
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcurrent base and silver(I) catalysis pulsed by fuel acid†
Isa
Valiyev‡
 ,
Amit
Ghosh‡
,
Indrajit
Paul
and
Michael
Schmittel
*
,
Amit
Ghosh‡
,
Indrajit
Paul
and
Michael
Schmittel
*
Center of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, Siegen D-57068, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 7th January 2022
Abstract
Treatment of a crown-ether receptor and a silver(I)-loaded cyclam derivative (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NetState-I) with a fuel acid reversibly afforded the protonated cyclam and the silver(I)-loaded crown ether (NetState-II). While NetState-I was catalytically OFF, a base-catalysed Michael addition and a silver(I)-catalysed oxime cyclisation reaction was pulsed under dissipative conditions in NetState-II.
NetState-I) with a fuel acid reversibly afforded the protonated cyclam and the silver(I)-loaded crown ether (NetState-II). While NetState-I was catalytically OFF, a base-catalysed Michael addition and a silver(I)-catalysed oxime cyclisation reaction was pulsed under dissipative conditions in NetState-II.
Multicellular organisms maintain life out of equilibrium through autonomous chemical networks that involve information handling1 and storage2 as well as energy transfer.3 In order to advance autonomy in artificial systems4 or precise control in the time domain,5 it is desirable to interconnect (by chemical communication) molecular ensembles and to operate them under dissipative conditions. Implementation of the time domain has recently stimulated the development of chemically fuelled systems.6–8 Based on fuel, a handful of systems involving either molecular communication,9,10 or catalysis have become known,11,12 however, dissipatively pulsed catalysis within a communicating network is unprecedented.
Building on communication protocols involving metal-ion pulsing,13 we demonstrate a fully reversible system with (i) fast signalling between interdependent components that allows (ii) concurrent orthogonal double catalysis, all under dissipative conditions. The design was based on two well-defined networked states (NetStates). NetState-I encompasses the anthracene-appended cyclam 1, the anthracene-appended azacrown ether 2 and silver(I) ions in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (Scheme 1) and requires an incomplete self-sorting14 affording [Ag(1)]+ and free 2. Upon acid addition, either TFA or a fuel acid (= an acid that, after its deprotonation and concomitant decarboxylation, recaptures the proton6), NetState-II is formed, which involves the protonated cyclam 1·H+ and the complex [Ag(2)]+. It is revealed that both species in NetState-II are catalytically active, whereas there is no catalysis operating in NetState-I.
:1:1 ratio (Scheme 1) and requires an incomplete self-sorting14 affording [Ag(1)]+ and free 2. Upon acid addition, either TFA or a fuel acid (= an acid that, after its deprotonation and concomitant decarboxylation, recaptures the proton6), NetState-II is formed, which involves the protonated cyclam 1·H+ and the complex [Ag(2)]+. It is revealed that both species in NetState-II are catalytically active, whereas there is no catalysis operating in NetState-I.
 | ||
| Scheme 1 Communication between two anthracene-appended aza crowns with chemical fuel. NetState-II has the potential to act as a double catalyst. | ||
Ligand 1 was readily synthesised from 9-(bromomethyl) anthracene and cyclam by a substitution reaction, whereas ligand 2 was available from former work.13 The cyclam-appended ligand 1 has been selected because it binds Ag+ (logK = 8.43 ± 0.41; ESI,† Fig. S34) much stronger than ligand 2 (logK = 3.66 ± 0.29).15
As foreseen, after addition of silver(I) to a mixture of ligands 1 and 2, an incomplete self-sorting was observed leading to [Ag(1)]+ + 2 denoted here as NetState-I. Upon addition of 1 equiv. of TFA to NetState-I, the 1H-NMR, (Fig. 1) showed complete translocation of Ag+ from [Ag(1)]+ to luminophore 2. Signals of protons a-H, b-H, and f-H of [Ag(1)]+ shifted from 8.47, 8.07, and 4.58 ppm to 8.56, 8.10, and 4.75 ppm, respectively, confirming formation of 1·H+. Association of Ag+ to 2 was monitored by the appearance of a new peak for proton 6-H at 7.05 ppm as well as the disappearance of the signal at 6.70 ppm. The ESI-MS proved presence of complex [Ag(2)]+ (ESI,† Fig. S28) by peaks at m/z = 682.2 (singly charged) and of 1·H+ by a signal at m/z = 391.5 (singly charged) (ESI,† Fig. S23). Upon addition of 1 equiv. of DBU to NetState-II, a clean reverse translocation of Ag+ from luminophore 2 to ligand 1 was achieved.
 | ||
| Fig. 1 Comparison of 1H NMR spectra (400 MHz, CDCl3, 298 K) of (a) NetState-I: [Ag(1)]+ + 2; (b) after addition of 1 equiv. of TFA; (c) after addition of 1 equiv. of DBU. | ||
The reversibility of the conversion of NetState-I ⇆ NetState-II was easily traceable by fluorescence spectroscopy, because the yellow emission of 2 (λ = 554 nm) shifted to a pale blue (λ = 472 nm), i.e. the band of [Ag(2)]+ (Fig. 2a). In addition, the intensity of the emission of 1 at λ = 420 nm increased 5-fold due to protonation. After the addition of DBU, the emission reversed back to λ = 554 nm. The emission changes between the two states were reproduced over three cycles (Fig. 2b) with a small decline in emission intensity, most likely due to the formation of protonated DBU.
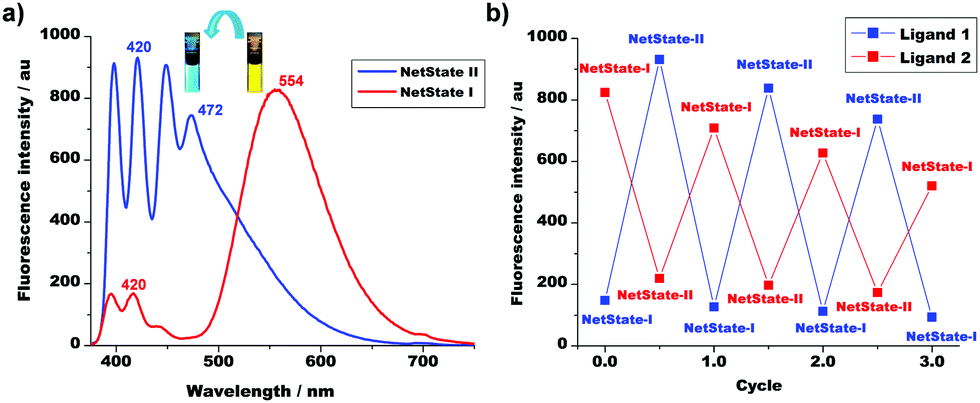 | ||
| Fig. 2 (a) Emission changes after addition of TFA to [Ag(1)]+ + 2 (c = 7.69 × 10−6 M). (b) Reversibility of the networked system in presence of TFA and DBU over three cycles monitored by fluorescence. Blue and red lines indicate the changes of fluorescence intensity of ligands 1 and 2 at λ = 420 and 554 nm, respectively. | ||
To probe the fuel-driven interconversion of [Ag(1)]+ + 2 ⇆ 1·H+ + [Ag(2)]+, an equimolar amount of 2-cyano-2-(4-methoxyphenyl)propanoic acid (3)16 was added to NetState-I. As elaborated by Di Stefano, acid 3 behaves as a chemical fuel because after deprotonation and concomitant decarboxylation of 4 the resultant anion 5 acts as a strong base thus reclaiming protons from the system to form 6 (Scheme 2).
For running the fuelled translocation, ligands 1 (c = 3.98 × 10−3 M), 2 and silver(I) ions (1:1:1) were mixed in 450 μL of CD2Cl2, then an equimolar amount of acid 3 was added. The disappearance of the 1H NMR signals at 7.46 and 6.85 ppm and the appearing peaks at 7.28 and 6.91 ppm are indicative of the conversion of 3 to 6. Five minutes after addition of 3, the 1H-NMR spectrum showed full conversion of NetState-I to NetState-II (ESI,† Fig. S33). For instance, the signals of protons f-H of [Ag(1)]+ and of 6-H of ligand 2 shifted from 4.58 to 4.75 ppm and from 6.70 to 7.05 ppm, respectively. Over the ensuing 120 min, complete disappearance of the 1H-NMR signals of NetState-II and return of the system back to NetState-I was monitored by the diagnostic peaks of 1·H+ at 4.75 ppm and of waste 6 at 6.91 ppm. The time dependence of the fuel-driven transformation was also followed by fluorescence (Fig. 3a). After the addition of one equiv. of fuel 3 (c = 1.54 × 10−3 M) to NetState-I (i.e.1:2:Ag+ = 1:1:1 ratio) at room temperature, the emission of 2 shifted within the first minute from λ = 554 to 472 nm. Thus, protonation of [Ag(1)]+ by acid 3 apparently culminated in the pulsed formation of [Ag(2)]+. Simultaneously, the fluorescence intensity at λ = 420 nm increased by 4.5-fold due to the conversion of [Ag(1)]+ to 1·H+. After decarboxylation of 4 and deprotonation of 1·H+ by 5, full back translocation of Ag+ was detected by the emission shifting back to the original position within ca. 140 min (Fig. 3a). In the course of three repetitive fuelled cycles, each started by the addition of 1 equiv. of acid 3, a sawtooth profile of 1·H+ was generated (Fig. 3b). As seen from the fluorescence, the intensity spiked after each fuel addition and then dropped slowly. With each cycle, the reversal to NetState-I slightly increased in time (140 vs. 150 and 158 min). Moreover, the relative top level of the emission slightly decreased, most probably owing to the formation of fuel waste 6 that acted as quencher (ESI,† Fig. S35 and Scheme 2).
 | ||
| Fig. 3 (a) Communication between both NetStates after addition of fuel monitored by fluorescence (c = 7.69 × 10−6 M). (b) Reversibility of the chemical network in presence of fuel over three cycles monitored by fluorescence intensity at λ = 420 nm. | ||
 | ||
| Scheme 2 The decarboxylation pathway of fuel 3 in presence of a base. | ||
Based on former work, we expected silver(I) to be active in catalysis when it is coordinated to the azacrown unit as in [Ag(2)]+,15 but importantly [Ag(1)]+ should be catalytically inactive. After a screening of possible reactions, we found that [Ag(2)]+ (10 mol%) catalysed the cyclisation of substrate 10 (100 mol%) in CD2Cl2:CD3CN = 50:1 (at room temp. within 15 min) affording the isoquinoline-2-oxide 11 in (70 ± 2)% (Scheme 3b), whereas in presence of [Ag(1)]+, 2, or 1·H+ (10 mol%) formation of 11 was completely off. Less expected was the fact that protonated cyclam 1·H+ proved active in a base-catalysed Michael addition. Rewardingly, when substrates 7 and 8 (100 mol%) were reacted with 1·H+ (10 mol%) in CD2Cl2:CD3CN = 50:1 at room temp. for 15 min, the substituted malononitrile 9 formed in (40 ± 2)% (Scheme 3a). In contrast, the Michael addition did not proceed in presence of 2, [Ag(2)]+ or [Ag(1)]+ (10 mol%) as potential catalysts.17
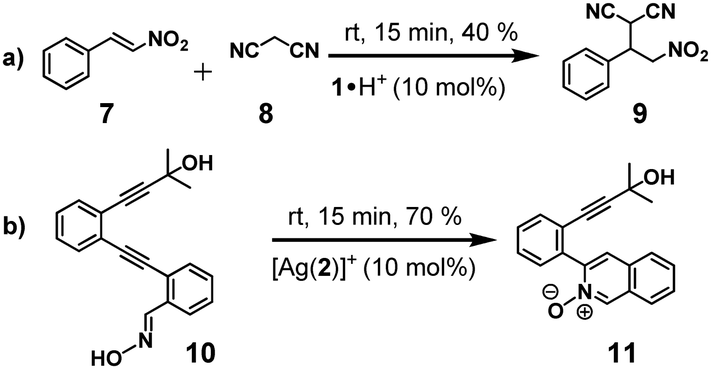 | ||
| Scheme 3 The two catalytic reactions used in this work. (a) Michael addition; (b) cyclisation reaction. | ||
The surprising catalysis of the Michael addition by 1·H+ might in principle be alternatively explained by the intermediate formation of the ketenimine NC–CHCNH, i.e. the “enol tautomer” of 8, and thus an acid catalysed reaction. To examine the possible involvement of acid catalysis we tested the reaction of compounds 7 and 8 (1:1 ratio, in CD2Cl2, 2 h, room temp.) in presence of various acids. Neither acid 3 nor TFA nor higher protonated states of cyclam 1 (Fig. 4) were active in the catalysis. Moreover, we were able to demonstrate that erratically formed spurious amounts of cyclam 1 were unable to exist in presence of [Ag(2)]+ because any free 1 immediately afforded [Ag(1)]+ (ESI,† Page S47 and S48). In sum, we conclude that 1·H+ despite carrying a proton, uses some of the remaining basic sites for a base catalysed Michael addition.
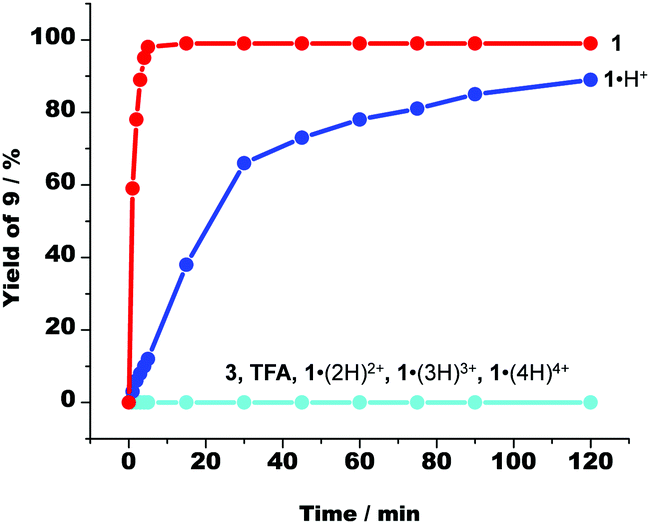 | ||
| Fig. 4 The time-dependent formation of Michael addition product 9 in presence of a variety of acids and of cyclam 1. 1·(2H)2+, 1·(3H)3+ and 1·(4H)4+ represent the multiply protonated cyclam 1. | ||
After successfully identifying the catalytic activity of 1·H+ and [Ag(2)]+, both catalytic processes were studied in the networked system. Firstly, ligands 1, 2, and silver(I) were mixed in 450 μL of CD2Cl2 in a 1:1:1 ratio in order to obtain NetState-I, then 10 equiv. of reactants 7, 8, and 10 were added. As expected, both the Michael addition and oxime cyclisation were shut down (OFF). The reactions are OFF, because silver(I) ions in [Ag(1)]+ are catalytically inactive and there is no free reactive NH unit available. After the addition of 1 equiv. of TFA to NetState-I, NetState-II was furnished with both complexes being in the catalytically active state. Remarkably, yields of both the Michael addition and the oxime cyclisation in NetState-II were close to those of the individual experiments. For instance, after 15 min, the cyclisation furnished 69 (±2)% of product 11, monitored by a new peak at 8.86 ppm (singlet) in the 1H-NMR spectrum, whereas the Michael addition yielded 38 (±2)% of product 9 as indicated by a multiplet at 5.06–4.93, a doublet at 4.47, and a multiplet at 4.17–4.10 ppm.
To examine the catalytic activity of both states in presence of chemical fuel over several cycles (Fig. 5), we started with 10 mol% of NetState-I and 100 mol% of reactants 7, 8, and 10. The mixture was first monitored by 1H-NMR spectroscopy at room temperature for 100 min, but no product formation was detected. After the addition of 3 (10 mol%), the system turned to NetState-II delivering 9% of both 9 and 11 after 5 min. After 10 min, the yields of 9 and 11, i.e. the Michael addition and cyclisation, were increased to 19% and 31%, respectively. After 20 min the speed of both processes decreased since some amount of chemical fuel had converted to 6; at this time 41% of 9 and 64% of product 11 had formed. Finally, after 50 min, the Michael addition and the cyclisation furnished 55% and > 99% yield, respectively. After 120 min, with all the fuel 3 being consumed, the system reversed to NetState-I and the Michael addition reaction showed a yield of 60%. For another fuelled cycle, the consumed amounts of reactants were supplemented. Monitoring for another 100 min at r.t. by 1H-NMR spectroscopy demonstrated the system to be completely in the OFF mode. In the second cycle, products 9 and 11 were obtained in 57% and >99% yield respectively. A small decrease in the yield of 9 (60% → 57% → 52%) was observed over three cycles.
 | ||
| Fig. 5 Time dependence of product formation in presence of fuel 3 monitored by 1H NMR spectroscopy (CD2Cl2, 298 K) with 1,3,5-trimethoxybenzene as an internal reference. Black and red dots represent formation of 9 and 11, respectively. | ||
In conclusion, we demonstrated a switchable multicomponent network for double catalysis that was based on the fully reversible communication between two anthracene-appended azacrowns via translocation of Ag(I) ions, a process requiring a high level of self-sorting.18 Interconversion between NetState-I and NetState-II was realised by addition of stoichiometric amounts of acid and base or alternatively of chemical fuel acid (over three cycles). The use of chemical fuel 3 allowed simultaneous triggering of two orthogonal reactions under dissipative conditions. Notably, acid addition led to both base and silver(I) catalysis in concert.
This paper is dedicated to Prof. Dr A. Maercker on the occasion of his 90th birthday. We are indebted to the Deutsche Forschungsgemeinschaft (Schm 647/22-1) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- U. Pischel, J. Andréasson, D. Gust and V. F. Pais, Information processing with molecules − Quo vadis?, ChemPhysChem, 2013, 14, 28–46 CrossRef CAS PubMed.
- C. J. Bashor, N. Patel, S. Choubey, A. Beyzavi, J. Kondev, J. J. Collins and A. S. Khalil, Complex signal processing in synthetic gene circuits using cooperative regulatory assemblies, Science, 2019, 364, 593–597 CrossRef CAS PubMed.
- A. I. Brown and D. A. Sivak, Theory of Nonequilibrium Free Energy Transduction by Molecular Machines, Chem. Rev., 2020, 120, 434–459 CrossRef CAS PubMed.
- R. Merindol and A. Walther, Materials learning from life, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC.
- M. A. Cardona, R. Chen, S. Maiti, I. Fortunati, C. Ferrante, L. Gabrielli, K. Das and L. J. Prins, Time-gated fluorescence signalling under dissipative conditions, Chem. Commun., 2020, 56, 13979–13982 RSC.
- C. Biagini and S. Di, Stefano, Abiotic Chemical Fuels for the Operation of Molecular Machines, Angew. Chem., Int. Ed., 2020, 59, 8344–8354 CrossRef CAS PubMed.
- K. Das, L. Gabrielli and L. J. Prins, Chemically Fueled Self-Assembly in Biology and Chemistry, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- L. S. Kariyawasam, M. M. Hossain and C. S. Hartley, The Transient Covalent Bond in Abiotic Nonequilibrium Systems, Angew. Chem., Int. Ed., 2021, 60, 12648–12658 CrossRef CAS PubMed.
- O. R. Maguire, A. S. Y. Wong, J. H. Westerdiep and W. T. S. Huck, Early warning signals in chemical reaction networks, Chem. Commun., 2020, 56, 3725–3728 RSC.
- (a) A. Goswami, S. Saha, P. Biswas and M. Schmittel, (Nano)mechanical Motion Triggered by Metal Coordination: from Functional Devices to Networked Multicomponent Catalytic Machinery, Chem. Rev., 2020, 120, 125–199 CrossRef CAS PubMed; (b) M. Schmittel, Dynamic Functional Molecular Systems: from Supramolecular Structures to Multi-component Machinery and to Molecular Cybernetics, Isr. J. Chem., 2019, 59, 197–208 CrossRef CAS.
- C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Dissipative Catalysis with a Molecular Machine, Angew. Chem., Int. Ed., 2019, 58, 9876–9880 CrossRef CAS PubMed.
- B. A. Grzybowski and W. T. S. Huck, The Nanotechnology of Lifeinspired Systems, Nat. Nanotechnol., 2016, 11, 585–592 CrossRef CAS PubMed.
- A. Ghosh, I. Paul and M. Schmittel, Time-Dependent Pulses of Lithium Ions in Cascaded Signaling and Out-of-Equilibrium (Supra)molecular Logic, J. Am. Chem. Soc., 2019, 141, 18954–18957 CrossRef CAS PubMed.
- (a) Z. He, W. Jiang and C. A. Schalley, Integrative self-sorting: a versatile strategy for the construction of complex supramolecular architecture, Chem. Soc. Rev., 2015, 44, 779–789 RSC; (b) M. L. Saha and M. Schmittel, Degree of molecular self-sorting in multicomponent systems, Org. Biomol. Chem., 2012, 10, 4651–4684 RSC; (c) W. Jiang and C. A. Schalley, Integrative self-sorting is a programming language for high level self-assembly, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10425–10429 CrossRef CAS PubMed.
- I. Paul, N. Mittal, S. De, M. Bolte and M. Schmittel, Catch-Release System for Dosing and Recycling Silver(I) Catalyst with Status of Catalytic Activity Reported by Fluorescence, J. Am. Chem. Soc., 2019, 141, 5139–5143 CrossRef CAS PubMed.
- C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. Di, Stefano, Variations in the fuel structure control the rate of the back and forth motions of a chemically fueled molecular switch, Chem. Sci., 2018, 9, 181–188 RSC.
- (a) R. Singha and T. Goswami, Acid catalyzed 1,2 Michael addition reaction: a viable synthetic route in designing fullerene core starlike macromolecule, J. Phys. Org. Chem., 2008, 21, 225–236 CrossRef; (b) T. C. Wabnitz and J. B. A. Spencer, General, Brønsted Acid-Catalyzed Hetero-Michael Addition of Nitrogen, Oxygen, and Sulfur Nucleophiles, Org. Lett., 2003, 5, 2141–2144 CrossRef CAS PubMed.
- M. Schmittel, From self-sorted coordination libraries to networking nanoswitches for catalysis, Chem. Commun., 2015, 51, 14956–14968 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterisations, spectral data, UV-vis titrations data. See DOI: 10.1039/d1cc06398g |
| ‡ Isa Valiyev and Amit Ghosh contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |