
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombating amyloid-induced cellular toxicity and stiffness by designer peptidomimetics†
Mouli
Konar‡
,
Debasis
Ghosh‡
,
Sourav
Samanta
and
Thimmaiah
Govindaraju
 *
*
Bioorganic Chemistry Laboratory, New Chemistry Unit and School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bengaluru 560064, Karnataka, India. E-mail: tgraju@jncasr.ac.in
First published on 23rd December 2021
Abstract
Amyloid beta (Aβ) aggregation species-associated cellular stress instigates cytotoxicity and adverse cellular stiffness in neuronal cells. The study and modulation of these adverse effects demand immediate attention to tackle Alzheimer's disease (AD). We present a de novo design, synthesis and evaluation of Aβ14-23 peptidomimetics with cyclic dipeptide (CDP) units at defined positions. Our study identified AkdNMC with CDP units at the middle, N- and C-termini as a potent candidate to understand and ameliorate Aβ aggregation-induced cellular toxicity and adverse stiffness.
Introduction
Aberrant aggregation of Aβ peptides and their extracellular deposition in the human brain are the major hallmarks of AD.1–3 Compelling evidence suggests that the accumulation of amyloid aggregation species in brain tissue induces significant stress on neurons through cell membrane interactions.4–7 This causes redox imbalance and reorganization of the cytoskeletal system of neuronal cells.4 Aβ aggregation species elicit endoplasmic reticulum (ER) stress and oxidative stress through the production of excessive reactive oxygen species (ROS).6,7 Although there are cellular pathways to reverse the stress, the adverse outcome of this pathogenic situation is cell apoptosis. Under the amyloid-induced stress conditions, the cells become rough and rigid due to the polymerization and stiffening of cytosolic actin filaments leading to the formation of stress fibres.8–11 Atomic force microscopy (AFM)-based advanced imaging techniques are some of the best methods to assess such amyloid-induced physio-mechanical changes in the cells.12–15 PeakForce Quantitative NanoMechanics-AFM (PF QNM-AFM) offers high resolution imaging of cells with real-time spatial resolution mapping of the nanoindentation parameter Derjaguin–Muller–Toporov (DMT) modulus (measure of stiffness).16,17 In this study, special emphasis is given to understanding the neuronal cells under amyloid-induced cellular stress conditions and their rescue by de novo designed peptidomimetics inhibitors employing confocal and PF QNM-AFM techniques.Natural peptide-based amyloid inhibitors offer numerous advantages over small molecule inhibitors18–21 owing to their biological origin, biocompatibility, target-specific binding, sequence variability and ease of synthesis.22–29 Short peptides with 16KLVFF20 derived from Aβ42 have been shown to inhibit Aβ aggregation.30–33 The propensity of natural peptides for proteolytic cleavage and self-aggregation has led to the development of peptidomimetics-based inhibitors such as peptoids and cyclic peptides.34–37
We hypothesized that the incorporation of rigid, proteolytically stable CDP units into Aβ14-23 at defined positions would overcome the limitations of linear peptides and the cytotoxicity and flexibility issues often associated with large cyclic peptide-based amyloid modulators. The exceptional intermolecular hydrogen bonding ability and biological activities of CDPs are anticipated to control Aβ aggregation and associated stress-induced cellular mechanical changes through interactions with Aβ monomers or aggregation species.38–40
We designed and synthesized a set of Aβ14-23 (I) peptidomimetics by incorporating cyclo(Lys–Asp)-based CDP–unnatural amino acid (kd) at the middle (AkdM, II), C-terminal (AkdC, III), N-terminal (AkdN, IV), and at all three positions (AkdNMC, V) (Fig. 1, detailed characterizations in ESI,† Tables S1 and S2). The resemblance between Aβ14-23 (I) and Aβ42 (the most toxic form of Aβ) in their pH-dependent aggregation, metal binding and cytotoxic properties32,41–43 prompted us to investigate the aggregation behaviour of I in the presence of II–V in pH 2.0 (glycine–HCl buffer, 10 mM) and pH 7.4 (PBS: phosphate buffer saline, 10 mM) conditions. First, we sought to identify a competitive in vitro inhibitor of Aβ14-23 aggregation from II–V. Thereafter, we aimed to evaluate the efficiency of the lead inhibitor to ameliorate cellular toxicity and adverse cell mechanics caused by Aβ42 amyloid-induced cellular stress.
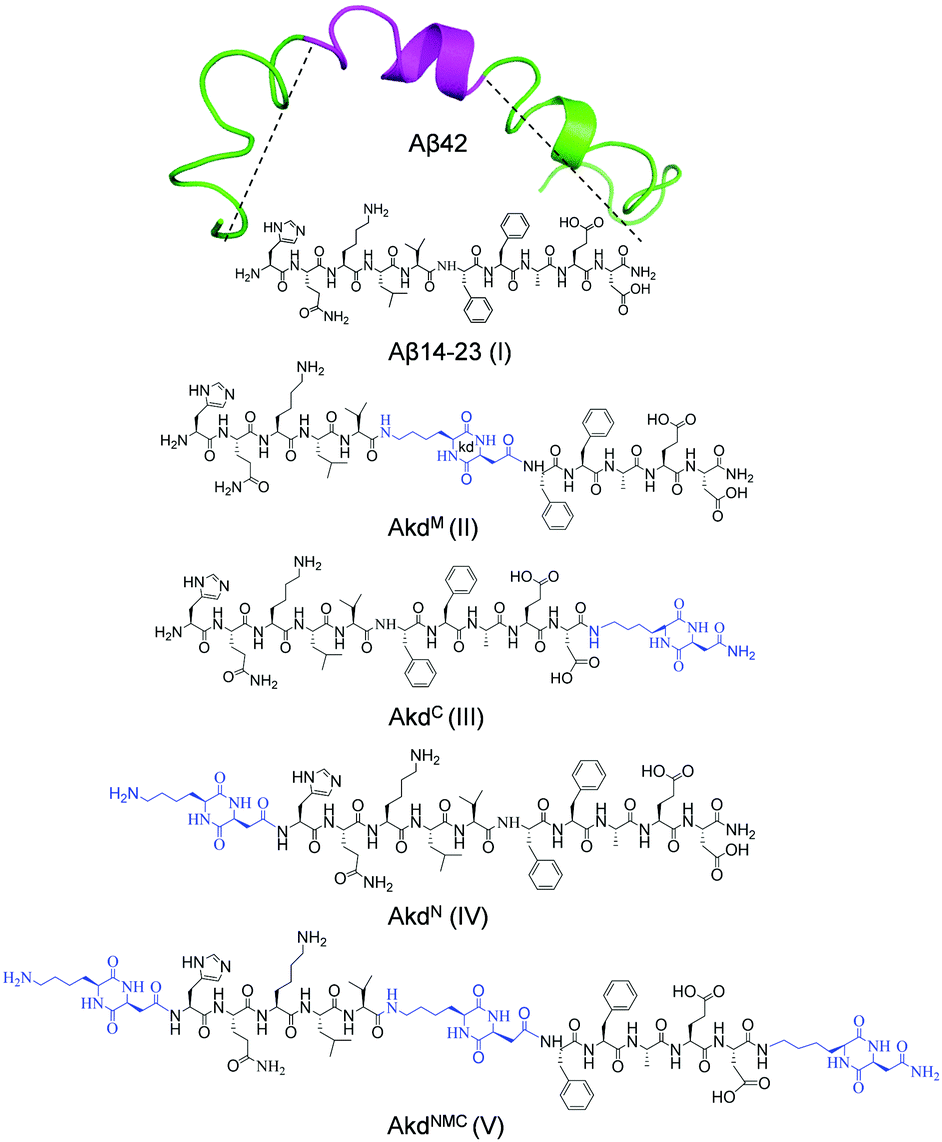 | ||
| Fig. 1 Structures of Aβ14-23 (I) derived from Aβ42 (PDB: 1Z0Q) and CDP (kd) inserted peptidomimetics (II–V). | ||
Results and discussion
A time-dependent thioflavin T (ThT) fluorescence assay was performed to assess the aggregation kinetics of Aβ14-23 and determine the aggregation rate constant (κ) and lag time (TLag) upon treatment with II–V for 20 days at two pH conditions (Fig. 2a–c). The aggregation of Aβ14-23 plateaued after 6 and 15 days under the pH 2.0 and 7.4 conditions, respectively. Aβ14-23 showed a faster aggregation of κ = 1.1 day−1 at pH 2.0 as compared to that at pH 7.4 (κ = 0.5 day−1). The co-incubation of Aβ14-23 with II–V distinctly influenced its aggregation propensity. Aβ14-23 treatment with V resulted in the longest TLag (199.2 and 316.8 h for pH 2.0 and 7.4, respectively) with low κ values (∼0.4 day−1). This data suggests the effective interaction of V with Aβ14-23 at the lag phase and stabilization of the monomeric state. The TLag values for the II (100.8 and 216.0 h in pH 2.0 and 7.4, respectively) and III (120.0 and 124.8 h in pH 2.0 and 7.4, respectively) treated Aβ14-23 samples suggest monomer stabilization albeit to a lesser extent as compared to V. Relatively better inhibitory efficiency was observed for II compared to III. Meanwhile, IV was found to enhance the aggregation rate of Aβ14-23 by shortening TLag (60.1 and 54.2 h in pH 2.0 and 7.4, respectively). The higher κ for IV-treated Aβ14-23 is attributed to the rapid aggregation of monomers into growth-directing critical nuclei at the lag phase. The kinetic analysis thus inferred that II, III, and V inhibited Aβ14-23 aggregation under both acidic and physiological conditions. The position of kd in peptidomimetics plays a crucial role in the modulation of Aβ14-23 aggregation. Peptidomimetics with single kd at the middle (II) or C-terminal (III) and three kd units at the middle, N- and C-termini (V) are effective modulators of Aβ14-23 aggregation with overall inhibitory efficiency in the order of V > II > III. In contrast, IV with kd at the N-terminal exhibited propensity for enhancing amyloid aggregation. | ||
| Fig. 2 Aβ14-23 (I) aggregation in the absence and presence of II, III, IV and V at pH 2.0 (left panel) and 7.4 (right panel) for 20 days. (a and b) Time-dependent ThT fluorescence (λem = 482 nm) assay data, (c) I aggregation parameters, κ and TLag, (d and e) CD spectra, and (f and g) deconvoluted FTIR spectra (amide I region: 1600–1700 cm−1, solid line) at pH 2.0 and 7.4, respectively. Dotted-line: α-helix, dotted-dashed line: random coil and dashed-line: β-sheet. | ||
Aβ undergoes aggregation through conformational changes from random coil to α-helix to β-sheet.41 We performed circular dichroism (CD) and Fourier transform infrared (FTIR) studies to understand the effect of II–V on Aβ14-23 aggregation-induced secondary conformations. The negative ellipticity around 218–220 nm (∼−4–4.2 mdeg) in the CD spectra of Aβ14-23 at both pH conditions confirmed its aggregation through the β-sheet structure (Fig. 2d and e). Upon incubation with II, III and V at pH 2.0, the CD band at 218 nm (−4.2 mdeg) was shifted to 200 nm (−3.4, −3.6, and −4.5 mdeg) suggesting the stabilization of Aβ14-23 monomers with the random coil conformation (Fig. 2d). The random coil and β-sheet features observed upon treatment with III indicated its moderate effect in stabilizing Aβ14-23 monomers. Further, the negative bands around 200 and 210 nm (around −4 and −3 mdeg, respectively) at pH 7.4 suggested the monomer stabilization of Aβ14-23 through random coil and α-helix conformations, respectively, in the presence of II, III and V (Fig. 2e). Meanwhile, the CD spectra of IV-treated Aβ14-23 showed broad negative bands around 216–218 nm (−3.9 mdeg), which confirmed the β-sheet-driven aggregation at both pH conditions. The weak positive CD signal (∼0.8 mdeg) observed for I at pH 2.0 is possibly attributed to the twisted nature of the aggregates as revealed by the transmission electron microscopy (TEM) study (Fig. S1, ESI†).44 The amide I region (1600–1700 cm−1) of the FTIR spectra confirmed the aggregation propensity of Aβ14-23 with characteristic features of parallel (1620 cm−1) and antiparallel (1620 and 1680 cm−1) β-sheet conformations at pH 2.0 and 7.4, respectively (Fig. 2f and g).45
Aβ14-23 samples incubated with II–IV showed significant β-sheet characteristics at both pH conditions (Fig. S2, ESI†). Under similar conditions, treatment with V induced either random coil (1640 and 1670 cm−1) or α-helix (1660 cm−1) features, confirming its potential to stabilize Aβ14-23 monomers. The 1H NMR study validated the stabilization of the monomeric state of I by V (Fig. S3, ESI†).
AFM data showed the formation of topographically and morphologically distinct aggregates by Aβ14-23 at two pH (2.0 and 7.4) conditions (Fig. 3). The short fibril-like compact aggregates (average height ∼3–5 nm) and typical oligomer-associated elongated fibrils (average height ∼1–2 nm) were observed at pH 2.0 and 7.4, respectively. TEM revealed that Aβ14-23 formed short twisted fibrils at pH 2.0 and elongated fibrils with oligomers at pH 7.4 (Fig. S1, ESI†). The formation of short fibril-like aggregates at lower pH is anticipated to follow the non-cooperative elongation mechanism.46 Inhibition of Aβ14-23 aggregation by II, III and V was confirmed by both AFM and TEM studies with inhibition efficacy in the order of V > II > III (Fig. 3 and Fig. S1, ESI†). The abundant fibrillar aggregates of the IV-treated Aβ14-23 sample (I + IV) reiterated the fibrillation promoting nature of IV.
 | ||
| Fig. 3 AFM images of pre-incubated Aβ14-23 (I) in the absence and presence of II–V. | ||
Next, we assessed the cytotoxicity of II–V in cultured neuronal cells (SH-SY5Y). The treatment of cells with II–V (10 μM) for 24 h showed cell viabilities of ∼93%, 99%, 99%, and 99%, respectively, compared to the PBS-treated (10 mM, pH 7.4) control (100%) (Fig. S4a and b, ESI†). This result suggests the excellent viability and biocompatibility of II–V to neuronal cells. To evaluate the efficacy of the peptidomimetics to rescue the cells from Aβ14-23-aggregation-induced cytotoxicity, the cells were treated with Aβ14-23 (10 μM) in the absence and presence of II–V at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratios. As expected, Aβ14-23 treatment showed reduced cell viability of ∼66%, which confirmed its neurotoxic nature. The cells treated with Aβ14-23 in the presence of II–V showed ∼90%, 83%, 69% and 97% cell viability, respectively, which corresponds to ∼24%, 17%, 3%, and 31% improved viability over the Aβ14-23-treated cells (Fig. S4c, ESI†). From these results, we identified II and V as potential inhibitors of Aβ14-23-induced in cellulo toxicity. The effects of I–V on Aβ42-induced toxicity were further assessed by treating the cells with Aβ42 (10 μM) with I–V at 1:1 ratios for 24 h (Fig. S4d, ESI†). Aβ42 alone showed cell viability of ∼64%, indicating its highly toxic nature towards SH-SY5Y cells. The cells incubated with Aβ42 in the presence of I–V showed viabilities of ∼47%, 72%, 68%, 67%, and 97%, respectively. Notably, Aβ42 + I showed severe cytotoxicity to the cells due to the high aggregation propensity of both I and Aβ42. Remarkably, V showed a strong neuronal rescue with ∼33% improved viability compared to the Aβ42-treated cells. These results motivated us to perform a detailed evaluation of the inhibitory potential of V against amyloid-induced alterations at cellular levels using confocal imaging-based immunocytochemistry, fluorescence imaging-based live/dead assay and modulation of ROS generation. The ThT, CD, AFM and TEM-based biophysical data of Aβ42 and Aβ42 + V are in good agreement with in cellulo studies and support V as a potential inhibitor of Aβ aggregation at pH 7.4 (Fig. S5, ESI†).
:1 stoichiometric ratios. As expected, Aβ14-23 treatment showed reduced cell viability of ∼66%, which confirmed its neurotoxic nature. The cells treated with Aβ14-23 in the presence of II–V showed ∼90%, 83%, 69% and 97% cell viability, respectively, which corresponds to ∼24%, 17%, 3%, and 31% improved viability over the Aβ14-23-treated cells (Fig. S4c, ESI†). From these results, we identified II and V as potential inhibitors of Aβ14-23-induced in cellulo toxicity. The effects of I–V on Aβ42-induced toxicity were further assessed by treating the cells with Aβ42 (10 μM) with I–V at 1:1 ratios for 24 h (Fig. S4d, ESI†). Aβ42 alone showed cell viability of ∼64%, indicating its highly toxic nature towards SH-SY5Y cells. The cells incubated with Aβ42 in the presence of I–V showed viabilities of ∼47%, 72%, 68%, 67%, and 97%, respectively. Notably, Aβ42 + I showed severe cytotoxicity to the cells due to the high aggregation propensity of both I and Aβ42. Remarkably, V showed a strong neuronal rescue with ∼33% improved viability compared to the Aβ42-treated cells. These results motivated us to perform a detailed evaluation of the inhibitory potential of V against amyloid-induced alterations at cellular levels using confocal imaging-based immunocytochemistry, fluorescence imaging-based live/dead assay and modulation of ROS generation. The ThT, CD, AFM and TEM-based biophysical data of Aβ42 and Aβ42 + V are in good agreement with in cellulo studies and support V as a potential inhibitor of Aβ aggregation at pH 7.4 (Fig. S5, ESI†).
Immunocytochemistry assays unambiguously evaluate the efficiency of an inhibitor to prevent the membrane-localization of Aβ42 aggregates by stabilizing the nontoxic monomers.47 For this study, the SH-SY5Y cells were incubated independently with Aβ42, Aβ42 + I, and Aβ42 + V at pH 7.4 for 3 h followed by successive staining with fibril-specific primary antibody (OC), fluorescent-labelled secondary antibody (λex = 633 nm and λem = 650 nm) and DAPI (nuclear staining dye). The confocal microscopy images showed the localization of Aβ aggregates in abundance on the cell membrane both in Aβ42 (12.3%, total area covered by red fluorescence-labelled Aβ aggregates) and Aβ42 + I (13.3%) treated cells, while Aβ42 + V displayed significant reduction of membrane-localized aggregates (1.5%) indicating the amyloid inhibitory potential of V under cellular conditions (Fig. 4a, Fig. S4e and f, ESI†). We also performed a live/dead assay to confirm the cytotoxicity of Aβ42 and neuronal rescue from amyloid-induced stress upon treatment with V. The SH-SY5Y cells were cultured in a 35 mm confocal dish and treated with calcein-AM (2 μM) and propidium iodide (4.5 μM) to stain the live (green) and dead (red) cells, respectively. The fluorescence images displayed a significant extent of dead cells present in the Aβ42 (10 μM) treated samples (Fig. 4b and Fig. S6, ESI†). Meanwhile, the samples treated with Aβ42 + V (10 μM, 1:1) showed an appreciable reduction in the number of dead cells. Quantitatively, the ImageJ analysis revealed that the Aβ42 treated samples contained ∼36% dead cells, which reduced to ∼4% in the presence of V compared to the PBS-treated controls. These findings have proved that V successfully attenuates Aβ42 aggregation-induced toxicity under in cellulo conditions.
 | ||
| Fig. 4 (a) Confocal images of Aβ42 (10 μM) and Aβ42 + V (10 μM, 1:1) treated SH-SY5Y cells in PBS (pH 7.4) immunostained with OC and secondary antibody (red). Blue: nuclear staining with DAPI. Scale bar: 20 μm. (b) Fluorescence microscopy images of SH-SY5Y cells upon treatment with Aβ42 (10 μM) and Aβ42 + V (1:1) for 24 h. Green: staining with calcein AM, red: staining with propidium iodide. (c) Intracellular ROS generation in SH-SY5Y cells incubated with Aβ42 (10 μM), CuII (10 μM), ascorbate (300 μm), in the absence and presence of V (10 μM). (d) ROS levels measured in SH-SY5Y cells upon incubating (4 h) with H2O2 in the absence and presence of V. ROS produced is quantified by measuring DCF fluorescence emission at 529 nm for a given time point. Each experiment was repeated three times (n = 3), and the error bars represent the standard deviation (SD) (*p < 0.0001). | ||
We have demonstrated the role of Aβ42 in excessive ROS production and oxidative stress in previous studies.18,24 Here, we attempted to evaluate the role of V in reducing ROS levels in Aβ42-induced amyloidogenic conditions. Intracellular ROS levels in SH-SY5Y cells (in the form of hydroxyl and peroxyl radicals) were investigated using DCFDA (2,7-dichlorofluorescein diacetate) fluorescence dye. In this study, Aβ42–CuII (10 μM), ascorbate (300 μM), and Aβ42–CuII + V (10 μM) were added to DCFDA-treated SH-SY5Y cells and the DCF fluorescence was monitored at 529 nm. Our data revealed that V significantly reduced the ROS level produced by the Aβ42–CuII + Asc system in cells to ∼38% (Fig. 4c). The cellular study also demonstrated that V effectively reduces the exogenously added H2O2 to ∼35% (Fig. 4d). Overall, V acts as an effective amyloid inhibitor and modulator of amyloid-induced excessive ROS generation and related cellular stress.
PF QNM-AFM was employed to gain insights into the changes in the physio-mechanical properties of the cell membrane due to Aβ42-induced cellular stress and the rescuing effect of V. From the PF QNM-AFM data, we evaluated parameters such as the height, peak force error (a measure of exact tip-surface interactions) and cell stiffness (DMT modulus) with spatial resolution for both single (Fig. 5a–c) and a colony of SH-SY5Y cells (Fig. S7, ESI†) treated with Aβ42 and Aβ42 + V for 24 h. Fig. 5a shows smooth topography (height 5.4 ± 0.6 μm) with low mechanical stiffness (30 ± 5 kPa) for the control cells, where no sign of stress fiber formation was observed. The smooth topography and mechanical stiffness of 30 ± 5 kPa indicate healthy cellular characteristics of the control cells.11 Upon Aβ42 treatment, the cells were deformed (height 1.6 ± 0.2 μm) with increased cell stiffness (417 ± 39 kPa) and the formation of networks of stress-fibers was observed (indicated by the arrows). The stress-fiber-containing deformed cellular features with rigid mechanics (high stiffness) indicate the emergence of stress due to Aβ–membrane interactions.9–11 Remarkably, Aβ42 + V treated cells showed cell morphology (height 6.4 ± 0.4 μm), stiffness (21 ± 9 kPa) and cytoskeletal organization comparable to the control cells. This affirmatively validated V as a potential candidate to combat amyloid-induced cellular stress and to maintain healthy cellular mechanics.
 | ||
| Fig. 5 PF QNM-AFM images of SH-SY5Y cells under healthy and amyloid-induced stress conditions in height, peak force error and DMT modulus modes. The cells treated with (a) PBS, pH 7.4 (control), Aβ42 and Aβ42 + V. Arrows in the Aβ42-treated cells indicate stress fibers. (b and c) Height and mechanical stiffness calculated from the PF QNM-AFM data. Number of experiments = 3, mean ± SD, *p < 0.001. (d) Representative docked conformation between Aβ42 (surface representation) and V (stick representation) (Left) and their possible interactions based on the hydrogen bonding donor–acceptor sites in the 14–23 region of Aβ42 (surface, Right). | ||
We performed molecular docking to investigate the binding interactions between Aβ42 and V. The docking results showed that V binds to Aβ42 with a binding energy of −5.2 kcal mole−1 (Table S3, ESI†). The data in Table S3 (ESI†) also indicate that KLVFF recognition plays an important role in the binding of V around the 14–23 region of Aβ42. Multiple hydrogen bonding and hydrophobic interactions via the His14, Leu17, Val18, Ala21 and Glu22 residues of Aβ42 and the kd residues of V are found to stabilize the hydrophobic 14–23 region (Fig. 5d). V-mediated spatial interactions with Aβ42 can segregate the Aβ42 monomers and thus inhibit amyloid aggregation inhibition, as revealed by the spectroscopy data. Further, the docking analyses revealed V as the most effective amyloid inhibitor, which stabilized Aβ14-23 monomers through KLVFF recognition (Fig. S8 and Table S4, ESI†). The innocuous inhibitor V was found to exhibit significant stability against the enzymatic (trypsin) degradation and non-self-aggregation property as confirmed by time-dependent AFM studies (Fig. S9 and S10, ESI†). It should be noted that IV exhibited self-aggregation (ThT fluorescence studies, Fig. S11, ESI†), which explains its aggregation-enhancing behaviour towards I.
Analyses of the experimental and theoretical results reveal that a single kd unit at the N-terminal (IV) is far away from the aggregation inducer region with FF, which explains the inability of IV to stop the self-aggregation of I; instead, it serves as an Aβ aggregation enhancer through the β-sheet structure (Fig. S8, ESI†). Interestingly, the incorporation of kd units close to FF (II, III and V) of I was found to effectively stabilize the Aβ monomers through multiple hydrogen bonding and other noncovalent interactions (Fig. 5d). The highest Aβ aggregation-inhibition of V is not only attributed to the presence of multiple kd units but also to their positions, especially at the middle and C-terminus (Tables S3 and S4, ESI†). The docked structures of Aβ42:V and I:V suggest that the binding sites of the middle and C-terminal kd units are suitably positioned to provide maximum interaction around KLVFF to stabilize the Aβ monomer, while the kd unit at the N-terminal offers supporting interactions.
In summary, Aβ14-23 peptidomimetics incorporated with CDP units (kd) effectively combat amyloid-induced membrane toxicity, excessive ROS production and adverse cellular mechanics under stress conditions. Our study demonstrated that Aβ14-23 peptidomimetics with three kd units at the N- and C-termini and middle (V) is a potent inhibitor of amyloid aggregation of both Aβ14-23 and Aβ42. In Aβ14-23, the incorporation of kd units at the middle and C-terminal (II and III, respectively) proximal to the FF region was found to impart effective aggregation-inhibition ability. Meanwhile, a single kd unit at the N-terminal (IV) away from the FF region turns out to be an aggregation enhancer due to its self-aggregation behaviour. The PF QNM-AFM study proved that the neuronal cells were grown with healthy cell-stiffness and other features in the presence of V under amyloid-induced stress conditions. The biocompatibility, enzymatic stability and non-self-aggregating properties combined with the effective modulation of Aβ aggregation and ROS generation thus demonstrated V as a potential candidate to ameliorate amyloid-induced toxicity and adverse cellular mechanics.
Experimental section
Aggregation kinetics study using ThT fluorescence
The aggregation kinetics of peptide I (Aβ14-23) were studied in the presence and absence of peptidomimetics (II–V) using the thioflavin T (ThT) fluorescent dye (λex = 442 nm and λem = 482 nm). The peptide I (25 μM) alone and with peptidomimetics (25 μM) was incubated with ThT (20 μM) for 20 days at 37 °C in pH 2.0 (glycine–HCl buffer, 10 mM) and pH 7.4 (PBS, 10 mM) under shaking conditions. ThT fluorescence was monitored in a time-dependent manner using a microplate reader (SpectrsMax i3x). The data were fitted to the following sigmoidal equation:Where, y denotes the fluorescence intensity at time t, y0 and ymax indicate the initial and maximum fluorescence intensities, respectively, t1/2 is the time required for half maximum of the fluorescence intensity (halfway from nuclei to fibrils), and κ is the apparent first-order rate constant of aggregation. The lag time (TLag) was determined by t1/2 − 2κ.
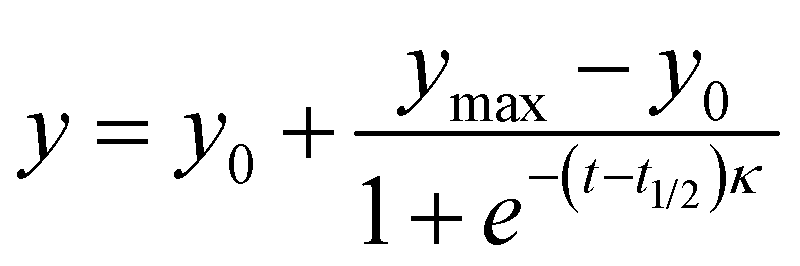
PF QNM-AFM imaging for SH-SY5Y cells
The SH-SY5Y cells were cultured in glass bottom Petri dishes (14026-20, TEM PELLA, Inc.) using Dulbecco's modified Eagle medium/nutrient mixture (DMEM/F-12) media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (PS) in a humidified CO2 (5%) incubator at 37 °C. The cells were treated with pre-incubated (48 h) Aβ42 (20 μM) alone and with AkdNMC (20 μM) for 24 h under cell growing conditions. Then, the cells were washed with PBS (10 mM, pH 7.4) and fixed with 4% paraformaldehyde (PFA) solution. PeakForce Quantitative NanoMechanics-AFM (PF QNM-AFM) was used to acquire the AFM images of neuronal cells along with the nanoindentation parameter DMT modulus using a Bruker BIOSCOPE Resolve AFM Instrument with the PeakForce Tapping Technique. For fluid imaging, a non-conductive, backside reflective Au-coated silicon nitride (MCLT-BIO, force constant: 0.01 N m−1) tip of length 0.55 μm and resonance frequency 7 kHz was used. Finally, the images were processed using the NanoScope 1.8 analysis software (Bruker, Inc.).Neuronal rescue studies
The ability of peptidomimetics (II–V) to rescue neuronal cells from amyloid-induced stress induced by Aβ14-23 (I) and Aβ42 was studied using the Alamar blue assay in the SH-SY5Y cell line. At first, the cells were cultured in a 96-well plate (15000 cells per well) using DMEM/F-12 medium (Gibco, Invitrogen) containing FBS (10%) and PS (1%). Then, the cell media was replaced with low serum (2% FBS) containing DMEM/F-12 media. Next, the cells were treated (24 h) with the peptide I (10 μM) and Aβ42 (10 μM), which were preincubated (48 h) at pH 7.4 in the absence and presence of peptidomimetics (10 μM). Finally, experimental cells were treated with Alamar blue solution for 2 h at 37 °C and the absorbance was measured at 570 nm using a microplate reader. The data were plotted and analysed using the GraphPad prism software (one-way ANOVA).
Membrane toxicity
To determine the modulation of Aβ42-induced plasma membrane toxicity by peptidomimetics, we performed immunocytochemistry in the SH-SY5Y cell line. The cells were cultured in a 35 mm confocal dish using DMEM/F-12 medium (Gibco, Invitrogen) containing FBS (10%) and PS (1%) and treated (3 h) with 10 μM Aβ42 (fibrils) alone and preincubated with AkdNMC (10 μM). The cells were washed with PBS (10 mM, pH 7.4) and fixed with 4% PFA solution and the cells were again washed with PBS (3 times), and blocked using 5% bovine serum albumin (BSA). Next, the cells were treated with Aβ42 fibril-specific primary antibody OC (1:250) for 16 h at 4 °C followed by treatment of red fluorescent-labelled (λex = 633 nm and λem = 650 nm) anti-mouse IgG secondary antibody (1:200). The excess antibody was washed using PBS and the cell nuclei were stained with DAPI for confocal imaging. The images were acquired using an OLYMPUS FLUOVIEW FV3000 confocal microscope and analysed using the cellSens software.
Author contributions
M. K. and D. G. contributed equally to performing the various experiments, data collection, analyses and preparation of the manuscript. D. G. performed the synthesis. D. G. and S. S. performed the cellular studies. M. K. performed the docking studies. T. G. designed and supervised the research and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank JNCASR, SwarnaJayanti Fellowship (DST/SJF/CSA-02/2015-2016), DST, India, and Shiva Prasad for the AFM study and Kavana for technical help.Notes and references
- Alzheimer's Disease Facts and Figures, Alzheimer's Dement., 2021, 17, 327–406 CrossRef PubMed.
- D. J. Selkoe and J. Hardy, The Amyloid Hypothesis of Alzheimer's Disease at 25 years, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, The Amyloid State and Its Association with Protein Misfolding Diseases, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- K. Rajasekhar, M. Chakrabarti and T. Govindaraju, Function and Toxicity of Amyloid Beta and Recent Therapeutic Interventions Targeting Amyloid Beta in Alzheimer's Disease, Chem. Commun., 2015, 51, 13434–13450 RSC.
- A. G. Kreutzer and J. S. Nowick, Elucidating the Structures of Amyloid Oligomers with Macrocyclic β-Hairpin Peptides: Insights into Alzheimer's Disease and Other Amyloid Diseases, Acc. Chem. Res., 2018, 51, 706–718 CrossRef CAS PubMed.
- P. H. Nguyen, et al., Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer's Disease, Parkinson's Disease, Type Ii Diabetes, and Amyotrophic Lateral Sclerosis, Chem. Rev., 2021, 121, 2545–2647 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Oxidative Stress and the Amyloid Beta Peptide in Alzheimer's Disease, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed.
- D. A. Fletcher and R. D. Mullins, Cell Mechanics and the Cytoskeleton, Nature, 2010, 463, 485–492 CrossRef CAS PubMed.
- V. Lulevich, C. C. Zimmer, H.-s. Hong, L.-w. Jin and G.-y. Liu, Single-Cell Mechanics Provides a Sensitive and Quantitative Means for Probing Aβ Peptide and Neuronal Cell Interactions, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13872–13877 CrossRef CAS PubMed.
- Q. Gao, Y. Fang, S. Zhang, H. S. H. Wong, Y. E. Chan, S. S. M. Wong, K. K. L. Yung and K. W. C. Lai, Dynamic Effect of Beta-Amyloid 42 on Cell Mechanics, J. Biomech., 2019, 86, 79–88 CrossRef PubMed.
- Y. Fang, C. Y. Y. Iu, C. N. P. Lui, Y. Zou, C. K. M. Fung, H. W. Li, N. Xi, K. K. L. Yung and K. W. C. Lai, Investigating Dynamic Structural and Mechanical Changes of Neuroblastoma Cells Associated with Glutamate-Mediated Neurodegeneration, Sci. Rep., 2014, 4, 7074 CrossRef PubMed.
- Q. Luo, D. Kuang, B. Zhang and G. Song, Cell Stiffness Determined by Atomic Force Microscopy and Its Correlation with Cell Motility, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 1953–1960 CrossRef CAS PubMed.
- T. M. Kruger, K. J. Bell, T. I. Lansakara, A. V. Tivanski, J. A. Doorn and L. L. Stevens, Reduced Extracellular Matrix Stiffness Prompts Sh-Sy5y Cell Softening and Actin Turnover to Selectively Increase Aβ(1–42) Endocytosis, ACS Chem. Neurosci., 2019, 10, 1284–1293 CrossRef CAS PubMed.
- X. Wang, R. Bleher, M. E. Brown, J. G. N. Garcia, S. M. Dudek, G. S. Shekhawat and V. P. Dravid, Nano-Biomechanical Study of Spatio-Temporal Cytoskeleton Rearrangements That Determine Subcellular Mechanical Properties and Endothelial Permeability, Sci. Rep., 2015, 5, 11097 CrossRef CAS PubMed.
- S. Corvaglia, B. Sanavio, R. P. Hong Enriquez, B. Sorce, A. Bosco, D. Scaini, S. Sabella, P. P. Pompa, G. Scoles and L. Casalis, Atomic Force Microscopy Based Nanoassay: A New Method to Study α-Synuclein-Dopamine Bioaffinity Interactions, Sci. Rep., 2014, 4, 5366 CrossRef CAS PubMed.
- P. K. Viji Babu, C. Rianna, U. Mirastschijski and M. Radmacher, Nano-Mechanical Mapping of Interdependent Cell and Ecm Mechanics by AFM Force Spectroscopy, Sci. Rep., 2019, 9, 12317 CrossRef PubMed.
- J. Hu, S. Chen, D. Huang, Y. Zhang, S. Lü and M. Long, Global Mapping of Live Cell Mechanical Features Using Peakforce QNM AFM, Biophys. Rep., 2020, 6, 9–18 CrossRef CAS.
- K. Rajasekhar, S. Samanta, V. Bagoband, N. A. Murugan and T. Govindaraju, Antioxidant Berberine-Derivative Inhibits Multifaceted Amyloid Toxicity, iScience, 2020, 23, 101005 CrossRef CAS PubMed.
- S. Samanta, K. Rajasekhar, M. Ramesh, N. A. Murugan, S. Alam, D. Shah, J. P. Clement and T. Govindaraju, Naphthalene Monoimide Derivative Ameliorates Amyloid Burden and Cognitive Decline in a Transgenic Mouse Model of Alzheimer's Disease, Adv. Ther., 2021, 4, 2000225 CrossRef CAS.
- J. Yao, X. Gao, W. Sun, T. Yao, S. Shi and L. Ji, Molecular Hairpin: A Possible Model for Inhibition of Tau Aggregation by Tannic Acid, Biochemistry, 2013, 52, 1893–1902 CrossRef CAS PubMed.
- A. J. Doig, M. P. del Castillo-Frias, O. Berthoumieu, B. Tarus, J. Nasica-Labouze, F. Sterpone, P. H. Nguyen, N. M. Hooper, P. Faller and P. Derreumaux, Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer's Disease?, ACS Chem. Neurosci., 2017, 8, 1435–1437 CrossRef CAS PubMed.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. Asok Kumar, E. M. Castaño and B. Frangione, β-Sheet Breaker Peptides Inhibit Fibrillogenesis in a Rat Brain Model of Amyloidosis: Implications for Alzheimer's Therapy, Nat. Med., 1998, 4, 822–826 CrossRef CAS PubMed.
- D. Maity, M. Howarth, M. C. Vogel, M. Magzoub and A. D. Hamilton, Peptidomimetic-Based Vesicles Inhibit Amyloid-B Fibrillation and Attenuate Cytotoxicity, J. Am. Chem. Soc., 2021, 143, 3086–3093 CrossRef CAS PubMed.
- S. Samanta, K. Rajasekhar, V. Babagond and T. Govindaraju, Small Molecule Inhibits Metal-Dependent and -Independent Multifaceted Toxicity of Alzheimer's Disease, ACS Chem. Neurosci., 2019, 10, 3611–3621 CrossRef CAS PubMed.
- B. Dai, et al., Tunable Assembly of Amyloid-Forming Peptides into Nanosheets as a Retrovirus Carrier, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2996–3001 CrossRef CAS PubMed.
- A. Frydman-Marom, R. Shaltiel-Karyo, S. Moshe and E. Gazit, The Generic Amyloid Formation Inhibition Effect of a Designed Small Aromatic β-Breaking Peptide, Amyloid, 2011, 18, 119–127 CrossRef CAS PubMed.
- T. Ikenoue, F. A. Aprile, P. Sormanni and M. Vendruscolo, Rationally Designed Bicyclic Peptides Prevent the Conversion of Aβ42 Assemblies into Fibrillar Structures, Front. Neurosci., 2021, 15, 623097 CrossRef PubMed.
- A. Abbott and E. Dolgin, Leading Alzheimer's Theory Survives Drug Failure, Nature, 2016, 540, 15–16 CrossRef CAS PubMed.
- N. Kokkoni, K. Stott, H. Amijee, J. M. Mason and A. J. Doig, N-Methylated Peptide Inhibitors of B-Amyloid Aggregation and Toxicity. Optimization of the Inhibitor Structure, Biochemistry, 2006, 45, 9906–9918 CrossRef CAS PubMed.
- A. Frydman-Marom, M. Convertino, R. Pellarin, A. Lampel, R. Shaltiel-Karyo, D. Segal, A. Caflisch, D. E. Shalev and E. Gazit, Structural Basis for Inhibiting B-Amyloid Oligomerization by a Non-Coded B-Breaker-Substituted Endomorphin Analogue, ACS Chem. Biol., 2011, 6, 1265–1276 CrossRef CAS PubMed.
- S. L. Bernstein, et al., Amyloid-β Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer's Disease, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Naslund, F. Lindqvist, J. Johansson, A. R. Karlstrom, J. Thyberg, L. Terenius and C. Nordstedt, Arrest of Beta Amyloid Fibril Formation by a Pentapeptide Ligand, J. Biol. Chem., 1996, 271, 8545–8548 CrossRef CAS PubMed.
- F. T. Senguen, N. R. Lee, X. Gu, D. M. Ryan, T. M. Doran, E. A. Anderson and B. L. Nilsson, Probing Aromatic, Hydrophobic, and Steric Effects on the Self-Assembly of an Amyloid-β Fragment Peptide, Mol. BioSyst., 2011, 7, 486–496 RSC.
- A. Henning-Knechtel, et al., Designed Cell-Penetrating Peptide Inhibitors of Amyloid-Beta Aggregation and Cytotoxicity, Cell Rep. Phys. Sci., 2020, 1, 100014 CrossRef.
- M. Richman, S. Wilk, M. Chemerovski, S. K. T. S. Wärmländer, A. Wahlström, A. Gräslund and S. Rahimipour, In Vitro and Mechanistic Studies of an Antiamyloidogenic Self-Assembled Cyclic D,L-A-Peptide Architecture, J. Am. Chem. Soc., 2013, 135, 3474–3484 CrossRef CAS PubMed.
- J. Luo and J. P. Abrahams, Cyclic Peptides as Inhibitors of Amyloid Fibrillation, Chemistry, 2014, 20, 2410 CrossRef CAS PubMed.
- J. Kaffy, et al., Designed Glycopeptidomimetics Disrupt Protein–Protein Interactions Mediating Amyloid β-Peptide Aggregation and Restore Neuroblastoma Cell Viability, J. Med. Chem., 2016, 59, 2025–2040 CrossRef CAS PubMed.
- C. Balachandra and T. Govindaraju, Cyclic Dipeptide-Guided Aggregation-Induced Emission of Naphthalimide and Its Application for the Detection of Phenolic Drugs, J. Org. Chem., 2020, 85, 1525–1536 CrossRef CAS PubMed.
- C. Balachandra, D. Padhi and T. Govindaraju, Cyclic dipeptide: A privileged molecular scaffold to derive structural diversity and functional utility, ChemMedChem, 2021, 16, 2558–2587 CrossRef CAS PubMed.
- A. D. Borthwick, 2,5-Diketopiperazines: Synthesis, Reactions, Medicinal Chemistry, and Bioactive Natural Products, Chem. Rev., 2012, 112, 3641–3716 CrossRef CAS PubMed.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, Chiral Metallohelical Complexes Enantioselectively Target Amyloid B for Treating Alzheimer's Disease, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS PubMed.
- M.-C. Hsieh, C. Liang, A. K. Mehta, D. G. Lynn and M. A. Grover, Multistep Conformation Selection in Amyloid Assembly, J. Am. Chem. Soc., 2017, 139, 17007–17010 CrossRef CAS PubMed.
- H. Abe, K. Kawasaki and H. Nakanishi, pH-Dependent Aggregate Forms and Conformation of Alzheimer Amyloid β-Peptide (12-24), J. Biochem., 2002, 132, 863 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–103 CrossRef CAS PubMed.
- G. Zandomeneghi, M. R. H. Krebs, M. G. McCammon and M. Fändrich, Ftir Reveals Structural Differences between Native Beta-Sheet Proteins and Amyloid Fibrils, Protein Sci., 2004, 13, 3314–3321 CrossRef CAS PubMed.
- R. Carrotta, M. Manno, D. Bulone, V. Martorana and P. L. San Biagio, Protofibril Formation of Amyloid β-Protein at Low Ph Via a Non-Cooperative Elongation Mechanism, J. Biol. Chem., 2005, 280, 30001 CrossRef CAS PubMed.
- S. Jin, N. Kedia, E. Illes-Toth, I. Haralampiev, S. Prisner, A. Herrmann, E. E. Wanker and J. Bieschke, Aβ(1-42) Aggregation Initiates Its Cellular Uptake and Cytotoxicity, J. Biol. Chem., 2016, 291, 19590–19606 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00235j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |