
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly constrained cis-dihydride platinum complex trapped by cooperative gold/platinum dihydrogen activation†
Nereida
Hidalgo‡
 a,
Felipe
de la Cruz-Martínez‡
a,
M. Trinidad
Martín
b,
M. Carmen
Nicasio
*b and
Jesús
Campos
*a
a,
Felipe
de la Cruz-Martínez‡
a,
M. Trinidad
Martín
b,
M. Carmen
Nicasio
*b and
Jesús
Campos
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla, Avenida Américo Vespucio 49, Sevilla 41092, Spain. E-mail: Spainjesus.campos@iiq.csic.es
bDepartamento de Química Inorgánica, Universidad de Sevilla, Aptdo 1203, Sevilla 41071, Spain. E-mail: mnicasio@us.es
First published on 19th July 2022
Abstract
A series of Au(I)/Pt(0) combinations that behave as bimetallic frustrated Lewis pairs activates dihydrogen in a cooperative manner. The steric bulk of the terphenyl phosphines that stabilize both fragments allows for the isolation of a rather unique and highly distorted cis-type dihydride platinum(II) structure.
In the last fifteen years, frustrated Lewis pair (FLP) systems have become a major research topic in main group chemistry and catalysis, mainly due to their ability to activate small molecules and perform a broad range of catalytic transformations.1 Although first developed by combining main group Lewis acids and Lewis bases with high steric profiles to prevent quenching by donor–acceptor interactions, the FLP concept has been progressively extended to transition metals due to the enormous structural and electronic diversity of their complexes. Inspired by pioneering work of Wass2 and Erker3 on zirconium/phosphine FLPs, we have pursued the design of frustrated systems in which the two components are based on transition metals.4
Among these studies, we have investigated in detail Au(I)/Pt(0) pairs5 comprised of [(PR2Ar′)Au(NTf2)] and Pt(PtBu3)2 fragments, where Ar′ stands for bulky terphenyl groups (C2H3-2,6-Ar2) and NTf2 for the weakly coordinating triflimide anion (see Fig. 1a). Since the activation of dihydrogen has become a benchmark reaction to gauge FLP reactivity,6 we exposed the aforesaid Au(I)/Pt(0) pairs to H2 gas, which led us to postulate the first genuine bimetallic FLP mechanism for dihydrogen cleavage.5c Besides, our studies indicated a strong influence of the gold fragment steric properties on the rate of bond activation and also on the resulting product distribution.
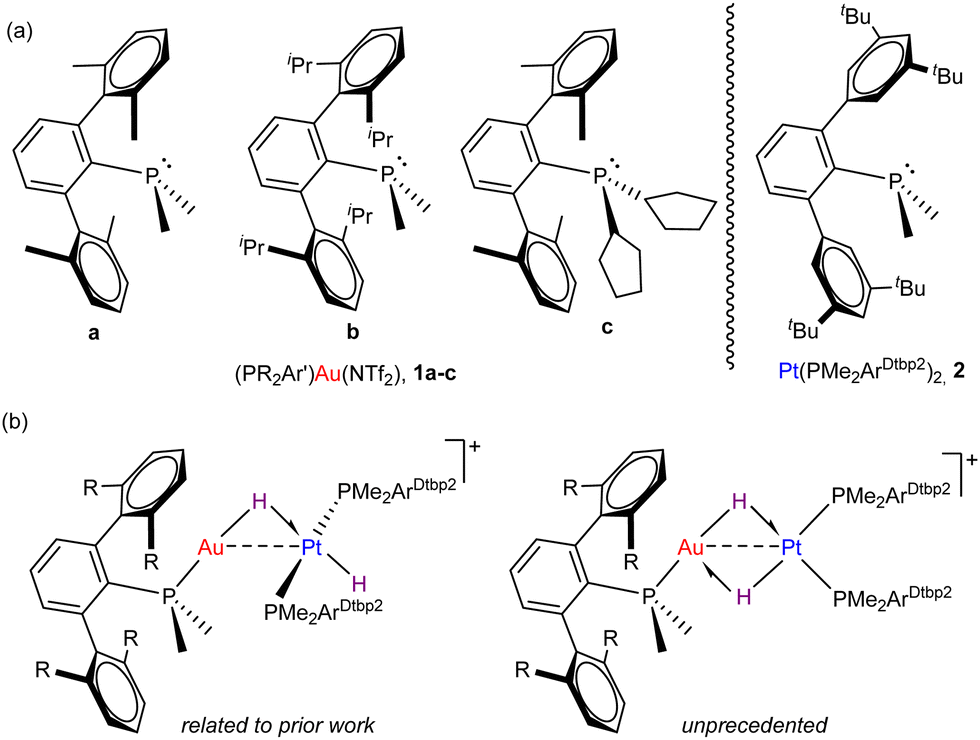 | ||
| Fig. 1 (a) Terphenyl phosphine ligands used in this work to stabilize Au(I) and Pt(0) compounds (NTf2− (triflimide) = [N(SO2CF3)2]−); (b) key bimetallic Au(I)/Pt(II) dihydride isomeric structures isolated and characterized herein. | ||
Encouraged by our recent access to the highly congested Pt(0) compound Pt(PMe2ArDtpb2)2 (Dtbp = 3,5-di-tert-butylphenyl) (Fig. 1a),7 and by the still lively debate on the precise mechanism of dihydrogen activation by FLPs,8 we report herein our studies on the contrasting reactivity offered by three Au(I)/Pt(PMe2ArDtpb2)2 pairs towards H2 (Fig. 1b). The combination of two metallic fragments stabilized by terphenyl phosphine ligands have allowed us to isolate unique models of cis–trans-isomerization in platinum(II) dihydrides, mostly associated to unhindered and highly reactive complexes.9
Before investigating the reactivity of the 1:2 pairs with dihydrogen, we first examined the potential formation of bimetallic adducts 3 characterized by a dative Pt → Au bond (Scheme 1). The existence of acid-base interactions in FLP systems has a profound impact on their cooperative reactivity. As such, the presence of Pt → Au bonds greatly diminishes the reactivity of Au(I)/Pt(0) pairs, though it does not quench their frustrated reactivity as they may behave as thermally induced FLPs.5c Thus, we examined the combination of Pt(0) compound 2 and Au(I) triflimide complexes 1a–c (Scheme 1) bearing phosphines PMe2ArXyl2 (1a), PMe2ArDipp2 (1b) and PCyp2ArXyl2 (1c) (Cyp = cyclopentyl) and using solvents of different polarity.
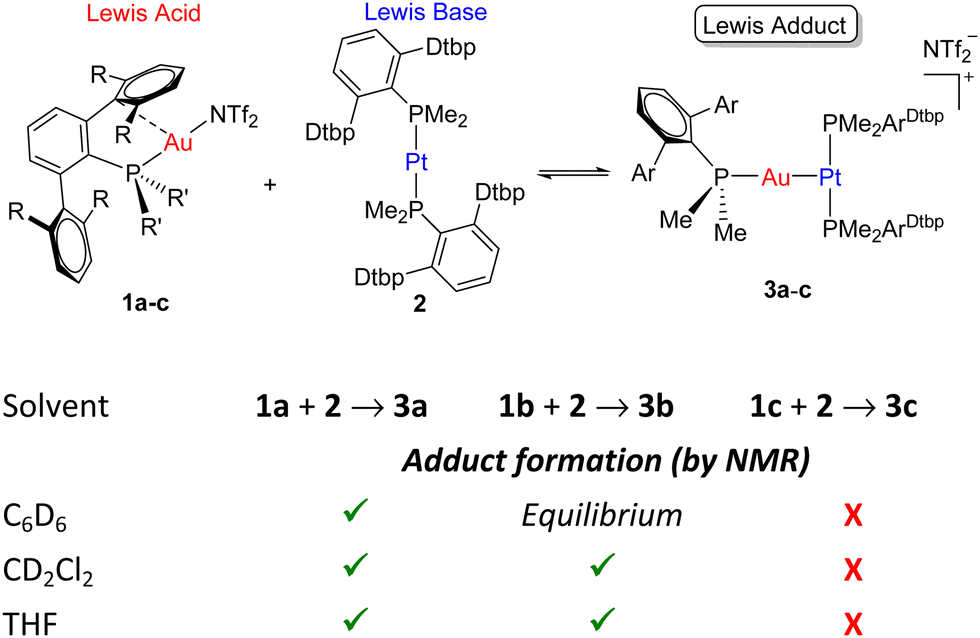 | ||
| Scheme 1 Bimetallic Lewis adduct formation in 1:2 systems. | ||
As anticipated, the steric hindrance of the phosphines coordinated to gold (PCyp2ArXyl2 > PMe2ArDipp2 > PMe2ArXyl2)10 is crucial to modulate the equilibrium between Lewis adduct formation and complete bimetallic frustration. The more congested 1c is unable to coordinate to compound 2, contrasting with related examples based on [Pt(0)(PR3)]2 species that recurrently led to the formation of metal-only Lewis pairs.11 Conversely, when compound 2 is combined with one equivalent of 1a, the corresponding bimetallic Lewis adduct 3a is formed. It exhibits 31P{1H} NMR signals at −23.2 and 13.5 ppm due to PMe2ArXyl2 and PMe2ArDtbp2, respectively. The former reveals a pronounced decreased of the 1JPPt coupling constant to 3077 Hz (cf. 3794 Hz in 2), while the latter signal is flanked by 195Pt satellites (2JPPt = 2060 Hz), both features indicative of formation of adduct 3a.
An interesting situation is found in the case of compound 1b bearing the intermediate size phosphine PMe2ArDipp2, which upon equimolar combination with 2 in C6D6 seems to be in equilibrium between adduct formation and monometallic fragments. We infer this from the corresponding 31P{1H} NMR spectrum, which shows broadened signals for the two monometallic species which sharpen upon cooling the NMR probe, a behaviour comparable to our prior studies with Au(I)/Pt(0).5c Solvent effects have an important influence in traditional FLP systems,12 and the same applies herein. Thus, in more polar solvents as CD2Cl2 or THF compound 3b cleanly forms (31P{1H} NMR, δ 11.5 (1JPPt = 3054 Hz), −25.4). It is likely that the higher propensity to solvate the departing triflimide anion upon adduct formation favours the formation of the latter.
Next, we investigated the cooperative reactivity of these Au(I)/Pt(0) pairs towards hydrogen. It is important to note that neither gold precursor 1 nor complex 2 could react with dihydrogen (1 bar) even under harsher reaction conditions (80 °C, up to 1 week) to those attempted with the bimetallic pairs. In contrast, pairs 1:2 rapidly reacted with dihydrogen, though the selectivity of the transformation was highly dependent on the terphenyl phosphine anchored to gold. First, addition of H2 under mild conditions (1 bar, 25 °C) to an equimolar mixture of 1a:2 in C6D6 produced the new heterobimetallic Au–Pt complex 4a (Scheme 2). The formulation of 4a was rapidly inferred from two distinctive low-frequency signals in its 1H NMR spectrum at −1.15 (2JHP = 100, 1JHPt = 585 Hz) and −6.93 (1JHPt = 1223 Hz) ppm, corresponding to a bridging and a terminal hydride, in analogy to the previously reported compound [(PMe2ArDipp2)Au(μ-H)Pt(PtBu3)2(H)].5a In contrast, the bulkier Au(I) precursors 1b and 1c led to a different spectroscopic pattern upon combination with 2 and exposure to dihydrogen (1 bar, 25 °C). In those cases, a single hydridic signal for two protons was recorded at −4.03 (2JHP = 57, 1JHPt = 581 Hz) and −4.28 ppm (2JHP = 51, 1JHPt = 564 Hz) for the pairs 1b:2 and 1c:2, respectively.
 | ||
| Scheme 2 Reaction of Au(I)/Pt(0) bimetallic pairs with H2 in toluene solution. | ||
We initially postulated a rapid equilibration between a terminal and bridging hydride as in 4a to account for the sole low-frequency signal recorded. In fact, we could investigate such a solution dynamic process for compound 4a through exchange spectroscopy (EXSY) experiments at variable temperature (from 30 to 55 °C, in tol-d8). An Eyring analysis led to the activation parameters ΔG‡298 = 17.6 kcal mol−1, ΔH‡ = 8.7 kcal mol−1 and ΔS‡ = −29.7 cal mol−1 (see Scheme S1 and Fig. S22 in the ESI†). We tentatively attribute the high negative value calculated for the entropic parameter to coordination of the triflimide anion to the electrophilic gold centre in the key transition state of the exchange process.
Nonetheless, we were unable to split the single hydride signal at ca. −4 ppm after hydrogenation of pairs 1b:2 and 1c:2 even at low temperature (−80 °C). This fact, along with their corresponding 31P{1H} spectra, which clearly differs from that of 4a (see ESI†), and also the appearance of distinctive infrared signals due to hydride ligands at 2144 (1b:2 + H2) and 2141 (1c:2 + H2) cm−1, dissimilar to that of 4a (2064 cm−1), led us to consider the formation of a rare cis-type dihydride structure with a Au(μ-H)2Pt core. Trogler and co-workers investigated in the 1980s the cis–trans isomerization of square-planar Pt(II) dihydride complexes of formula Pt(PR3)2(H)2,9,13 demonstrating that only in the case of particularly small phosphines the cis-isomer can be detected (e.g. 20% and 3% of cis-isomer in toluene solution for PMe3 and PEt3, respectively, while completely undetected for bulkier ligands).9 Though electronic arguments (trans influence) seems to support the preference for the cis-isomer, trans-dihydrides are thermodynamically favoured,14 in part due to the dominance of steric effects. In fact, the only available information on these elusive isomers derive from the use of chelating phosphine ligands that geometrically constrain the cis-conformation.15 However, no other unconstrained cis-dihydrides complexes have been reported since the early work of Trogler,16 despite widespread examples of square planar Pt(II) trans-dihydrides.17
Our assumption of a bis-(μ-H) core is consistent with our 1H NMR data compared to the aforesaid limited examples of Pt(II) cis-dihydrides and also fits with our simulation using gnmr software (Fig. S15, ESI†). However, it seems rather surprising, even more considering an additional interaction with the bulky [(PCyp2ArXyl2)Au]+ fragment, that a cis-type dihydride isomer may exist for such an extremely crowded environment.
X-Ray diffraction studies allowed us to confirm the contrasting reactivity between the pairs 1a:2 and 1c:2. While the former leads to the heterobimetallic trans-dihydride 4a (Fig. 2), the latter indeed activates H2 to yield the unique cis-type dihydride 5c (Fig. 2; and thereby 1b:2 towards cis-dihydride 5b). Compounds 4a and 5c were crystallized from their concentrated toluene solutions at 25 °C. Compound 4a is characterized by a Pt–Au bond distance of 2.7628(7) Å, comparable to other Pt(μ-H)Au species,18 and further confirming the presence of a bridging hydride. The trans-disposition of the Pt(II) fragment is illustrated by an almost ideal P–Pt–P angle of 175.76(10)°. In stark contrast, the analogous angle in compound 5c is drastically decreased to 107.53(3)°, similar to Pt(II) cis-dihydride structures geometrically constrained by chelating phosphines.15 This structural disposition entails a high proximity between the two PMe2ArDtbp2 phosphines, which is partly compensated by a slightly elongated Pt–P bond in 5c (P1–Pt: 2.3178(7) Å) compared to 4a (2.292(2) and 2.296(2) Å). The Pt centre thus adopts an almost ideal trigonal arrangement with respect to the phosphines and gold fragment (P1–Pt–Au, 125.654(19)°; P2–Pt–Au, 126.32(2)°). This could be rationalized as the coordination of the electrophilic [LAu]+ fragment to an electron rich Pt(II) cis-dihydride, as previously proposed by Venanzi for other related heterobimetallic species.18a
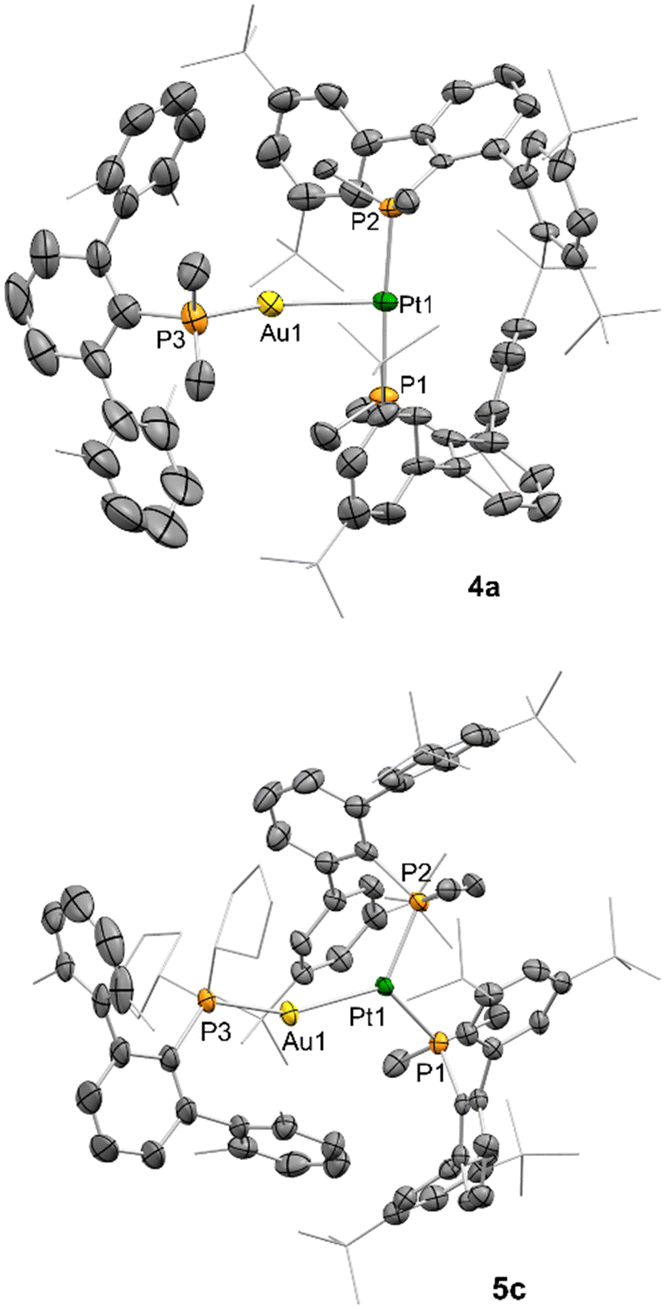 | ||
| Fig. 2 ORTEP diagrams for compounds 4a and 5c. Thermal ellipsoids are drawn at the 50% probability. Metal hydrides could not be located in the difference Fourier electron density map. Counterion and hydrogen atoms omitted and tert-butyl, cyclopentyl and methyl groups from aryl rings in wireframe format for clarity. | ||
We decided to investigate the effect of using THF as a polar solvent, which has been shown in the past to drastically affect selectivity in cis–trans isomerization.9,13 While the outcome of hydrogenation for the pair 1c:2 was not affected by solvent, dissimilar results were found in the case of 1a:2 and 1b:2. In contrast, using 1,2-difluorobenzene did not offer any difference compared to toluene. For the pair 1a:2, carrying out the reaction in THF resulted in the appearance of an additional minor Au/Pt species (4a′, ca. 25%) with multinuclear NMR resonances very similar to those of 4a. Two new hydride signals were recorded at −2.63 and −8.87 ppm, which appear as broad, as well as those of 4a. Cooling down the NMR probe sharpened the two pairs of signals, while coalescence was reached at 30 °C (see Fig. S18, ESI†). Evaporation of volatiles and addition of toluene reverses the mixture towards clean formation of 4a. Kinetic parameters for this dynamic process were obtained from lineshape analysis in the temperature interval from −20 to 60 °C, leading to ΔH‡ = 16.1 kcal mol−1 and ΔS‡ = 31.6 cal mol K−1, which correspond to a ΔG‡298 of 6.7 kcal mol−1 (see ESI†). Once more the strong entropic contribution is associated to weak coordination of triflimide to access 4a′, which in this case would entail desolvation of THF. More interesting is the solvent effect on the pair 1b:2, where the selectivity was inverted and a mixture of 4b and 5b in an approximate 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was recorded by 1H NMR monitoring (Fig. S17, ESI†). In fact, drying under vacuum a toluene solution of pure cis-dihydride 5b and redissolving the residue in THF leads to the same 9:1 ratio of 4b and 5b (Scheme 3). This result is opposed to prior studies by Trogler, where shifting to more polar solvents tend to favour cis-dihydride species.9
:1 ratio was recorded by 1H NMR monitoring (Fig. S17, ESI†). In fact, drying under vacuum a toluene solution of pure cis-dihydride 5b and redissolving the residue in THF leads to the same 9:1 ratio of 4b and 5b (Scheme 3). This result is opposed to prior studies by Trogler, where shifting to more polar solvents tend to favour cis-dihydride species.9
 | ||
| Scheme 3 Isomerization of 5b into 4b promoted by THF as solvent. | ||
To complete these studies and offer a first hint of the bimetallic H2 cleavage mechanism, we carried out some preliminary studies to determine the kinetic isotope effect (KIE). Those were run in duplicates for the pairs 1a:2 and 1b:2 in both toluene-d8 and THF-d8 at 0 °C to facilitate kinetic analysis (see Fig. S23–S26, ESI†). Interestingly, we calculated inverse KIEs for the reactions carried out in toluene, which account for 0.74 ± 0.04 for the formation of 4a and an even stronger inverse KIE of 0.49 ± 0.09 associated to the appearance of 5b. These values correlate well with our prior studies on hydrogen splitting by 1a–b:[Pt(PtBu3)2] pairs (1a, 0.46 ± 0.04; 1b, 0.50 ± 0.02), where a genuine bimetallic FLP mechanism could be stated based on a thorough experimental/computational approach.5c On the contrary, kinetic studies performed herein in THF led to normal KIEs of 1.33 ± 0.05 and 2.11 ± 0.55 due to 1a:2 and 1b:2, respectively. We are yet unsure on the precise nature of the contrasting KIEs as a function of solvent, but we have already gathered some relevant information that set the basis of our future mechanistic studies. First, similar and distinctive KIEs depend on solvent and not on product distribution, which speaks in favour of a common mechanism for H2 cleavage. Also, the medium size phosphine seems to provide an ideal scenario to freeze the cis–trans isomerization, which cannot be rate-limiting as it occurs very rapidly (or never does occur for the more hindered 5c). The strong effect of THF on the KIEs likewise suggests an active participation of the triflimide anion, which is expected to be strongly solvated only by the ethereal solvent. It probably has an impact as well on the availability of the electrophilic [Au(PR2Ar’)]+ fragment, which agrees with the moderately higher rates of H2 splitting in THF (see ESI† for details). In addition, the absence of H/D scrambling when exposing compounds 1 to H2/D2 atmosphere (1:1) in either toluene or THF rules out the active participation of the ethereal solvent or triflimidate as basic partners for H–H bond cleavage.
In conclusion, we report the bimetallic activation of dihydrogen by three Au(I)/Pt(0) pairs whose monometallic fragments are all stabilized by sterically crowded terphenyl phosphines. Cooperation between the two metals is evinced by complete absence of reactivity for the individual complexes. Besides, having two metallic fragments of great steric bulk allowed us to isolate a unique structure characterized by a Au(μ-H)2Pt core. It represents virtually the only example of a cis-dihydride group 10 d8 complex, of relevance for a broad spectrum of catalytic reactions, and in which isomerization is not geometrically quenched by using chelating scaffolds or favoured by particularly small phosphines.
N. H. and F. C. M. synthesised and characterised all new complexes and carried out reactivity studies. M. T. M. synthesized the platinum precursor. M. C. N. and J. C. supervised the overall project. N. H. and F. C. M. wrote the original draft and all authors contributed to review and editing.
This work was supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575) and Grants PID2019-110856GA-I00 and PID2020-113797R funded by MCIN/AEI/10.13039/501100011033. We also thank US/JUNTA/FEDER, UE (Grants US-1380849 and US-1262266). F. C. M. acknowledges the Spanish Ministry of Universities for a Margarita Salas fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- See for example: (a) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed; (b) M. Navarro, J. J. Moreno and J. Campos, in Comprehensive Organometallic Chemistry IV (COMC-IV), ed. K. Meyer, D. O’Hare and G. Parkin, Elsevier, Amsterdan, 2022 Search PubMed.
- (a) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826–8829 CrossRef CAS PubMed; (b) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463–18478 CrossRef CAS PubMed.
- (a) X. Xu, R. Fröhlich, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2012, 48, 6109–6111 RSC; (b) X. Xu, G. Kehr, C. G. Daniliuc, G. Kehr and G. Erker, J. Am. Chem. Soc., 2013, 135, 6465–6476 CrossRef CAS PubMed.
- M. Navarro and J. Campos, in Adv. Organomet. Chem., ed. P. J. Pérez, Elsevier, Amsterdam, 2021, ch. 3, vol. 75, pp. 95–148 Search PubMed.
- (a) J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed; (b) N. Hidalgo, S. Bajo, J. J. Moreno, C. Navarro-Gilabert, B. Q. Mercado and J. Campos, Dalton Trans., 2019, 48, 9127–9138 RSC; (c) N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 5982–5993 CrossRef CAS PubMed; (d) N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Organometallics, 2020, 39, 2534–2544 CrossRef CAS PubMed; (e) N. Hidalgo, C. Romero-Pérez, C. Maya, I. Fernández and J. Campos, Organometallics, 2021, 40, 1113–1119 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- M. T. Martín, M. Marín, R. J. Rama, E. Álvarez, C. Maya, F. Molina and M. C. Nicasio, Chem. Commun., 2021, 57, 3083 RSC.
- (a) S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed; (b) T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef CAS PubMed; (c) D. Yepes, P. Jaque and I. Fernández, Chem. – Eur. J., 2016, 22, 18801–18809 CrossRef CAS PubMed; (d) G. Skara, F. De Vleeschouwer, P. Geerlings, F. De Proft and B. Pinter, Sci. Rep., 2017, 7, 16024 CrossRef PubMed; (e) I. Fernández, Chem. Commun., 2022, 58, 4931–4940 RSC.
- R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 1138–1140 CrossRef CAS.
- M. Marín, J. J. Moreno, M. M. Alcaide, E. Álvarez, J. López-Serrano, J. Campos, M. C. Nicasio and E. Carmona, J. Organomet. Chem., 2019, 896, 120–128 CrossRef.
- See for examples: (a) H. Braunschweig, K. Radacki and K. Schwab, Chem. Commun., 2010, 46, 913–915 RSC; (b) M. Ma, A. Sidiropoulos, L. Ralte, A. Stasch and C. Jones, Chem. Commun., 2013, 49, 48–50 RSC; (c) B. R. Barnett, C. E. Moore, P. Chandrasekaran, S. Sproules, A. L. Rheingold, S. DeBeerde and J. S. Figueroa, Chem. Sci., 2015, 6, 7169–7178 RSC.
- L. X. Dang, G. K. Schenter, T.-M. Chang, S. M. Kathmann and T. Autrey, J. Phys. Chem. Lett., 2012, 3, 3312–3319 CrossRef CAS.
- (a) D. L. Packett, C. M. Jensen, R. L. Cowan, C. E. Strouse and W. C. Trogler, Inorg. Chem., 1985, 24, 3578–3583 CrossRef CAS; (b) D. L. Packett and W. C. Trogler, J. Am. Chem. Soc., 1986, 108, 5036–5038 CrossRef CAS; (c) D. L. Packett and W. C. Trogler, Inorg. Chem., 1988, 27, 1768–1775 CrossRef CAS.
- J. O. Noell and P. J. Hay, J. Am. Chem. Soc., 1982, 104, 4578–4584 CrossRef CAS.
- See for example: (a) C. J. Moulton and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1976, 365–366 RSC; (b) F. Novio, P. González-Duarte, A. Lledós and R. Mas-Ballesté, Chem. – Eur. J., 2007, 13, 1047–1063 CrossRef CAS PubMed; (c) R. Friedemann and K. Seppelt, Eur. J. Inorg. Chem., 2013, 1197–1206 CrossRef CAS.
- A somewhat related [Pt(μ-H)2Pt] structure has been described by dimerization of two monohydride Pt complexes: A. Koppaka, V. Yempally, L. Zhu, G. C. Fortman, M. Temprado, C. D. Hoff and B. Captain, Inorg. Chem., 2016, 55, 307–321 CrossRef CAS PubMed.
- See for example: (a) A. B. Goel and S. Goel, Inorg. Chim. Acta, 1982, 65, L77 CrossRef CAS; (b) E. Prack, C. A. O’Keefe, J. K. Moore, A. Lai, A. J. Lough, P. M. Macdonald, M. S. Conradi, R. W. Schurko and U. Fekl, J. Am. Chem. Soc., 2015, 137, 13464–13467 CrossRef CAS PubMed.
- (a) A. Albinati, H. Lehner, L. M. Venanzi and M. Wolfer, Inorg. Chem., 1987, 26, 3933–3939 CrossRef CAS; (b) M. Crespo, J. Sales and X. J. Solans, J. Chem. Soc., Dalton Trans., 1989, 1089–1092 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data of new compounds, NMR spectra, variable temperature and kinetic experiments and crystal data. CCDC 2175895 and 2175896. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03089f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |