
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChromonic nematic liquid crystals in a room-temperature ionic liquid†
Jose Rodrigo
Magana
a,
Adria
Pérez-Calm
ab and
Carlos
Rodriguez-Abreu
 *ab
*ab
aInstitute for Advanced Chemistry of Catalonia (IQAC), Spanish National Research Council (CSIC), Jordi Girona 18-26 08034, Barcelona, Spain. E-mail: carlos.rodriguez@iqac.csic.es
bNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Jordi Girona 18-26 08034, Barcelona, Spain
First published on 4th January 2022
Abstract
Planar multiaromatic molecules hierarchically and selectively arrange into nematic chromonic liquid crystals in the room temperature ionic liquid 2-hydroxyethylammonium formate. In a proof of concept, these liquid crystals were used as reaction media to produce mesostructured silica materials under mild biomimetic conditions. Several other applications are envisaged.
Lyotropic chromonic liquid crystals (LCLCs) are hierarchical self-assemblies formed in a solvent in which planar, multi-aromatic molecules with solubilizing groups at their periphery spontaneously arrange by non-covalent interactions into higher-order nanostructures.1,2 Potential applications of LCLCs include polarizers,3,4 biosensors,5 or scaffolds to produce ordered (in)organic (nano)materials.6–9 LCLCs are particularly interesting as alternatives to thermotropic liquid crystals as they require considerably lower mesogen content and their properties can be additionally tuned by varying the concentration.3,10–14 Despite their potential interest, most LCLCs reported so far exist only in aqueous media and some limited organic solvents.2,15–17 This results in poor conductivity and/or solvent evaporation, which might hamper their application in e.g., electronics.
In the present work, we envision a new type of LCLC using a room-temperature ionic liquid (IL) as a solvent. ILs have emerged as an alternative to conventional polar solvents due to their environmentally benign nature, along with their superb physicochemical properties, such as high conductivity, thermal stability, and low vapor pressure.18 While surfactants and block copolymers are known to self-assemble in ILs and find applications in a wide range of fields,19–22 to our knowledge, there are no reports in the literature on the occurrence of lyotropic chromonic columnar mesophases in ILs. These ionic liquid-based LCLCs would behave basically as thermotropic mesophases due to the low vapor pressure of ILs, but with much lower mesogen content, which is beneficial in terms of cost and processability. Advantageously, the structural and electronic properties can be tuned without the need for complex chemical modifications by just varying the concentration in the system.
Finding suitable aggregation media for LCLC mesogens is a challenging task. For an LCLC to form, the chosen solvent must, on the one hand, be able to dissolve an appropriate amount of mesogen, whereas, on the other hand, it also should promote adequate intermolecular interactions, i.e., induce long–range order. Note that although some empirical rules govern the formation of these aggregates (i.e. flat aromatic molecules with solubilizing groups at the periphery), predicting LCLC occurrence based solely on the molecular architecture remains elusive. Thus, to initially assess the occurrence of LCLCs, we screened several dyes and known chromonic mesogens in three room-temperature ILs, namely, ethyl ammonium nitrate (EAN), 2-hydroxyethylammonium formate (HEAF), and 1-ethyl-3-methylimidazolium acetate (EMIAc) (Table S1, ESI†). For this purpose, we monitored birefringence changes when contacting the ionic liquid and the mesogen in solid forms by polarized optical microscopy (POM). We found that only some cyanine dyes (e.g. pinacyanol and cationic thiacarbocyanines) and perylene diimide chloride (PDI) formed liquid crystalline structures in HEAF (Fig. S1; for a complete list of molecules tested see Table S1, ESI†). In the present case, we attribute the LCLC occurrence to HEAF's superior solubilization capacity, inferred from its higher dielectric constant (about 60) compared to EAN and EMIAc (26.2 and 20, respectively).23 Electrostatic interactions generally increase in solvents with low dielectric constants, facilitating stronger π–π interactions and leading to poor solubilization/crystallization as in EAN and EMIAc.
The cohesive energy density, expressed in terms of the Gordon parameter, G, is also a useful indicator of solvent quality. High G solvents, such as water (G = 2.74 J mol1/3 m−3), enable the formation of multiple liquid crystalline phases in surfactant systems. The same applies to a wide range of ionic liquids. Herewith, the formed LCLCs matched this trend as HEAF had a higher G value (1.45 J mol1/3 m−3) when compared to EAN (1.06 J mol1/3 m−3) and EMIAc (0.87 J mol1/3 m−3).‡,24–26 This seems to indicate that LCLC occurrence not only depends on weak π–π interactions but requires sufficient solvophobic interactions between the aromatic core and the solvent.
LCLCs formed with the cyanine dye pinacyanol (either with an acetate or chloride anion) were metastable and crystalized into a solid after minutes of preparation (Fig. S2, ESI†). PDI, on the other hand, formed stable LCLCs at room temperature. Thus, to gain insight into the aggregation mechanism, we further investigated the assembly behavior of PDI in HEAF. First, we studied PDI samples at low concentrations. Because the aggregation process of LCLCs is considered isodesmic, the calculated properties can also be useful to describe systems at higher concentrations and liquid crystalline phases. Advantageously, the self-assembly of chromophores, such as PDI, can be monitored by UV-Vis absorption spectroscopy. Typically, molecularly dissolved PDI chromophores show three bands corresponding to 0 → 2, 0 → 1 and 0 → 0 electronic singlet transitions at 465 nm, 496 nm, and 532 nm, respectively. In water, PDIs generally show a hypsochromic shift with aggregation caused by electron delocalization, indicating the formation of H-aggregates, namely, face-to-face columnar stacks.27 The optical properties of the stacks are mildly influenced by the solvent as long as it does not change the PDI's packing arrangement. PDI solutions in HEAF showed an absorption reduction in the 532 nm band and an increase in the 496 nm and 465 nm bands with concentration indicating that the aggregation is similar to that in water (Fig. 1).
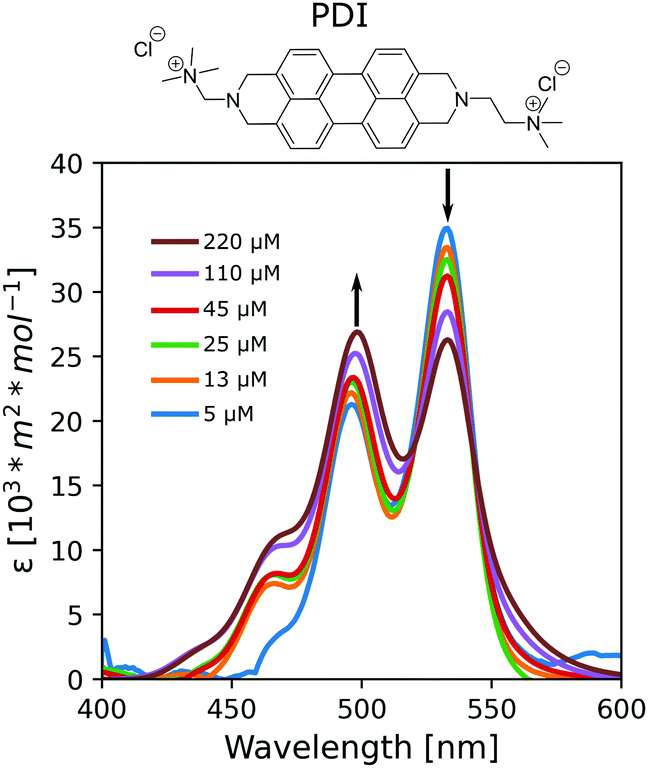 | ||
| Fig. 1 Absorption spectra at 25 °C of PDI in HEAF at different concentrations ranging from 5 μM (0.0005 wt%) to 220 μM (0.01 wt%). | ||
The aggregation constants, KD, were calculated from the UV/Vis data sets at different compositions according to the isodesmic model, which assumes identical KD values for all binding events independently of the size of the aggregates. The KD values for the PDI/IL samples (KD = 5.2·103 M−1, ΔG = −21 kJ mol−1) were comparable with values in the literature for non-ionic PDIs with peripheral polar substituents in polar acetonitrile, butanol, and propanol or thiacarbocyanines in water.7,28 In contrast, the KD for PDI samples in water was tenfold higher (KD = 1.4·105 M−1, ΔG = −29 kJ mol−1), indicating the more significant tendency of PDIs to aggregate.
The monomer band (located at 532 nm) also increases in intensity with temperature for both water and HEAF suggesting dissociation of aggregates (Fig. S3, ESI†). Moreover, the estimated energetic barriers (Ea) for aggregation, calculated from the temperature-dependent UV-Vis spectra using Arrhenius plots, were 7.7 kJ mol−1 and 12.2 kJ mol−1 for water and IL, respectively (Fig. S4 and for a full overview of the thermodynamic parameters refer to Table S2, ESI†). These Ea values match our initial assumption that PDI aggregation in water is more energetically favorable than in HEAF. We rationalize this as HEAF is a better solvent for monomeric PDI. The higher solvation forces of the IL, compared to water, are less favorable for aggregation.
The collected fluorescence spectra were consistent with those of already reported PDIs in water. Samples excited at the maximum absorption wavelength of the monomeric PDI (λexc = 530 nm) displayed a band and a shoulder located at 550 nm and 590 nm, respectively (Fig. 2a). The maximum fluorescence intensity showed an expected increase with concentration of up to 45 μM. The band ratios were constant at all concentrations studied. Moreover, increasing PDI concentration results in aggregation-induced quenching due to electron delocalization. Above 110 μM, the fluorescence is practically negligible, indicating that most of the PDI molecules are aggregated.
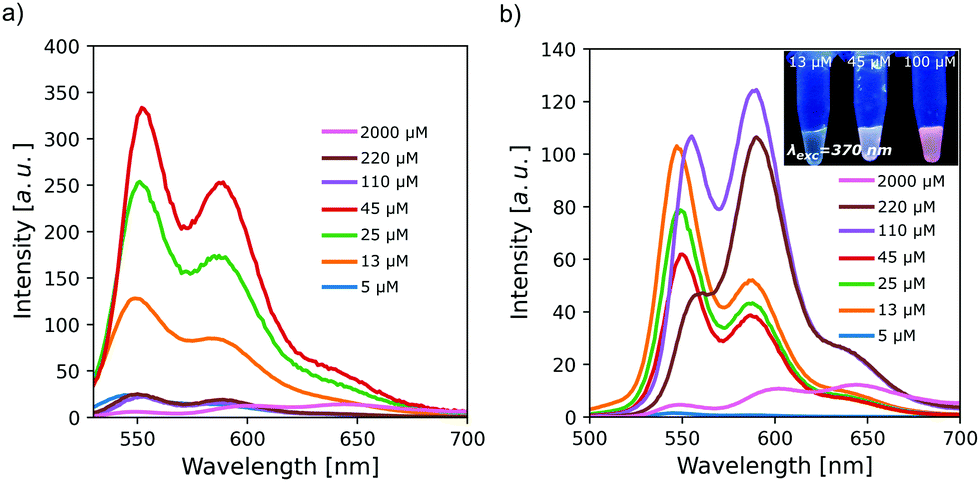 | ||
| Fig. 2 Fluorescence spectra of PDI solutions for concentrations from 5 μM (0.0005 wt%) to 2000 μM (0.094 wt%) in HEAF at 25 °C excited at (a) λexc = 530 nm and (b) λexc = 490 nm. The inset shows the bulk fluorescence of different samples (λexc = 370 nm). | ||
Excitation at the dimeric band wavelength (λexc = 490 nm) produced spectra similar to those obtained with excitation at 530 nm at concentrations below 45 μM (Fig. 2b). Above this concentration, where most molecules are aggregated, there was an abrupt change in the spectra, as a new band appears at 590 nm. The pronounced bathochromic shift is attributed to an excimer-type excited state in PDIs.29 This effect allows the fluorescence emission to go from yellow to red (inset in Fig. 2b).
As the concentration increases, there is an onset of the interaction between the PDI molecular stacks, which indicates the formation of columnar liquid crystals in an appropriate solvent. We found that PDI generated liquid crystals only in HEAF from about 30 wt%. PDIs in water are reported to form chromonic liquid crystals at room temperature and concentrations as low as 6 wt%.13,30 We attribute this stark contrast to several reasons. First, the IL screens the charges of the quaternary amines and consequently reduces the effective volume fraction in solution; second, HEAF seems to be a better solvent for the perylene cores; thus, the average length of the PDI aggregates is shorter compared to water.
Small/wide-angle X-ray scattering (SWAXS) of samples of up to 40 wt% of PDI in HEAF featured a relatively broad peak in the SAXS region (small scattering vector q) ascribed to the intercolumnar distance, d, in the nematic phase (Fig. 3). The intercolumnar distances were calculated to be about 30 Å. The cross-sectional area, assuming a loose hexagonal packing, was estimated as 230 Å2. If this value is compared with the theoretical molecular area (160 Å2), it suggests that the stacks have virtually a unimolecular cross-section. Above 40 wt% several sharp peaks appeared, indicating the presence of PDI solid crystals. All patterns displayed a broad peak in the WAXS region (large scattering vector q) with a maximum at 1.8 Å−1, corresponding to the intermolecular stacking distance, dπ–π, (about 3.4 Å). Optical textures under polarized light of 30 wt% and 40 wt% were consistent with a chromonic nematic phase (see Fig. 4b and Fig. S5, ESI†).
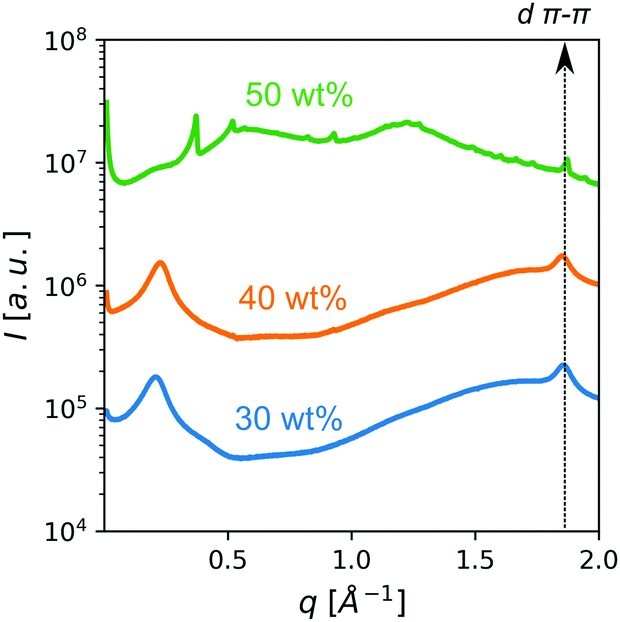 | ||
| Fig. 3 Concentration-dependent SWAXS patterns of PDI samples at room temperature in HEAF. | ||
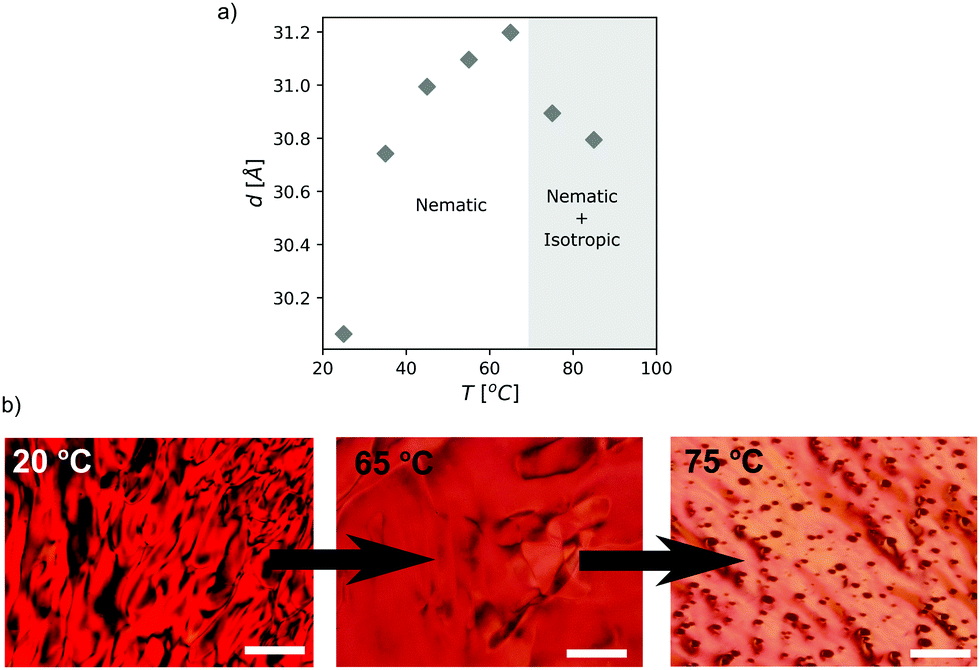 | ||
| Fig. 4 (a) Intercolumnar distance in a 30 wt% PDI/HEAF nematic phase as a function of temperature. (b) Representative POM micrographs of the same sample during the phase transition (scale bar 100 μm). | ||
As the temperature increases, the dπ–π distance remains unchanged, whereas the intercolumnar distance, d, drops at temperatures higher than 65 °C (Fig. S6, ESI† and Fig. 4a). Polarized optical microscopy at these temperatures confirmed a transition from a nematic columnar phase to a mixture of an isotropic liquid and a nematic liquid crystal (Fig. 4b). The reduction in d indicates that the longer columns remain in the nematic phase, while the shorter ones segregate in the isotropic liquid.
 | ||
| Fig. 5 SEM micrograph of the calcined silica material obtained by PDI scaffolding in HEAF (scale bar 10 μm). | ||
To demonstrate the potential applications of these systems, we investigated the feasibility of using LCLCs as scaffolds for the templated synthesis of inorganic materials. The proton transfer from the acid to the base is crucial for protic ionic liquids, such as HEAF. Ideally, this transfer is completed after preparing the protic ionic liquid, so that the individual species in the liquid are the anion and the cation forms. This assumption, however, is proved to be unlikely as the neutral species also coexist in the ionic liquid.31 Taking advantage of these IL properties, we decided to investigate whether it was possible to use acidic or basic neutral species to catalyze the hydrolysis of tetra-alkoxy silanes and further condensate those over the surface of the PDI aggregates. Note that polyaminated surfaces, such as those generated in the surface of the PDI columnar aggregates generally promote the condensation of silicic acid at room temperature and neutral pH.6 Briefly, we mixed 0.5 mL of a 0.5 μM solution of PDI in HEAF with 50 μL of tetraethyl orthosilicate (TEOS). In theory, complete hydrolysis of this small TEOS quantity would require trace amounts of water in HEAF, which is possible, as the samples were prepared under contact with humid air. Karl Fischer titrations revealed up to 3 wt% of water content in the neat HEAF. Remarkably, the PDI/silica materials sediment after 12 hours of reaction, leaving the HEAF almost colorless, i.e., PDI-free. A control sample containing only TEOS in HEAF did not produce any sediment suggesting that PDI is required to condensate silica. This behavior is in line with studies regarding catalytic condensation of silicic acid with macromolecules bearing pendant amines.6 SEM observation of the calcined sample revealed the presence of fibers (Fig. 5), further confirming the successful scaffolding of the PDI aggregates. The calcined SiO2 fibers had a BET surface area, calculated from the N2 adsorption curve (Fig. S7, ESI†), of 530 m2 g−1, almost twice that of silica/PDI materials obtained in water.32
In summary, we discovered the formation of LCLCs in a room-temperature IL. We found that among the ILs tested, only HEAF allowed the formation of LCLCs at room temperature using cyanines and PDI molecules as mesogens. Studies on PDI assembly in HEAF showed that the aggregation constant, KD, is lower in HEAF compared to water. This reduction in KD is attributed to the higher solubilization capacity of HEAF towards the aromatic cores compared with water. Temperature-dependent UV-Vis and fluorescence spectroscopy further support this evidence. The supramolecular aggregates arrange into nematic LCLCs at PDI contents higher than 30 wt%. These concentrations at which LCLCs exist are beyond those found in water (ca. 6 wt%). Finally, we exploited these systems to produce mesostructured oxides under mild conditions (i.e., room temperature and without added acid or base). Applications in materials synthesis and dye-sensitized solar cells are envisaged.
Jose Rodrigo Magana: formal analysis, investigation, validation, visualization, writing – original draft, and writing – review and editing. Adria Pérez-Calm: investigation, writing – review and editing. Carlos Rodriguez-Abreu: conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, supervision, validation, writing – review and editing, and funding acquisition.
We are grateful to the Spanish Ministry of Science and Innovation (CTQ2017-84998-P project), Agencia Estatal de Investigación and European Regional Development Fund for financial support. Spectroscopy, Optical Microscopy and SEM observations have been carried out at the Nanostructured Liquid Characterization Unit, affiliated to the NANBIOSIS ICTS of the Biomedical Networking Research Center (CIBER-BBN).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Lydon, J. Mater. Chem., 2010, 20, 10071–10099 RSC.
- S. W. Tam-Chang and L. Huang, Chem. Commun., 2008, 1957–1967 RSC.
- I. K. Iverson, S. M. Casey, W. Seo, S.-W. Tam-Chang and B. A. Pindzola, Langmuir, 2002, 18, 3510–3516 CrossRef CAS.
- I. K. Iverson and S. W. Tam-Chang, J. Am. Chem. Soc., 1999, 121, 5801–5802 CrossRef CAS.
- S. L. Helfinstine, O. D. Lavrentovich and C. J. Woolverton, Lett. Appl. Microbiol., 2006, 43, 27–32 CrossRef CAS PubMed.
- J. R. Magana, B. Gumí-Audenis, R. P. Tas, L. Gascoigne, D. L. Atkins and I. K. Voets, Chem. – Eur. J., 2020, 26, 15330–15336 CrossRef CAS PubMed.
- J. R. Magana, C. Solans, L. M. Salonen, E. Carbó-Argibay, J. Gallo, G. J. T. Tiddy and C. Rodríguez-Abreu, J. Taiwan Inst. Chem. Eng., 2018, 92, 134–142 CrossRef CAS.
- S. Wang, D. P. Maruri, J. M. Boothby, X. Lu, L. K. Rivera-Tarazona, V. D. Varner and T. H. Ware, J. Mater. Chem. B, 2020, 8, 6988–6998 RSC.
- J. R. Magana, Y. V. Kolen’Ko, F. L. Deepak, C. Solans, R. G. Shrestha, J. P. Hill, K. Ariga, L. K. Shrestha and C. Rodriguez-Abreu, ACS Appl. Mater. Interfaces, 2016, 8, 31231–31238 CrossRef CAS PubMed.
- F. Scarpelli, L. Ricciardi, M. La Deda, E. Brunelli, A. Crispini, M. Ghedini, N. Godbert and I. Aiello, J. Mol. Liq., 2021, 334, 116187 CrossRef CAS.
- C. Rodríguez-Abreu, Y. V. Kolen’ko, K. Kovnir, M. Sanchez-Dominguez, R. G. Shrestha, P. Bairi, K. Ariga and L. K. Shrestha, Phys. Chem. Chem. Phys., 2020, 22, 23276–23285 RSC.
- R. K. Gupta and A. A. Sudhakar, Langmuir, 2018, 35, 2455–2479 CrossRef PubMed.
- A. Pérez-Calm, J. Esquena, L. M. Salonen and C. Rodríguez-Abreu, J. Mol. Liq., 2021, 325, 114657 CrossRef.
- N. Usol’tseva, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 288, 201–210 CrossRef.
- Y.-J. Bae, H.-J. Yang, S.-H. Shin, K.-U. Jeong and M.-H. Lee, J. Mater. Chem., 2011, 21, 2074–2077 RSC.
- H. Eichhorn, D. W. Bruce and D. Wöhrle, Adv. Mater., 1998, 10, 419–422 CrossRef CAS.
- B. Donnio, Curr. Opin. Colloid Interface Sci., 2002, 7, 371–394 CrossRef CAS.
- S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038 CrossRef CAS.
- M. U. Araos and G. G. Warr, J. Phys. Chem. B, 2005, 109, 14275–14277 CrossRef CAS PubMed.
- Y. He, Z. Li, P. Simone and T. P. Lodge, J. Am. Chem. Soc., 2006, 128, 2745–2750 CrossRef CAS PubMed.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- R. D. Noble and D. L. Gin, J. Membr. Sci., 2011, 369, 1–4 CrossRef CAS.
- M.-M. Huang and H. Weingärtner, ChemPhysChem, 2008, 9, 2172–2173 CrossRef CAS PubMed.
- M. C. Castro, A. Arce, A. Soto and H. Rodríguez, J. Chem. Eng. Data, 2016, 61, 2299–2310 CrossRef CAS.
- T. L. Greaves, A. Weerawardena, C. Fong and C. J. Drummond, Langmuir, 2007, 23, 402–404 CrossRef CAS PubMed.
- E. C. Wijaya, F. Separovic, C. J. Drummond and T. L. Greaves, Phys. Chem. Chem. Phys., 2016, 18, 24377–24386 RSC.
- T. Heek, C. Fasting, C. Rest, X. Zhang, F. Würthner and R. Haag, Chem. Commun., 2010, 46, 1884–1886 RSC.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 RSC.
- R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS PubMed.
- S. W. Tam-Chang, J. Helbley and I. K. Iverson, Langmuir, 2008, 24, 2133–2139 CrossRef CAS PubMed.
- D. R. MacFarlane, J. M. Pringle, K. M. Johansson, S. A. Forsyth and M. Forsyth, Chem. Commun., 2006, 1905–1917 RSC.
- C. Rodríguez-Abreu, C. Aubery-Torres, C. Solans, A. López-Quintela and G. J. T. Tiddy, ACS Appl. Mater. Interfaces, 2011, 3, 4133–4141 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc05800b |
| ‡ The Gordon parameter for 1-ethyl-3-methylimidazolium acetate (EMIAc) was estimated using data reported in22 using G = γ/Vm1/3, where γ and Vm are the surface tension and molar volume, respectively. |
| This journal is © The Royal Society of Chemistry 2022 |