
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative profiling of PTM stoichiometry by resolvable mass tags†
Ying
Chen‡
acde,
Baiyi
Quan‡
acde,
Yuanpei
Li‡
acde,
Yuan
Liu
acde,
Wei
Qin
bcde and
Chu
Wang
 *abcde
*abcde
aCollege of Chemistry and Molecular Engineering, Peking University, Beijing, China. E-mail: chuwang@pku.edu.cn
bPeking-Tsinghua Center for Life Science, Peking University, Beijing, China
cBeijing National Laboratory for Molecular Sciences, Peking University, Beijing, China
dSynthetic and Functional Biomolecules Center, Peking University, Beijing, China
eKey Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, Peking University, Beijing, China
First published on 5th October 2022
Abstract
Post-translational modifications (PTMs) play important roles in modulating the biological functions of proteins. Stoichiometry, which quantifies the modification percentage, is a critical factor for any given PTM. In this work, we developed a chemoproteomic strategy called “STO-MS” to systematically quantify the PTM stoichiometry in complex biological samples. This strategy employs a resolvable mass tag to differentiate proteoforms with different numbers of modifications and utilizes liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) techniques to measure PTM stoichiometry at the proteomic level. As a proof-of-concept, we successfully determined the stoichiometry of 197 proteins modified by 4-hydroxynonenal (HNE), a well-characterized lipid-derived electrophile and biomarker for oxidative stress. Our work expands the toolbox for quantification of PTM stoichiometry and sheds light on understanding the biological significance of PTMs in oxidative stress.
Post-translational modifications (PTMs) play important roles in regulating protein structures, activities and functions. A large number of proteomic and chemoproteomic methods have been developed to identify the protein targets and exact sites modified by a variety of PTMs, such as glycosylation,1 lipidation2 and phosphorylation.3 In recent years, emerging studies have focused on measuring the stoichiometry of a given PTM, which is defined as the percentage of the modified fraction. Until now, stoichiometry has been determined for ubiquitination,4 lysine acetylation,5 phosphorylation6 and S-sulfenylation7 in a site-specific manner. Meanwhile, the labeling stoichiometry on cysteines can be indirectly determined by comparing low versus high probe concentrations using isoTOP-ABPP,8 whose competitive version can also be used to quantify cysteine modifications by lipid-derived electrophiles.9 Alternatively, resolvable mass tags have enabled the direct quantification of the modification stoichiometry of endogenous O-GlcNAcylation10 and S-fatty acylation11 on specific proteins (Fig. 1A). However, global quantification of the PTM stoichiometry at a proteomic level by resolvable mass tags remains unexplored. In this study, we aim to develop a chemoproteomic strategy called “STO-MS” (short for “quantitative profiling of STOichiometry by Mass Shift”) to globally quantify PTM stoichiometry in complex biological samples using resolvable mass tags.
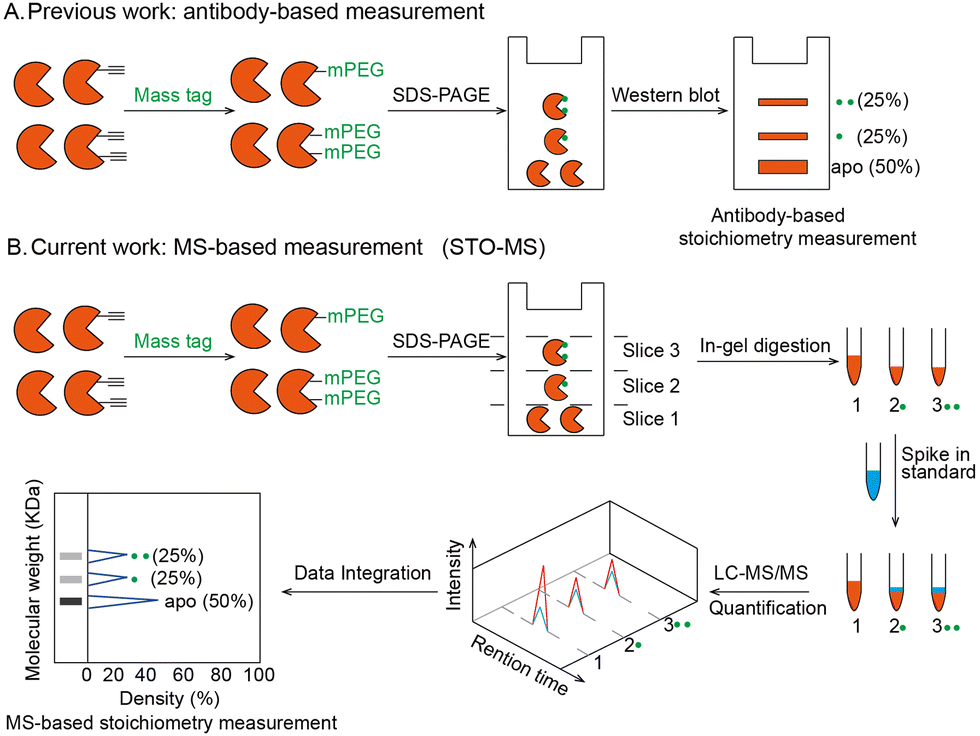 | ||
| Fig. 1 Strategy to quantify stoichiometry at the individual protein level or global level. (A) Workflow of the antibody-based stoichiometry measurement. Proteins bearing alkyne handles were clicked with a mass resolvable tag and analyzed by SDS-PAGE. After transfer, the stoichiometry of an individual protein was detected by its antibody. (B) Workflow of the MS-based stoichiometry measurement (STO-MS) in the case of a single protein. Proteins bearing alkyne handles were clicked with a resolvable mass tag and analyzed by SDS-PAGE. Then the gel was cut into slices and subjected to in-gel trypsin digestion, respectively. Afterwards, each fraction was spiked in an internal standard and analyzed by LC-MS/MS. After data integration, the stoichiometry can be obtained for the protein. | ||
The workflow of STO-MS is constituted by five major steps (Fig. 1B). Firstly, probe labeled proteins are clicked with a resolvable mass tag with a defined molecular weight. Secondly, the proteins are resolved by SDS-PAGE. Since proteoforms with different numbers of probe modifications will carry different numbers of mass tags, they will migrate with different paces in the gel, forming a mass ladder. Thirdly, the gel is cut into multiple slices according to the resolution of the mass tag, and each slice is subjected to in-gel trypsin digestion. After digestion, all fractions are spiked in with a SILAC12 internal standard. Fourthly, each of the mixed samples is analyzed by LC-MS/MS and quantified ratios are collected to generate a distribution curve across all fractions. Lastly, peaks are detected in the curve, each of which corresponds to a proteoform of the target protein with a certain number of modifications. The modification stoichiometry is eventually calculated using the following formula:
| Rmodified/(Rmodified + Rnative) |
As a proof-of-concept, we developed and optimized the STO-MS strategy using 4-hydroxynonenal (HNE) as a model system. HNE is a typical lipid-derived electrophile which is broken down from polyunsaturated fatty acids when cells are under oxidative stress.13 Structurally, HNE is an α,β-unsaturated aldehyde that can covalently modify the nucleophilic residues such as cysteines, lysines and histidines via Michael addition to form one specific class of PTMs termed “carbonylations”.14 Emerging pieces of evidence have shown that protein carbonylations are involved in diverse pathological conditions such as ferroptosis,15 neurodegeneration,16 inflammation17 and cancer.18 Chemoproteomic strategies have been applied to identify and quantify the sites of HNE modifications in proteomes.15,19,20 However, methods to globally quantify the HNE modification stoichiometry are still lacking.
One of the key factors for precisely measuring stoichiometry by STO-MS is the efficiency of mass tag incorporation. As a result, we first optimized the yield of mass tag incorporation in a purified protein system by using HNEyne, a commonly used bioorthogonal probe for mimicking HNE modification and N3-mPEG-2000, a resolvable mass tag with a molecular weight of 2.0 kDa (Fig. 2A). The model protein, BSA, was labeled with 100 μM of HNEyne (Fig. 2B), conjugated with N3-mPEG-2000 (Fig. 2C) via copper-catalyzed azide–alkyne cycloaddition (CuAAC) and finally resolved by SDS-PAGE. The mass-tag incorporation efficiency is estimated by measuring the band shift observed on the gel. Temperature- and time-dependent labeling showed that 37 °C and 4 h are the optimal temperature and reaction time for incorporating the mass tag, respectively, under which there are clear band shifts from the unlabeled control sample (Fig. 2D).
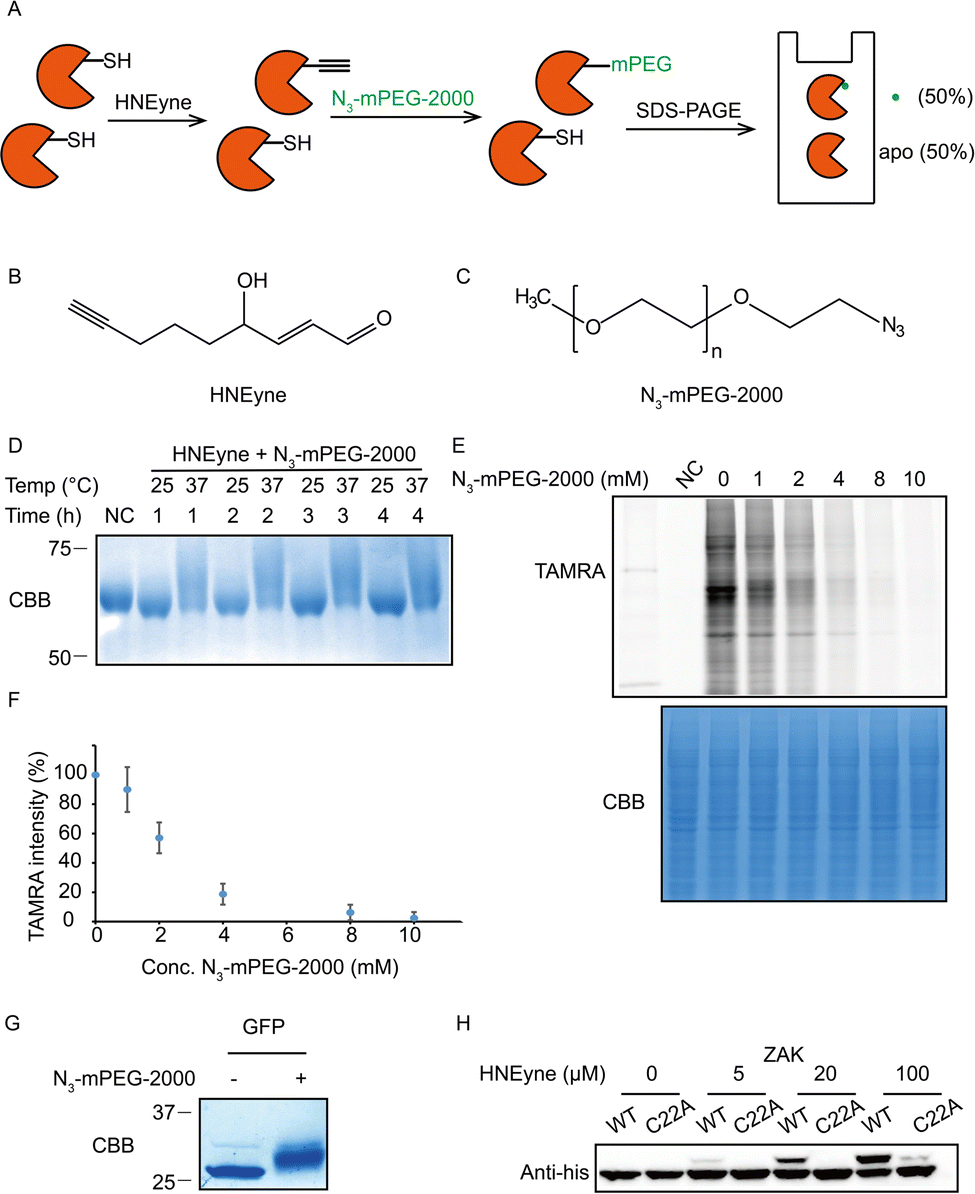 | ||
| Fig. 2 Optimization of the efficiency of mass resolvable tag incorporation. (A) Platform of optimizing the yield of the mass tag incorporation reaction. Purified BSA proteins were reacted with the HNEyne probe and clicked with N3-mPEG-2000, and the samples were analyzed by SDS-PAGE. As a result, the yield of mass tag incorporation can be obtained by the percentage of mass shift. (B) Chemical structure of the HNEyne probe. (C) Chemical structure of the N3-mPEG-2000 mass tag. (D) Optimizing the temperature and time of click chemistry between HNEyne modification and the resolvable mass tag. (E) The efficiency of N3-mPEG-2000 click chemistry was quantified by a competition assay using N3-TAMRA. (F) Statistical analysis of the efficiency of the mass resolvable tag incorporated from three biological replicates. (G) A 100% mass shift was observed in the model of unnatural amino acid inserted GFP proteins using optimized click conditions. (H) The stoichiometry of the ZAK kinase was dose-dependent on HNEyne, and the mass shift disappeared after Cys22 was mutated to alanine, except at the highest probe concentration tested. | ||
We then evaluated the efficiency of the click reaction in cell lysates by a competitive in-gel fluorescence assay. HNEyne-labeled cell lysates were first reacted with N3-mPEG-2000 at 37 °C for 4 h, and excessive click reagents were removed by methanol–chloroform precipitation. We then initiated a second click reaction to conjugate N3-TAMRA with any remaining HNEyne modifications bearing free alkyne handles in the lysates. Thus, the efficiency of incorporating N3-mPEG-2000 can be evaluated by measuring its competition on the fluorescent signal of TAMRA, with a lower fluorescent signal indicating a higher percentage of N3-mPEG-2000 incorporation. As expected, the fluorescent signals decreased with the increasing concentration of N3-mPEG-2000 (Fig. 2E) and the percentage of mass tag incorporation reaches nearly 100% when 10 mM of N3-mPEG-2000 was used (Fig. 2F).
To validate the condition of the click reaction for maximizing the mass tag incorporation, we inserted an alkyne-functionalized unnatural amino acid (Fig. S1A, ESI†) into a green fluorescent protein (GFP) via the genetic code expansion technique and obtained a purified protein sample with 100% “alkyne labeling”. We then initiated the click reaction under the optimized conditions (37 °C, 4 h, and 10 mM mass tag) to conjugate N3-mPEG-2000 onto GFP and observed a clear-cut band shift in the gel stained by Coomassie Blue (Fig. 2G). A similar shift was also observed when the alkyne-functionalized GFP was premixed with cell lysates and then clicked with the mass tag (Fig. S1B, ESI†). All these results suggest that, under these optimized click conditions, the resolvable mass tag is applicable in both purified proteins and complex cell lysates and it should also work in principle with STO-MS for globally profiling the stoichiometry of HNE modifications in proteomes.
We first attempted to determine the stoichiometry of HNE modification on ZAK, a mitogen-activated protein triple kinase that was reported to be modified by HNE at its Cys22,9 by using the resolvable mass tags. Lysates from HEK293T cells which stably overexpress 6xhis-ZAK WT or C22A were labeled with various concentrations of HNEyne before conjugation with N3-mPEG-2000. Immunoblotting with the anti-6xhis antibody showed that the stoichiometry of HNEyne modification increased dose-dependently and reached 48.4% when 100 μM of HNEyne was used for labeling. As the negative control, the ZAK C22A mutant did not show a visible band shift, except at the highest probe concentration tested. These results confirmed that Cys22 is the major site modified by HNEyne, suggesting our approach can indeed differentiate proteoforms with or without the modification at the active site (Fig. 2H). Similarly, the stoichiometry of HNEyne modification on another target protein, adenylate kinase 2 (AK2), was determined as 49.5% at a HNEyne concentration of 100 μM (Fig. S2, ESI†).
Next, we proceeded to systematically quantify the stoichiometry of HNEyne modification at the proteomic level by STO-MS according to the scheme shown in Fig. 1B. The experiments were performed in two biological replicates and proteins that have the same number of modifications in both replicates were kept for further analysis. For stoichiometry calculations, in brief, the light-to-heavy ratios in each of the 23 fractions are quantified and normalized from zero to one. All the normalized ratios that are smaller than 0.1 are eliminated from the analysis. For each protein, an estimated fraction is calculated according to the molecular weight of the apo protein. All the proteins without a ratio larger than 0.1 within 2 fraction range of the estimated fraction are eliminated from the analysis because of an unmatched molecular weight. All the proteins that pass the data filter are subjected to a peak-searching algorithm starting from 2 fractions under the estimated fraction. In the algorithm, peaks are separated by “saddle points” which are defined as the fraction with a ratio lower than the ratios of adjacent fractions plus 0.2 (Fig. S3, ESI†). The peak-searching ends when three continuous fractions generate no new peak to minimize the influence caused by endogenous modifications, such as ubiquitylation. The area under each peak is subsequently calculated and normalized to obtain the final stoichiometry values. To improve the modification assignment in the HNEyne-treated samples, data from the samples omitting HNEyne treatment are utilized as the negative control.
In total, the stoichiometry of 197 proteins was successfully quantified (Supplementary table, ESI†), which is distributed in a wide range from 15% to 86% (Fig. 3A). In comparison, the vast majority of HNEyne modified proteins lack a mass shift in the negative control (Fig. S4, ESI†). The presence of five false positive proteins might be due to endogenous modifications (e.g. phosphorylation, lipidation and ubiquitylation). Gene Ontology analysis reveals that these heavily modified proteins (stoichiometry >0.6) are significantly enriched in biological processes including Rab protein signal transduction and translation (Fig. S5, ESI†). For instance, EIF3J, one of the eukaryotic translation initiation factors, has the highest modification stoichiometry by HNEyne. EIF3J has been previously reported to be modified by endogenous HNE20 and the modification event was recently found to occur at Cys207.15 According to the quantification by STO-MS, EIF3J is mono-modified by HNEyne with a stoichiometry of 86% (Fig. 3B).
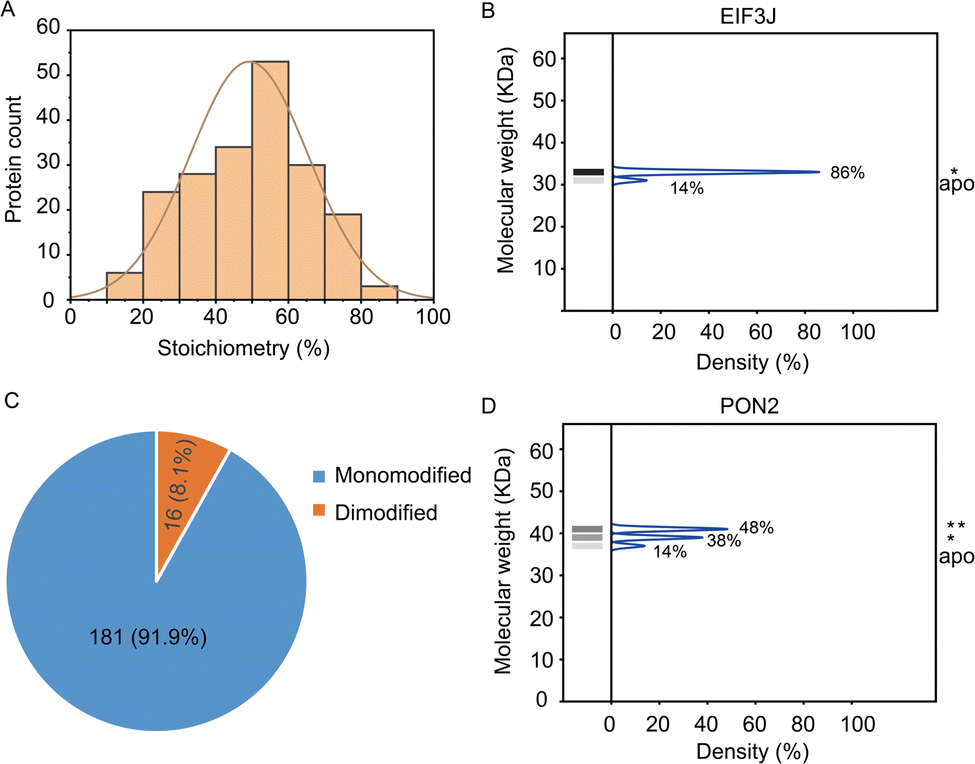 | ||
| Fig. 3 Analysis of the stoichiometry of HNE modification which is determined through the STO-MS strategy. (A) The stoichiometry of HNE modification is widely distributed from 15 to 86%. (B) The eukaryotic translation initiation factor EIF3J has a maximum stoichiometry of 86% corresponding to the mono-modified status. (C) Modification status distribution of HNEyne modified proteins shows that 181 proteins are mono-modified and 16 proteins are dimodified. (D) PON2 was quantified with a stoichiometry of 48% and 38% corresponding to the mono-modified and di-modified populations. | ||
In STO-MS, proteins modified with multiple bioorthogonal probes will result in ladders in SDS-PAGE that could be quantified to obtain information on the stoichiometry for each of the proteoforms with different numbers of modifications per protein. In the HNEyne profiling data, we classified proteins as “mono-”, “di-” and “tri-”modified based on the number of modifications detected. In total, 181 and 16 proteins were determined to be mono-modified and di-modified, respectively (Fig. 3C). For example, PON2, a serum paraoxonase/arylesterase, was determined to have two modified populations, with a quantified stoichiometry of 38% and 48% corresponding to the mono-modified and di-modified populations, respectively (Fig. 3D).
Last, we validated the measured stoichiometry of select targets by independent assays. FBL is an rRNA 2-O-methyltransferase fibrillarin that is involved in the pre-rRNA process, and the protein is quantified with a mono-modified population with 36% stoichiometry (Fig. 4A). To confirm this result, cell lysates were incubated with HNEyne, reacted with N3-mPEG-2000, resolved by SDS-PAGE and immunoblotted against an FBL antibody. Measurement of immunoblotting signals shows that FBL has one modified population with a stoichiometry of 30% (Fig. 4B). Another target we tested is GAPDH, a glyceraldehyde-3-phosphate dehydrogenase which plays an important role in the glycolysis process (Fig. S6, ESI†). The Cys152 and Cys247 of GAPDH have been previously identified as the HNE modification sites; however, their stoichiometry is unknown.15 Using STO-MS, we found that the mass shift completely disappeared when Cys152 was mutated, while no significant difference was observed when Cys247 was mutated (Fig. 4C). The result indicates that Cys152 is the major HNE-modified site in GAPDH, which correlates with its higher reactivity according to a previous study of cysteinome profiling.15
 | ||
| Fig. 4 Validation of the stoichiometry of HNE modification on individual proteins. (A) FBL is mono-modified by HNE with 36% stoichiometry in the STO-MS measurement. (B) The stoichiometry of FBL is determined to be 30% in an independent biochemical experiment. (C) C152 is the dominantly modified site in the case of GAPDH. In detail, compared to wildtype proteins, the mass shift totally disappears when C152 is mutated. However, no significant difference is observed when C247 is mutated. (D) Stoichiometry quantification of endogenous carbonylation on ZAK in arachidonic acid-treated cells. | ||
In order to investigate the capability of STO-MS to analyze endogenous PTMs, an aldehyde-directed chemical probe, m-APA,15 was utilized to label natural HNE modification (Fig. S7A and B, ESI†). The m-APA labeling can be further conjugated with N3-mPEG-2000 via click chemistry, which reveals the stoichiometry of HNE modification on GAPDH in HNE-treated cell lysates (Fig. S7C, ESI†). Using this approach, we also determined the stoichiometry of endogenous carbonylation on ZAK upon the treatment of arachidonic acid that can generate HNE via lipid peroxidation (Fig. 4D). These data highlight the potential of our method to profile the stoichiometry of endogenous PTMs at the proteomic level.
Conclusions
In conclusion, we have combined the resolvable mass tags with mass spectrometry to establish a chemoproteomic platform, STO-MS, to systematically quantify the PTM stoichiometry at a proteomic level. Using HNE modification as a model system, the efficiency of the mass tag conjugation was optimized, and the stoichiometry of bioorthogonal HNE probe modifications on 197 proteins was successfully determined by STO-MS. The method has the resolution to differentiate proteoforms with different numbers of modifications and is in principle compatible with any PTMs that can be labeled by reactive-capture, metabolic-labeling or chemoenzymatic-labeling probes.21 However, a mass tag of 2.0 kDa in the current STO-MS strategy may not be the best choice for resolving large proteins on SDS-PAGE which leads to the absence of stoichiometry information on proteins with a molecular weight >60 kDa. Also, if a modified proteoform spans across multiple fractions or overlap with another proteoform with a different type of modification, the resulting stoichiometry might be less accurately quantified. Moreover, we cannot exclude the possibility that a 100% (mono-) modified proteoform exists when only a single peak is detected, and further experiments are required to differentiate such cases from a 100% unmodified proteoform. Nevertheless, with STO-MS, researchers can add the new dimension of stoichiometry to PTM proteomic profiling and focus more on target proteins with higher stoichiometry for functional validation. It is also possible to establish new links between the modification stoichiometry and the functional implication of a certain PTM on the target protein, such as degradation,22 transport and localization.23 Collectively, we envision that STO-MS will serve as a valuable tool for enhancing the resolution of PTM profiling and facilitate functional studies of PTMs in future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly appreciate Dr Xiao Xie in Prof. Peng R Chen's group for help with purification of UAA inserted GFP. We thank the Computing Platform of the Center for Life Science for supporting the proteomic data analysis. This work was supported by the National Natural Science Foundation of China (91953109, 92153301 and 21925701).Notes and references
- I. Bagdonaite, S. A. Malaker, D. A. Polasky, N. M. Riley, K. Schjoldager, S. Y. Vakhrushev, A. Halim, K. F. Aoki-Kinoshita, A. I. Nesvizhskii, C. R. Bertozzi, H. H. Wandall, B. L. Parker, M. Thaysen-Andersen and N. E. Scott, Nat. Rev. Methods Primers, 2022, 2, 48 CrossRef CAS.
- H. C. Hang and M. E. Linder, Chem. Rev., 2011, 111, 6341–6358 CrossRef CAS PubMed.
- J. V. Olsen, B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen and M. Mann, Cell, 2006, 127, 635–648 CrossRef CAS PubMed.
- Y. Li, J. Evers, A. Luo, L. Erber, Z. Postler and Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 537–541 CrossRef CAS PubMed.
- B. K. Hansen, R. Gupta, L. Baldus, D. Lyon, T. Narita, M. Lammers, C. Choudhary and B. T. Weinert, Nat. Commun., 2019, 10, 1055 CrossRef PubMed.
- J. V. Olsen, M. Vermeulen, A. Santamaria, C. Kumar, M. L. Miller, L. J. Jensen, F. Gnad, J. Cox, T. S. Jensen, E. A. Nigg, S. Brunak and M. Mann, Sci. Signaling, 2010, 3, ra3 CrossRef PubMed.
- Y. Shi, L. Fu, J. Yang and K. S. Carroll, Nat. Chem., 2021, 13, 1140–1150 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef CAS PubMed.
- C. Wang, E. Weerapana, M. M. Blewett and B. F. Cravatt, Nat. Methods, 2014, 11, 79–85 CrossRef CAS.
- J. E. Rexach, C. J. Rogers, S. H. Yu, J. Tao, Y. E. Sun and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2010, 6, 645–651 CrossRef CAS PubMed.
- A. Percher, S. Ramakrishnan, E. Thinon, X. Yuan, J. S. Yount and H. C. Hang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4302–4307 CrossRef CAS PubMed.
- S. E. Ong, B. Blagoev, I. Kratchmarova, D. B. Kristensen, H. Steen, A. Pandey and M. Mann, Mol. Cell. Proteomics, 2002, 1, 376–386 CrossRef CAS.
- K. S. Fritz and D. R. Petersen, Chem. Res. Toxicol., 2011, 24, 1411–1419 Search PubMed.
- Y. Chen, W. Qin and C. Wang, Curr. Opin. Chem. Biol., 2016, 30, 37–45 CrossRef CAS PubMed.
- Y. Chen, Y. Liu, T. Lan, W. Qin, Y. Zhu, K. Qin, J. Gao, H. Wang, X. Hou, N. Chen, J. P. Friedmann Angeli, M. Conrad and C. Wang, J. Am. Chem. Soc., 2018, 140, 4712–4720 CrossRef CAS PubMed.
- T. T. Reed, Free Radicals Biol. Med., 2011, 51, 1302–1319 CrossRef CAS PubMed.
- M. Delmastro-Greenwood, B. A. Freeman and S. G. Wendell, Annu. Rev. Physiol., 2014, 76, 79–105 CrossRef CAS PubMed.
- D. A. Butterfield, L. Gu, F. Di Domenico and R. A. Robinson, Mass Spectrom. Rev., 2014, 33, 277–301 CrossRef CAS PubMed.
- J. Yang, K. A. Tallman, N. A. Porter and D. C. Liebler, Anal. Chem., 2015, 87, 2535–2541 CrossRef CAS PubMed.
- Y. Chen, Y. Cong, B. Quan, T. Lan, X. Chu, Z. Ye, X. Hou and C. Wang, Redox Biol., 2017, 12, 712–718 CrossRef CAS PubMed.
- W. Qin, F. Yang and C. Wang, Curr. Opin. Chem. Biol., 2020, 54, 28–36 CrossRef CAS.
- K. N. Swatek and D. Komander, Cell Res., 2016, 26, 399–422 CrossRef CAS PubMed.
- Y. Wang, H. Lu, C. Fang and J. Xu, Adv. Exp. Med. Biol., 2020, 1248, 399–424 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00179a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |