
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe chemistry and biology of natural ribomimetics and related compounds
Takeshi
Tsunoda
 ,
Samuel
Tanoeyadi
,
Philip J.
Proteau
and
Taifo
Mahmud
*
,
Samuel
Tanoeyadi
,
Philip J.
Proteau
and
Taifo
Mahmud
*
Department of Pharmaceutical Sciences, Oregon State University, Corvallis, OR 97331, USA. E-mail: Taifo.Mahmud@oregonstate.edu
First published on 7th April 2022
Abstract
Natural ribomimetics represent an important group of specialized metabolites with significant biological activities. Many of the activities, e.g., inhibition of seryl-tRNA synthetases, glycosidases, or ribosomes, are manifestations of their structural resemblance to ribose or related sugars, which play roles in the structural, physiological, and/or reproductive functions of living organisms. Recent studies on the biosynthesis and biological activities of some natural ribomimetics have expanded our understanding on how they are made in nature and why they have great potential as pharmaceutically relevant products. This review article highlights the discovery, biological activities, biosynthesis, and development of this intriguing class of natural products.
 Takeshi Tsunoda | Takeshi Tsunoda received his BSc in 2013 from Kitasato University under the supervision of Prof. Tohru Nagamitsu. Subsequently, he received his MSc in 2015 from Tokyo Institute of Technology (under Prof. Tadashi Eguchi) and PhD in 2019 from The University of Tokyo (under Profs Yasuo Ohnishi and Yohei Katsuyama). In 2019, he joined Prof. Taifo Mahmud's laboratory at Oregon State University as a postdoctoral scholar. His current research concerns biochemical studies of natural product biosynthetic enzymes, genome mining-based natural products discovery, and investigation of the ecological role of natural products. |
 Samuel Tanoeyadi | Samuel Tanoeyadi received his BS degree in Chemistry with Biochemistry as its major option in 2020 from Oregon State University (OSU). He then joined the OSU College of Pharmacy to pursue a PhD in Pharmaceutical Sciences under the supervision of Dr Taifo Mahmud. His current research focuses on the biochemistry of enzymes involved in the production of secondary metabolites, particularly sugar-related compounds and sugar mimetics. He is also interested in understanding how those compounds impact the producing organisms and/or other organisms in nature. |
 Philip J. Proteau | Philip Proteau received his PhD degree in 1993 from Oregon State University, with William Gerwick as his mentor. After a postdoctoral research experience with Dale Poulter at the University of Utah, he joined the faculty in the College of Pharmacy at Oregon State University in late 1995. In 2002 he was promoted to Associate Professor. After serving as an Associate Editor of the Journal of Natural Products since late 2008, he became Editor-in-Chief in 2020. His current research interests include cyanobacterial natural products and isoprenoid biosynthesis. |
 Taifo Mahmud | Taifo Mahmud is a Professor in the College of Pharmacy and an Adjunct Professor in the Department of Chemistry at Oregon State University (OSU). Prior to coming to OSU, he was a Research Assistant Professor and Postdoctoral Research Associate (with Heinz G. Floss) at the University of Washington. He received his PhD and MSc from Osaka University (with Isao Kitagawa and Motomasa Kobayashi). His research interests are broadly in drug discovery and development. His group employs a multidisciplinary approach that utilizes cutting-edge technologies in molecular genetics, enzymology, and chemistry to produce novel pharmaceuticals. |
1. Introduction
Glycomimetics, also known as pseudosugars, are chemical entities that are structurally similar to sugars. As sugars play important roles in various structural and physiological aspects of living organisms, many glycomimetics have significant biological activities.1 Consequently, some of them have been developed as drugs to treat diseases in humans, animals, and plants. Six-membered ring pseudosugar compounds such as the aminoglycoside antibiotics (e.g., streptomycin (1), gentamicin, and neomycin) have been used for more than a half century for the treatment of various infectious diseases.2 The pseudo-oligosaccharide acarbose (2) as well as the iminosugar miglitol (3) (a synthetic derivative of deoxynojirimycin) have been used to treat patients with type 2 diabetes (Fig. 1).3,4 Two other synthetic iminosugars, miglustat (4) and migalastat (5), are used to treat Gaucher's disease type 1 and Fabry disease, respectively.5,6 Furthermore, the pseudo-oligosaccharide validamycin A (6) is used as a crop protectant against fungal infections.7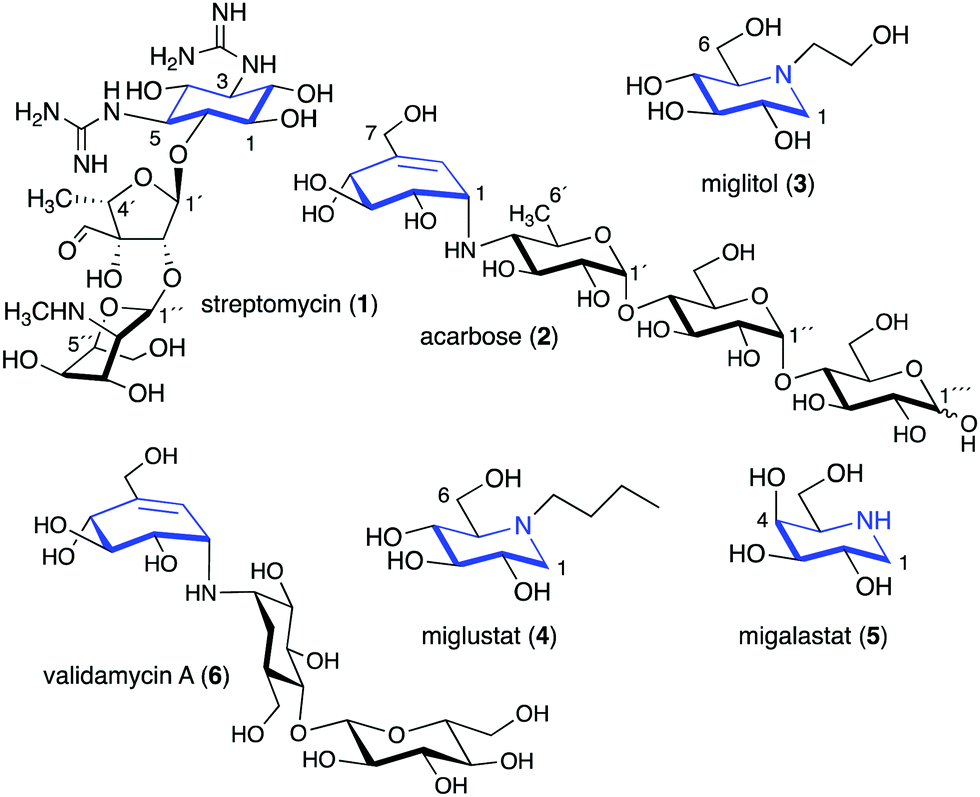 | ||
| Fig. 1 Chemical structures of six-membered ring pseudosugars. | ||
In addition to the six-membered pseudosugars, many natural and synthetic compounds contain five-membered pseudosugar units, known as ribomimetics. Structurally, they resemble ribose, deoxyribose, or arabinose, or contain a highly modified ribose, which contributes to their various biological activities. A number of synthetic ribomimetics have also made their way to clinical use, e.g., the anti-influenza drug peramivir (7), the anti-hepatitis B drug entecavir (8), and the HIV nucleoside reverse transcriptase inhibitors lamivudine (9) and abacavir (10) (Fig. 2). In addition, many natural ribomimetics have attracted significant attention from scientists due to their promising biological activities.
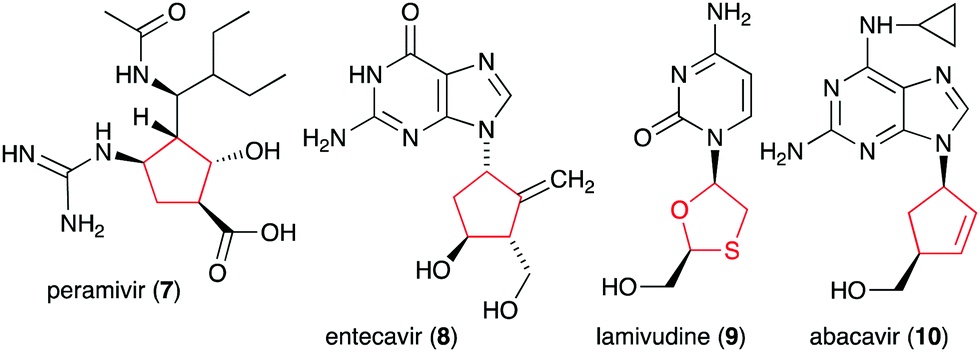 | ||
| Fig. 2 Chemical structures of clinically used ribomimetics. | ||
Naturally occurring ribomimetics and related compounds may be classified into two groups. The first group consists of ribose-like compounds in which the ring oxygen of ribose, deoxyribose, arabinose and related five-membered ring sugars has been replaced by a sulphur, a nitrogen, or a carbon. Accordingly, they are classified as S-, N-, and C-ribomimetics. Members of this family of natural products include the albomycins (11–13), 1,4-dideoxy-1,4-iminoarabinitol (DAB-1, 14), aristeromycin (15) and pactamycin (16) (Fig. 3). The second group of ribomimetics consists of nucleotide/nucleoside antibiotics that still contain a ribose backbone, but with a highly modified C-5. This group of ribomimetics includes nikkomycin (17), caprazamycin B (18), and tunicamycin (19) (Fig. 3). A number of outstanding review articles describing the latter compounds (the nucleotide/nucleoside antibiotics) have been published recently;8–10 therefore, they are excluded from this article, which focuses on the discovery, biological activities, biosynthesis, and development of the S-, N-, and C-ribomimetics and related compounds.
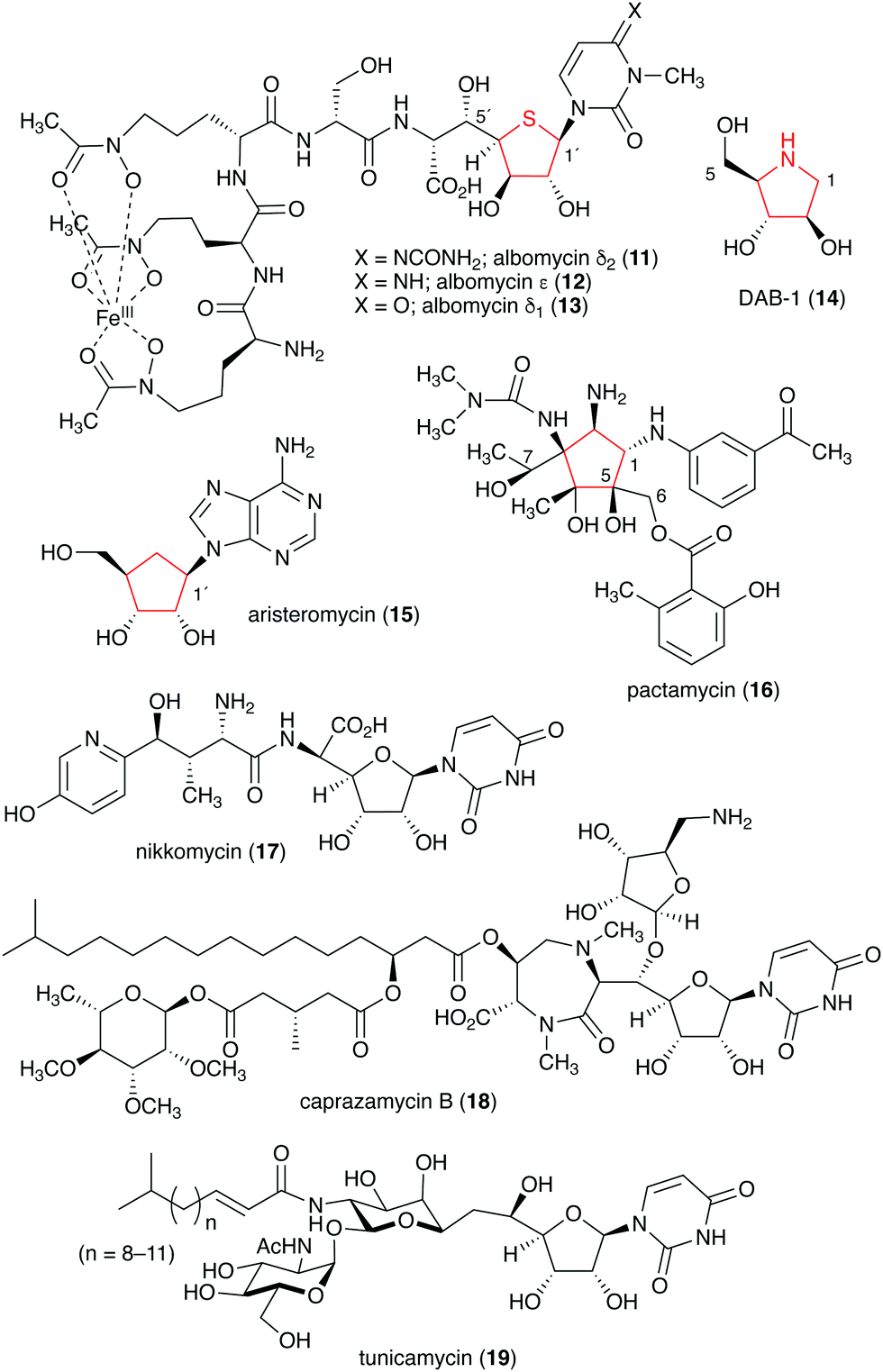 | ||
| Fig. 3 Chemical structures of natural ribomimetics. | ||
| Name of compound | Organism source | Bioactivity or function | Key reference(s) |
|---|---|---|---|
| Albomycin δ2 (11) | Streptomyces pneumoniae | Bacterial seryl-tRNA synthetases inhibitor | 12–16, 18–20 and 31 |
| Albomycin ε (12) | S. pneumoniae | Bacterial seryl-tRNA synthetases inhibitor | 12–16, 18–20 and 31 |
| Albomycin δ1 (13) | S. pneumoniae | Bacterial seryl-tRNA synthetases inhibitor | 12–16, 18–20 and 31 |
| Salacinol (20) | Salacia reticulata | α-Glucosidase inhibitor | 21 and 30 |
| Neosalacinol (21) | S. reticulata | α-Glucosidase inhibitor | 22, 23 and 30 |
| Kotalanol (22) | S. reticulata, Salacia oblonga | α-Glucosidase inhibitor | 24, 25 and 30 |
| Neokotalanol (23) | S. reticulata, S. oblonga | α-Glucosidase inhibitor | 26 and 30 |
| Ponkoranol (24) | S. reticulata, S. oblonga | α-Glucosidase inhibitor | 27 and 30 |
| Neoponkoranol (25) | S. reticulata, S. oblonga | α-Glucosidase inhibitor | 28 and 30 |
| Salaprinol (26) | S. oblonga | α-Glucosidase inhibitor | 29 and 30 |
| Neosalaprinol (27) | S. oblonga | α-Glucosidase inhibitor | 28 and 30 |
2. S-Ribomimetics and related compounds
Natural S-ribomimetics, also known as thiofuranoses, have been found in bacteria and plants (Table 1). On the basis of the known natural thiofuranoses identified so far, they may be classified into two structural groups, the albomycins (11–13) and the salacinols (20–27) (Fig. 4). The albomycins are a group of peptidyl nucleoside analogues containing a peptide-derived siderophore unit and a 6′-amino-4′-thioheptose nucleoside moiety connected by an amide bond (Fig. 3).11 The salacinols, including kotalanol (22), ponkoranol (24), salaprinol (26), and other related compounds, are a group of plant-derived thiosugar sulfonium compounds, some of which contain sulfonium sulfate inner salt structures.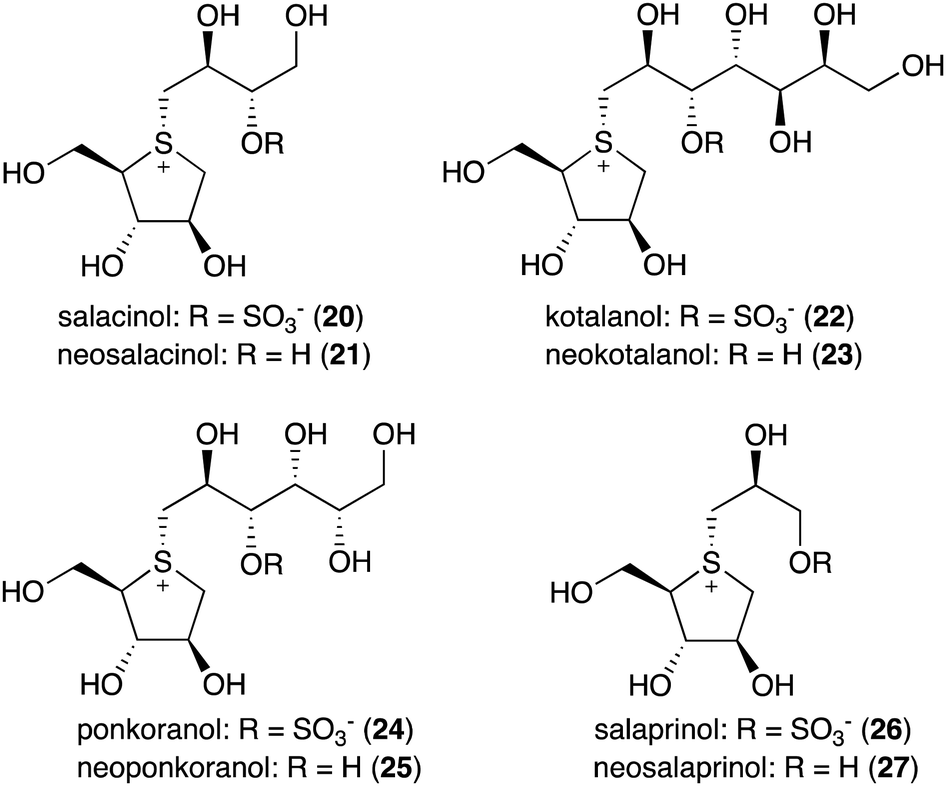 | ||
| Fig. 4 Chemical structures of natural S-ribomimetics. | ||
2.1. Discovery and biological activities of S-ribomimetics
