. Cyr, L. Dinkelborg, T. Stellfeld, A. Srinivasan, U. Voigtmann and U. Klar, Angew. Chem., Int. Ed., 2008, 47, 4922–4925 CrossRef CAS PubMed ; for a review on their biological applications, see: ; (d) L. Gower-Fry, T. Kronemann, A. Dorian, Y. Pu, C. Jaworski, C. Wängler, P. Bartenstein, L. Beyer, S. Lindner, K. Jurkschat, B. Wängler, J. J. Bailey and R. Schirrmacher, Pharmaceuticals, 2021, 14, 701–719 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed ; for selected reviews, see: ; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (d) J. E. Hein and W. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- P. B. Koswatta and C. J. Lovely, Tetrahedron Lett., 2009, 50, 4998–5000 CrossRef CAS.
- P. S. Donelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459–2461 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and copies of 1H, 13C, 29Si and 19F NMR NMR spectra for new compounds. Radio- and UV-HPLC traces for [18F]compounds. CCDC 2169890. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03258a |
| ‡ Crystallographic data for this structure has been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2169890. |
| § Calculated by hypothesising a 100% grafting in the 6- > PS-7 step and multiplying by the mass ratios at each step. |
| ¶ The side-product [18F]8 was recovered with [18F]11a (see ESI†). |
| || An indirect labelling was attempted for comparison, i.e. synthesis of [18F]8 and consecutive CuAAC conjugation with 9a, but product [18F]11a was isolated in only 2% RCY (see ESI†). |
| This journal is © The Royal Society of Chemistry 2022 |