
Switching photorelease to singlet oxygen generation by oxygen functionalization of phenothiazine photocages†
Mamata
Ojha
a,
Moumita
Banerjee
b,
Souvik
Ray
a,
Amit Kumar
Singh
a,
Anakuthil
Anoop
 *b and
N. D. Pradeep
Singh
*a
*b and
N. D. Pradeep
Singh
*a
aDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: ndpradeep@chem.iitkgp.ac.in
bDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: anoop@chem.iitkgp.ac.in
First published on 28th January 2022
Abstract
A phenothiazine-based photoremovable protecting group (PRPG) for single and dual release of carboxylic acids was developed. The change in the oxidation state of the sulfur atom of the phenothiazine PRPG resulted in singlet oxygen generation, rather than photorelease. The difference in the photochemistry between oxygen-free and oxygen-functionalized phenothiazine was investigated and supported by DFT calculations.
Photoremovable protecting groups (PRPGs) are used to achieve non-invasive spatiotemporal control over the release of biologically active molecules on exposure to light.1 For this reason, PRPGs have gained considerable importance, particularly in drug delivery systems (DDSs).2,3 At present, the applications of PRPGs are no longer restricted to the release of a single bioactive molecule of interest. PRPGs with dual release ability have become more important than those with single release ability in drug delivery as they reduce drug resistance,4 therapeutic doses,5 and side effects. In the literature, a few PRPGs for the dual release of active molecules are reported, including carboline,6 carbazole,7o-nitrobenzyl,8 and bimane9 derivatives. Hence, there is a real need to develop a fluorescent PRPG for the dual release of the active molecules with moderate to high photochemical quantum yield in the visible wavelength region.
Phenothiazine (PTZ) is an active component in push–pull systems because of its strong electron-donating ability. The literature shows that the photophysical properties and the HOMO–LUMO energy levels of PTZ derivatives could be modulated by having substituents at the nitrogen and the 3,7-positions of the phenothiazine unit.10 PTZ and its derivatives are used in various applications, from pharmacological to biological fields. Phenothiazine derivatives have gained considerable importance among nitrogen-containing heterocyclic compounds, mainly because of their attractive properties, such as (i) exhibition of diverse biological activities,11 (ii) large Stokes shifts,12 (iii) flexible functionalization on the parent skeleton, (iv) intramolecular charge transfer (ICT)13 and (v) acting as a photoinitiator.14
Recently, Carlotti and co-workers reported an effective strategy to tailor the photophysics of phenothiazine by utilizing oxygen functionalization on PTZ based push–pull systems.15 Using the above strategy, they demonstrated that oxidizing the sulfur atom in the PTZ moiety leads to converting intramolecular charge transfer (PTZ derivatives) to a highly efficient emission process (phenothiazine-oxide and dioxide derivatives). In the case of oxygen-functionalized PTZ, the highly fluorescent excited state gets populated due to planar intramolecular charge transfer (PICT). On the other hand, a twisted intramolecular charge transfer (TICT) state is produced upon photoexcitation of the oxygen-free PTZ derivatives, leading to significant fluorescence quenching. They also showed that phenothiazine-5-oxide and phenothiazine-5,5-dioxide acted as weak electron donors relative to phenothiazine for a naphthalimide acceptor. The above exciting strategy of changing TICT to PICT by modulating the electron-donating ability of the phenothiazine chromophore on oxidizing the sulfur atom prompted us to develop phenothiazine as a PRPG.
In this study, we developed for the first time phenothiazine as a fluorescent PRPG for the single and dual release of carboxylic acids and an anticancer drug (valproic acid) on exposure to UV (λ ≥ 365 nm) and visible light (λ ≥ 410 nm) irradiation. Interestingly phenothiazine PRPG showed singlet oxygen generation rather than photorelease on changing the oxidation state of the sulfur atom (S(II) to S(IV)) of the thiazine ring. The difference in the photochemistry between the oxygen-free phenothiazine and oxygen-functionalized phenothiazine PRPGs was investigated and supported by DFT calculations.
Single- and dual-arm phenothiazine based caged esters were synthesized as shown in Scheme 1. First, commercially available 10H-phenothiazine was N-alkylated and used as a precursor for synthesizing the single- and dual-arm (same and different) caged esters. To synthesize single-arm caged esters (4a–e), first PRPG 3 was obtained by Vilsmeier–Haack formylation of 1 with one equivalent of POCl3, followed by NaBH4 reduction. Then, esterification of 3 with one equivalent of the corresponding carboxylic or amino acids by EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) coupling generated the desired single-arm caged esters (4a–e). For the synthesis of dual-arm (same) caged esters (8a–c), PRPG 7 was obtained by Vilsmeier–Haack formylation of 1 with ten equivalents of POCl3, followed by NaBH4 reduction. Compound 7 was then esterified by EDC coupling with three equivalents of the corresponding carboxylic or amino acids to afford the final dual-arm (same) caged esters (8a–c). Finally, dual-arm (different) caged esters (12a and 12b) were synthesized, first by esterifying 7 with one equivalent of anisic acid to obtain 11, and further treatment of 11 with toluic acid (1.0 equiv.) and phenylacetic acid (1.0 equiv.), separately, afforded dual-arm caged esters 12a and 12b.
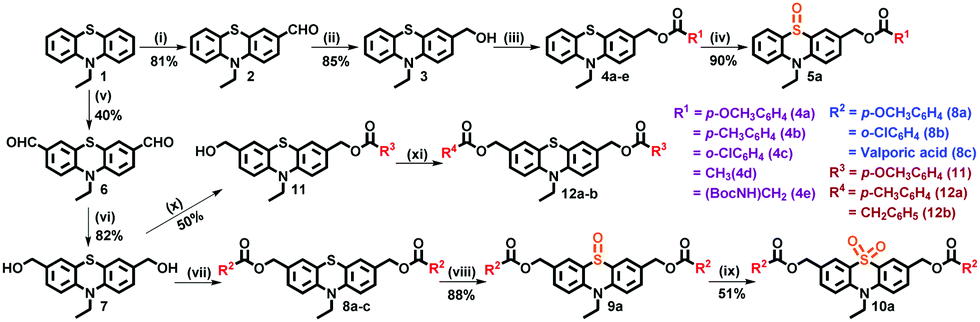 | ||
| Scheme 1 Synthesis of single- and dual (same and different)-arm caged esters and oxidized caged ester of phenothiazine. Reagents and conditions: POCl3 (1.1 equiv.), DMF, 60 °C, 12 h; (ii) NaBH4, MeOH, r.t., 3 h; (iii) EDC (1 equiv.), R1COOH (1 equiv.), DMAP, dry DCM, 3 h; (iv) m-CPBA, CH2Cl2; (v) POCl3 (10 equiv.), DMF, chlorobenzene, 110 °C, 6 h; (vi) NaBH4, MeOH, r.t., 3 h; (vii) EDC (3 equiv.), R2COOH (3 equiv.), DMAP, dry DCM, 3 h; (viii) m-CPBA, CH2Cl2; (ix) H2O2, acetic acid; (x) EDC (1 equiv.), R3COOH (1 equiv.), DMAP, dry DCM, 30 min; (xi) EDC (1 equiv.), R4COOH (1 equiv.), DMAP, dry DCM, 3 h. | ||
The mono-oxidized phenothiazine (mono-OPTZ) caged ester 5a was synthesized by oxidizing compound 4a with m-CPBA (meta-chloroperoxybenzoic acid). Similarly, the di-oxidized phenothiazine (di-OPTZ) caged ester 10a was synthesized by oxidizing compound 8a with m-CPBA to form 9a, and then further oxidation of 9a with H2O2 in the presence of acetic acid afforded compound 10a. The products obtained in each step were characterized by 1H NMR, 13C NMR, and HRMS (Fig. S1–S30, ESI†).
The absorption and emission spectra of all the phenothiazine-based caged esters (10−5 M) were recorded in acetonitrile solution. The absorption spectra of single and dual-arm caged esters 4a and 8a exhibited absorption maxima (λabs) at 307 nm and 308 nm, respectively, which correspond to the localized π–π* transitions (Fig. S31a, ESI†). On the other hand, the absorption spectra of oxidized phenothiazine caged esters 5a (mono-OPTZ) and 10a (di-OPTZ) showed absorption maxima at 340 and 335 nm, respectively, which correspond to the n–π* transitions.
In the emission spectrum, the emission maximum of single (4a) and dual-arm (8a) caged esters were observed at 451 and 456 nm, respectively (Fig. 1a). We found that all single- and dual-arm caged esters showed large Stokes shifts (140 and 148 nm) and exhibited green fluorescence. On the other hand, the emission spectra of oxidized phenothiazine caged esters 5a (mono-OPTZ) and 10a (di-OPTZ) exhibited blue-shifted emission maxima at 453 and 350 nm, respectively.
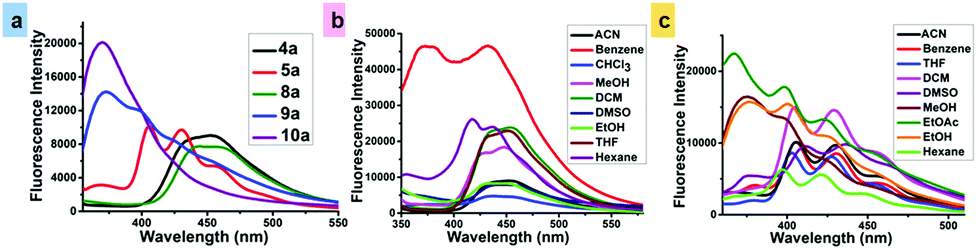 | ||
| Fig. 1 Emission spectra of (a) single-arm (4a), single-arm mono-OPTZ (5a), dual-arm (8a), dual-arm mono-OPTZ (9a) and dual-arm di-OPTZ (10a) caged esters in CAN (10−5 M), and (b) single-arm (4a) and (c) mono-OPTZ (5a) caged esters in different solvents (10−5 M). | ||
The solvent effects on the absorption and emission spectra were investigated for single-arm caged ester 4a and mono-oxidized caged ester 5a (mono-OPTZ). The effects of different solvents on the absorption spectra of 4a and 5a were found to be insignificant (Fig. S31b and c, ESI†), whereas we found a pronounced solvent effect on the emission spectra of 4a and 5a. It was observed that, in nonpolar solvents (benzene, hexane), the fluorescence intensity of 4a was comparatively higher, with an emission maximum at 427 nm, whereas in polar solvents (e.g., acetonitrile, tetrahydrofuran, methanol, etc.), it exhibited a comparatively lower fluorescence intensity along with a red-shifted emission maximum in the range of 439–460 nm (Fig. 1b). The above results indicate that the excited states of caged esters are stabilized in polar solvents compared to their ground states.
However, the n–π* transition due to the thiocarbonyl group of the oxidized phenothiazine caged ester 5a shifted towards a shorter wavelength (blue-shift) as the polarity of the solvent was increased (Fig. 1c). The reason is that the non-bonding electrons of oxygen interact strongly (hydrogen bonding) with the hydrogen of polar protic solvents like EtOH, MeOH, EtOAc, etc. Hence, the non-bonding electrons are easily promoted to π*, increasing the energy gap between n–π*.16
The absorption and emission maxima, Stokes shifts, and fluorescence quantum yields of all the single-(4a–e) and dual-arm caged esters (8a–c, 12a and b), mono-oxidized caged esters (5a, 9a), and di-oxidized caged ester (10a) are summarized in Table S1 (ESI†). The fluorescence quantum yields (Φf) of all the caged esters in acetonitrile at room temperature were in the range of 0.026 ≤ Φf ≤ 0.038. The fluorescence quantum yields of all the caged esters were calculated using 9,10-diphenylanthracene as a standard (Φf = 0.95 in ethanol).17
Considering our main interest in studying the application of phenothiazine as a PRPG, we irradiated all the caged esters (1 × 10−4 M) individually in ACN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) using a 125 W medium pressure Hg lamp at two different irradiation wavelengths (≥365 and ≥410 nm). We found that the single- and dual-arm (same and different) caged esters released the corresponding carboxylic acids in high chemical (93–96%) and good photochemical quantum (Φp = 0.14–0.20) yields at 365 nm and 410 nm (Table 1). However, in the case of 410 nm, the caged esters required a longer irradiation time to release their corresponding carboxylic acids. The photochemical quantum yield (Φp) was calculated using potassium ferrioxalate as an actinometer18 (see page 39 in the ESI†). Interestingly, we observed insignificant photorelease for the oxidized phenothiazine 5a (mono-OPTZ) and 10a (di-OPTZ) caged esters.
:1 v/v) using a 125 W medium pressure Hg lamp at two different irradiation wavelengths (≥365 and ≥410 nm). We found that the single- and dual-arm (same and different) caged esters released the corresponding carboxylic acids in high chemical (93–96%) and good photochemical quantum (Φp = 0.14–0.20) yields at 365 nm and 410 nm (Table 1). However, in the case of 410 nm, the caged esters required a longer irradiation time to release their corresponding carboxylic acids. The photochemical quantum yield (Φp) was calculated using potassium ferrioxalate as an actinometer18 (see page 39 in the ESI†). Interestingly, we observed insignificant photorelease for the oxidized phenothiazine 5a (mono-OPTZ) and 10a (di-OPTZ) caged esters.
:1 v/v) solvent
| Caged ester | ≥365 nm | ≥410 nm | ||
|---|---|---|---|---|
| % of deprotectiona | Quantum yieldb (Φp) | % of deprotectionc | Quantum yieldd (Φp) | |
| a % of deprotection of the caged ester with respect to the initial concentration after 30 min of irradiation time. b Photochemical quantum yield at λ ≥ 365 nm (error limit within ±5%). c % of deprotection of the caged ester with respect to initial concentration after 240 min of irradiation time. d Photochemical quantum yield at λ ≥ 410 nm (error limit within ±5%). | ||||
| 4a | 95 | 0.205 | 80 | 0.152 |
| 4b | 94 | 0.202 | 78 | 0.148 |
| 4c | 96 | 0.207 | 75 | 0.142 |
| 4d | 93 | 0.200 | 77 | 0.146 |
| 4e | 94 | 0.202 | 75 | 0.142 |
| 5a | <5 | — | <5 | — |
| 8a | 96 | 0.207 | 76 | 0.144 |
| 8b | 95 | 0.205 | 74 | 0.140 |
| 8c | 92 | 0.201 | 72 | 0.139 |
| 9a | <5 | — | <5 | — |
| 10a | <5 | — | <5 | — |
| 12a | 95 | 0.205 | 74 | 0.140 |
| 12b | 96 | 0.207 | 75 | 0.142 |
The photorelease ability of single-arm caged ester 4a was monitored by reverse-phase (RP) HPLC at different irradiation intervals (Fig. 2a). The HPLC chromatogram showed a gradual decrease of the peak corresponding to 4a at the retention time (tR) 4.65 min, with an increase in irradiation time, indicating the photodecomposition of caged ester 4a. In addition, we also noted a gradual increase of two new peaks at tR 2.77 and 3.91 min, which correspond to released carboxylic acids, i.e., p-anisic acid and the photoproduct 3, (10-ethyl-10H-phenothiazin-3-yl)methanol, respectively. The corresponding photoproduct (3) and released anisic acid were confirmed by injecting authentic samples and isolation and characterization using 1H NMR analysis (Fig. S32, ESI†). Furthermore, we monitored the photorelease of our single-arm caged ester 4a using fluorescence spectroscopy (Fig. 2b). Also, we monitored the photorelease of dual-arm (same) caged ester 8a (1 × 10−4 M) in ACN/H2O (1:1 v/v) at 365 nm by 1H NMR (Fig. S33, ESI†) and HRMS (Fig. S34, ESI†) spectroscopy at different irradiation times (0–30 min).
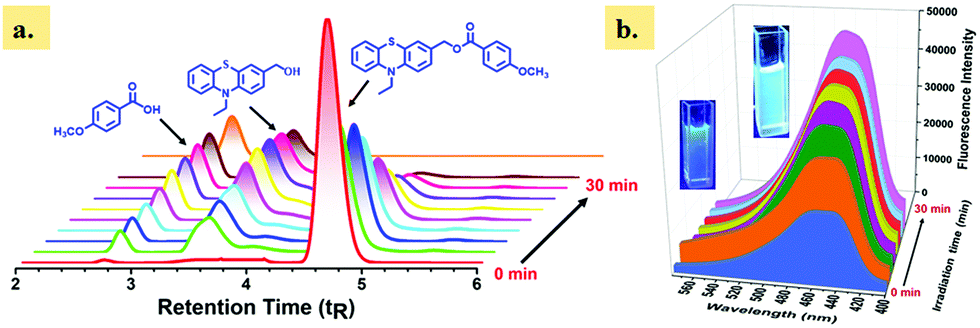 | ||
| Fig. 2 (a) HPLC profiles for the photolysis of the caged ester 4a (1 × 10−4 M) in ACN/H2O (1:1 v/v) at different intervals of time (0–30 min). Irradiation wavelength: λ ≥ 365 nm. (b) Change in the fluorescence spectral profiles of 4a with increasing irradiation time (excitation wavelength 310 nm). | ||
Photoirradiation of dual-arm (different) caged ester 12a for 20 min resulted in the release of two different carboxylic acids, i.e., p-anisic acid and toluic acid, and the formation of photoproduct 7, which was monitored by HRMS spectroscopy (Fig. S35, ESI†). HRMS analysis found that all the possible intermediates (11, 7) for the stepwise mechanism were present in the reaction mixture along with released carboxylic acids.
The hydrolytic stability of all the caged esters was also tested by keeping them individually in the dark in acetonitrile:water (1:1 v/v) for 30 days. We observed only 8–10% decomposition of the caged esters (Table S2, ESI†).
Based on the literature,19 and photolysis quenching experiments (Fig. S36, ESI†), we suggest a possible stepwise mechanism for the photolysis of phenothiazine caged esters (Scheme 2a). As a representative example, irradiation of dual-arm (different) caged ester 12a using λ ≥365 nm light in aqueous ACN leads to its singlet excited state (S1), which then undergoes heterolysis of the benzylic C–O bond to produce a tight ion pair. The cation part reacts with the solvent to form photoproduct 11, and the anion part abstracts a proton to form the released carboxylic acid (p-anisic acid). The photoproduct (11) formed in situ then absorbs light, and gets excited to its singlet state and releases the second carboxylic acid (toluic acid) along with the formation of the final photoproduct (7) by following a similar mechanism like the first release.
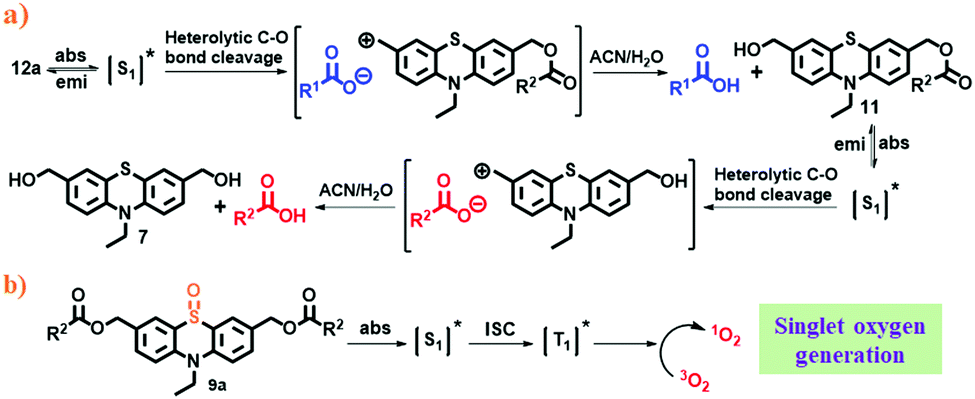 | ||
| Scheme 2 (a) Possible photorelease mechanism of PTZ caged esters and (b) mechanism for singlet oxygen generation of mono-OPTZ. | ||
On the other hand, mono-oxidized phenothiazine caged ester (9a) gets excited to its singlet state upon irradiation. It then undergoes ISC to its triplet excited state and transfers its energy to molecular oxygen, resulting in the generation of singlet oxygen (Scheme 2b).
TD-DFT calculations were carried out (for details, see page no. 39–42 in the ESI†) to rationalize the difference in the photochemistry between an oxygen-free phenothiazine caged ester (4d) and its oxidized caged ester (mono-OPTZ of 4d).
We noticed that the electron density of the LUMO of the mono-OPTZ of 4d has more distribution onto the thiocarbonyl group, while the electron density of the LUMO of the single-arm caged ester (4d) is dispersed towards the ester group (Fig. S39 and S40, ESI†). Thus, mono-OPTZ of 4d exhibits a relatively larger overlap between the HOMO and LUMO, leading to stronger oscillator strength (f) for the S0–S1 transition and a larger ΔEST (Table S3, ESI†). In the case of the single-arm caged ester (4d), the energy gap between S1 and the nearest lower energy triplet state (T3) is considerably small (ΔES1–T3 = 0.04 eV); thus, this similar energy level leads to an effective RISC channel from T3 to S1 (Fig. 3a).20 On the other hand, an ISC is expected to be favorable from the S1 state of the oxidized phenothiazine caged ester of 4d to T3 because the ΔES1–T3 gap is only 0.32 eV (Fig. 3b). Thus, our computations support photocleavage from the singlet excited state and singlet oxygen generation from the triplet excited state. Also, we calculated the energy barriers for photorelease after implementing solvation energies in the SMD model, which are 11.36 kcal mol−1 for single-arm caged ester 4d and 24.49 kcal mol−1 for the mono-oxidized caged ester of 4d. The lower barrier height (13.13 kcal mol−1 lower) in the case of 4d indicates a faster photorelease.
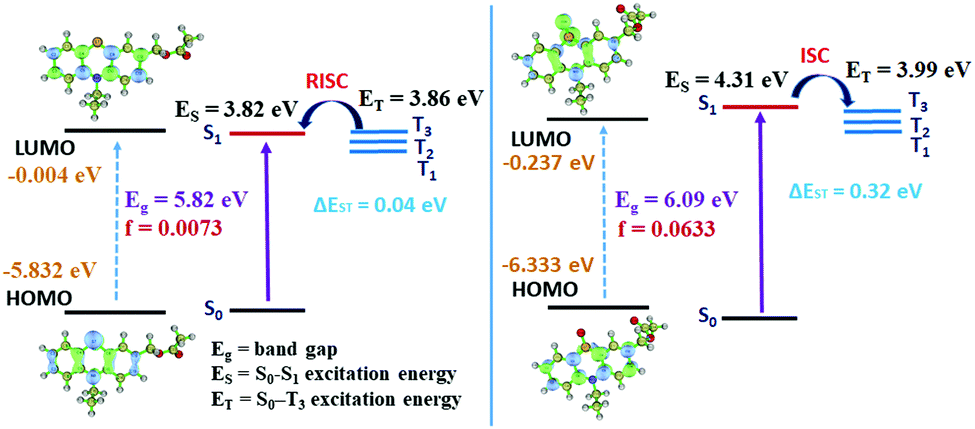 | ||
| Fig. 3 The energy levels of the singlet and triplet excited states of PTZ and mono-OPTZ caged esters and the exciton decay process after photoexcitation. | ||
To support the photochemistry of oxidized phenothiazine caged esters occurring from the triplet excited state, we checked their singlet oxygen generation ability by performing DPBF photodegradation. We observed that the decrease in the absorption maximum of DPBF at 415 nm with an increase in irradiation time indicated singlet oxygen generation by our mono-oxidized phenothiazine caged ester (5a) and di-oxidized phenothiazine caged ester (10a), respectively. The quantum yields for singlet oxygen generation of 5a (mono-OPTZ) and 10a (di-OPTZ) were calculated as 0.28 and 0.24, respectively (rose bengal was used as a reference with a ΦΔ of 0.53)21 (Fig. S37, ESI†).
In summary, we demonstrated phenothiazine as an efficient PRPG for the single and dual (same and different) release of carboxylic acids from the singlet excited state in the visible wavelength region. The phenothiazine caged esters exhibited green fluorescence with a large Stokes shift. Interestingly the oxidized phenothiazine caged esters generated singlet oxygen rather than photorelease due to the thiocarbonyl group resulting in efficient ISC to its triplet excited state, supported by DFT calculations.
We thank DST SERB (Grant No. DIA/2018/000019) for the financial support and DST (SR/FST/CSII-026/2013) for the 400 MHz NMR spectrometer. We acknowledge the Supercomputing Facility “PARAM Shakti” at IIT Kharagpur established under National Supercomputing Mission (NSM), Government of India. M. Ojha is thankful to UGC-New Delhi for the fellowship and M. Banerjee acknowledges IIT Kharagpur for the fellowship.
Conflicts of interest
There are no conflicts to declare.References
- (a) R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135 CrossRef CAS PubMed; (b) D. Kand, P. Liu, M. X. Navarro, L. J. Fischer, L. R. Noori, D. F. Morvinski, A. H. Winter, E. W. Miller and R. Weinstain, J. Am. Chem. Soc., 2020, 142, 4970 CrossRef CAS PubMed.
- S. S. Agasti, A. Chompoosor, C. C. You, P. Ghosh, C. K. Kim and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 5728 CrossRef CAS PubMed.
- N. C. Fan, F. Y. Chenge, J. A. Ho and C. S. Yeh, Angew. Chem., Int. Ed., 2012, 51, 8806 CrossRef PubMed.
- C. M. J. Hu and L. Zhang, Biochem. Pharmacol., 2012, 83, 1104 CrossRef CAS PubMed.
- Y. Li, T. Thambi and D. S. Lee, Adv. Healthcare Mater., 2018, 7, 1700886 CrossRef PubMed.
- Y. Venkatesh, H. K. Srivastava, S. Bhattacharya, M. Mehra, P. K. Datta, S. Bandyopadhyay and N. D. P. Singh, Org. Lett., 2018, 20, 2241 CrossRef CAS PubMed.
- P. T. Wong, S. Tang, J. Cannon, J. Mukherjee, D. Isham, K. Gam, M. Payne, S. A. Yanik, J. R. Baker and S. K. Choi, ChemBioChem, 2017, 18, 126 CrossRef CAS PubMed.
- A. Chaudhuri, Y. Venkatesh, K. K. Behara and N. D. P. Singh, Org. Lett., 2017, 19, 1598 CrossRef CAS PubMed.
- A. Sikder, M. Banerjee, T. Singha, S. Mondal, P. K. Datta, A. Anoop and N. D. P. Singh, Org. Lett., 2020, 22, 6998 CrossRef CAS PubMed.
- (a) Y. Hua, S. Chang, D. Huang, X. Zhou, X. Zhu, J. Zhao, T. Chen, W. Y. Wong and W. K. Wong, Chem. Mater., 2013, 25, 2146 CrossRef CAS; (b) Y. Lu, H. Song, X. Li, H. Ågren, Q. Liu, J. Zhang, X. Zhang and Y. Xie, ACS Appl. Mater. Interfaces, 2019, 11, 5046 CrossRef CAS PubMed.
- (a) B. Varga, Á. Csonka, A. Csonka, J. Molnár, L. Amaral and G. Spengler, Anticancer Res., 2017, 37, 5983 CAS; (b) K. Pluta, M. Jeleń, B. M. Młodawska, M. Zimecki, J. Artym, M. Kocięba and E. Zaczyńska, Eur. J. Med. Chem., 2017, 138, 774 CrossRef CAS PubMed.
- (a) J. Gai, C. Chen, J. Huang, J. Sheng, W. Chen and X. Song, Tetrahedron Lett., 2020, 61, 152038 CrossRef CAS; (b) X.-Y. Qi, S.-J. Liu, Y.-Q. Hao, J.-W. Sun and S. Chen, Spectrochim. Acta, Part A, 2020, 227, 117675 CrossRef PubMed.
- W. Qiu, X. Cai, M. Li, L. Wang, Y. He, W. Xie, Z. Chen, M. Liua and S.-J. Su, J. Mater. Chem. C, 2021, 9, 1378 RSC.
- X. Ma, R. Gu, L. Yu, W. Han, J. Li, X. Li and T. Wang, Polym. Chem., 2017, 8, 6134 RSC.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933 CrossRef CAS PubMed.
- A. I. Adeogun, N. W. Odozi, N. O. Obiegbedi and O. S. Bello, Afr. J. Biotechnol., 2008, 7, 2736 CAS.
- J. V. Morris, M. A. Mahaney and J. R. Huberr, J. Phys. Chem., 1976, 80, 969 CrossRef CAS.
- J. N. Dema, W. D. Bowman, E. F. Zalewsk and R. A. Velapold, J. Phys. Chem., 1981, 85, 2766 CrossRef.
- R. Schmidt, D. Geissler, V. Hagen and J. Bendig, J. Phys. Chem. A, 2007, 111, 5768 CrossRef CAS PubMed.
- D. Hu, L. Yao, B. Yang and Y. Ma, Philos. Trans. R. Soc., A, 2015, 373, 0318 CrossRef PubMed.
- N. E. Elezcanoa, V. M. Martínez, E. P. Cabrerab, C. F. A. G. Duránb, I. L. Arbeloaa and S. Lacombe, RSC Adv., 2016, 6, 41991 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc06950k |
| This journal is © The Royal Society of Chemistry 2022 |