The peptidyl nucleoside analogue albomycins have been isolated from several actinomycetes.12,13 They showed antibiotic activity against Gram-positive and Gram-negative bacteria,14–16 most likely by inhibiting bacterial seryl-tRNA synthetases.17 The thioheptose unit of the compounds is believed to be important for this activity, whereas the siderophore unit is important for the compounds to get into bacterial cells by utilizing the bacterial iron uptake system. After getting into the bacterial cells, the amide bond is hydrolysed by endogenous peptidases releasing the active thioheptose nucleoside unit (Fig. 5). Due to this unique mechanism, the albomycins have been branded as “trojan horse” antibiotics.18–20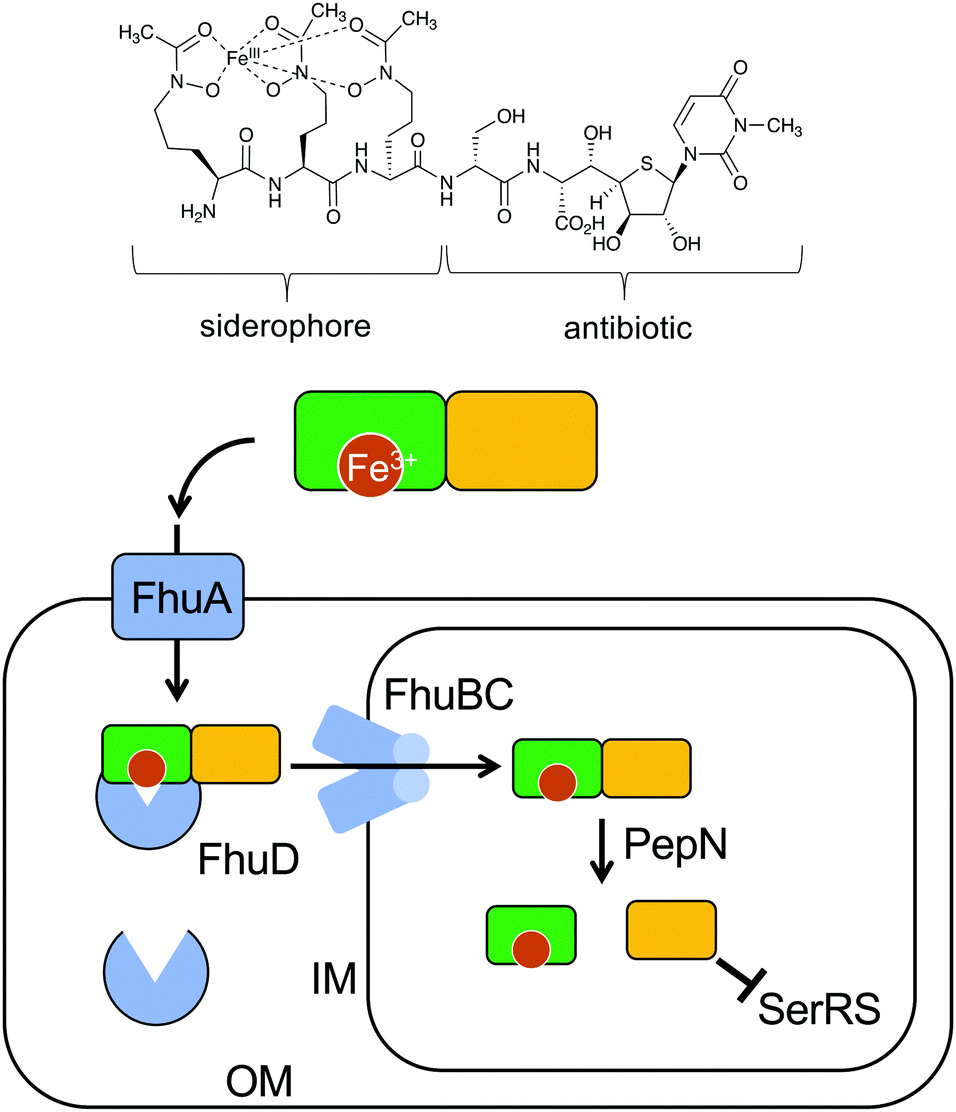 | ||
| Fig. 5 Albomycin enters a bacterial cell using the bacterial iron uptake system. The siderophore unit is important for the compound to get into bacterial cells by utilizing the bacterial iron uptake system. First, the ferric siderophore passes the outer membrane through the ferrichrome outer membrane transporter FhuA. In the periplasm, the ferrichrome binds FhuD, which is then transported into the cytoplasm through the membrane channel FhuBC. In the cytoplasm, the amide bond is hydrolysed by the endogenous peptidase PepN releasing the active thioheptose nucleoside unit, which inhibits seryl-tRNA synthetase (SerRS). OM, outer membrane; IM, inner membrane. | ||
The plant-derived S-ribomimetic compound salacinol (20) was first isolated from the roots and stems of Salacia reticulata, which is traditionally used in Sri Lanka for the treatment of diabetes.21 Consistent with the empirical use of the plant as an anti-diabetic, the active compound salacinol does have strong α-glucosidase inhibitory activity.21 In addition to salacinol, several of its analogues, e.g., neosalacinol (21),22,23 kotalanol (22),24,25 neokotalanol (23),26 ponkoranol (24),27 neoponkoranol (25),28 salaprinol (26),29 and neosalaprinol (27),28 were also isolated from S. reticulata or a related species, Salacia oblonga (Fig. 4). All of them share the same S-ribomimetic core structure, but with different polyalcohol side chains with various chain lengths (C3–C7), either sulfonated or non-sulfonated. The antidiabetic effects of Salacia plants were reviewed very recently, describing the basic chemical and pharmacological research of the salacinols, as well as their applications as functional food ingredients.30
2.2. Biosynthesis of S-ribomimetics
Despite their interesting chemical structures, no biosynthetic studies of the salacinols have been reported in the literature. On the other hand, studies on albomycin biosynthesis have been conducted by several groups. The biosynthetic gene cluster (BGC) of albomycin (the abm cluster) was first reported from Streptomyces sp. ATCC 700974, along with a related gene cluster (the ctj cluster) postulated for an albomycin analogue from another strain of Streptomyces (Fig. 6).31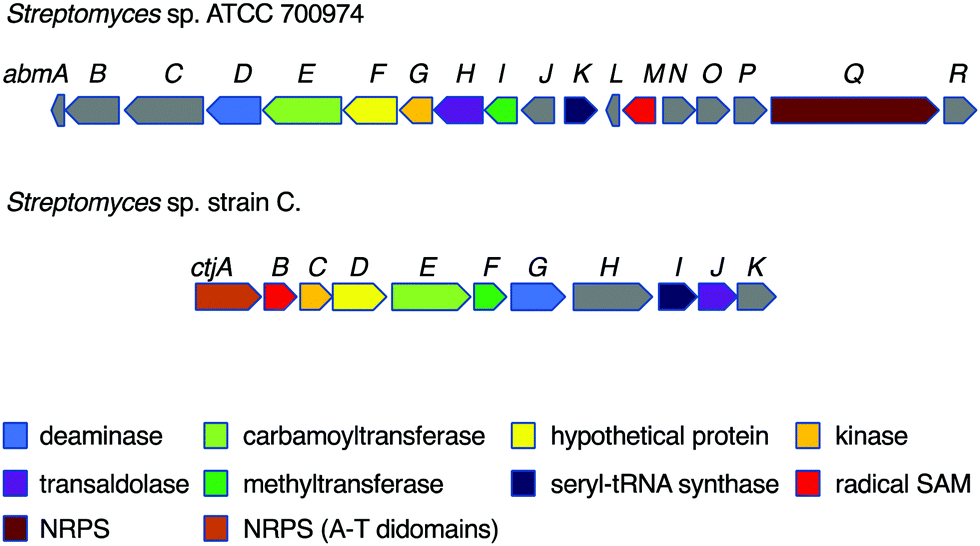 | ||
| Fig. 6 Biosynthetic gene clusters of the albomycins and a putative albomycin analogue. | ||
While the formation of the thiofuranose core unit has not been fully determined, it has been proposed that the key reaction that incorporates sulfur into the sugar moiety may adopt a radical-mediated mechanism analogous to the biotin synthase BioB.32 In fact, in the albomycin biosynthetic gene cluster, the gene abmM encodes a radical SAM enzyme containing both [4Fe–4S] and [2Fe–2S] cluster binding motifs, indicating that AbmM may be involved in the formation of the thiofuranose core unit in albomycin biosynthesis. However, no biochemical data are available to support this notion.
On the other hand, gene inactivation and in vitro biochemical assays of several other enzymes from the albomycin pathway did shed some light on the overall biosynthesis of the antibiotics (Fig. 7). The PLP-dependent transaldolase homologue AbmH was found to be responsible for the incorporation of L-threonine into the thionucleoside core unit, and the PLP-dependent epimerase AbmD catalyses the epimerization at C-6′ (Fig. 7).33,34 Inactivation of the abmE and abmI genes revealed the catalytic functions of AbmE and AbmI to be a N-carbamoyltransferase and a SAM-dependent methyltransferase that catalyse the N-carbamoylation and the methylation of the nucleobase, respectively. AbmI converted cytidine to 3-N-methylcytidine in an in vitro assay, indicating that AbmI is indeed involved in the modification of the cytidine moiety of albomycin.31 However, the order of these biotransformations is still unclear.
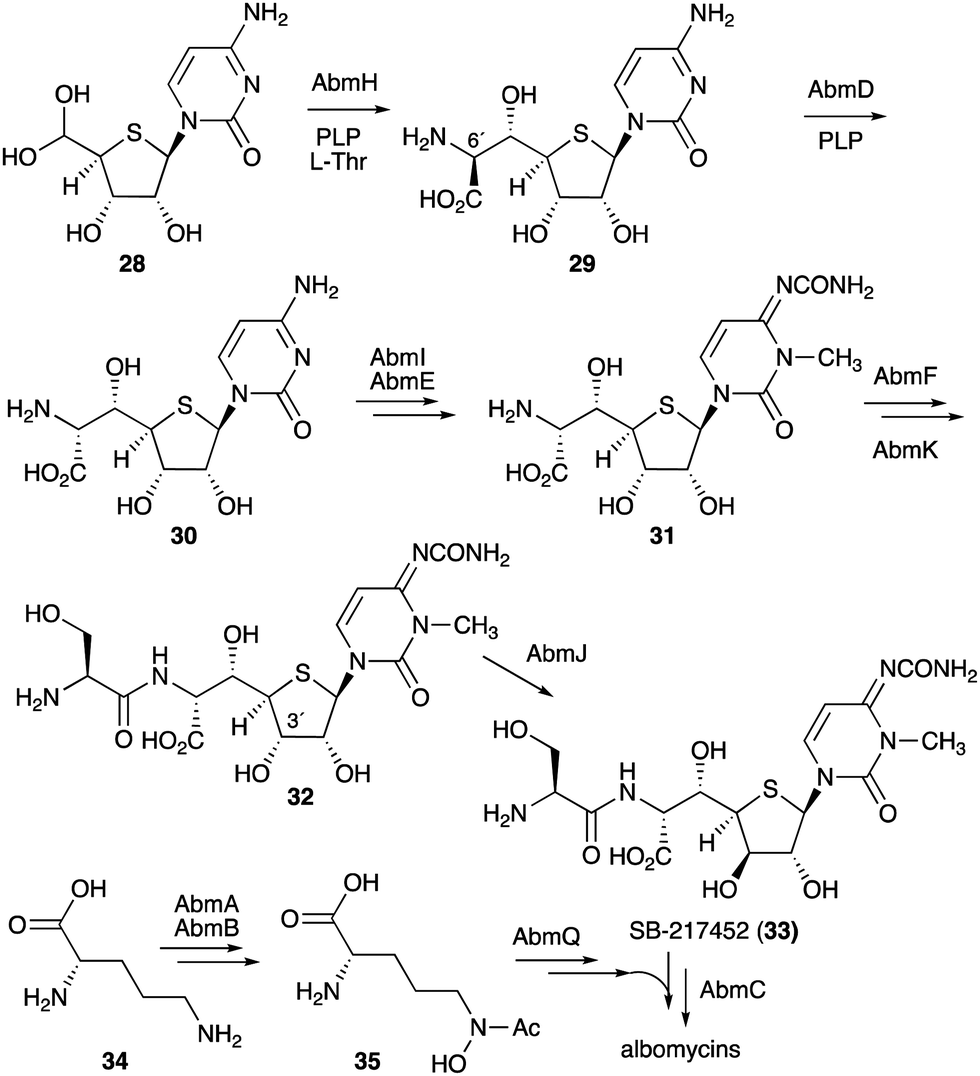 | ||
| Fig. 7 Biosynthetic pathway to albomycins. | ||
The hypothetical protein AbmF, which is similar to aminoacyl-tRNA synthetases, has been shown to catalyse the extension of the AbmE/AbmI product with L-serine, whereas the serine adenylase AbmK has been shown to play a role in supplying the amino acid for this reaction. The product (compound 32) is then epimerized at C-3′ by the radical S-adenosyl-L-methionine (SAM) enzyme AbmJ to give SB-217452 (33). This sequence of reactions was biochemically confirmed, as the recombinant AbmF/AbmK proteins incubated with L-Ser and ATP can only use the AbmI/AbmE product (31), but not its 3′ epimer,34 whereas AbmJ can only epimerize the product of AbmF (32), but not the product of AbmI/AbmE (31).
The formation of the siderophore (ferrichrome) moiety and the latter steps of albomycin biosynthesis have also been investigated.34 The ferrichrome building block N5-acetyl-N5-hydroxy-L-ornithine (35) is biosynthesized from L-ornithine (34) catalysed by the N-acetyltransferase AbmA and the flavin-dependent monooxygenase AbmB (Fig. 7), whereas the assembly of the tripeptide moiety is catalysed by the NRPS protein AbmQ. Interestingly, AbmQ, which contains two condensation (C) domains, lacks a C-terminal thioesterase (TE) domain, suggesting that no free siderophore is released from AbmQ. It was also hypothesized that AbmC, which shows low similarity to ATP-dependent proteases/ligases, is responsible for off-loading the tripeptide and directly transferring it to the corresponding intermediate SB-217452 (33). Inactivation of the abmC gene in the albomycin producing strain indeed abolished the production of albomycin in this mutant, and 33 accumulated in the culture, indicating that AbmC is responsible for the ligation of the ferrichrome moiety to SB-217452 (33).
2.3. Development of S-ribomimetics
Besides being used as anti-diabetic medications in Ayurvedic medicine, not many S-ribomimetics are used as clinically approved medications. Nevertheless, lamivudine (9), which contains both oxygen and sulfur atoms within the five-membered ring, has been widely used as an anti-HIV and anti-hepatitis B medication. Several salacinol analogues have been synthesized and tested for their human maltase and glucoamylase inhibitory activity and anti-infective activity.35 In addition, a number of thiofuranoses have been developed as nucleic acid-based therapeutics.36 The replacement of ribose with S-ribose has been shown to increase serum stability and enhance silencing longevity of siRNAs.Many deoxynucleoside analogues have also been developed as antineoplastic/anticancer drugs, as they interfere with DNA replication in cells to ultimately cause DNA damage and trigger apoptosis.37 Among them is thiarabine (4′-thio-β-D-arabinofuranosylcytosine or T-araC) (37), an analogue of the anticancer drug cytarabine (ara-C, 36) that contains thiofuranose instead of arabinose (Fig. 8).38,39
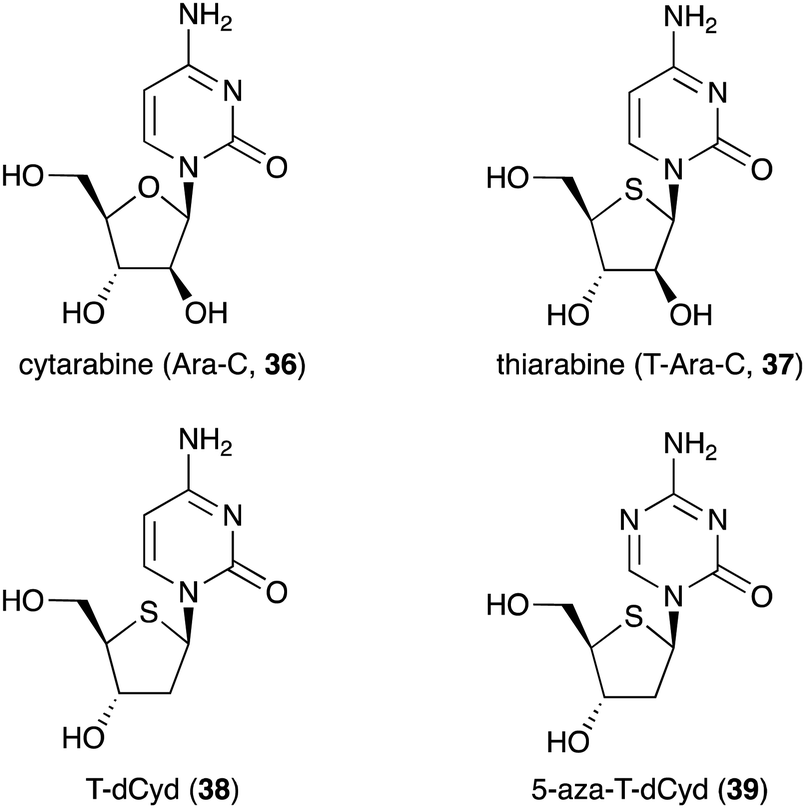 | ||
| Fig. 8 Chemical structures of cytarabine and its thiofuranose analogues. | ||
Despite the similarity of their structures and mechanisms of action, T-Ara-C (37) shows dramatically higher antitumour activity than Ara-C (36) when tested against human tumour cells in mice.40 This may be due to quantitative differences in biochemical or metabolic activity of T-Ara-C (37) versus Ara-C (36).40 T-Ara-CTP (a triphosphate version of thiarabine) has ∼10 times longer half-life in solid tumour cells compared to Ara-C (36),41 demonstrating how a simple ring heteroatom replacement of a sugar moiety can alter biological activity.
Other thiofuranoses that are currently in the clinical development are 4′-thio-2′-deoxycytidine (T-dCyd, 38) and 5-aza-T-dCyd (39) (Fig. 8).42 T-dCyd (38) depletes DNA methyltransferase 1 (DNMT1), both in vitro and in vivo in tumour cells. In non-small lung NCI-H23 xenograft studies, T-dCyd (38) inhibited tumour growth with concomitant depletion of DNMT1 at well tolerated doses.42 Unfortunately, the therapeutic index of 38 is relatively narrow, leading to the suspension of its clinical study. However, its analogue, 5-aza-T-dCyd (39), appears to be more promising, as it is 3- to 4-fold more potent than 38 in various human tumour cells with a larger therapeutic index in mice bearing CEM xenografts. Results from an on-going phase I clinical trial suggest that 5-aza-T-dCyd (39) is safe and well tolerated with a toxicity profile similar to currently approved hypomethylating agents.43 It is important to note that the therapeutic index of 39 is much larger than those of the currently approved drugs 5-aza-dCyd and 5-aza-Cyd, which are normally administered for short periods followed by a long recovery period. This suggests that substitution of oxygen for sulfur in the furanose moiety of nucleosides and deoxynucleosides is beneficial for improving their pharmacological properties.
3. N-Ribomimetics and related compounds
3.1. Discovery and bioactivities of N-ribomimetics
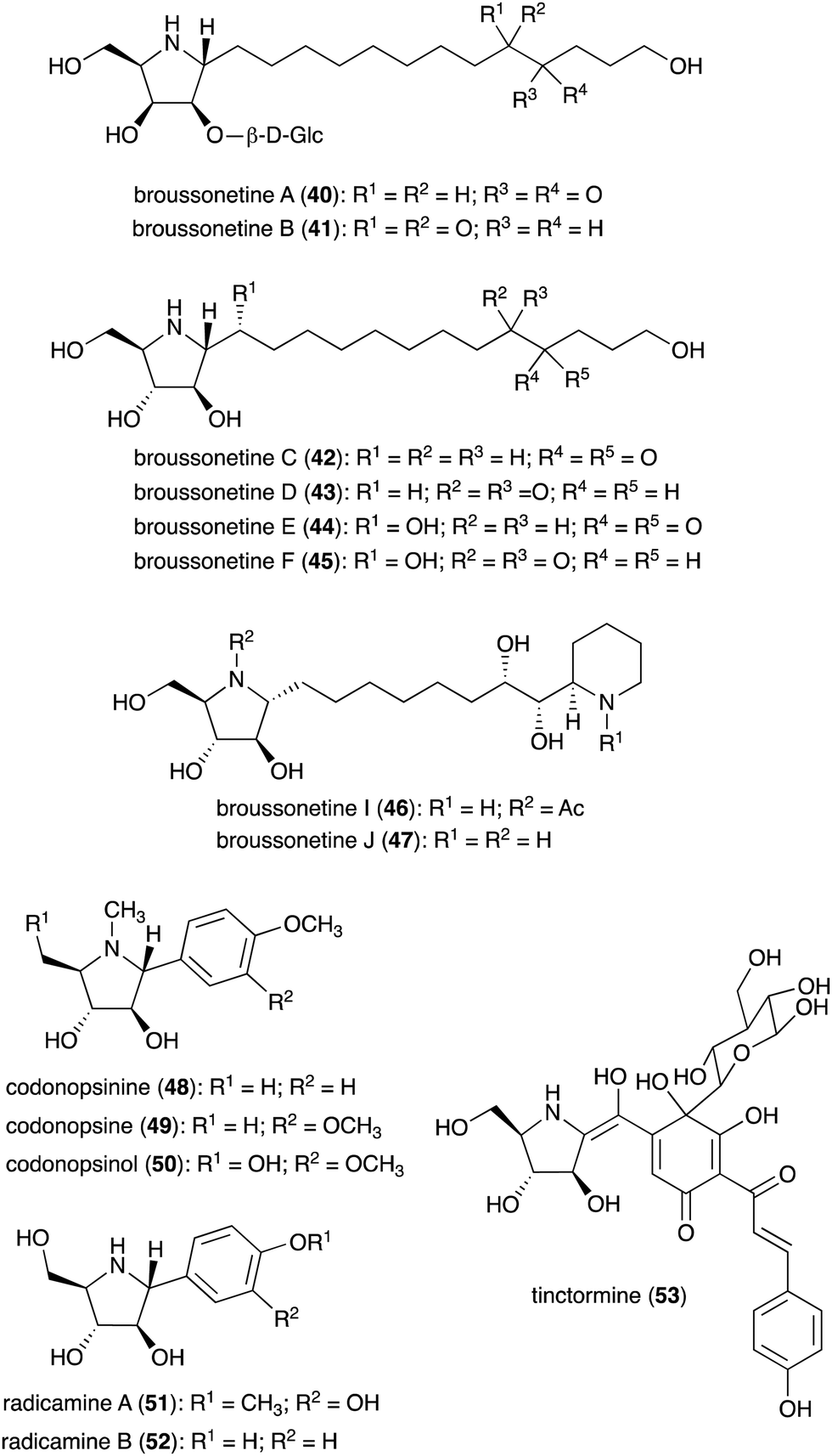
N-Ribomimetics are defined as ribose-like compounds in which the ring oxygen atom has been replaced by a nitrogen. Here, we define N-ribomimetics as nitrogen-containing five-membered rings that bear one or more hydroxy groups – to discriminate them from other pyrrolidine alkaloids. Compared to S-ribomimetics, N-ribomimetics appear to be more common in nature. Many N-ribomimetics have been isolated from a variety of sources including bacteria, fungi, and plants (Table 2). Among them are the broussonetines (40–47) (Fig. 9), which were isolated from the moraceous plant Broussonetia kazinoki.44–48 Broussonetines have been shown to have inhibitory activity against various glycosidases.49,50| Name of compound | Organism source | Bioactivity or function | Key reference(s) |
|---|---|---|---|
| Broussonetine A (40) | Broussonetia kazinoki | Glycosidase inhibitory activity | 48 |
| Broussonetine B (41) | B. kazinoki | Glycosidase inhibitory activity | 48 |
| Broussonetine C (42) | B. kazinoki | Glycosidase inhibitory activity | 48 |
| Broussonetine D (43) | B. kazinoki | Glycosidase inhibitory activity | 48 |
| Broussonetine E (44) | B. kazinoki | Glycosidase inhibitory activity | 48 |
| Broussonetine F (45) | B. kazinoki | Glycosidase inhibitory activity | 48 |
| Broussonetine I (46) | B. kazinoki | Glycosidase inhibitory activity | 93 |
| Broussonetine J (47) | B. kazinoki | Glycosidase inhibitory activity | 45, 82 and 93 |
| Codonopsinine (48) | Codonopsis clematidea | Glycosidase inhibitory activity | 51, 52 and 54 |
| Codonopsine (49) | C. clematidea | Glycosidase inhibitory activity | 52 and 54 |
| Codonopsinol (50) | C. clematidea | Glycosidase inhibitory activity | 52 and 54 |
| Radicamine A (51) | Lobelia chinensis | Glycosidase inhibitory activity | 54–56 |
| Radicamine B (52) | L. chinensis | Glycosidase inhibitory activity | 54–56 |
| Tinctormine (53) | Carthamus tinctorius | Ca2+ channel antagonistic activity | 57 |
| DAB-1 (14) | Angylocalyx spp., Arachniodes standishii, Morus bombycis, Eugenia spp., Hyacinthoides non-scripta, Scilla campanulate, Adenophora triphylla var. japonica | Glycosidase inhibitory activity | 58, 64, 65 and 84 |
| 2-epi-DAB-1 (54) | Morus spp. | Glycosidase inhibitory activity | 58 and 59 |
| CYB-1 (55) | Castanospermum australe | Glycosidase inhibitory activity | 58 and 59 |
| N-Hydroxyethyl-2-deoxy-DAB-1 (56) | C. australe | Glycosidase inhibitory activity | 6 and 58 |
| FR900483 (nectrisine, 57) | Nectria lucida F-4490 | Glycosidase inhibitory activity, immunomodulator activity | 58, 62 and 63 |
| 2,5-Dideoxy-2,5-imino-D-mannitol (DMDP, 58) | Streptomyces sp. KSC-5791, Morus alba, Derris elliptica, Lonchocarpus spp., Endospermum sp., Omphalea diandra, Nephthytis poissoni, Aglaonema spp., Hyacinthoides non-scripta, Campanula rotundifolia, Hyacinthus orientalis, Scilla campanulate, Adenophora spp. | Inhibitory activity against various glycosidases | 58 |
| 6-Deoxy-DMDP (59) | Angylocalyx spp. | Glycosidase inhibitory activity | 58 and 66 |
| HomoDMDP (60) | H. non-scripta, H. orientalis, Scilla campanulate, Muscari armeniacum | Glycosidase inhibitory activity | 58 and 67 |
| 6-Deoxy-homo-DMDP (61) | H. orientalis, M. armeniacum | Glycosidase inhibitory activity | 58, 68 and 69 |
| 2,5-Imino-2,5,6-trideoxy-D-gulo-heptitol (62) | H. orientalis | Glycosidase inhibitory activity | 58, 70 and 71 |
| DMDP N-acetic acid (63) | Stevia rebaudiana | Glycosidase inhibitory activity | 72 |
| Collemin A (64) | Collema cristatum | Sunscreen activity | 73 |
| Pramanicin (65) | Stagonospora sp. ATCC 74235 | Antifungal activity toward Candida albicans KF1 and Cryptococcus neoformans | 74 and 75 |
| Pramanicin A (66) | Stagonospora sp. ATCC 74235 | Antifungal activity toward Candida albicans KF1 and Cryptococcus neoformans | 74 |
| Virgaricin (67) | Virgaria sp. FKI-4860 | Antibacterial activity toward Bacillus subtilis KB 211 | 77 |
| Virgaricin B (68) | Virgaria boninensis FKI-4958 | Antibacterial activity against E. coli KB 213 and B. subtilis KB 211 | 78 |
| TMC-260 (69) | Acremonium kiliense Grutz TC 1703 | Potential candidate for interleukin-4 inhibitors | 76 |
| Anisomycin (70) | Streptomyces roseochromogenes, S. griseolus, S. hygrospinosus | Toxicity toward pathogenic protozoa, fungi, and human tumour cell lines | 79 and 88 |
| Gualamycin (71) | Streptomyces sp. NK1168 | Acaricidal activity | 80 and 81 |
 | ||
| Fig. 9 Chemical structures of plant-derived N-ribomimetics. | ||
Some other plants also produce N-ribomimetics. For example, the Asian bell flower plant Codonopsis clematidea produces codonopsinine (48), codonopsine (49), and codonopsinol (50),51–53 all of which showed good glycosidase inhibitory activity.54 Codonopsinine (48) and codonopsine (49) also have antibacterial and anti-hypertensive activities.52,54 Radicamines (51–52), analogues of codonopsinine, are produced by the traditional Chinese medicinal plant Lobelia chinensis.55,56 Similar to codonopsinine, the radicamines also show inhibitory activity against glycosidases.54 A related metabolite, tinctormine (53), is produced by the asteraceous plant Carthamus tinctorius, which is also known as safflower. Tinctormine showed Ca2+ channel antagonistic activity.57
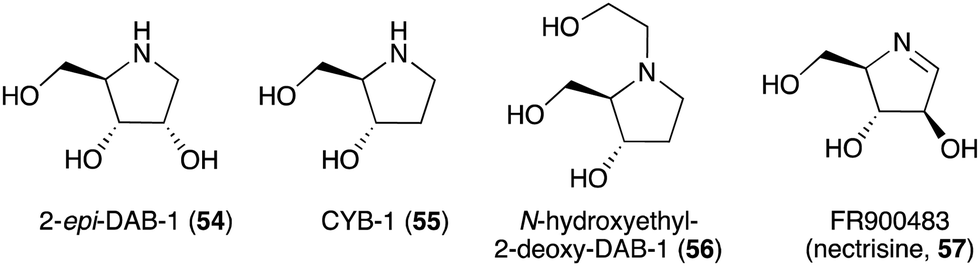
The broad-spectrum glycosidase inhibitor 1,4-dideoxy-1,4-iminoarabinitol (DAB-1, 14) is a widespread plant secondary metabolite that inhibits several glycosidases including α-glucosidases, α-D-arabinosidase, β-glucosidases and α-mannosidases.58 This simple N-ribomimetic natural product has been isolated from both tropical and temperate plants.58 More recently, DAB-1 was also identified in the bacterium Chitinophaga pinensis DSM 2588. In addition, several DAB-1 analogues, e.g., 2-epi-DAB-1 (54), 2-deoxy-DAB-1 (CYB3, 55), and N-hydroxyethyl-2-deoxy-DAB-1 (56) (Fig. 10), have been isolated from Morus spp.59 and Castanospermum australe.60,61 FR900483 (57), an unsaturated-DAB (Fig. 10), was isolated from the fungus Nectria lucida F-4490.62 The latter compound, a.k.a. nectrisine, showed immunomodulator activity in proliferatively suppressed lymphocytes and immunosuppressed mice.62 It also showed potent α-glucosidase inhibitory activity.63
 | ||
| Fig. 10 Chemical structures of DAB-1 analogues. | ||
Crystal structures of Bombyx mori α-glucosidase (BmSUH) in complex with DAB-1 or sucrose, as well as those of α-L-arabinofuranosidase in complex with DAB-1 or arabinose showed that DAB-1 binds to the active site of both enzymes and functions as a competitive inhibitor of the enzymes (Fig. 11).64,65
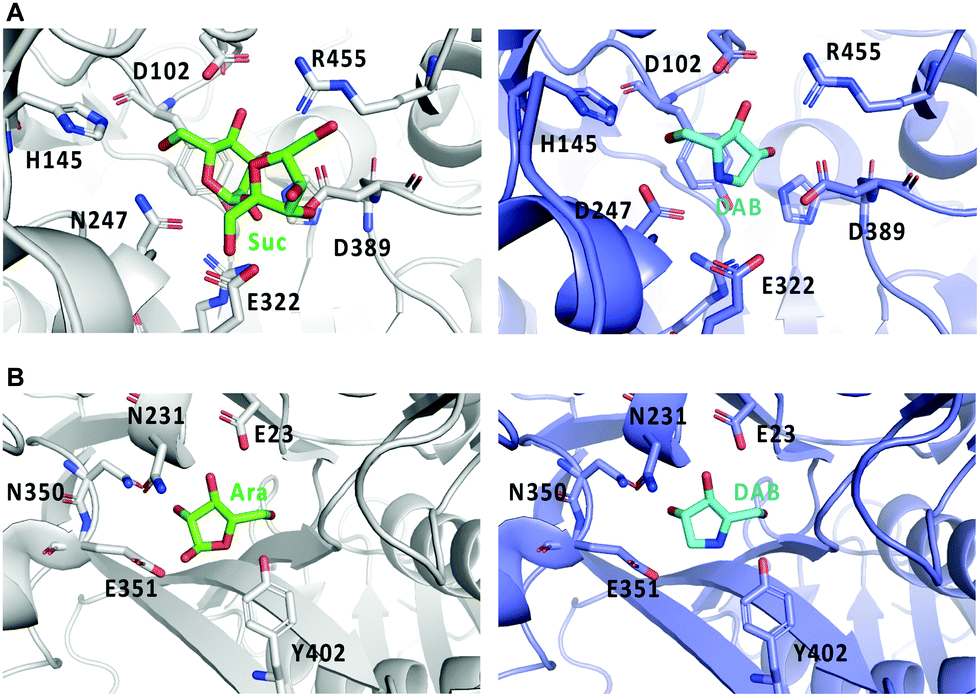 | ||
| Fig. 11 Crystal structures of glycosidases and DAB-1. (A) binding model of sucrose (Suc) with BmSUH (left; PDB code, 6LGF) and DAB-1 (DAB) with BmSUH (right; PDB code, 6LGD); (B) binding of arabinose (Ara) with α-L-arabinofuranosidase (left; PDB code, 6zpy) and DAB-1 with α-L-arabinofuranosidase (right; PDB code, 6zq1). | ||
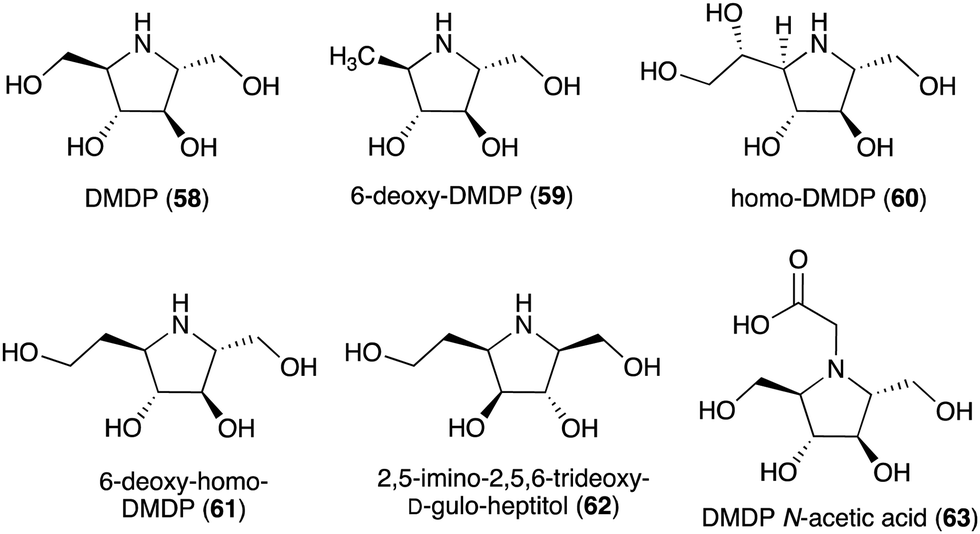
Some plants and bacteria also produce 2,5-dideoxy-2,5-imino-D-mannitol (DMDP, 58) and related compounds, e.g., 6-deoxy-DMAP (2,5-imino-1,2,5-trideoxy-D-mannitol, 59),66 homo-DMAP (2,5-dideoxy-2,5-imino-DL-glycero-D-manno-heptitol, 60),67 3,6-deoxy-homo-DMDP (61),68,69 and 2,5-imino-2,5,6-trideoxy-D-gulo-heptitol (62) (Fig. 12).58,70,71 This class of compounds is widely distributed in nature as they have been isolated from unrelated families of both tropical and temperate plants as well as from strains of Streptomyces soil bacteria.58 Consistent with their chemical structures, they showed inhibitory activity against various glycosidases including glucosidases, mannosidases, galactosidases, and fucosidases.58 More recently, DMDP N-acetic acid (63) was isolated from Stevia rebaudiana, a commercially important plant – a source of the natural sweetener steviol. Although such derivatives with acetic and propionic acids have been detected by GC-MS in a broad range of plants, it was the first isolated natural amino acid derived from an iminosugar.72 Compared to DMDP (58), which showed strong inhibitory activity against β-galactosidases, DMDP N-acetic acid (63) is more active against α-galactosidases.72
 | ||
| Fig. 12 Chemical structures of DMDP and related compounds. | ||
Besides plants, fungi have also been known as producers of N-ribomimetics. The mycosporine compound collemin A (64), which possesses sunscreen activity, was isolated from the lichenized ascomycete Collema cristatum (Fig. 13).73 Collemin A (64) absorbs UV-B and prevents UV-B induced cell destruction and erythema when applied to the skin prior to UV-B irradiation.73 Other fungal-derived N-ribomimetics include the pramanicins (65 and 66) and the virgaricins (67 and 68) (Fig. 13). Although their core structure is effectively a γ-lactam ring, due to a high degree of hydroxylation, their structures fall into our definition of N-ribomimetics. Thus, they are included in this review.
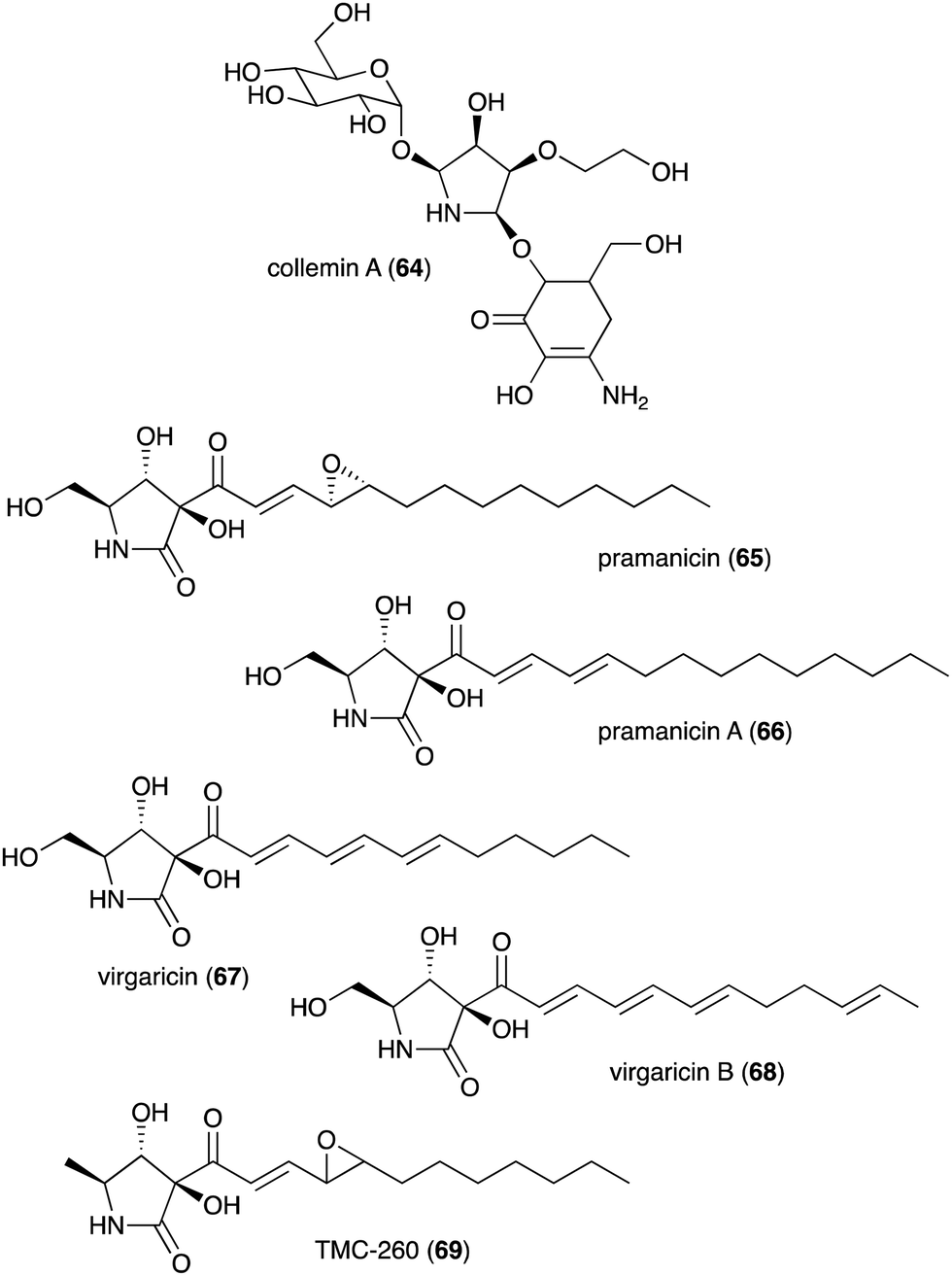 | ||
| Fig. 13 Chemical structures of fungal-derived N-ribomimetics. | ||
The pramanicins, which contain a long alkyl or alkenyl side chain, were isolated from the fungus Stagonospora sp. ATCC 74235. They showed antifungal activity toward Candida albicans KF1 and Cryptococcus neoformans.74,75 An analogue of pramanicin, TMC-260 (69), was identified from the broth of Acremonium kiliense Grutz TC 1703, as part of a screening effort to find interleukin-4 inhibitors.76 Virgaricin (67), another analogue of pramanicin, was isolated from Virgaria sp. FKI-4860. This compound showed modest antibacterial activity toward Bacillus subtilis KB 211.77 Virgaricin B (68), which was isolated from Virgaria boninensis FKI-4958, also showed antibacterial activity against E. coli KB 213 and B. subtilis KB 211.78
The bacterial-derived N-ribomimetic anisomycin (70) was isolated from several Streptomycetes including Streptomyces roseochromogenes, S. griseolus, and S. hygrospinosus (Fig. 14).79 Anisomycin (70) showed various biological activities including toxicity toward pathogenic protozoa, fungi, and human tumour cell lines, as well as activation of the mitogen-activated protein kinase signalling pathways. Another bacterial N-ribomimetic, gualamycin (71), was isolated from the broth of Streptomyces sp. NK1168. This gulosamine- and galactose-containing glycoside showed potent acaricidal activity with no phytotoxicity to kidney bean (Fig. 14).80,81
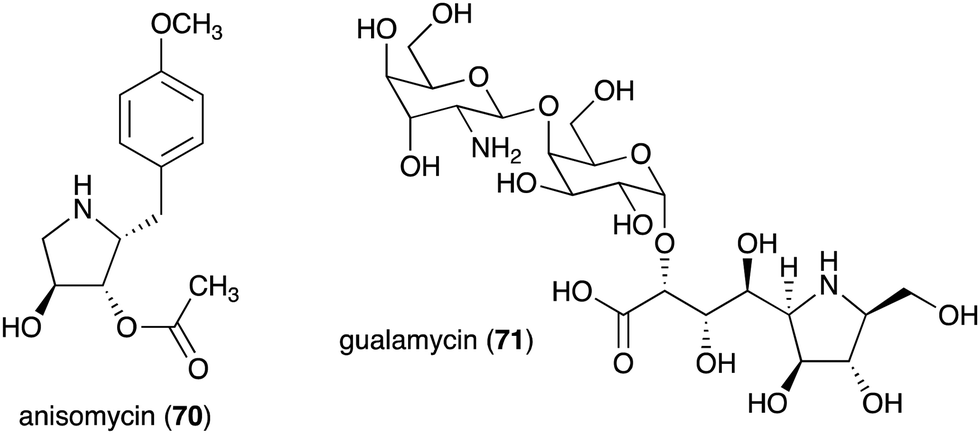 | ||
| Fig. 14 Chemical structures of bacterial N-ribomimetics. | ||
3.2. Biosynthesis of N-ribomimetics
Known natural N-ribomimetics have been shown to derive from different precursors or pathways, e.g., the acetate pathway, the pentose phosphate pathway, or the glycolytic pathway. The nitrogen atoms generally originate from amino acids, either incorporated intact or functioning as a source of nitrogen through the catalysis of an aminotransferase.Incorporation of 13C atoms into the 1, 4, 6, 8, 10, 12, 14, 16 and 18 positions, as determined by NMR, indicated that the C-4–C-16 of broussonetine J originates from palmitoyl-CoA (77), which may be synthesized from glucose via the acetate-malonate pathway. The result also suggested that the C-1–C-3 unit of broussonetine J is derived from serine (76), as C-3 of serine normally originates from C-1 of glucose (Fig. 15). However, no incubation experiments using isotopically labelled serine have been performed to confirm this finding.
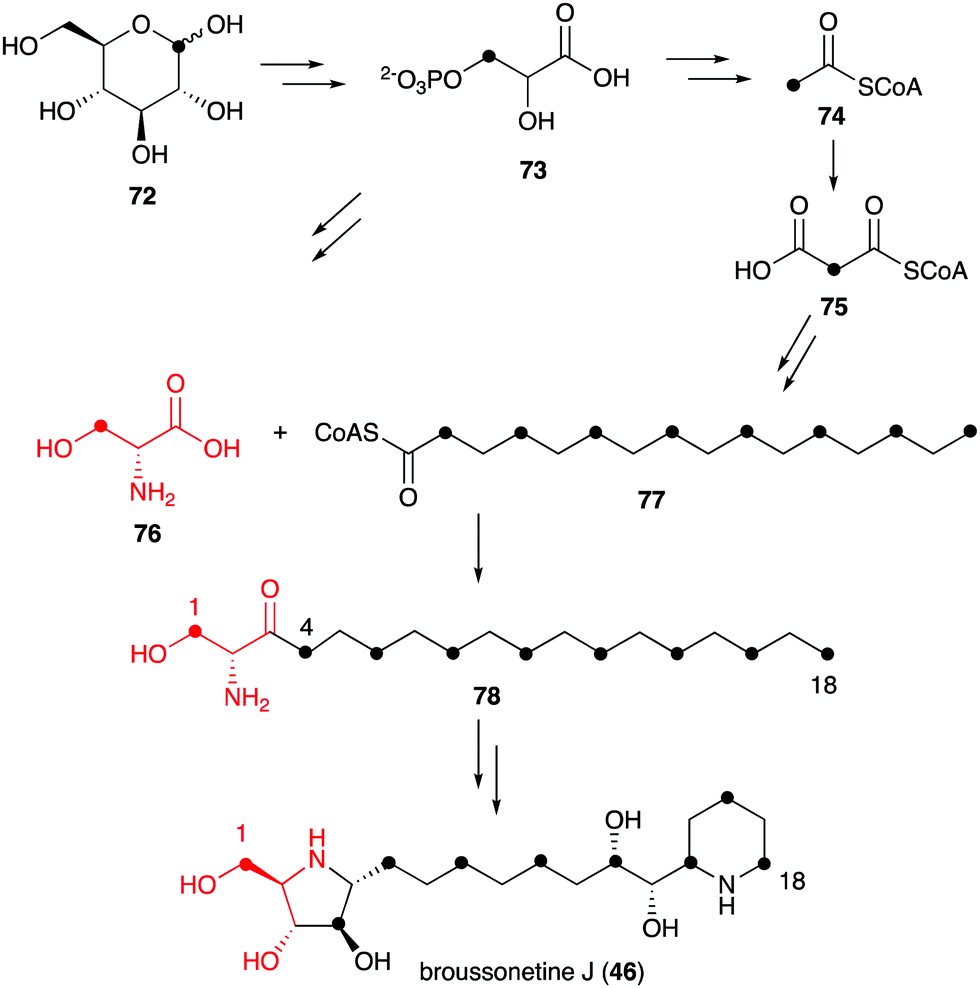 | ||
| Fig. 15 Biosynthetic origin of broussonetine J. | ||
However, in the biosynthetic studies of pramanicin (65) in Stagonospora sp. ATCC 74235, both [1,2-13C2]acetate (80) and DL-[1-13C]serine were tested, and good enrichment of 13C was observed when either one of them was added to the culture of the producing fungus.83 Consistent with the labelling observed in broussonetine biosynthesis, supplementation with [1,2-13C2]acetate resulted in the incorporation of 13C into the 2–3, 7–8, 9–10, 11–12, 13–14, 15–16, 17–18 and 19–20 positions, whereas incubation with DL-[1-13C]serine showed 13C incorporation into the C-4 position. These results indicated that the alkenyl side chain is biosynthesized by seven acetate units and the ribomimetic moiety is formed from acetate and serine (Fig. 16).
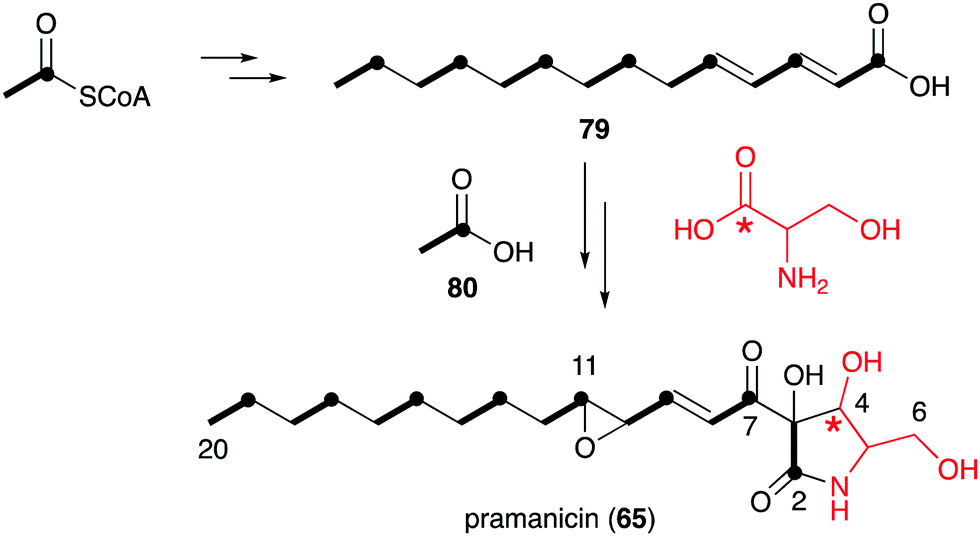 | ||
| Fig. 16 Biosynthetic origin of pramanicin. | ||
While no biochemical studies of the biosynthetic enzymes have been conducted, the biosynthetic pathway to DAB-1 (14) is believed to resemble that of deoxynojirimycin.85,86 Xylulose 5-phosphate (81), which is an intermediate of the pentose phosphate pathway, may be converted to 4-amino-4-deoxyarabinitol (83) by the GabT1 orthologue and the phosphatase (Fig. 17). Subsequently, the compound is converted to 4-deoxy-4-imino-D-arabinitol (85) by the GutB1 orthologue and reduced to DAB-1 by an unknown enzyme/mechanism. It is important to note that while bacteria produce DAB-1 from xylulose 5-phosphate, plants may use a different mechanism to produce it. Thus, how this compound is made in plants remains an open question.
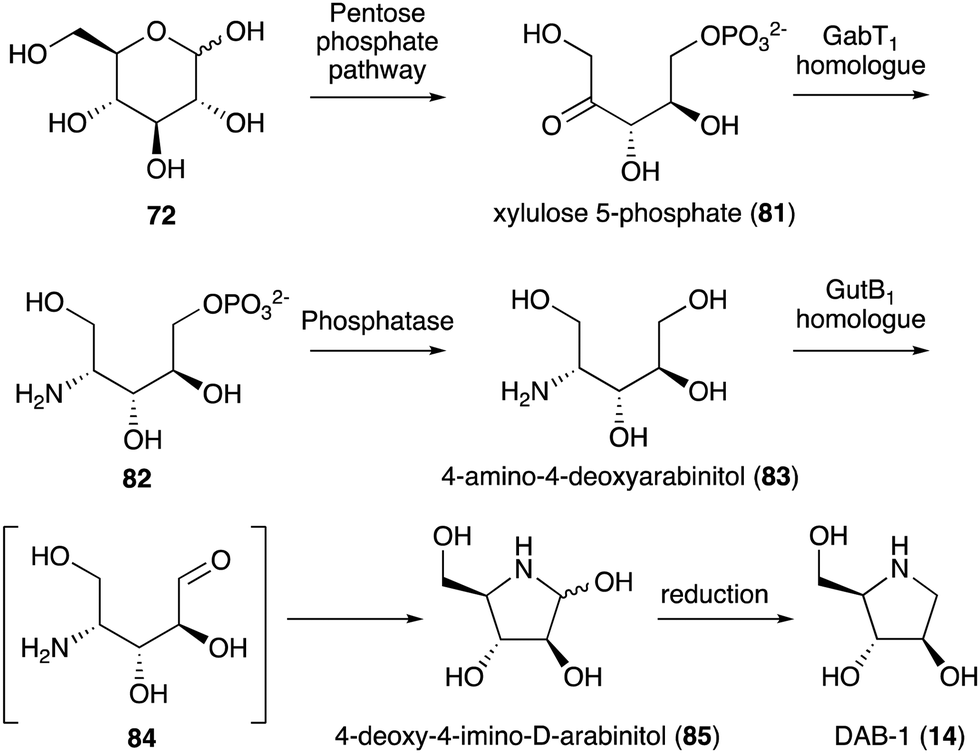 | ||
| Fig. 17 Proposed biosynthetic pathway to DAB-1. | ||
On the basis of gene inactivation, labelling experiments, and/or biochemical studies, the biosynthetic pathway to anisomycin (70) is proposed to initiate by deamination of L-tyrosine to 4-hydroxyphenylpyruvate (HPP, 86) catalysed by the aminotransferase AniQ (Fig. 18).88 This is followed by a coupling reaction of the product with glyceraldehyde 3-phosphate catalysed by the transketolase AniP. The product (DHPP, 87) is then glucosylated by the glycosyltransferase AniO and converted to the glycosylated hydroxyphenyl-aminopentitol (Glc-HPAP, 89) by AniQ. As aniQ is the only aminotransferase gene in the anisomycin BGC, the gene product is hypothesized to work twice in the pathway, the deamination of L-tyrosine and the amination of the glycosylated ketone Glc-DHPP (88). In fact, in the presence of PLP and α-ketoglutarate the recombinant AniQ can convert 89 back to 88.
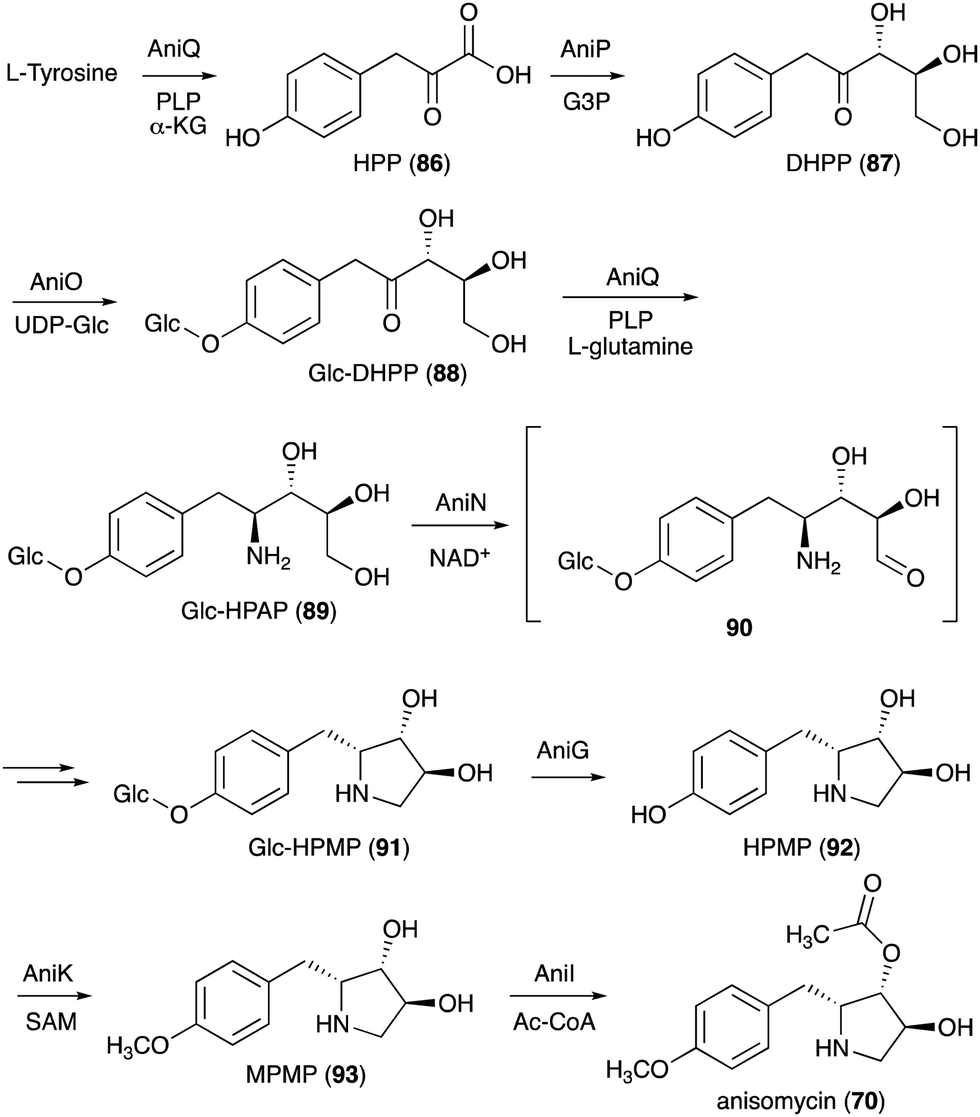 | ||
| Fig. 18 Biosynthesis of anisomycin. | ||
Oxidation of Glc-HPAP by the short-chain dehydrogenase AniN would give an aldehyde intermediate (90), which then undergoes Schiff base formation and reduction to give the N-ribomimetic-containing product Glc-HPMP (91). This step is also confirmed by gene inactivation, as the ΔaniN mutant only produces the glycosylated amino intermediate. Glc-HPMP (91) is hypothesized to undergo deglycosylation by the putative glucosidase AniG before being modified by the methyltransferase AniK and the O-acetyltransferase AniI to give anisomycin (70). The attachment of glucose to the AniP product 87 and the removal of the sugar moiety later in the pathway by AniG is somewhat unusual and puzzling, as in principle the biosynthesis may proceed without these glucosylation–deglucosylation steps. However, the presence of both genes in the anisomycin BGC and the identification of the glucosylated intermediates in the AniG mutant strongly suggest that the glucosylation–deglucosylation steps are involved. The timing of the glycosylation step in the pathway and the selectivity of the glycosyltransferase enzyme were confirmed by in vitro enzymatic assay where the recombinant AniO could only glycosylate the AniP product DHPP (87) and not any other intermediates. In addition, the transamination reaction by AniQ took place more efficiently with a glucosylated substrate. While there is no biochemical study on the glucosidase AniG, deglycosylation must precede the O-methylation on the phenol moiety. Interestingly, the methyltransferase AniK can methylate both HPMP (92) and its acetylated derivative. Therefore, while disruption of the putative O-acetyltransferase gene aniI resulted in a strain that accumulated only deacetylanisomycin, it is still possible that acetylation takes place before O-methylation in the pathway.
3.3. Development of N-ribomimetics
Similar to the S-ribomimetics, the development of N-ribomimetics has been done mostly by chemical synthesis. One significant example of this effort is the development of purine nucleoside analogue (PNA)-based immunosuppressive agents that can inhibit purine nucleoside phosphorylase (PNP), leading to the inhibition of human lymphocyte proliferation.89 Among the PNAs developed, forodesine (BCX1777 or immucillin H) (94) (Fig. 19) was found to have human PNP inhibitory activity. Subsequent preclinical and clinical evaluation of 94 showed its efficacy in treating patients with B-cell acute lymphoblastic leukemia.90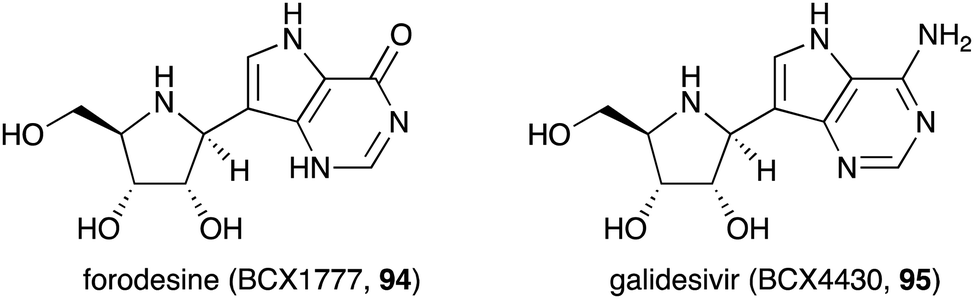 | ||
| Fig. 19 Chemical structures of forodesine and galidesivir. | ||
Another useful nucleoside analogue structurally similar to forodesine is galidesivir (BCX4430, 95) (Fig. 19). This compound is being developed as an antiviral drug. Galidesivir (95) is active in vitro against several pathogenic RNA viruses, such as MERS-CoV, SARS-CoV, and other lethal viruses (Ebola virus, Marburg virus, and Yellow Fever virus).91,92 Similar to the mechanism of action of other nucleoside-based antiviral drugs, 95 is phosphorylated by cellular kinases to form a triphosphate version of the compound that resembles ATP. This ATP mimic is then used by the viral RNA polymerase as one of the building blocks, incorporating galidesivir monophosphate into the growing RNA chain, leading to premature chain termination. The fact that forodesine (94) and galidesivir (95) are structurally very similar to each other but their biological activities are very different is remarkable. Therefore, it is expected that many more analogues of N-ribomimetics will be developed, and more exciting biological activities will emerge from this class of compounds.
4. C-Ribomimetics and related compounds
4.1. Discovery and bioactivities of C-ribomimetics
The C-ribomimetics (carbafuranoses) are among naturally occurring sugar mimetics that have been studied more extensively, both for their biological activities and biosynthesis. Most, if not all, of the known C-ribomimetics are of bacterial origin (Table 3). Among them is bacteriohopanetetrol (BHT) cyclitol ether (96) (Fig. 20), which was first identified as a component of glycolipids in the Gram-negative bacterium Zymomonas mobilis.| Name of compound | Organism source | Bioactivity or function | Key reference(s) |
|---|---|---|---|
| BHT cyclitol ether (96) | Zymomonas mobilis | Component of glycolipids in Zymomonas mobilis | 145 |
| Aristeromycin (15) | S. citricolor | Antitumour and antibacterial activities (S-adenosyl-L-homocysteine hydrolase inhibitor) | 94–99, 151–159, 161 and 162 |
| Neplanocin A (97) | Ampullariella regularis A11079 | Antitumour and antibacterial activities (S-adenosyl-L-homocysteine hydrolase inhibitor) | 94–98, 151–158 and 160–162 |
| Adecypenol (98) | Streptomyces sp. OM-3223 | Calf intestinal adenosine deaminase inhibitor | 100 |
| Pactamycin (16) | S. pactum | Antibiotic, antiplasmodial, and antitumour activity | 105–109, 114, 146–150 and 163–168 |
| 7-Deoxypactamycin (99) | S. pactum | More active than pactamycin (16) | 110–113 |
| Pactamycate (100) | S. pactum | Less active than pactamycin (16) | 110–112, 114 and 115 |
| Pactalactam (101) | S. pactum | Less active than pactamycin (16) | 110–112, 114 and 115 |
| Jogyamycin (102) | S. pactum | Antitrypanosomal activity | 112 |
| Allosamidin (103) | Streptomyces sp. no. 1713 | Chitinase inhibitor, stress tolerance enhancement, inhibition of moulting, inhibition of yeast cell separation, inhibition of malarial ookinete transmission, inhibition of encystment of Entamoeba invadens, promotion of chitinase production in Streptomyces, suppression of airway inflammation in mammals | 116–129 |
| Trehazolin (105) | Micromonospora coriacea (SANK 62192) | Trehalase inhibitor | 131, 132 and 140 |
| Caryose (107) | Pseudomonas caryophylli | Lipid membrane constituents of P. caryophylli | 133 and 134 |
| Calditol (108) | Sulfolobus acidocaldarius | Thermoacidophilic archaebacterial lipid membrane constituent | 135, 136 and 141–144 |
| Queuosine (115) | Certain bacteria and eukaryotes | Enhances translation fidelity | 137–139 |
| Epoxyqueuosine (114) | Certain bacteria and eukaryotes | Enhances translation fidelity | 137–139 |
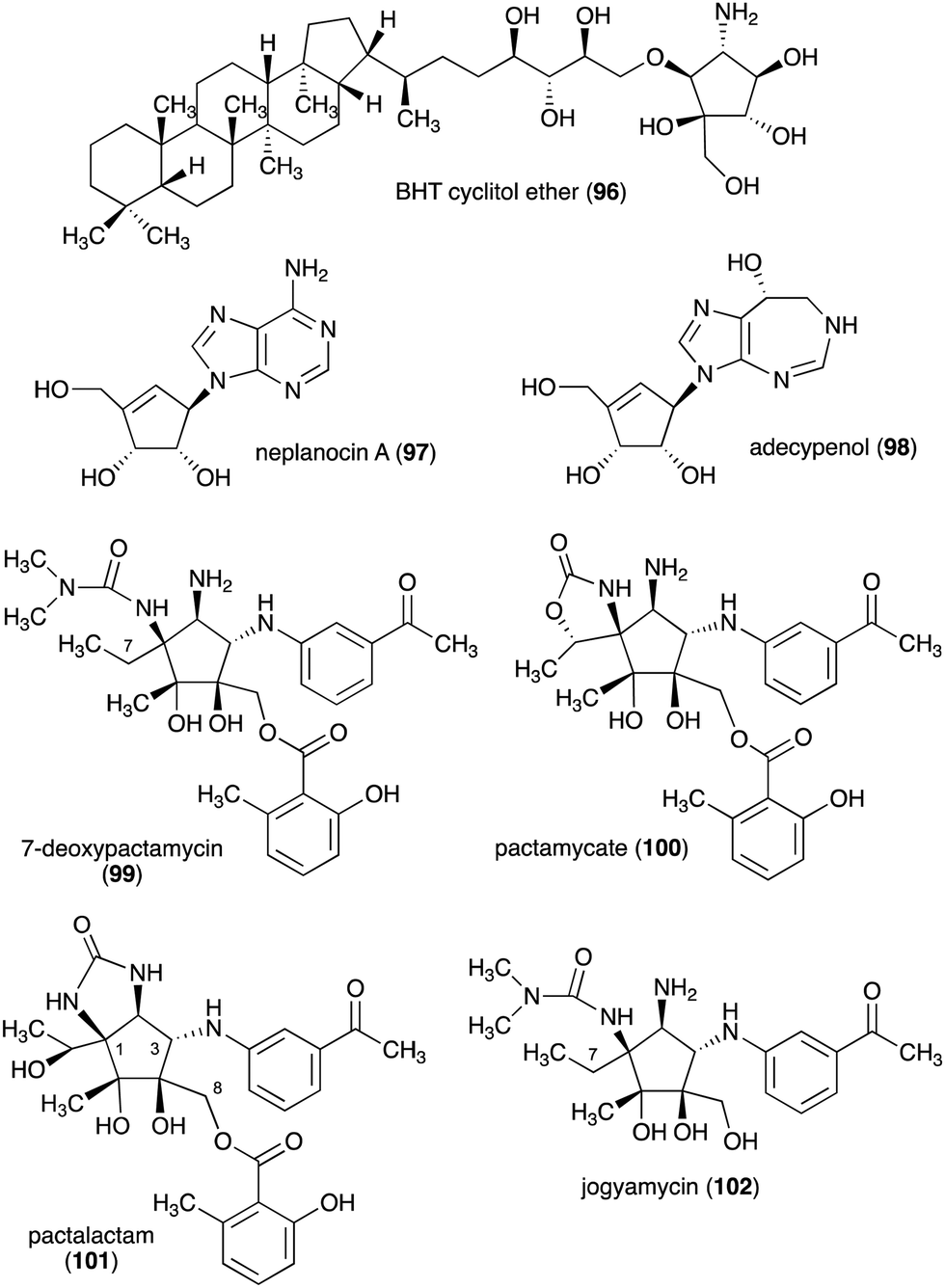 | ||
| Fig. 20 Chemical structures of C-ribomimetics. | ||
Other C-ribomimetics, aristeromycin (15) (Fig. 3) and neplanocin A (97) (Fig. 20), were isolated from the cultures of Streptomyces citricolor and Ampullariella regularis A11079, respectively.94,95 These compounds exhibited significant antitumour and antibacterial activities, as well as inhibition of vaccinia virus multiplication in monolayer cultures of mouse L-cells.96,97 These antibiotics inhibit the activity of the S-adenosyl-L-homocysteine hydrolase (SAHH) enzyme, which is involved in the activated methyl cycle. This activity was also postulated to account for the observed antiviral activity of the compounds, as SAM-dependent macromolecule methylation reactions are essential to the production of new virus particles.97,98
Crystal structures of SAHH in complex with 3′-keto-aristeromycin (3KA) indicates that 3KA binds to SAHH at the same position as adenosine, which is one of the substrates for SAHH in the formation of S-adenosyl-L-homocysteine (SAH) (Fig. 21).99 Docking studies of SAHH with aristeromycin (15) or 3KA showed that the distance between the cofactors (NAD+ or NADH) and the 3′-position of the corresponding substrate (aristeromycin or 3KA, respectively) changed from 3.2 Å in NAD+-aristeromycin to 3.6 Å in NADH-3KA, suggesting that the oxidation of aristeromycin by NAD+ occurs more readily than the reduction of 3KA back to aristeromycin by NADH. Therefore, 3KA appears to be more thermodynamically stable than aristeromycin in SAHH, making it a better inhibitor for the enzyme. Thus, it is postulated that after competing with the natural substrate adenosine for SAHH, aristeromycin is subsequently oxidized to 3KA.
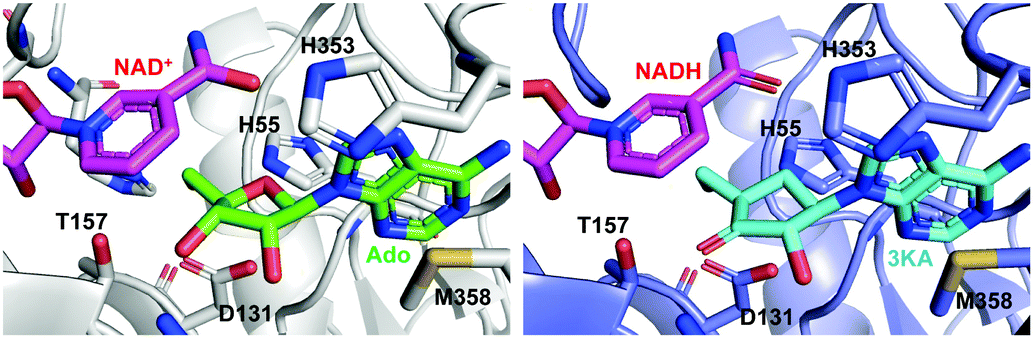 | ||
| Fig. 21 Structures of the mammalian SAHH complexed with NAD+ and adenosine (Ado) (left; PDB code, 5AXA) and with NADH and 3KA (right; PDB code, 5AXC). | ||
Adecypenol (98), a homologue of neplanocin A (97) in which the adenine moiety has been modified, was isolated from the culture broth of Streptomyces sp. OM-3223.100 It contains a unique homopurine chromophore, 8(R)-3,6,7,8-tetrahydroimidazo[4,5-d]-[1,3]diazepin-8-ol, similar to that found in adechlorin, coformycin, and deoxycoformycin (pentostatin).101–103 However, in contrast to the latter compounds, which have potent and tight-binding inhibitory activity against adenosine deaminase, adecypenol is a semi tight-binding type inhibitor against calf intestinal adenosine deaminase.104 In contrast to neplanocin A, adecypenol lacks antimicrobial activity against various bacteria and fungi.
The antitumour antibiotic pactamycin (16) (Fig. 3) was identified in the culture medium of Streptomyces pactum.105 Pactamycin inhibits protein synthesis by interacting with the 16S rRNA of the 30S ribosomal subunit of bacteria or the equivalent small ribosomal subunit in other organisms. Analysis of the crystal structures of the Thermus thermophilus 30S ribosomal subunit in complex with pactamycin and 16S rRNA and in vitro assays suggest that pactamycin interacts with the ribosomal E-site and prevents tRNA-mRNA complex relocation into the E site (Fig. 22).106–108 Despite its potent biological activity, the development of pactamycin as a clinically useful drug was hampered by its broad-spectrum cytotoxicity.109
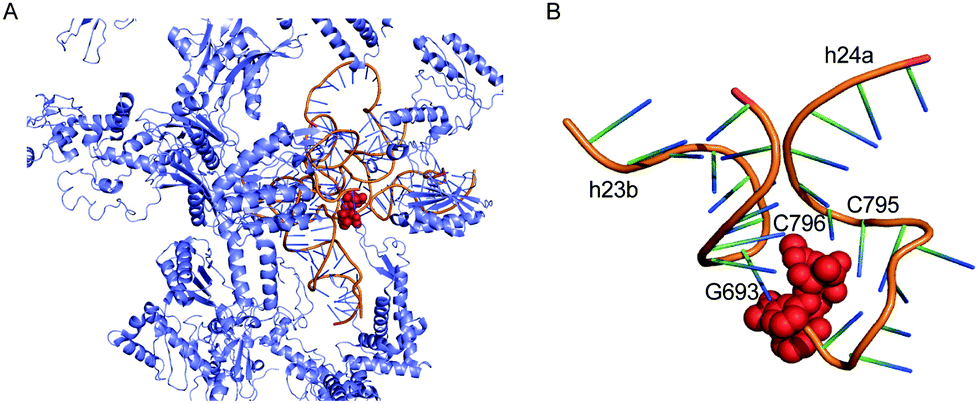 | ||
| Fig. 22 Structure of the T. thermophilus ribosome with bound pactamycin (PDB code, 1HNX). The red spheres show pactamycin. | ||
In addition to pactamycin, a number of pactamycin analogues, e.g., 7-deoxypactamycin (99), pactamycate (100), pactalactam (101), and jogyamycin (102), have been isolated from various strains of S. pactum (Fig. 20).110–112 7-Deoxypactamycin (99), which lacks a hydroxy group at C-7, showed superior activities compared to pactamycin in a number of biological assays.113 On the other hand, pactamycate (100) and pactalactam (101) are significantly less active than pactamycin against bacteria, Plasmodium falciparum, and several cancer cell lines,114,115 indicating the importance of the free C-2 amino group and the dimethylurea moiety for their activities. On the other hand, jogyamycin has been shown to have better antitrypanosomal activity compared to pactamycin.112
Other interesting C-ribomimetic-containing natural products include the chitinase inhibitor allosamidin (103) and the trehalase inhibitor trehazolin (105) (Fig. 23). Allosamidin (103), from Streptomyces sp. no. 1713,116 is a pseudotrisaccharide consisting of a ribomimetic moiety (allosamizoline, 104), and two units of N-acetyl-D-allosamine. The carbafuranose structure of allosamizoline is linked with a dimethylaminooxazoline ring, resembling the oxazolium ion intermediate of chitin hydrolysis mediated by chitinases from family 18. Allosamidin has various biological activities including stress tolerance enhancement in plants,117 inhibition of moulting in insects,116,118–120 inhibition of yeast cell separation,121 inhibition of malarial ookinete transmission,122–124 inhibition of encystment of Entamoeba invadens,125 promotion of chitinase production in Streptomyces,126–128 and suppression of airway inflammation in mammals.129
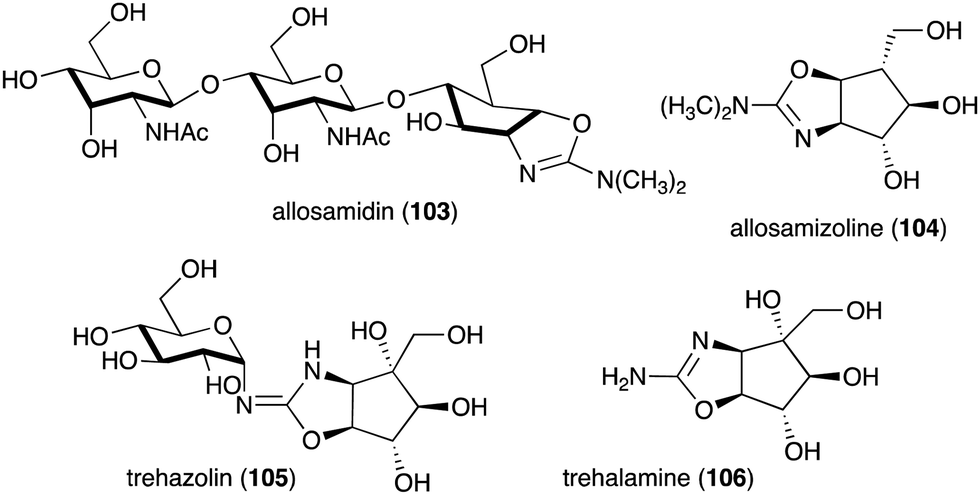 | ||
| Fig. 23 Structures of allosamidin, trehazolin and their ribomimetic moieties. | ||
Analogues of allosamidin include methylallosamidin, demethylallosamidin, glucoallosamidin A, glucoallosamidin B, methyl-N-demethylallosamidin, and didemethylallosamidin.130 All of them can also bind to family 18 chitinases and inhibit their catalytic activity. Trehazolin (105), from Micromonospora coriacea (SANK 62192),131 is a pseudodisaccharide with an N-glycosidic linkage between the glucose residue and the C-ribomimetic unit trehalamine (106) (Fig. 23).132 Similar to allosamizoline, trehalamine consists of a C-ribomimetic structure fused with an aminooxazoline ring.
Although most natural ribomimetics are secondary metabolites, there are examples of C-ribomimetics that are essential for the survival of the producing microorganisms. Caryose (4,8-cyclo-3,9-dideoxy-L-erythro-D-ido-nonose) (107), a C9-ribomimetic (Fig. 24), was found alongside caryophyllose (3,6,10-trideoxy-4-C-[(R)-1-hydroxyethyl]-D-erythro-D-gulo-decose) in the lipopolysaccharide (LPS) fractions of the phytopathogenic bacterium Pseudomonas caryophylli (Burkholderia caryophylli), which is responsible for the wilting of carnations. This interesting C-ribomimetic can be found as a standalone monosaccharide as well as a homopolymer, known as caryan, with a (1 → 7)-glycosidic linkage.133,134
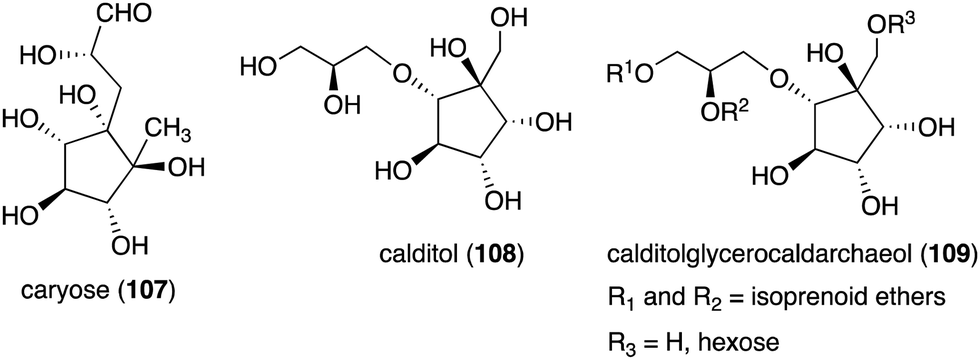 | ||
| Fig. 24 Structures of caryose and calditol found in lipopolysaccharides or archaeal lipids. | ||
Another important membrane-related C-ribomimetic is calditol (108) (Fig. 24), which was found in the membrane lipids of a subset of thermoacidophilic archaea, e.g., Sulfolobus acidocaldarius.135 This C-ribomimetic unit is ether-linked directly to the glycerol backbone of glycerol dibiphytanyl glycerol tetraethers (GDGTs) or related compounds, such as in calditolglycerocaldarchaeol (109), to form the components of the membrane lipids. These lipids are thought to be essential for archaebacteria to preserve their membrane functions under extreme conditions, such as high temperatures and strong acidity.136 Deletion of calditol synthase (Cds) in S. acidocaldarius resulted in mutants that are sensitive to extremely low pH, implying that the C-ribomimetic moiety is important for protecting archaeal cells from environmental stress.136
A C-ribomimetic unit was also found in modified nucleosides, queuosine and epoxyqueuosine, from certain bacterial and eukaryotic tRNAs. Queuosine and epoxyqueuosine are conserved products of the post-translational processing of tRNA in bacteria and eukaryotes, specifically at position 34 in the anticodons of tRNAs coding for the amino acids aspartic acid, asparagine, histidine, and tyrosine.137 While bacteria are capable of producing these compounds de novo, eukaryotes may obtain queousine from their diet or intestinal flora.137 In higher vertebrates, including humans, some queousine is glycosylated to form galactosyl- and mannosyl-queousine.138 The biological significance of these hypermodified RNA nucleosides is still elusive.
4.2. Biosynthesis of C-ribomimetics
Studies on the biosynthesis of several C-ribomimetics revealed that they are derived from different precursors employing distinct catalytic mechanisms. The C-ribomimetic units in queuosine (115) and epoxyqueuosine (114) are derived from S-adenosylmethionine catalysed by a unique enzyme, S-adenosylmethionine:tRNA ribosyltransferase-isomerase (QueA) (Fig. 25),137,139 whereas the C-ribomimetic units in BHT cyclitol ether (96), pactamycin (16), and allosamidin (103) are derived from N-acetylglucosamine. Interestingly, the C-ribomimetic unit of trehazolin (105), which is highly similar to that of allosamidin, is derived from glucose, instead of N-acetylglucosamine.140 In contrast to allosamidin, the two nitrogen atoms of trehazolin originate from L-arginine.140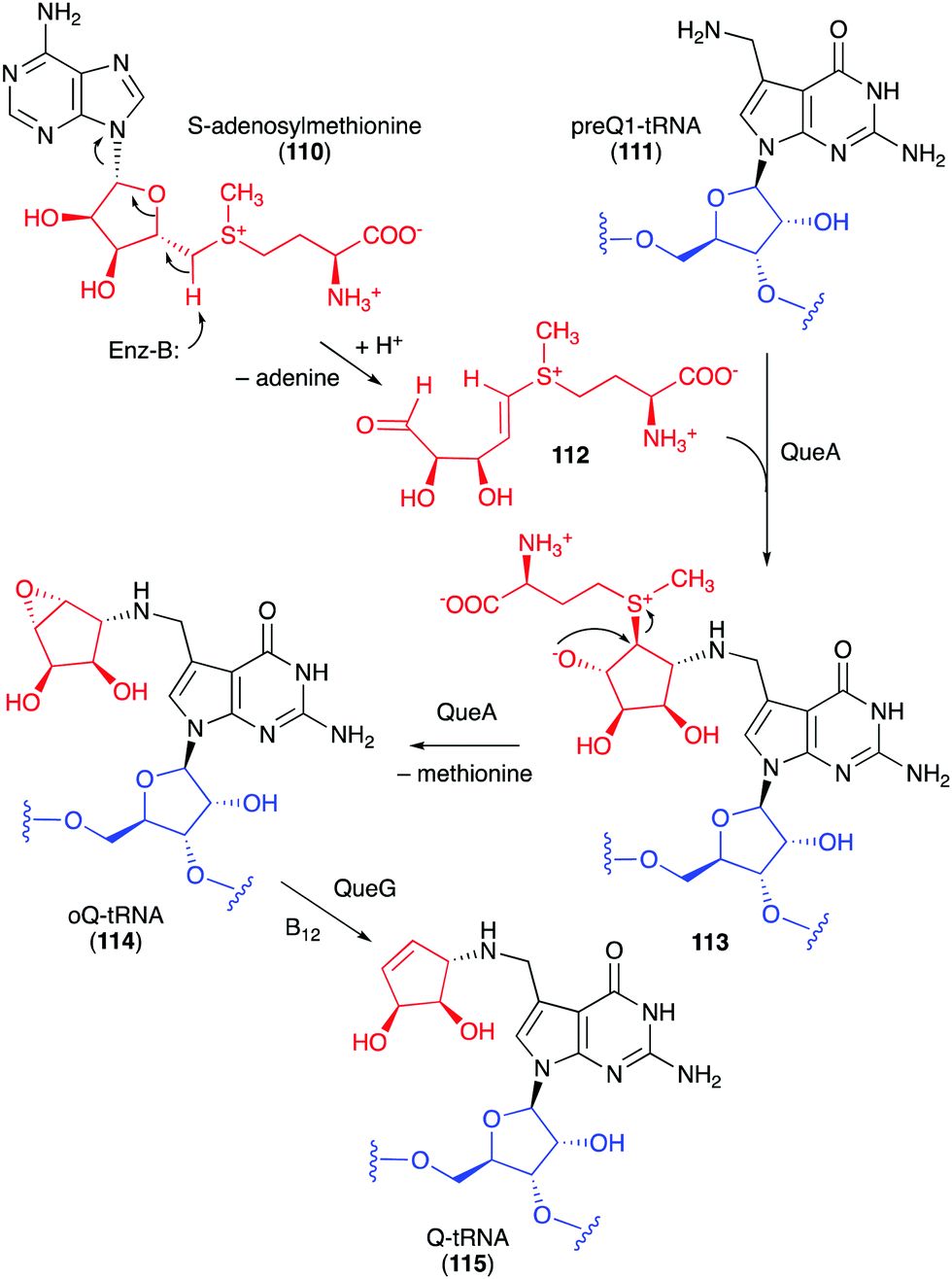 | ||
| Fig. 25 Formation of the C-ribomimetics of epoxyqueuosine and queuosine in modified tRNA (oQ-tRNA and Q-tRNA, respectively). | ||
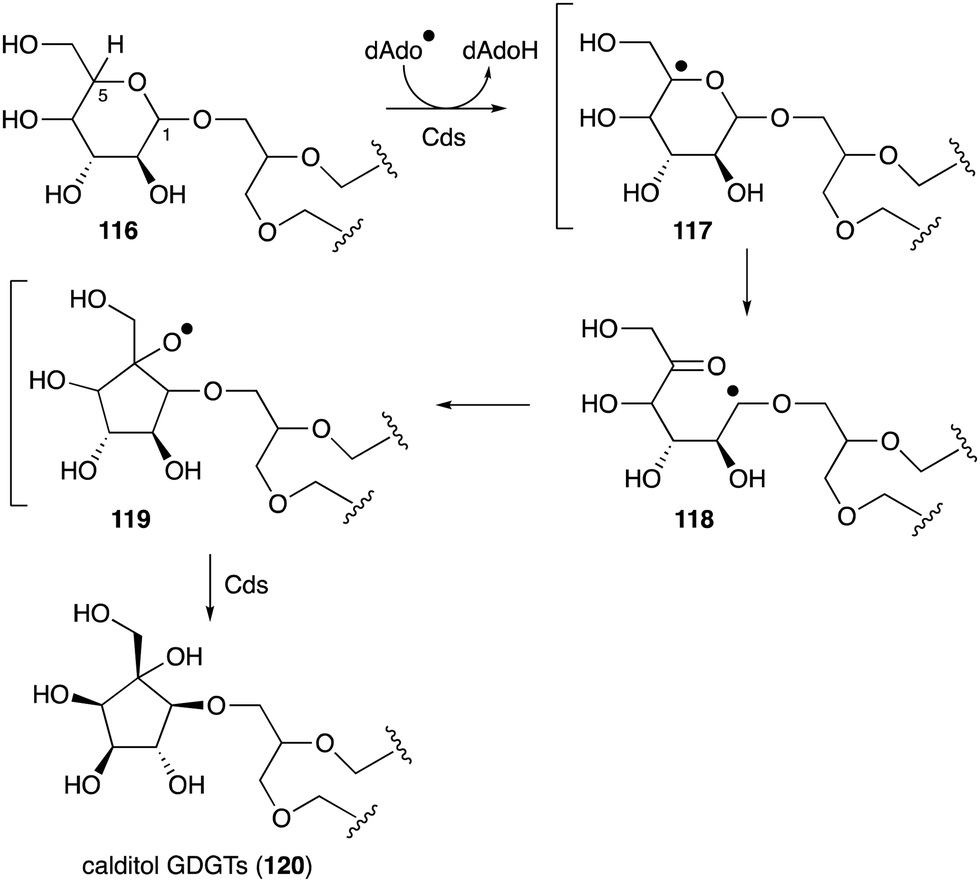 | ||
| Fig. 26 Proposed calditol formation from a hexose via a radical mechanism. | ||
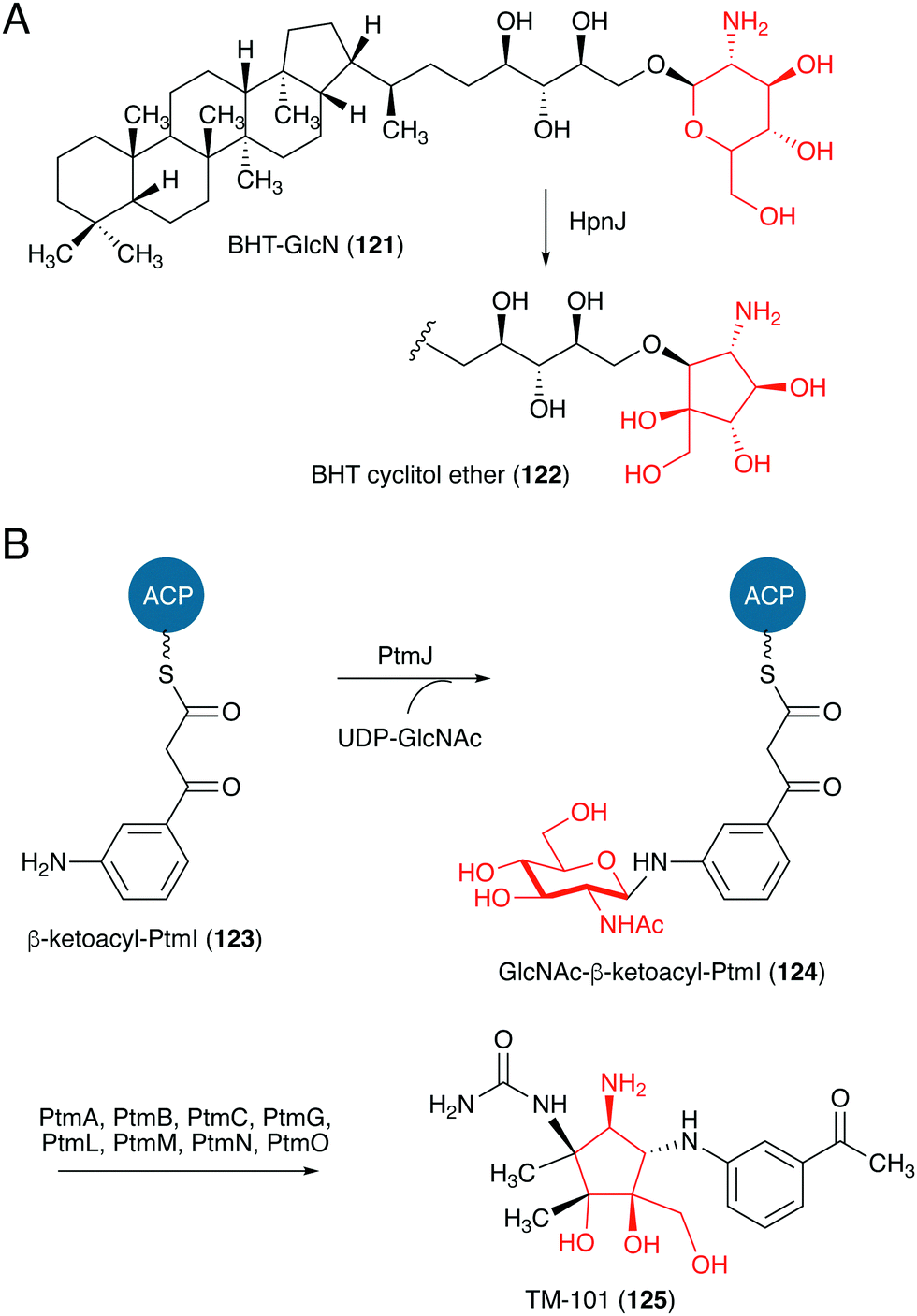 | ||
| Fig. 27 Proposed formation of the C-ribomimetics of BHT cyclitol ether and pactamycin. ACP, acyl carrier protein. | ||
TM-101 (125) and the subsequent downstream intermediates in the pactamycin pathway were identified through a series of gene inactivation experiments.149 Based on these experiments the radical SAM-dependent C-methyltransferase PtmH is believed to catalyse the conversion of TM-101 to TM-102 (126), which is then converted to jogyamycin (102) by the O-methyltransferase PtmD (Fig. 28).149 The KASIII-like protein PtmR has been shown to catalyse the transfer of 6-methylsalicylyl moiety from the iterative type I PKS PtmQ to the primary alcohol of jogyamycin.150 The product, 7-deoxypactamycin (99), is then hydroxylated to the final product pactamycin, most likely by the cytochrome P450 protein PtmY.109
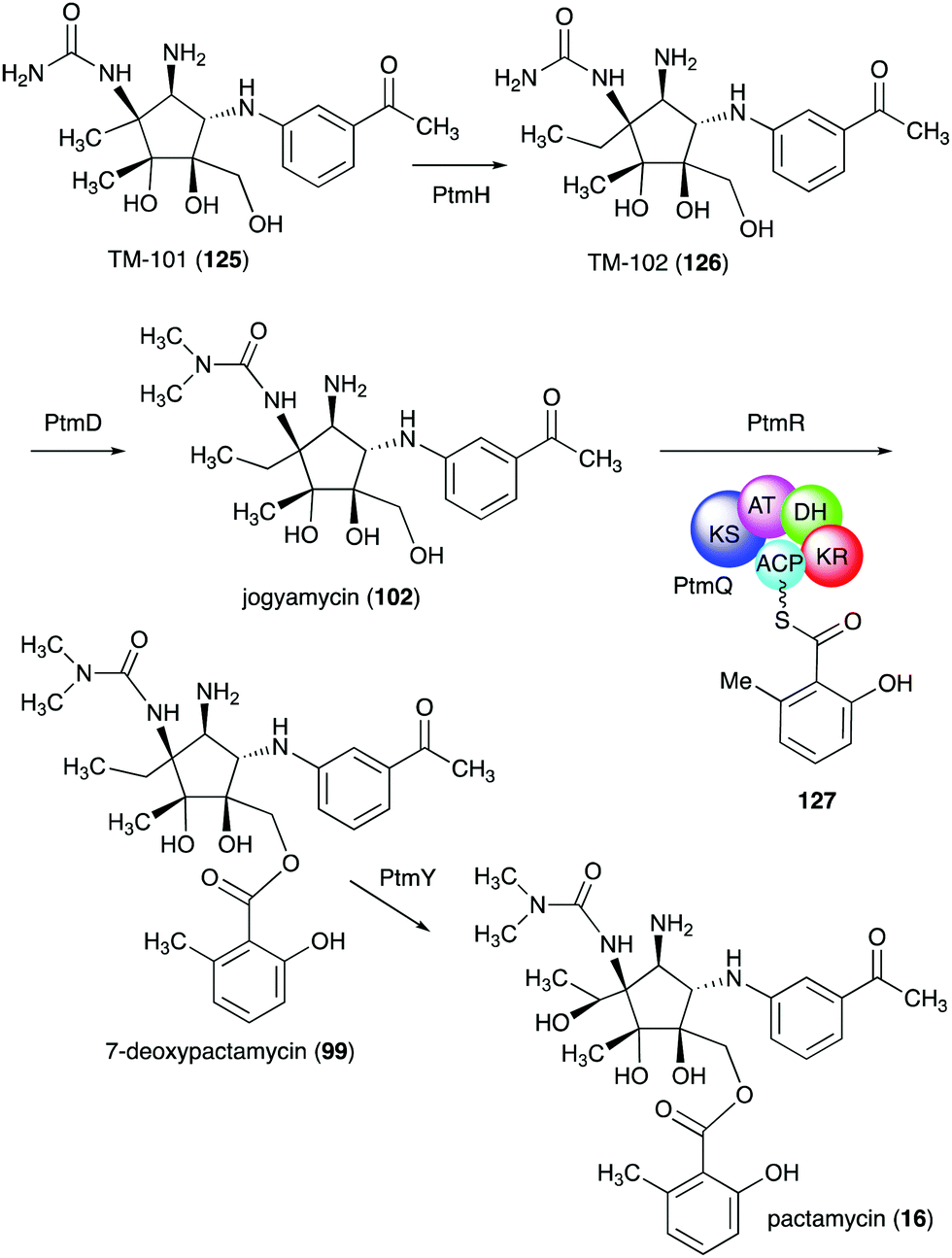 | ||
| Fig. 28 Proposed tailoring steps in pactamycin biosynthesis. KS, ketosynthase; AT, acyltransferase; DH, dehydratase; KR, ketoreductase; ACP, acyl carrier protein. | ||
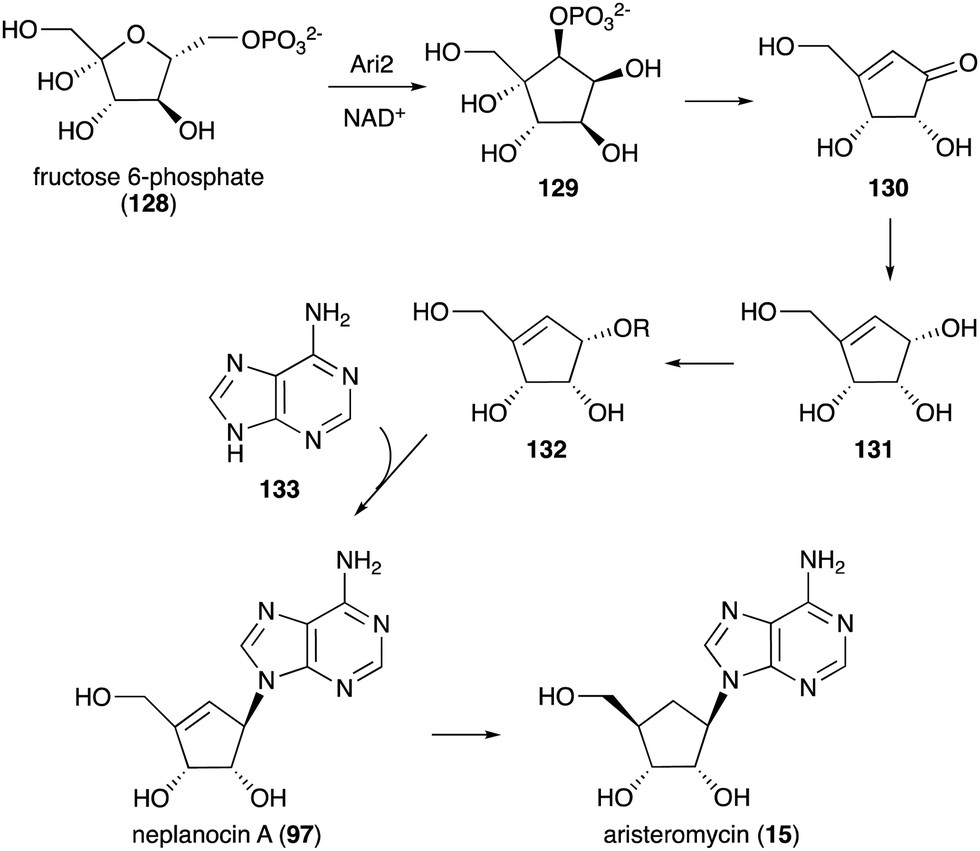 | ||
| Fig. 29 Biosynthesis of neplanocin A and aristeromycin. | ||
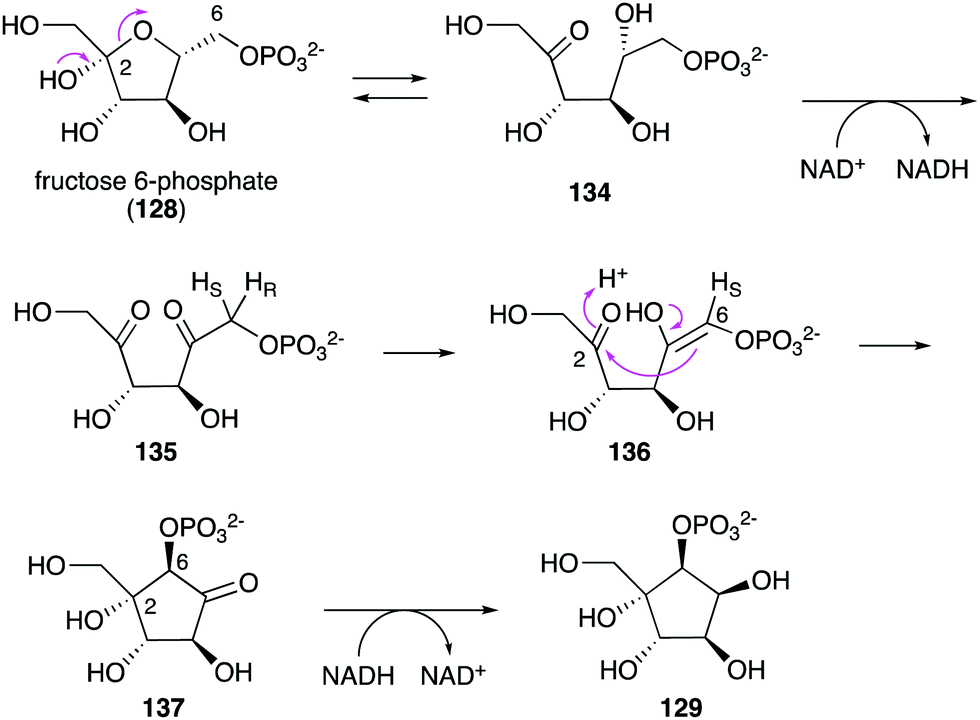 | ||
| Fig. 30 Catalytic mechanism of Ari2. | ||
4.3. Development of C-ribomimetics
As described earlier, ribomimetics are privileged chemical scaffolds, as structurally they resemble sugars that play critical roles in living organisms. A number of synthetic C-ribomimetics have been used to treat patients with influenza, hepatitis B, and HIV infections. In addition, analogues of natural C-ribomimetics, e.g., aristeromycin (15), neplanocin A (97), and pactamycin (16), possess great potential to be developed as clinically useful drugs. Aristeromycin and neplanocin A showed good antiviral activity,97,155 but their significant cytotoxicity against mammalian cells has hampered their clinical development as novel antiviral or anticancer drugs.156,157 A number of structure–activity relationship studies of aristeromycin and neplanocin A have been done, and several derivatives were found to have enhanced biological activities.158 Of particular interest are the C-6 fluorinated aristeromycin and neplanocin A, as they have increased antiviral or antitumour activities, respectively. 6,6-Difluoroaristeromycin showed greater activity against several viruses including MERS-CoV, SARS-CoV, ZIKA, and CHIKV, with 3–100 times more activity compared to aristeromycin.159 Similarly, 6′-fluoroneplanocin A showed more potent antitumour activity than neplanocin A against several human tumour cell lines.160 While the mechanism of action of neplanocin A for its antitumour activity is still unclear, it is suggested that the toxicity results from the induction of signalling pathways leading to apoptosis.161,162Analogues of pactamycin have also been produced by precursor-directed biosynthesis, mutasynthesis, genetic engineering, and by chemoenzymatic and chemical synthesis. Earlier incorporation studies by adding alternative precursors to the cultures of S. pactum resulted in fluorinated pactamycins, albeit in low yields. A mutasynthetic study with a ptmT knockout mutant of S. pactum ATCC 27456 also resulted in a number of fluorinated pactamycins.163 Analogues of pactamycin have also been produced by generating mutant strains of S. pactum ATCC 27456, in which one or more of the putative tailoring genes have been inactivated.114 Some of the products, e.g., TM-025 and TM-026, showed better selectivity against the malarial parasite Plasmodium falciparum with significantly less cytotoxicity and antibacterial activity.115 These new pactamycin analogues also induce p53 dependent cell-cycle arrest at S-phase in human head and neck cell carcinoma cells.164 The highly promiscuous KASIII-like protein PtmR has also been used to produce analogues of pactamycin with different C-6 ester moieties.150 This enzyme recognizes various N-acetylcysteamine (NAC)-thioesters as acyl-donors and TM-025 as an acyl acceptor.
The Structure–Activity Relationship (SAR) studies of pactamycin have also been done by chemical synthesis during the past decade, revealing the role of functional groups within the compound in its biological activities (Fig. 31).165–167 Modifications of the urea moiety of pactamycin generally led to a loss or reduction of antibacterial, cytotoxic, and antimalarial activities.166 Replacement of the 3-aminoacetophenone moiety with several aniline derivatives also affects their biological activities, more prominently on cytotoxicity and antimalarial activity.166,167
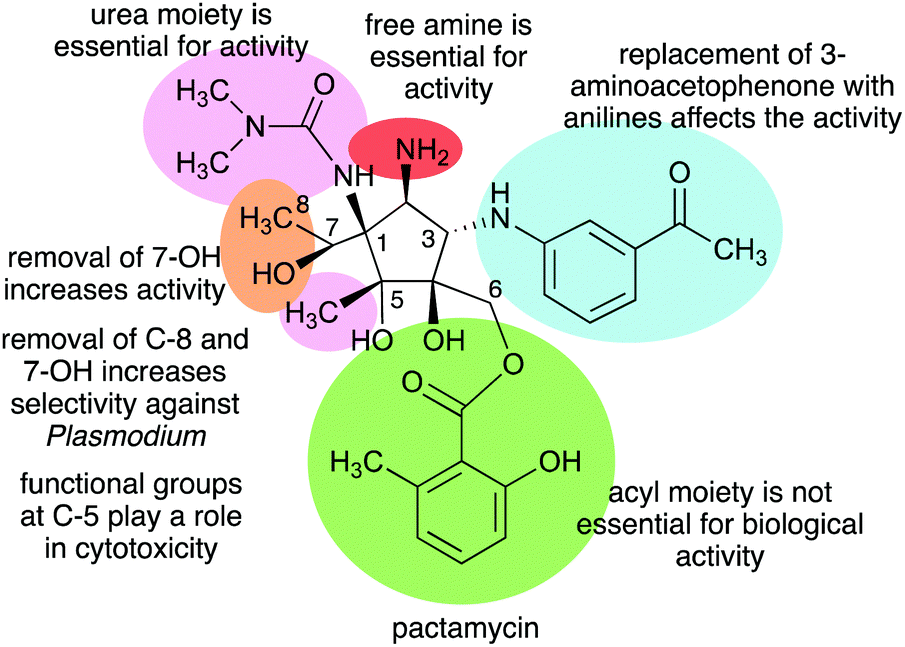 | ||
| Fig. 31 Structure–activity relationships of pactamycin. | ||
The C-5 position appears to play a significant role in the cytotoxicity of the compound. Replacing the C-5 methyl with hydrogen resulted in an analogue that is more cytotoxic than the parent compound. This was proposed to be due to the change in lipophilicity.167 On the other hand, replacement of the C-5 methyl with longer-chain alkanes, i.e., ethyl or hexyl groups, resulted in a loss of cytotoxicity against several human tumour cell lines.167
The C-6 ester moiety does not appear to be essential for the biological activity of pactamycin. Diversification of this side chain did not result in any significant differences in biological activity compared to pactamycin, implying the limited role of the C-6 ester moiety in the compound binding interactions with the 30S ribosomal subunit.166,168 A number of C-6 and C-7 bis-acylated derivatives have also been synthesized.167 The results showed a linear decrease in activity as the steric hindrance of the C-7 ester group enlarges, hinting that the hydroxy group at C-7 plays a more important role in the bioactivity compared to that at C-6.167
5. Conclusions
Due to their high resemblance to ribose, deoxyribose, or arabinose, which play significant roles in the structural, physiological, and/or reproductive functions of living organisms, many ribomimetics and related compounds have potent biological activities. Consequently, they have a tremendous potential to be developed as drug leads. In fact, some of them have been used to treat viral infections. Natural ribomimetics are produced by many different organisms including plants, fungi, and bacteria. They include ribose-like compounds in which the ring oxygen atom of ribose or related five-member ring sugars has been replaced by a sulphur (S-ribomimetics), a nitrogen (N-ribomimetics), or a carbon (C-ribomimetics) atom. With the exception of some N-ribomimetics, most of them are derived from a simple sugar such as glucose, fructose, or glucosamine. However, little is known about why they are produced or what benefits they provide to the producing organisms. Therefore, future studies should not only focus on the potential use of natural ribomimetics as pharmaceutically useful compounds but also include investigations on their physiological and/or ecological functions in nature.Author contributions
T. Tsunoda: writing – original draft. S. Tanoeyadi: writing – original draft. P. J. Proteau: writing – review and editing. T. Mahmud: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grant AI129957 (to T. M.) from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not represent the official views of the National Institute of Allergy and Infectious Diseases, or the National Institutes of Health (NIH). Takeshi Tsunoda is indebted to the financial support by the Uehara Memorial Foundation.References
- P. M. Flatt and T. Mahmud, Nat. Prod. Rep., 2007, 24, 358–392 RSC.
- F. Kudo and T. Eguchi, Chem. Rec., 2016, 16, 4–18 CrossRef CAS PubMed.
- L. J. Scott and C. M. Spencer, Drugs, 2000, 59, 521–549 CrossRef CAS PubMed.
- T. Mahmud, Nat. Prod. Rep., 2003, 20, 137–166 RSC.
- P. L. McCormack and K. L. Goa, Drugs, 2003, 63, 2427–2434 CrossRef CAS PubMed.
- E. H. McCafferty and L. J. Scott, Drugs, 2019, 79, 543–554 CrossRef CAS PubMed.
- N. Asano, T. Yamaguchi, Y. Kameda and K. Matsui, J. Antibiot., 1987, 40, 526–532 CrossRef CAS PubMed.
- T. Shiraishi and T. Kuzuyama, J. Antibiot., 2019, 72, 913–923 CrossRef CAS PubMed.
- J. M. Thomson and I. L. Lamont, Front. Microbiol., 2019, 10, 952 CrossRef PubMed.
- H. Osada, J. Antibiot., 2019, 72, 855–864 CrossRef CAS PubMed.
- G. Benz, T. Schröder, J. Kurz, C. Wünsche, W. Karl, G. Steffens, J. Pfitzner and D. Schmidt, Angew. Chem., Int. Ed. Engl., 1982, 21, 527–528 CrossRef.
- D. M. Reynolds and S. A. Waksman, J. Bacteriol., 1948, 55, 739–752 CrossRef CAS PubMed.
- E. O. Stapley, Appl. Microbiol., 1958, 6, 392–398 CrossRef CAS PubMed.
- G. F. Gause, Br. Med. J., 1955, 2, 1177–1179 CrossRef CAS PubMed.
- A. Pramanik, U. H. Stroeher, J. Krejci, A. J. Standish, E. Bohn, J. C. Paton, I. B. Autenrieth and V. Braun, Int. J. Med. Microbiol., 2007, 297, 459–469 CrossRef CAS PubMed.
- Z. Lin, X. Xu, S. Zhao, X. Yang, J. Guo, Q. Zhang, C. Jing, S. Chen and Y. He, Nat. Commun., 2018, 9, 3445 CrossRef PubMed.
- A. L. Stefanska, M. Fulston, C. S. Houge-Frydrych, J. J. Jones and S. R. Warr, J. Antibiot., 2000, 53, 1346–1353 CrossRef CAS PubMed.
- A. D. Ferguson, V. Braun, H. P. Fiedler, J. W. Coulton, K. Diederichs and W. Welte, Protein Sci., 2000, 9, 956–963 CrossRef CAS PubMed.
- T. E. Clarke, V. Braun, G. Winkelmann, L. W. Tari and H. J. Vogel, J. Biol. Chem., 2002, 277, 13966–13972 CrossRef CAS PubMed.
- A. Pramanik and V. Braun, J. Bacteriol., 2006, 188, 3878–3886 CrossRef CAS PubMed.
- M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G. Tanabe and O. Muraoka, Tetrahedron Lett., 1997, 38, 8367–8370 CrossRef CAS.
- G. Tanabe, K. Yoshikai, T. Hatanaka, M. Yamamoto, Y. Shao, T. Minematsu, O. Muraoka, T. Wang, H. Matsuda and M. Yoshikawa, Bioorg. Med. Chem., 2007, 15, 3926–3937 CrossRef CAS PubMed.
- G. Tanabe, W. Xie, A. Ogawa, C. Cao, T. Minematsu, M. Yoshikawa and O. Muraoka, Bioorg. Med. Chem. Lett., 2009, 19, 2195–2198 CrossRef CAS PubMed.
- M. Yoshikawa, T. Murakami, K. Yashiro and H. Matsuda, Chem. Pharm. Bull., 1998, 46, 1339–1340 CrossRef CAS PubMed.
- O. Muraoka, W. Xie, S. Osaki, A. Kagawa, G. Tanabe, M. F. A. Amer, T. Minematsu, T. Morikawa and M. Yoshikawa, Tetrahedron, 2010, 66, 3717–3722 CrossRef CAS.
- O. Muraoka, W. Xie, G. Tanabe, M. F. A. Amer, T. Minematsu and M. Yoshikawa, Tetrahedron Lett., 2008, 49, 7315–7317 CrossRef CAS.
- M. Yoshikawa, F. Xu, S. Nakamura, T. Wang, H. Matsuda, G. Tanabe and O. Muraoka, Heterocycles, 2008, 75, 1397–1405 CrossRef CAS.
- W. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T. Minematsu, M. Yoshikawa, X. Wu and O. Muraoka, Bioorg. Med. Chem., 2011, 19, 2015–2022 CrossRef CAS PubMed.
- G. Tanabe, M. Sakano, T. Minematsu, H. Matusda, M. Yoshikawa and O. Muraoka, Tetrahedron, 2008, 64, 10080–10086 CrossRef CAS.
- T. Morikawa, K. Ninomiya, G. Tanabe, H. Matsuda, M. Yoshikawa and O. Muraoka, J. Nat. Med., 2021, 75, 449–466 CrossRef CAS PubMed.
- Y. Zeng, A. Kulkarni, Z. Yang, P. B. Patil, W. Zhou, X. Chi, S. Van Lanen and S. Chen, ACS Chem. Biol., 2012, 7, 1565–1575 CrossRef CAS PubMed.
- C. I. Lin, R. M. McCarty and H. W. Liu, Chem. Soc. Rev., 2013, 42, 4377–4407 RSC.
- R. Ushimaru and H. W. Liu, J. Am. Chem. Soc., 2019, 141, 2211–2214 CrossRef CAS PubMed.
- R. Ushimaru, Z. Chen, H. Zhao, P. H. Fan and H. W. Liu, Angew. Chem., Int. Ed., 2020, 59, 3558–3562 CrossRef CAS PubMed.
- M. J. Kiefel, in Microbial Glycobiology, ed. O. Holst, P. J. Brennan, M. V. Itzstein and A. P. Moran, Academic Press, San Diego, 2010, pp. 915–932 DOI:10.1016/B978-0-12-374546-0.00047-X.
- J. Bramsen and J. Kjems, Front. Genet., 2012, 3, 154 CAS.
- R. Vardanyan and V. Hruby, in Synthesis of Best-Seller Drugs, ed. R. Vardanyan and V. Hruby, Academic Press, Boston, 2016, pp. 495–547 DOI:10.1016/B978-0-12-411492-0.00028-6.
- R. L. Whistler, L. W. Doner and U. G. Nayak, J. Org. Chem., 1971, 36, 108–110 CrossRef CAS PubMed.
- N. Ototani and R. L. Whistler, J. Med. Chem., 1974, 17, 535–537 CrossRef CAS PubMed.
- W. B. Parker, W. R. Waud and J. A. Secrist Iii, Curr. Med. Chem., 2015, 22, 3881–3896 CrossRef CAS PubMed.
- W. B. Parker, S. C. Shaddix, L. M. Rose, W. R. Waud, D. S. Shewach, K. N. Tiwari and J. A. Secrist, Biochem. Pharmacol., 2000, 60, 1925–1932 CrossRef CAS PubMed.
- W. B. Parker and J. V. Thottassery, J. Pharmacol. Exp. Ther., 2021, 379, 211–222 CrossRef CAS PubMed.
- D. J. Slamon, P. Neven, S. K. L. Chia, G. H. M. Jerusalem, M. D. Laurentiis, S.-A. Im, K. Petrakova, G. V. Bianchi, M. Martin, A. Nusch, G. S. Sonke, L. D. L. Cruz-Merino, J. T. Beck, C. Wang, U. Deore, A. Chakravartty, J. P. Zarate, T. Taran and P. A. Fasching, J. Clin. Oncol., 2021, 39, 1001 Search PubMed.
- D. Tsukamoto, M. Shibano, R. Okamoto and G. Kusano, Chem. Pharm. Bull., 2001, 49, 492–496 CrossRef CAS PubMed.
- D. Tsukamoto, M. Shibano and G. Kusano, Chem. Pharm. Bull., 2001, 49, 1487–1491 CrossRef CAS PubMed.
- M. Shibano, D. Tsukamoto, R. Fujimoto, Y. Masui, H. Sugimoto and G. Kusano, Chem. Pharm. Bull., 2000, 48, 1281–1285 CrossRef CAS PubMed.
- M. Shibano, S. Nakamura, N. Akazawa and G. Kusano, Chem. Pharm. Bull., 1998, 46, 1048–1050 CrossRef CAS PubMed.
- M. Shibano, S. Kitagawa, S. Nakamura, N. Akazawa and G. Kusano, Chem. Pharm. Bull., 1997, 45, 700–705 CrossRef CAS PubMed.
- H. Zhao, A. Kato, K. Sato, Y. M. Jia and C. Y. Yu, J. Org. Chem., 2013, 78, 7896–7902 CrossRef CAS PubMed.
- Q. K. Wu, K. Kinami, A. Kato, Y. X. Li, Y. M. Jia, G. W. J. Fleet and C. Y. Yu, Molecules, 2019, 24, 3712 CrossRef CAS PubMed.
- S. F. Matkhalikova, V. M. Malikov and S. Y. Yunusov, Chem. Nat. Compd., 1969, 5, 24–25 CrossRef.
- H. Iida, N. Yamazaki, C. Kibayashi and H. Nagase, Tetrahedron Lett., 1986, 27, 5393–5396 CrossRef.
- S. Ishida, M. Okasaka, F. Ramos, Y. Kashiwada, Y. Takaishi, O. K. Kodzhimatov and O. Ashurmetov, J. Nat. Med., 2008, 62, 236–238 CrossRef CAS PubMed.
- E. L. Tsou, S. Y. Chen, M. H. Yang, S. C. Wang, T. R. Cheng and W. C. Cheng, Bioorg. Med. Chem., 2008, 16, 10198–10204 CrossRef CAS PubMed.
- M. Shibano, D. Tsukamoto, A. Masuda, Y. Tanaka and G. Kusano, Chem. Pharm. Bull., 2001, 49, 1362–1365 CrossRef CAS PubMed.
- C. Y. Yu and M. H. Huang, Org. Lett., 2006, 8, 3021–3024 CrossRef CAS PubMed.
- M. R. Meselhy, S. Kadota, Y. Momose, M. Hattori and T. Namba, Chem. Pharm. Bull., 1992, 40, 3355–3357 CrossRef CAS PubMed.
- A. A. Watson, G. W. J. Fleet, N. Asano, R. J. Molyneux and R. J. Nash, Phytochemistry, 2001, 56, 265–295 CrossRef CAS PubMed.
- N. Asano, K. Oseki, E. Tomioka, H. Kizu and K. Matsui, Carbohydr. Res., 1994, 259, 243–255 CrossRef CAS.
- R. J. Nash, E. A. Bell, G. W. J. Fleet, R. H. Jones and J. M. Williams, J. Chem. Soc., Chem. Commun., 1985, 738–740, 10.1039/C39850000738.
- R. J. Molyneux, Y. Pan, J. E. Tropea, M. Benson, G. Kaushal and A. D. Elbein, Biochemistry, 1991, 30, 9981–9987 CrossRef CAS.
- T. Shibata, O. Nakayama, Y. Tsurumi, M. Okuhara, H. Terano and M. Kohsaka, J. Antibiot., 1988, 41, 296–301 CrossRef CAS PubMed.
- E. Tsujii, M. Muroi, N. Shiragami and A. Takatsuki, Biochem. Biophys. Res. Commun., 1996, 220, 459–466 CrossRef CAS PubMed.
- T. Miyazaki and E. Y. Park, J. Biol. Chem., 2020, 295, 8784–8797 CrossRef CAS PubMed.
- N. G. S. McGregor, J. P. Turkenburg, K. B. R. Morkeberg Krogh, J. E. Nielsen, M. Artola, K. A. Stubbs, H. S. Overkleeft and G. J. Davies, Acta Crystallogr., Sect. D: Struct. Biol., 2020, 76, 1124–1133 CrossRef CAS PubMed.
- R. J. Molyneux, Y. T. Pan, J. E. Tropea, A. D. Elbein, C. H. Lawyer, D. J. Hughes and G. W. J. Fleet, J. Nat. Prod., 1993, 56, 1356–1364 CrossRef CAS PubMed.
- A. A. Watson, R. J. Nash, M. R. Wormald, D. J. Harvey, S. Dealler, E. Lees, N. Asano, H. Kizu, A. Kato, R. C. Griffiths, A. J. Cairns and G. W. J. Fleet, Phytochemistry, 1997, 46, 255–259 CrossRef CAS.
- N. Asano, A. Kato, M. Miyauchi, H. Kizu, Y. Kameda, A. A. Watson, R. J. Nash and G. W. J. Fleet, J. Nat. Prod., 1998, 61, 625–628 CrossRef CAS PubMed.
- A. Kato, I. Adachi, M. Miyauchi, K. Ikeda, T. Komae, H. Kizu, Y. Kameda, A. A. Watson, R. J. Nash, M. R. Wormald, G. W. J. Fleet and N. Asano, Carbohydr. Res., 1999, 316, 95–103 CrossRef CAS PubMed.
- M. Sekar and K. J. R. Prasad, J. Nat. Prod., 1998, 61, 294–296 CrossRef CAS PubMed.
- A. Welter, J. Jadot, G. Dardenne, M. Marlier and J. Casimir, Phytochemistry, 1976, 15, 747–749 CrossRef CAS.
- R. F. Martínez, S. F. Jenkinson, S. Nakagawa, A. Kato, M. R. Wormald, G. W. J. Fleet, J. Hollinshead and R. J. Nash, Amino Acids, 2019, 51, 991–998 CrossRef PubMed.
- A. Torres, M. Hochberg, I. Pergament, R. Smoum, V. Niddam, V. M. Dembitsky, M. Temina, I. Dor, O. Lev, M. Srebnik and C. D. Enk, Eur. J. Biochem., 2004, 271, 780–784 CrossRef CAS PubMed.
- R. E. Schwartz, G. L. Helms, E. A. Bolessa, K. E. Wilson, R. A. Giacobbe, J. S. Tkacz, G. F. Bills, J. M. Liesch, D. L. Zink, J. E. Curotto, B. Pramanik and J. C. Onishi, Tetrahedron, 1994, 50, 1675–1686 CrossRef CAS.
- P. H. M. Harrison, P. A. Duspara, S. I. Jenkins, S. A. Kassam, D. K. Liscombe and D. W. Hughes, J. Chem. Soc., Perkin Trans. 1, 2000, 4390–4402, 10.1039/B006007K.
- M. Sakurai, H. Hoshino, J. Kohno, M. Nishio, N. Kishi, T. Okuda, K. Kawano and T. Ohnuki, J. Antibiot., 2003, 56, 787–791 CrossRef CAS PubMed.
- T. Ishii, K. Nonaka, M. Iwatsuki, R. Masuma, S. Ōmura and K. Shiomi, J. Antibiot., 2012, 65, 139–141 CrossRef CAS PubMed.
- T. Ishii, K. Nonaka, A. Sugawara, M. Iwatsuki, R. Masuma, T. Hirose, T. Sunazuka, S. Ōmura and K. Shiomi, J. Antibiot., 2015, 68, 633–637 CrossRef CAS PubMed.
- B. A. Sobin and F. W. Tanner, J. Am. Chem. Soc., 1954, 76, 4053 CrossRef CAS.
- K. Tsuchiya, S. Kobayashi, T. Harada, T. Kurokawa, T. Nakagawa, N. Shimada and K. Kobayashi, J. Antibiot., 1995, 48, 626–629 CrossRef CAS PubMed.
- K. Tsuchiya, S. Kobayashi, T. Kurokawa, T. Nakagawa, N. Shimada, H. Nakamura, Y. Iitaka, M. Kitagawa and K. Tatsuta, J. Antibiot., 1995, 48, 630–634 CrossRef CAS PubMed.
- M. Shibano, D. Tsukamoto, T. Inoue, Y. Takase and G. Kusano, Chem. Pharm. Bull., 2001, 49, 504–506 CrossRef CAS PubMed.
- P. H. M. Harrison, D. W. Hughes and R. William Riddoch, Chem. Commun., 1998, 273–274, 10.1039/A706799B.
- C. Nunez and N. A. Horenstein, J. Nat. Prod., 2019, 82, 3401–3409 CrossRef CAS PubMed.
- Y. Wu, J. Arciola and N. Horenstein, Protein Pept. Lett., 2014, 21, 10–14 CrossRef CAS PubMed.
- J. M. Arciola and N. A. Horenstein, Biochem. J., 2018, 475, 2241–2256 CrossRef CAS PubMed.
- N. M. Joye, R. V. Lawrence and L. J. Gough, J. Org. Chem., 1966, 31, 317–320 CrossRef.
- X. Zheng, Q. Cheng, F. Yao, X. Wang, L. Kong, B. Cao, M. Xu, S. Lin, Z. Deng, Y. H. Chooi and D. You, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4135–4140 CrossRef CAS PubMed.
- G. A. Kicska, L. Long, H. Hörig, C. Fairchild, P. C. Tyler, R. H. Furneaux, V. L. Schramm and H. L. Kaufman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4593–4598 CrossRef CAS PubMed.
- K. Balakrishnan, F. Ravandi, S. Bantia, A. Franklin and V. Gandhi, Clin. Lymphoma, Myeloma Leuk., 2013, 13, 458–466 CrossRef CAS PubMed.
- R. Taylor, P. Kotian, T. Warren, R. Panchal, S. Bavari, J. Julander, S. Dobo, A. Rose, Y. El-Kattan, B. Taubenheim, Y. Babu and W. P. Sheridan, J. Infect. Public Health, 2016, 9, 220–226 CrossRef PubMed.
- T. K. Warren, J. Wells, R. G. Panchal, K. S. Stuthman, N. L. Garza, S. A. Van Tongeren, L. Dong, C. J. Retterer, B. P. Eaton, G. Pegoraro, S. Honnold, S. Bantia, P. Kotian, X. Chen, B. R. Taubenheim, L. S. Welch, D. M. Minning, Y. S. Babu, W. P. Sheridan and S. Bavari, Nature, 2014, 508, 402–405 CrossRef CAS PubMed.
- H. Zhao, A. Kato, K. Sato, Y. M. Jia and C. Y. Yu, J. Org. Chem., 2013, 78, 7896–7902 CrossRef CAS PubMed.
- T. Kusaka, H. Yamamoto, M. Shibata, M. Muroi and T. Kishi, J. Antibiot., 1968, 21, 255–263 CrossRef CAS PubMed.
- S. Yaginuma, N. Muto, M. Tsujino, Y. Sudate, M. Hayashi and M. Otani, J. Antibiot., 1981, 34, 359–366 CrossRef CAS PubMed.
- A. Hoshi, M. Yoshida, M. Iigo, R. Tokuzen, K. Fukukawa and T. Ueda, J. Pharmacobio-Dyn., 1986, 9, 202–206 CrossRef CAS PubMed.
- R. T. Borchardt, B. T. Keller and U. Patel-Thombre, J. Biol. Chem., 1984, 259, 4353–4358 CrossRef CAS.
- A. Guranowski, J. A. Montgomery, G. L. Cantoni and P. K. Chiang, Biochemistry, 1981, 20, 110–115 CrossRef CAS PubMed.
- Y. Kusakabe, M. Ishihara, T. Umeda, D. Kuroda, M. Nakanishi, Y. Kitade, H. Gouda, K. T. Nakamura and N. Tanaka, Sci. Rep., 2015, 5, 16641 CrossRef CAS.
- S. Ōmura, H. Ishikawa, H. Kuga, N. Imamura, S. Taga, Y. Takahashi and H. Tanaka, J. Antibiot., 1986, 39, 1219–1224 CrossRef PubMed.
- Ō. Satoshi, N. Imamura, H. Kuga, H. Ishikawa, Y. Yamazaki, K. Okano, K. Kimura, Y. Takahashi and H. Tanaka, J. Antibiot., 1985, 38, 1008–1015 CrossRef PubMed.
- H. Nakamura, G. Koyama, Y. Iitaka, M. Ohno, N. Yagisawa, S. Kondo, K. Maeda and H. Umezawa, J. Am. Chem. Soc., 1974, 96, 4327–4328 CrossRef CAS PubMed.
- K. Kodama, H. Kusakabe, H. Machida, Y. Midorikawa, S. Shibuya, A. Kuninaka and H. Yoshino, Agric. Biol. Chem., 1979, 43, 2375–2377 CAS.
- S. Ōmura, H. Ishikawa, H. Kuga, N. Imamura, S. Taga, Y. Takahashi and H. Tanaka, J. Antibiot., 1986, 39, 1219–1224 CrossRef PubMed.
- B. K. Bhuyan, Appl. Microbiol., 1962, 10, 302–304 CrossRef CAS PubMed.
- L. Felicetti, B. Colombo and C. Baglioni, Biochem. Biophys. Acta, 1966, 119, 120–129 CAS.
- G. Dinos, D. N. Wilson, Y. Teraoka, W. Szaflarski, P. Fucini, D. Kalpaxis and K. H. Nierhaus, Mol. Cell, 2004, 13, 113–124 CrossRef CAS PubMed.
- J. Egebjerg and R. A. Garrett, Biochimie, 1991, 73, 1145–1149 CrossRef CAS PubMed.
- A. A. Eida and T. Mahmud, Appl. Microbiol. Biotechnol., 2019, 103, 4337–4345 CrossRef CAS PubMed.
- T. R. Hurley, T. A. Smitka, J. H. Wilton, R. H. Bunge, G. C. Hokanson and J. C. French, J. Antibiot., 1986, 39, 1086–1091 CrossRef CAS PubMed.
- D. D. Weller, A. Haber, K. L. Rinehart, Jr. and P. F. Wiley, J. Antibiot., 1978, 31, 997–1006 CrossRef CAS PubMed.
- M. Iwatsuki, A. Nishihara-Tsukashima, A. Ishiyama, M. Namatame, Y. Watanabe, S. Handasah, H. Pranamuda, B. Marwoto, A. Matsumoto, Y. Takahashi, K. Otoguro and S. Ōmura, J. Antibiot., 2012, 65, 169–171 CrossRef CAS PubMed.
- K. Otoguro, M. Iwatsuki, A. Ishiyama, M. Namatame, A. Nishihara-Tukashima, S. Shibahara, S. Kondo, H. Yamada and S. Ōmura, J. Antibiot., 2010, 63, 381–384 CrossRef CAS PubMed.
- T. Ito, N. Roongsawang, N. Shirasaka, W. Lu, P. M. Flatt, N. Kasanah, C. Miranda and T. Mahmud, ChemBioChem, 2009, 10, 2253–2265 CrossRef CAS PubMed.
- W. Lu, N. Roongsawang and T. Mahmud, Chem. Biol., 2011, 18, 425–431 CrossRef CAS PubMed.
- S. Sakuda, A. Isogai, S. Matsumoto and A. Suzuki, J. Antibiot., 1987, 40, 296–300 CrossRef CAS PubMed.
- Y. Takenaka, S. Nakano, M. Tamoi, S. Sakuda and T. Fukamizo, Biosci., Biotechnol., Biochem., 2009, 73, 1066–1071 CrossRef CAS PubMed.
- R. Blattner, R. H. Furneaux and G. P. Lynch, Carbohydr. Res., 1996, 294, 29–39 CrossRef CAS PubMed.
- P. J. Somers, R. C. Yao, L. E. Doolin, M. J. McGowan, D. S. Fukuda and J. S. Mynderse, J. Antibiot., 1987, 40, 1751–1756 CrossRef CAS PubMed.
- B. P. Filho, F. J. Lemos, N. F. Secundino, V. Pascoa, S. T. Pereira and P. F. Pimenta, Insect Biochem. Mol. Biol., 2002, 32, 1723–1729 CrossRef CAS PubMed.
- S. Sakuda, Y. Nishimoto, M. Ohi, M. Watanabe, S. Takayama, A. Isogai and Y. Yamada, Agric. Biol. Chem., 1990, 54, 1333–1335 CAS.
- M. Shahabuddin, T. Toyoshima, M. Aikawa and D. C. Kaslow, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4266–4270 CrossRef CAS PubMed.
- J. M. Vinetz, S. K. Dave, C. A. Specht, K. A. Brameld, B. Xu, R. Hayward and D. A. Fidock, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14061–14066 CrossRef CAS PubMed.
- S. Takeo, D. Hisamori, S. Matsuda, J. Vinetz, J. Sattabongkot and T. Tsuboi, Parasitol. Int., 2009, 58, 243–248 CrossRef CAS PubMed.
- J. C. Villagomez-Castro, C. Calvo-Mendez and E. Lopez-Romero, Mol. Biochem. Parasitol., 1992, 52, 53–62 CrossRef CAS PubMed.
- S. Suzuki, E. Nakanishi, T. Ohira, R. Kawachi, Y. Ohnishi, S. Horinouchi, H. Nagasawa and S. Sakuda, J. Antibiot., 2006, 59, 410–417 CrossRef CAS PubMed.
- S. Suzuki, E. Nakanishi, T. Ohira, R. Kawachi, H. Nagasawa and S. Sakuda, J. Antibiot., 2006, 59, 402–409 CrossRef CAS PubMed.
- S. Suzuki, E. Nakanishi, K. Furihata, K. Miyamoto, H. Tsujibo, T. Watanabe, Y. Ohnishi, S. Horinouchi, H. Nagasawa and S. Sakuda, Int. J. Biol. Macromol., 2008, 43, 13–19 CrossRef CAS PubMed.
- T. Matsumoto, H. Inoue, Y. Sato, Y. Kita, T. Nakano, N. Noda, M. Eguchi-Tsuda, A. Moriwaki, O. K. Kan, K. Matsumoto, T. Shimizu, H. Nagasawa, S. Sakuda and Y. Nakanishi, Biochem. Biophys. Res. Commun., 2009, 390, 103–108 CrossRef CAS PubMed.
- S. Sakuda, H. Inoue and H. Nagasawa, Molecules, 2013, 18, 6952–6968 CrossRef CAS PubMed.
- O. Ando, H. Satake, K. Itoi, A. Sato, M. Nakajima, S. Takahashi, H. Haruyama, Y. Ohkuma, T. Kinoshita and R. Enokita, J. Antibiot., 1991, 44, 1165–1168 CrossRef CAS PubMed.
- O. Ando, M. Nakajima, K. Hamano, K. Itoi, S. Takahashi, Y. Takamatsu, A. Sato, R. Enokita, T. Okazaki and H. Haruyama, et al. , J. Antibiot., 1993, 46, 1116–1125 CrossRef CAS PubMed.
- C. De Castro, A. Evidente, R. Lanzetta, P. Lavermicocca, E. Manzo, A. Molinaro and M. Parrilli, Carbohydr. Res., 1998, 307, 167–172 CrossRef CAS.
- C. De Castro, A. Molinaro, R. Lanzetta, O. Holst and M. Parrilli, Carbohydr. Res., 2005, 340, 1802–1807 CrossRef CAS PubMed.
- B. Nicolaus, A. Trincone, E. Esposito, M. R. Vaccaro, A. Gambacorta and M. De Rosa, Biochem. J., 1990, 266, 785–791 CAS.
- Z. Zeng, X.-L. Liu, J. H. Wei, R. E. Summons and P. V. Welander, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12932–12937 CrossRef CAS PubMed.
- R. K. Slany, M. Bosl, P. F. Crain and H. Kersten, Biochemistry, 1993, 32, 7811–7817 CrossRef CAS PubMed.
- M. Hillmeier, M. Wagner, T. Ensfelder, E. Korytiakova, P. Thumbs, M. Müller and T. Carell, Nat. Commun., 2021, 12, 7123 CrossRef CAS PubMed.
- S. G. Van Lanen and D. Iwata-Reuyl, Biochemistry, 2003, 42, 5312–5320 CrossRef CAS PubMed.
- Z.-Y. Zhou, S. Sakuda and Y. Yamada, J. Chem. Soc., Perkin Trans. 1, 1992, 1649–1652, 10.1039/P19920001649.
- A. Gambacorta, G. Caracciolo, D. Trabasso, I. Izzo, A. Spinella and G. Sodano, Tetrahedron Lett., 2002, 43, 451–453 CrossRef CAS.
- Y. Noriaki, K. Norisuke and U. Hideyoshi, Chem. Lett., 2006, 35, 1230–1231 CrossRef.
- Y. Noriaki, U. Hideyoshi, K. Norisuke and M. Tatsushi, Bull. Chem. Soc. Jpn., 2004, 77, 771–778 CrossRef.
- Y. Sugiyama, H. Nagasawa, A. Suzuki and S. Sakuda, J. Antibiot., 2002, 55, 263–269 CrossRef CAS PubMed.
- C. L. Schmerk, P. V. Welander, M. A. Hamad, K. L. Bain, M. A. Bernards, R. E. Summons and M. A. Valvano, Environ. Microbiol., 2015, 17, 735–750 CrossRef CAS PubMed.
- F. Kudo, Y. Kasama, T. Hirayama and T. Eguchi, J. Antibiot., 2007, 60, 492–503 CrossRef CAS PubMed.
- A. A. Eida, M. E. Abugrain, C. J. Brumsted and T. Mahmud, Nat. Chem. Biol., 2019, 15, 795–802 CrossRef CAS PubMed.
- A. Hirayama, J. Chu, E. Goto, F. Kudo and T. Eguchi, ChemBioChem, 2018, 19, 126–130 CrossRef CAS PubMed.
- M. E. Abugrain, W. Lu, Y. Li, J. D. Serrill, C. J. Brumsted, A. R. Osborn, A. Alani, J. E. Ishmael, J. X. Kelly and T. Mahmud, ChemBioChem, 2016, 17, 1585–1588 CrossRef CAS PubMed.
- M. E. Abugrain, C. J. Brumsted, A. R. Osborn, B. Philmus and T. Mahmud, ACS Chem. Biol., 2017, 12, 362–366 CrossRef CAS PubMed.
- F. Kudo, T. Tsunoda, M. Takashima and T. Eguchi, ChemBioChem, 2016, 17, 2143–2148 CrossRef CAS PubMed.
- F. Kudo, T. Tsunoda, K. Yamaguchi, A. Miyanaga and T. Eguchi, Biochemistry, 2019, 58, 5112–5116 CrossRef CAS PubMed.
- J. M. Hill, G. N. Jenkins, C. P. Rush, N. J. Turner, A. J. Willetts, A. D. Buss, M. J. Dawson and B. A. M. Rudd, J. Am. Chem. Soc., 1995, 117, 5391–5392 CrossRef CAS.
- R. J. Parry and Y. Jiang, Tetrahedron Lett., 1994, 35, 9665–9668 CrossRef CAS.
- M. S. Wolfe and R. T. Borchardt, J. Med. Chem., 1991, 34, 1521–1530 CrossRef CAS PubMed.
- L. L. Bennett, P. W. Allan, L. M. Rose, R. N. Comber and J. A. Secrist, Mol. Pharmacol., 1986, 29, 383–390 Search PubMed.
- L. L. Bennett, B. J. Bowdon, P. W. Allan and L. M. Rose, Biochem. Pharmacol., 1986, 35, 4106–4109 CrossRef CAS PubMed.
- M. Guinan, C. Benckendorff, M. Smith and G. J. Miller, Molecules, 2020, 25, 2050 CrossRef CAS PubMed.
- J.-s. Yoon, G. Kim, D. B. Jarhad, H.-R. Kim, Y.-S. Shin, S. Qu, P. K. Sahu, H. O. Kim, H. W. Lee, S. B. Wang, Y. J. Kong, T.-S. Chang, N. S. Ogando, K. Kovacikova, E. J. Snijder, C. C. Posthuma, M. J. van Hemert and L. S. Jeong, J. Med. Chem., 2019, 62, 6346–6362 CrossRef CAS PubMed.
- G. Chandra, Y. W. Moon, Y. Lee, J. Y. Jang, J. Song, A. Nayak, K. Oh, V. A. Mulamoottil, P. K. Sahu, G. Kim, T.-S. Chang, M. Noh, S. K. Lee, S. Choi and L. S. Jeong, J. Med. Chem., 2015, 58, 5108–5120 CrossRef CAS PubMed.
- L. Zhang, L. Deng, F. Chen, Y. Yao, B. Wu, L. Wei, Q. Mo and Y. Song, Oncotarget, 2014, 5, 10665–10677 CrossRef PubMed.
- E. K. W. Tam, T. M. Nguyen, C. Z. H. Lim, P. L. Lee, Z. Li, X. Jiang, S. Santhanakrishnan, T. W. Tan, Y. L. Goh, S. Y. Wong, H. Yang, E. H. Q. Ong, J. Hill, Q. Yu and C. L. L. Chai, ChemMedChem, 2015, 10, 173–182 CrossRef CAS PubMed.
- K. H. Almabruk, W. Lu, Y. Li, M. Abugreen, J. X. Kelly and T. Mahmud, Org. Lett., 2013, 15, 1678–1681 CrossRef CAS PubMed.
- G. Guha, W. Lu, S. Li, X. Liang, M. F. Kulesz-Martin, T. Mahmud, A. K. Indra and G. Ganguli-Indra, PLoS One, 2015, 10, e0125322 CrossRef PubMed.
- N. C. Gerstner, K. A. Nicastri and J. M. Schomaker, Angew. Chem., Int. Ed., 2021, 60, 14252–14271 CrossRef CAS PubMed.
- S. Hanessian, R. R. Vakiti, A. K. Chattopadhyay, S. Dorich and C. Lavallée, Bioorg. Med. Chem., 2013, 21, 1775–1786 CrossRef CAS PubMed.
- R. J. Sharpe, J. T. Malinowski, F. Sorana, J. C. Luft, C. J. Bowerman, J. M. DeSimone and J. S. Johnson, Bioorg. Med. Chem., 2015, 23, 1849–1857 CrossRef CAS PubMed.
- D. S. Tourigny, I. S. Fernández, A. C. Kelley, R. R. Vakiti, A. K. Chattopadhyay, S. Dorich, S. Hanessian and V. Ramakrishnan, J. Mol. Biol., 2013, 425, 3907–3910 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |