
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel screening strategy to identify histone methyltransferase inhibitors reveals a crosstalk between DOT1L and CARM1†
Yang
Si
a,
Corentin
Bon
 ab,
Magdalena
Barbachowska
ab,
Veronique
Cadet-Daniel
a,
Corinne
Jallet
a,
Laura
Soresinetti
a,
Mikaël
Boullé
c,
Magalie
Duchateau
d,
Mariette
Matondo
d,
Fabrice
Agou
c,
Ludovic
Halby
a and
Paola B.
Arimondo
*a
ab,
Magdalena
Barbachowska
ab,
Veronique
Cadet-Daniel
a,
Corinne
Jallet
a,
Laura
Soresinetti
a,
Mikaël
Boullé
c,
Magalie
Duchateau
d,
Mariette
Matondo
d,
Fabrice
Agou
c,
Ludovic
Halby
a and
Paola B.
Arimondo
*a
aEpigenetic Chemical Biology, Department of Structural Biology and Chemistry, Institut Pasteur, UMR3523 CNRS, 75015 Paris, France. E-mail: paola.arimondo@cnrs.fr
bEcole Doctorale MTCI, Université de Paris, Sorbonne Paris Cité, Paris, France
cChemogenomics and Biology Screening platform, Institut Pasteur, 75015 Paris, France
dProteomic Platform, Mass spectrometry for Biology, CNRS USR 2000, Institut Pasteur, 75015 Paris, France
First published on 22nd February 2022
Abstract
Epigenetic regulation is a dynamic and reversible process that controls gene expression. Abnormal function results in human diseases such as cancer, thus the enzymes that establish epigenetic marks, such as histone methyltransferases (HMTs), are potentially therapeutic targets. Noteworthily, HMTs form multiprotein complexes that in concert regulate gene expression. To probe epigenetic protein complexes regulation in cells, we developed a reliable chemical biology high-content imaging strategy to screen compound libraries simultaneously on multiple histone marks inside cells. By this approach, we identified that compound 4, a published CARM1 inhibitor, inhibits both histone mark H3R2me2a, regulated also by CARM1, and H3K79me2, regulated only by DOT1L, pointing out a crosstalk between CARM1 and DOT1L. Based on this interaction, we combined compound 4 and DOT1L inhibitor EPZ-5676 resulting in a stronger inhibition of cell proliferation and increase in apoptosis, indicating that our approach identifies possible effective synergistic drug combinations.
Introduction
Epigenetics is the study of the alterations of gene expression without modifications in the DNA sequence.1 The epigenetic regulation is a dynamic and reversible process that not only establishes normal cellular phenotypes but also contributes to human diseases.2 Because, differently from genetic mutations, epigenetic alterations are dynamic and reversible, the enzymes that are in charge of establishing and maintaining epigenetic marks together with the proteins reading the marks have become targets for disease treatments.3 This is the case of the histone methyltransferases (HMTs). Direct and indirect evidence has proven that in cancer,4 inflammation,5 neurological6 and metabolic diseases,7 when HMTs are mutated or abnormally expressed, they influence gene expression leading or even driving to dysfunctional cellular behaviours resulting in diseases.8,9 Thus selective small molecules inhibiting HMTs have become a promising new approach in drug discovery10 and the first HMT inhibitor, TazverikTM directed against EZH2,11 has received FDA approval in 2020 for two indications, advanced epithelioid sarcoma and follicular lymphoma.12Importantly, HMTs form multiprotein complexes that regulate the crosstalk among different histone marks and DNA modifications.13 Abnormally activation or inhibition of the HMTs complexes leads to dysfunctional coordinated modifications of histone marks in response to a complex cellular stimulus. Thus, highly selective and sensitive assays to probe epigenetic complexes directly in cells are expected to be very important in understanding the epigenetic regulation and speeding up the discovery of new epigenetic inhibitors for treatments.
Multiple techniques are available to evaluate the activities of small molecules as inhibitors of epigenetic methyltransferases.14 The most widely-used are robust and sensitive in vitro biochemical assays, such as radioactive assays including filter-binding assay15,16 and scintillation proximity assay.17–19 Their limitations are the throughput capacity and their cost. AlphaLISA assays20,21 provide a fast, highly sensitive assay compatible with automatization, but are expensive. In cells, the most widely used are antibody-based assays such as western blot or ELISA that detect the global cellular levels of epigenetic modifications after treatment of cells with the compounds to study.16,22 However, they are low-throughput and semi-quantitative. As secondary assay, affinity-based assays such as SPR20,21,23 and ITC24,25 provide binding affinity in vitro, while CETSA (Cellular Thermal Shift Assay)22 provides interaction information in cells but is highly time-consuming and has moderate sensitivity. Finally, mass spectrometry (MS) based proteomics approaches are developed but are expensive and have moderate throughput.26–29
Herein, we present a novel approach based on high-content imaging to provide a reliable tool for screening compounds for their effect on each histone mark directly in cells. The strategy is validated using different techniques (western blot, FACS and mass spectrometry). This approach has three major advantages: (1) it highlights novel HMT inhibitors that target the histone mark directly in cells; (2) it determines the specificity in cells for the histone mark since several marks are studied simultaneously; and (3) it brings new insights into the HMTs crosstalk in cells. We used this approach to screen home-made compounds on multiple histone marks in U2OS human osteosarcoma cells. We identified that compound 4, a known CARM1 (Coactivator-associated arginine methyltransferase 1) inhibitor,30 inhibits both histone mark H3R2me2a, regulated also by CARM1/PRMT4 (Protein arginine N-methyltransferase 4), and H3K79me2, regulated only by DOT1L (Disruptor of Telomeric Silencing 1-Like Histone Lysine Methyltransferase), revealing a cooperative interaction between CARM1 and DOT1L. We explored the therapeutic application of this crosstalk by a combined treatment of compound 4 and EPZ-5676 (reference inhibitor of DOT1L), resulting in an increased apoptosis and cell proliferation inhibition. The assay constitutes a fast and reliable chemical biology tool to identify new inhibitors of histone methylation marks that are active in cells, crosstalk interactions between the marks and the HMTs, and highlight novel synergistic drug combinations.
Results and discussion
Development of the high content screening (HCS) assay
A high-content screening assay capable of reporting simultaneously the effect of the compounds on several histone methylation modifications was developed (detailed in Fig. S1, ESI†). The assay aims to evaluate the global methylation state of selected histone marks after a long-term incubation time with the compound (10 days). The U2OS human osteosarcoma cell line was selected because chosen HMTs such as DOT1L and CARM1 are over-expressed in this cancer cell line.31 Technically, this cell line shows strong adherence, which well tolerates the fixation process required to measure the staining of the histone mark on the histone bodies and the automated immunofluorescence analysis.To set-up and validate the screening, two selective and cell-active HMTs inhibitors were used: UNC0642 and EPZ-5676. UNC0642 inhibits histone methyltransferases G9a (EHMT2), which catalyses the di/trimethylation of lysine 9 of histone 3 (H3K9me2/3), with an IC50 of 106 nM in MDA MB231 cells.32 Pinometostat (EPZ-5676) is a potent inhibitor of HMT DOT1L and inhibits the di/trimethylation of lysine 79 on histone 3(H3K79me2/3) in MV4-11 cells (MLL-rearranged biphenotypic leukemia) with an IC50 of 3.5 nM.33
U2OS cells in as little number as 250 cells were firstly seeded into a 96-well plate and treated with UNC0642 or EPZ-5676 at 1 μm for 10 days, as at these doses no cytotoxicity was observed.32,34 After treatment, cells were fixed and three different histone marks (H3K9me2, H3K79me2, H3K4me3) were labelled with the corresponding antibody using primary and secondary antibodies (fluorophore conjugated) and the immunofluorescence is measured automatically by using an Olympus IX83 fully-motorized microscope (Fig. S1A, ESI†). Multiple images were processed for quantitative analysis to evaluate the compound activity on the chosen histone mark (Fig. S1B, ESI†).
In Fig. 1A, a time-dependent experiment shows a clear decrease of H3K9 di-methylation (H3K9me2) after exposure to compound UNC0642 treatment compared to DMSO: 14% after 4 days, 27% after 8 days and 37% after 10 days, respectively (Fig. 1A). After 10 days of UNC0642 treatment, the integrated fluorescence intensity (IFI) of each cell from multiple images was analysed (Fig. 1B). When looking at each individual cell (dots in Fig. 1B), the main population of cells treated with UNC0642 has a lower fluorescence intensity compared to the control treated with DMSO (Fig. 1B). A similar inhibition effect was observed for the mark H3K79me2 in cells treated with EPZ-5676 at 1 μm: 10 days treatment reduced H3K79me2 to 70% of the original level (Fig. 1C). In agreement with reported data, UNC0642 and EPZ-5676 were ineffective at reducing H3K4me3 methylation levels in U2OS, comforting the specificity of the assay. The assay allows not only high throughput to screen epigenetic drugs on multiple histone marks simultaneously, but also allows to start with a very small number of cells and thus follow the effect of long-term treatments.
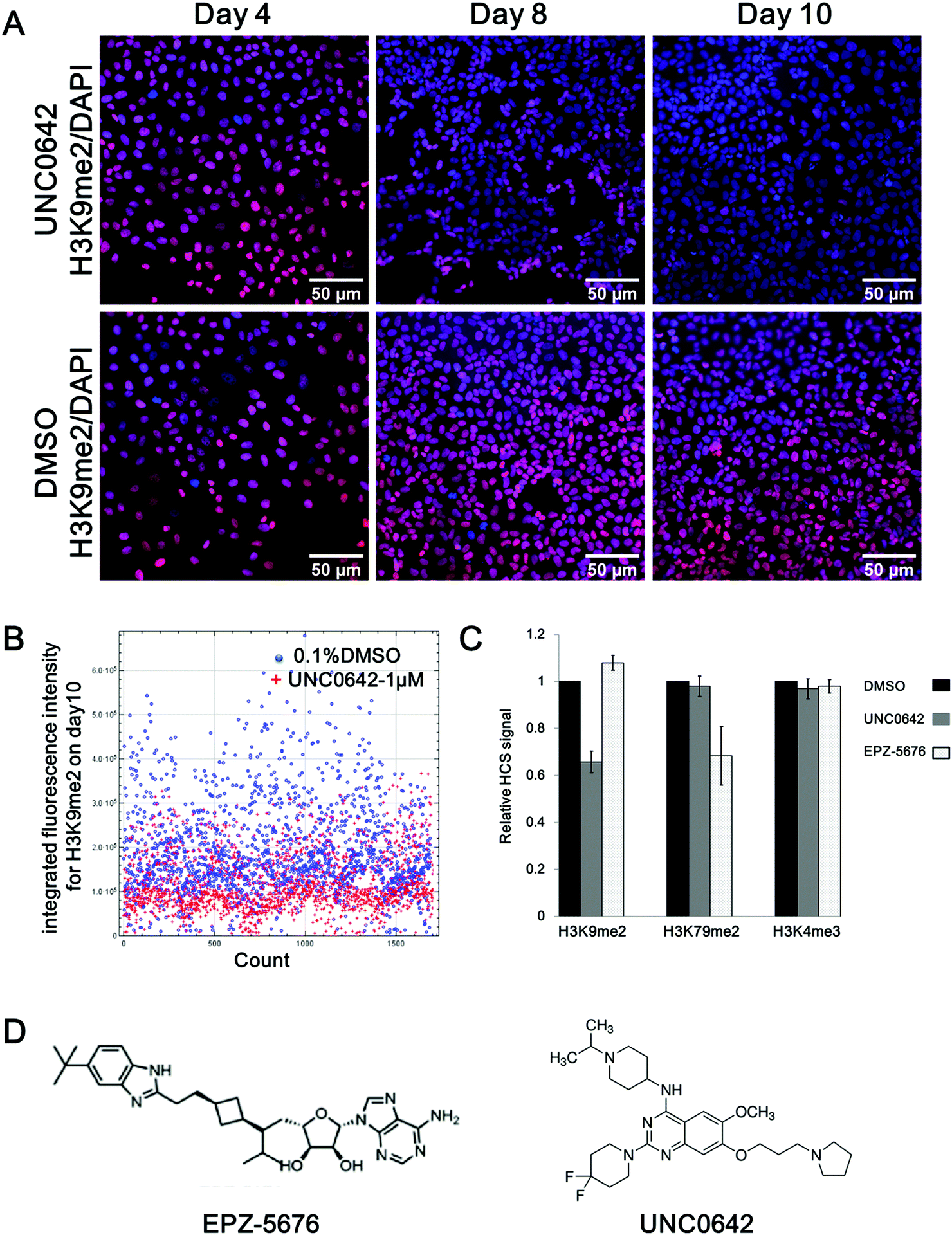 | ||
| Fig. 1 High-content screening assay development. (A) Chemical structure of EPZ-567633 and UNC0642.32 (B) Stain of H3K9me2 (red) merged with the staining of the nuclei (DAPI, blue) in U2OS cells treated by compound UNC0642 (1 μm, upper panel) and DMSO control (0.1%, bottom panel) over time (day 4, 8, 10). (C) HCS analysis of the H3K9 di-methylation (H3K9me2) on day 10 comparing treatment with UNC0642 (1 μm, red dots) to DMSO control (0.1%, blue dots). Each dot corresponds to the integrated fluorescence intensity (IFI) of each cell being analyzed. (D) Relative HCS signal for histone marks H3K9me2, H3K79me2 and H3K4me3 in U2OS cells on day 10 upon treatment with UNC0642 (1 μm, grey) or EPZ-5676 (1 μm, white) compared with DMSO control (0.1%, black). The error bars indicate the standard deviation between two biological replicates. (E) Chemical structure of EPZ-567633 and UNC0642.32 | ||
HCS screening of in-house compounds
Next we applied the HCS platform to screen a in-house compound set of bisubstrate and transition state analogues published previously (Fig. S2, ESI†)30,35 against seven different histone marks (H3K79me2, H3R2me2a, H3K4me3, H3K9me1, H3K9me2, H3R2me2s and H4K20me2/3) in U2OS cells.All analogues were evaluated in the HCS assay at a single dose (1 μm) with EPZ-5676 and UNC0642 as controls. In order to assess the cytotoxic effect of compounds, U2OS cell growth was compared in the presence or absence of compounds by cell number counting after DAPI staining of the nuclei in each well of the 96-wells plate (Fig. S3, ESI†). Over 10 days of incubation, the number of U2OS cells treated or not was essentially the same, implying that the compounds are not cytotoxic.
After a 10 days treatment, no compound showed significant activity on the 7 tested marks (Fig. S4, ESI†), except compound 4. As shown in Fig. 2A, compound 4 induced a decrease in the fluorescence intensity profile for the cross section of the image compared to treat with DMSO (in grey) on both H3R2me2a and H3K79me2 marks (Fig. 2A right panel). A 25% decrease of H3R2 asymmetric di-methylation (H3R2me2a) was observed. H3R2me2a was firstly discovered to be catalysed by PRMT4, also called coactivator-associated arginine methyltransferase 1 (CARM1).36 This was confirmed more recently by global mapping37 and by in vitro methylation assay.38 In addition, studies show that H3R2me2a is a major mark deposited by PRMT6,39,40 thus it is likely that H3R2me2a is regulated by both CARM1 and PRMT6.41 We previously reported that compound 4 inhibits CARM1, with an IC50 value of 1.5 μm30 and shows no inhibition against protein arginine methyltransferases PRMT6 and PRMT1 and little against PRMT7.30 Therefore, the decrease of H3R2me2a observed here is most likely linked to the inhibition of CARM1 by compound 4. Compound 7, structurally related to compound 4 (Fig. S2, ESI†), showed little inhibitory effect against CARM1 (25% at 10 μm30), and correspondingly shows here no inhibition of H3R2me2a (Fig. S4, ESI†). Surprisingly, compound 4 also reduced H3K79me2 (regulated by DOT1L) to 55% of the original level (Fig. 2B).
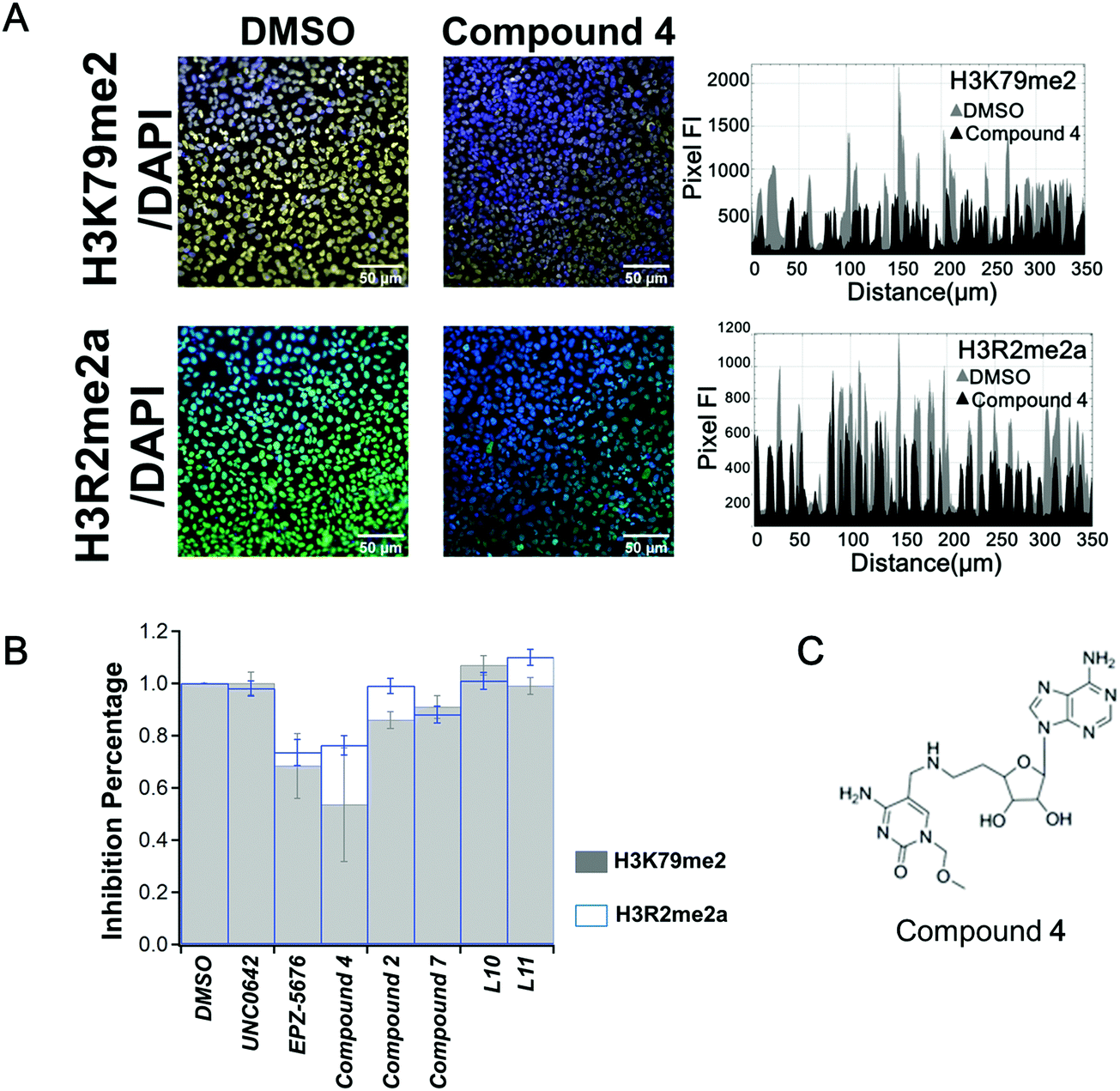 | ||
| Fig. 2 HCS screening of in-house compounds. (A) Stain of H3K79me2 (yellow) or H3R2me2a (green) merged with the staining of the nuclei (by DAPI, blue) in U2OS cells on day 10 treated with compound 4 (1 μm, middle panel) and DMSO control (0.1%, left panel). Fluorescence intensity profiles from the cross section of the images are shown (right panel). (B) HCS screening of the chemical compound sets on H3K79me2 (grey histogram) and H3R2me2a (blue histogram) in U2OS cells upon 10 days of treatment at the concentration of 1 μm. The error bars indicate the standard deviation between three biological replicates. (C) Chemical structure of bisubstrate and transition state analogue compound 4 (nomenclature as in ref. 30). | ||
Interestingly, the same decrease of both marks was observed when the cells were treated with EPZ-5676, the specific and potent inhibitor of DOT1L (Fig. 2B), which has been reported to be inactive against CARM1.33 Compound 4 and EPZ-5676 show no activity on the other five marks that have been tested (Fig. S4 and S5, ESI†).
In order to confirm these observations, we analysed the impact of compound 4 on the two histone marks by western blotting. Incubation of the U2OS cells in the presence of compound 4 at 3.2 μm for eight days led to a decrease in global cellular H3K79me2 (41% decrease) and H3R2me2a (38% decrease) levels as measured by immunoblot analysis of extracted histones with an antibody specific for H3K79me2 and H3R2me2a, respectively (Fig. 3A).
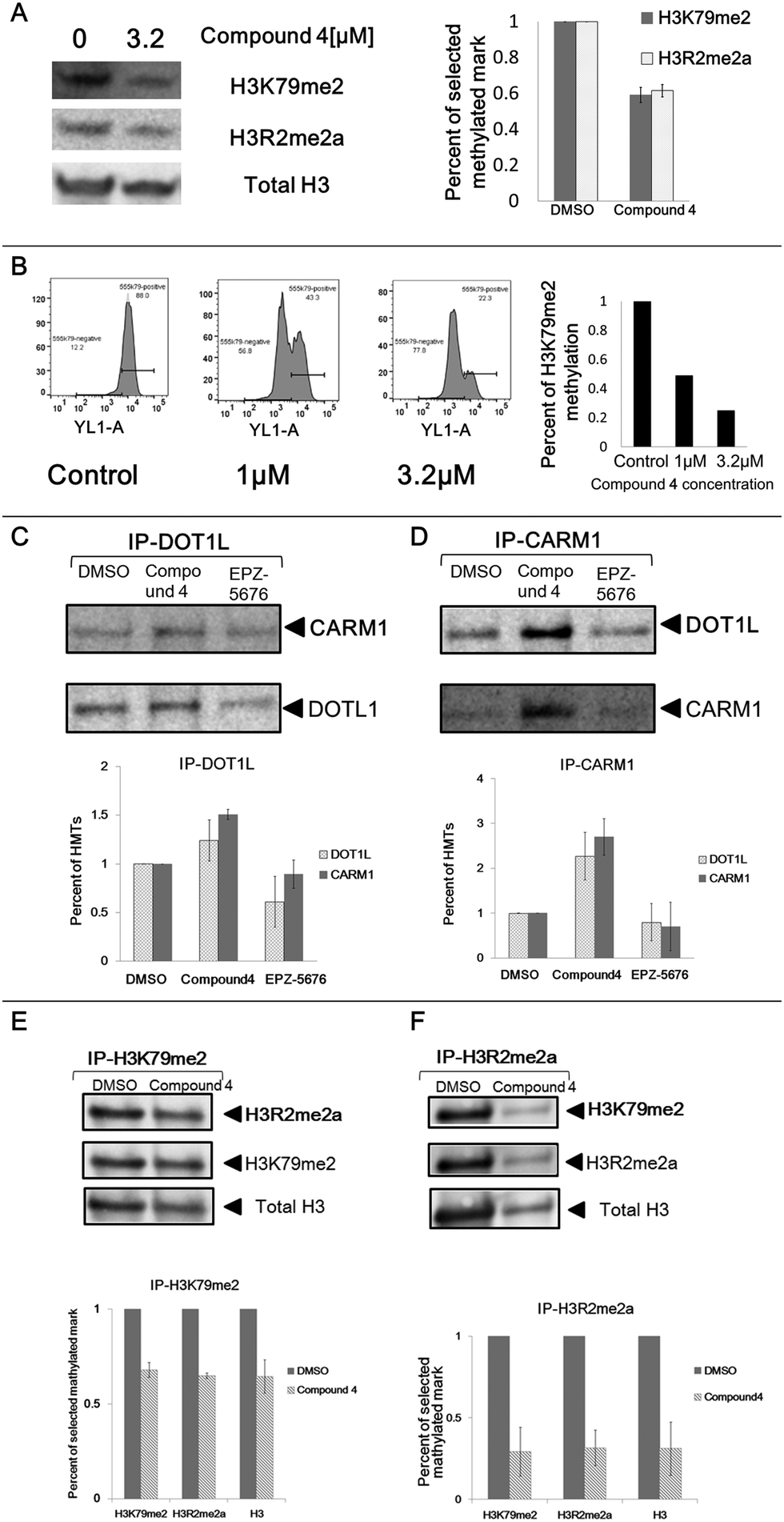 | ||
| Fig. 3 Secondary validation. (A) Effects of compound 4 (3.2 μm) on mark H3K79me2 and H3R2me2a in U2OS cells evaluated by western blotting assay following eight days of treatment. The error bars indicate the standard deviation between two biological replicates. (B) Effects of compound 4 on mark H3K79me2 in U2OS cells at 1 μm, 3.2 μm, respectively, measured by FACS after ten days of treatment. (C), (D) Co-immunoprecipitation of endogenous DOT1L (C) and CARM1 (D) in U2OS cells treated by compound 4 (3.2 μm) and EPZ-5676 (3.2 μm) after eight days treatment. The graph represents the intensity of the selected HMTs of the treated cells normalised to DMSO. The error bars indicate the standard deviation between two biological replicates. (E), (F) Co-immunoprecipitation of histone mark H3K79me2 (E) and H3R2me2a (F) in U2OS cells treated with compound 4 (3.2 μm) after eight days treatment. Cells treated with an equal amount of DMSO were used as negative control. The graph represents the intensity of the mark or histone H3 of the treated cells normalised to DMSO. The error bars indicate the standard deviation between two biological replicates. | ||
To further confirm this effect, we measured the compound 4-mediated cellular depletion of H3K79me2 by flow cytometry. After incubation of U2OS cells with compound 4 at 1 and 3.2 μm for ten days, cells were fixed and labelled with H3K79me2 antibody and subjected to flow cytometry analysis. A decrease of H3K79me2 was observed from 56% (1 μm) to 77% (3.2 μm) in a dose-dependent manner (Fig. 3B). This associated variation between H3K79me2 and H3R2me2a, firstly observed by HCS, confirmed by western blotting and flow cytometry, points out a crosstalk between DOT1L and CARM1.
Validation by mass spectrometry
We further validated the screening assay by analysis of the histone modifications by mass spectrometry (MS). This technique measures directly the abundance of histone proteins without use of antibodies that can present problems of specificity.42 U2OS cells were incubated with compound 4 or EPZ-5676 at 3.2 μm for 8 days or with DMSO as negative control. Leukaemia MV4-11 cells were also treated and analysed as they are particularly sensitive to DOT1L inhibition.33 Then, histones were extracted and separated by gel electrophoresis (Fig. S6, ESI†). Histones H3/H2A were collected and free amine groups on the N-termini and ε-amino group of unmodified or endogenously monomethylated lysines were derivatized with propionic anhydride to form propionyl amides before a digestion with trypsin. As a result all lysine residues are blocked and trypsin digestion induces proteolysis only at the C-terminal of arginine residues that are not methylated.43 In addition, the generated peptides are highly charged ions (2+, 3+, etc.), which favours their ionization and detection by ESI-MS and makes the MS/MS spectrum much easier to interpret. This methylation of arginine, when not followed by prolines, generates a missed cut and a longer peptide allowing identification of the methylation pattern. Another propionylation step was followed to modify the free N-termini, as propionylation reduces the charges on treated peptides, rendering the histone peptides less hydrophilic that can thus be easily separated by reverse-phase liquid chromatography (RP-HPLC) and produce peptides with length compatible with MS analysis and their masses can be predicted.Considering that all histones from treated and not treated cells react similarly to the propionylation and trypsin digestion, the ionization pattern is comparable in all samples and the ratio of the area under the peaks of each peptide is directly proportional to the abundance of each peptide. This analysis allowed to measure the change of peptides/proteins abundance induced by the treatment on each studied mark29 (Table 1).
| U2OS | MV4-11 | |||||
|---|---|---|---|---|---|---|
| Compound 4/3.2 μm | EPZ-5676/3.2 μm | Presence in DMSO control | Compound 4/3.2 μm | EPZ-5676/3.2 μm | Presence in DMSO control | |
| a Relative methylation ratio to DMSO control. b N.A. – ratio not analysable. | ||||||
| H3R2me1 | 0.56a | 0.57 | Yes | 0.36 | 0.27 | Yes |
| H3R17me1 | 0.79 | 0.75 | Yes | 0.8 | 0.6 | Yes |
| H3K9me2 | 0.98 | 1.09 | Yes | — | — | No |
| H3K79me1 | 0.68 | 0.39 | Yes | 0.44 | 0.28 | Yes |
| H3K79me2 | N.A.b | N.A. | Yes | N.A. | N.A. | Yes |
| H3K79me3 | N.A. | N.A. | Yes | N.A. | N.A. | Yes |
H3K79me2 and me3 were under the limit of detection to be quantified in the treated samples, but were present in the control DMSO samples, confirming the ability of the compounds to inhibit these methylation marks. H3K9me2 was used as control and it did not change in abundance upon treatment with compound 4 or EPZ-5676 in agreement with the results obtained by HCS. In the same samples, compound 4 treatment at 3.2 μm of U2OS cells led to a decrease of H3K79me1 (32% decrease) and H3R2me1 (44% decrease) levels. An even stronger effect was observed in MV4-11 cells, which are known to be dependent on DOT1L methylation.33 The same decrease was observed upon treatment with the reference DOT1L inhibitor EPZ-5676. These results strongly validate the HCS assay. Moreover, the results also confirmed the association of the methylation of H3K79 and H3R2, firstly observed by HCS.
Effect on DOT1L enzymatic activity
In order to study this concomitant inhibition in cells, we tested whether compound 4, which inhibits CARM1 with an IC50 of 1.5 μm,30 inhibits also the enzymatic activity of DOT1L responsible of H3K79me2. Compound 4 was tested at 0.1, 1, 10 μm in an AlphaLISA enzymatic assay with recombinant DOT1L,44 EPZ-5676 was used as reference drug. EPZ-5676 shows an IC50 of 17.4 nM, while compound 4 does not show any inhibition even at 10 μm (Fig. S7, ESI†), indicating that compound 4 does not inhibit directly the enzymatic activity of recombinant DOT1L.Study of the interplay between DOT1L and CARM1 by co-immunoprecipitation
Next, we addressed the functional interplay between DOT1L and CARM1 protein complexes by co-Immunoprecipitation (Co-IP). After treatment of U2OS cells with compound 4 or EPZ-5676 at 3.2 μm for 8 days, cells were lysed and the DOT1L protein was precipitated from the lysis with the corresponding antibody (Fig. 3C). Endogenous CARM1 was bound to the affinity-purified DOT1L protein complex as shown in Fig. 3C, confirming the interplay between DOT1L and CARM1 protein complexes. Moreover, the protein quantity of co-precipitated CARM1 from compound 4 treated samples is 1.5 times higher than the DMSO control sample (Fig. 3C bottom panel). This is in agreement with the strong binding of compound 4 with CARM1 observed previously in the co-crystal structure, in which it displaces all S-adenosyl-L-methionine (SAM) molecules from the catalytic pockets of the tetrameric crystal structure.30 The similar phenomenon was observed when CARM1 was precipitated from the cell lysis: DOT1L was bound to affinity-purified CARM1 (Fig. 3D). Again higher amounts of protein were recovered from cells treated with compound 4.These findings suggest that treatment with compound 4 increases the CARM1/DOT1L protein complex resulting in a decrease in the global H3K79me2 and H3R2me2a cellular levels. To confirm this, we analysed the interplay between H3K79me2 and H3R2me2a by probing the histone marks upon treatment with compound 4. After an 8 days incubation of U2OS cells with compound 4, histones were extracted and histone marks H3K79me2 (Fig. 3E) and H3R2me2a (Fig. 3F) were precipitated from the extraction from equal amounts of histones. The level of H3R2me2a and H3K79me2 were both decreased around 32–35% from the precipitated histone H3K79me2 complex (Fig. 3E), indicating that mark H3R2me2a was strongly associated with H3K79me2. A similar result was observed when H3R2me2a was precipitated (Fig. 3F), confirming our hypothesis.
Synergistic drug combination of compound 4 and EPZ-5676
Our findings show that compound 4 binds to CARM1, interacts with CARM1/DOT1L complex and inhibits both H3K79me2 and H3R2me2a marks, pointing out a synergistic interaction between CARM1 and DOT1L. In order to explore the therapeutic application of this synergistic interaction, we chose the MV4-11 cells, a MLL-rearranged leukaemia cell line known to be sensitive to DOT1L inhibition,33 as treatment with compound 4 and EPZ5676 induced no apoptosis in U2OS cells (data not shown). MV4-11 cells were treated with a combination of compound 4 and DOT1L inhibitor EPZ-5676. After a 15 days treatment, we performed flow cytometry experiments to measure the synergistic effects of compound 4/EPZ-5676 on the apoptosis in MV4-11 cells by Annexin V staining (Fig. 4).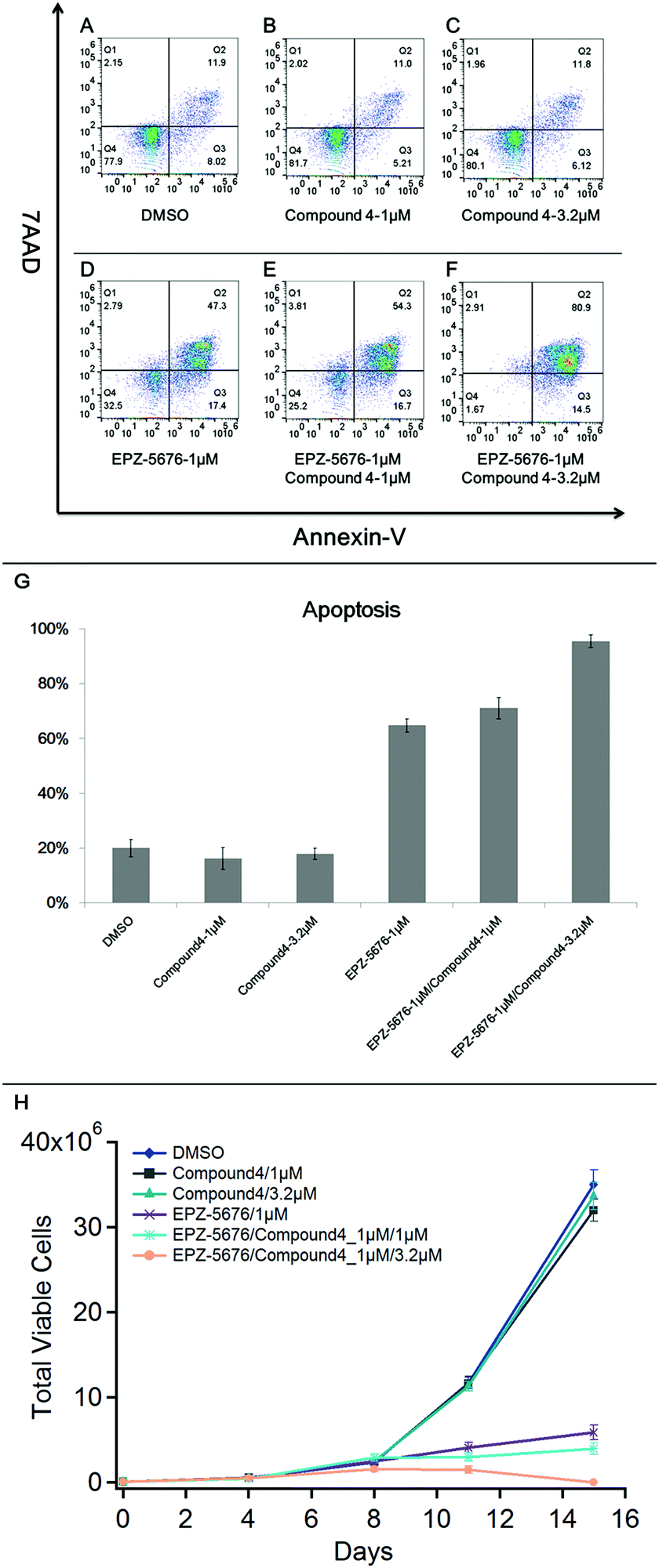 | ||
| Fig. 4 Cell apoptosis analysis. (A)–(F) Apoptosis ratio detection by Annexin V-APC/7-AAD double staining assay, analyzed by flow cytometry in MV4-11 cells treated with compound 4 (B, 1 μm), compound 4 (C, 3.2 μm), EPZ-5676 (D, 1 μm), EPZ-5676/compound 4 (E, 1/1 μm), EPZ-5676/compound 4 (F, 1/3.2 μm) after 15 days of treatment. Cells treated with DMSO (A) were used as negative control. The Q1 area represents damaged cells appearing in the process of cell collection, the Q2 area represents necrotic/late period apoptotic cells, the Q3 area represents early apoptotic cells, and the Q4 area represents normal cells. (G) Apoptosis analysis of MV4-11 cells treated with the combination of compound 4 and EPZ-5676 after 15-days of treatment. The error bars are calculated from two biological replicates. (H) Inhibition of the proliferation of MV4-11 cells following a 15 days treatment with the indicated concentrations of EPZ-5676 and compound 4. Cells treated with DMSO were used as negative control. The error bars indicate the standard deviation between two biological replicates. | ||
As shown in Fig. 4, treatment of MV4-11 cells with compound 4 alone at 1 or 3.2 μm had no significant effect on the apoptosis. As expected, treatment of MV4-11 cells with EPZ-5676 alone at 1 μm (Fig. 4D) showed an Annexin V positive cells with an apoptosis ratio of 64%. Interestingly, the synergistic effect is observed, in a dose-dependent manner, using EPZ-5676 at 1 μm associated with compound 4 at 1 μm (Fig. 4E, apoptotic: 71%) or 3.2 μm (Fig. 4F, apoptotic: 95%).
These observations are in agreement with the proliferation assay analysis as shown in Fig. 4H. The growth of cells treated with compound 4 was practically unaffected compared with control DMSO, while EPZ-5676 at 1 μm strongly inhibited the proliferation. Moreover, the number of viable MV4-11 cells was dramatically reduced by treated with EPZ-5676 at 1 μm combined with compound 4 at 3.2 μm, almost all cells died at day 15 indicating the highly increased potency of the drug combination.
These results demonstrate that, based on the crosstalk between DOT1L and CARM1, the combination of compound 4 and DOT1L inhibitor EPZ-5676 induces a stronger cell apoptosis than EPZ-5676 alone. Thus, compound 4 in combination with EPZ-5676 could improve the potency of the latter against MV4-11 cells and potentially against MLL-rearranged leukaemia.
Conclusions
In conclusion, a novel approach applying a chemical biology high-content imaging strategy was developed. The assay is based on the use of selective antibodies for the methylation state of specific histone lysine or arginine to analyse drug activity simultaneously on several histone marks in cells. The methodology was validated (1) with known inhibitors, EPZ5676 of DOT1L and UNC0642 of G9a, (2) using western blot and flow cytometry and (3) mass spectrometry analysis. This assay allows starting with a very small number of cells and thus following long-term treatments. The assay has several advantages: (1) it allows to screen chemical libraries and identify compounds that modulate histone methylation directly in cells; (2) it assesses directly the drug specificity in cells as it analyses several histone marks simultaneously; (3) it reveals crosstalk between histone marks and HMTs; and (4) it highlights new potential drug combination for treatment.We have used this approach to screen five in-house bisubstrate/transition state analogues of methyltransferases on multiple histone marks in U2OS human osteosarcoma cells. We identified that compound 4, a published CARM1 inhibitor,30 inhibits in cells both H3R2me2a, regulated also by CARM1, and H3K79me2, regulated only by DOT1L. Compound 4 does not inhibit in vitro the enzymatic activity of DOT1L suggesting a crosstalk between the HMTs in cells. The same behaviour was observed with EPZ-5676 that inhibits specifically DOT1L. Similar data were obtained with the commercial compounds SGC2085 and MS049, which are specific inhibitors of CARM1 and of CARM1 and PRMT6, respectively (Fig. S8, ESI†). This association between H3K79me2 and H3R2me2a in cells was further confirmed by western blotting, flow cytometry, mass spectrometry and co-immunoprecipitation pointing out a crosstalk between CARM1 and DOT1L. This crosstalk between DOT1L and CARM1 confirms the fact that HMTs often combine together to form multiprotein complexes13,45–47 and our results are in agreement with the CRISPR-based genetic screening13 that revealed an interaction between CARM1 and DOT1L. The authors introduced a CRISPR-based double knockout (CDKO) system to identify the genetic interactions from CRISPR-deleted gene pairs including DOT1L and CARM1 genes. They found double-knockout phenotype deviations away from Safe_Safe controls indicating a synergistic genetic interaction between DOT1L and CARM1.13,48 In addition, since when combined in MV4-11 cells EPZ-5676 and compound 4 resulted in a more severe lethality, our observation corroborates the negative genetic interaction between DOT1L and CARM1 as identified on the biogrid website.49
The assay described here does not involve alterations in protein expression as it does not require genetic manipulations, but it is based on the use of chemical compounds which effect is reversible. The strategy can be extended to all histone marks for which there are selective antibodies. The co-IP experiments confirmed the findings as they showed a strong binding of compound 4 with CARM1, in agreement with the crystallography results.30 These experiments suggest also that the presence of compound 4 increases the interaction between CARM1 and DOT1L, consequently, DOT1L activity is inhibited resulting in an inhibition of both H3K79me2 and H3R2me2a marks in cells. Most recently, we have identified a new nanomolar inhibitor of DOT1L active in cells. By using the described HCS assay, we observed that it also decreases the two marks in cells as EPZ5676.56
Moreover, the interplay between DOT1L and CARM1 prompted us to test the effect of the combination of compound 4 and DOT1L inhibitor EPZ-5676. The combination induced a more severe cell apoptosis in MV4-11 cells than EPZ-5676 alone, showing a synergistic effect. The identification of novel drug combination therapy is important and time-consuming for direct screening.13,50,51 Interestingly, our approach provides a novel strategy to identify the potential crosstalks between HMTs and thus to propose drug combinations.
Compared to other assays, the methodology developed here allows screening chemical libraries for histone mark inhibition directly in cells and simultaneously on different marks. In addition, it serves as a fast and sensitive tool to reveal potential crosstalk between histone marks and interactions between different HMTs. It can be applied to all types of chemical libraries. Follow-up studies based on these findings may reveal novel useful pathways and drug targets in the design of epigenetic drugs, and effective synergistic drug combinations.
Experimental methods
Cell culture
All cell lines were cultured at 37 °C, 5% CO2 in a humidified incubator. Human U2OS Osteosarcoma cells were obtained from ATCC (USA) and cultured in DMEM medium (Gibco) supplemented with 4.5 g L−1D-glucose and 10% foetal bovine serum (PAN-Biotech). Human leukaemia cell lines MV4-11 cells were from ATCC (USA) and cultured in RPMI medium (Gibco) supplemented with 1% L-glutamine (Gibco) and 10% foetal bovine serum (PAN-Biotech). All cell lines have been verified to be mycoplasma free on a regular basis by a PCR-based test (Takara), and no contamination has been occurred during this study.High content screening (HCS) assay
Exponentially growing U2OS cells were seeded at 250 cells per well in 100 μL in a 96-well clear bottom plate (Greiner 655090) and incubated at 37 °C and in 5% CO2 for 2 h. Compounds were added to reach a 1 μm concentration and cells were incubated for 10 days, the control was treated with 0.1% DMSO, each plate contained three technical repeats for each concentration. Culture media and drugs were replaced every 2–3 days. After incubation, an optimized conventional immunofluorescence staining protocol was performed. In brief, culture media was removed, cells were washed two times with room temperature PBS before fixed with 4% Paraformaldehyde (PFA) for 20 minutes at room temperature. After fixation, cells were washed one time with room temperature PBS and then the aldehyde groups were quenched with 50 mM NH4Cl in PBS at room temperature for 5 minutes. Cells were then permeabilized with 0.2% Triton X100 in PBS at room temperature for 10 minutes. Afterwards, cells were washed two times with PBS and blocked with 1% BSA for 1 h at room temperature. For the staining, cells were then incubated for either 1 h at room temperature or overnight at 4 °C in four pools of primary antibody in 1% BSA. The first pool contains AlexaFluor555-conjugated Histone H3 dimethyl K79 (H3K79me2) antibody (Bioss bsm-33093M-A555), AlexaFluor647-conjugated Histone H3 dimethyl K9 (H3K9me2) antibody (Abcam ab203851) and Anti-Histone H3 symmetric dimethyl R2 (H3R2me2s) antibody (Abcam ab194684); the second pool contains Anti-Histone H3 asymmetric dimethyl R2 (H3R2me2a) antibody (Abcam ab175007) and Anti-Histone H4 dimethyl K20, tri methyl K20(H4K20me2–3) antibody (Abcam ab78517); the third pool AlexaFluor555-conjugated trimethyl-Histone H3 Lys4 (H3K4me3) antibody (Cellsignal #11960); the fourth pool contains Anti-Histone H3 monomethyl K9 (H3K9me1) antibody (Abcam ab176880). After staining, cells were washed for 2 times with PBS. When primary antibody without conjugated fluorophore was used, the cells were then incubated for 1 h at room temperature with the corresponding secondary antibody (AlexaFluor488 anti-Rabbit Secondary Antibody (Invitrogen # A-11034); Goat AlexaFluor647 anti-Mouse Secondary Antibody (Invitrogen # A-21240). Finally, all cells were incubated for nuclear staining with DAPI at 300 nM and washed three times with 200 μL of PBS. Multiple images were acquired on a high content screening microscope (Olympus ix83 inverted microscope), and image analysis (FIJI) was performed to measure the integrated fluorescence intensity (IFI) of H3K79me2, H3K9me2, H3R2me2s, H3R2me2a, H4K20me2–3, H3K4me3, H3K9me1 signal per cell. A total of ∼3000 cells for each mark were analysed, each drug test was repeated at least two times for biological replicates, drug-treated sample were compared to DMSO control for each mark.Western blotting analysis
Exponentially growing U2OS cells were seeded at 6500 cells per well in 2 mL in a 6-well plate (Costar 3516) and incubated at 37 °C and in 5% CO2 for 2 hours. Compound 4 was added to reach a 3.2 μm concentration and cells were incubated for 8 days, the control was treated with 0.1% DMSO, each plate contained two technical repeats of each concentration. Culture media and drugs were replaced every 2–3 days. After incubation, cells were collected and lysed on ice in nuclear extraction buffer (10 mM Tris–HCl, 10 mM MgCl2, 25 mM KCl, 1% Triton X-100, 8.6% Sucrose and Roche protease inhibitor tablet 1836145), followed by centrifugation at 800 × g, 5 minutes at 4 °C. Pellets were collected and histones were extracted for 1 h with 0.4 N cold sulfuric acid. Histones were quantified using the BCA protein assay (Pierce 23225) and processed for western blotting. 15 μg of total histones were loaded and western blots were run following typical procedures. In brief, protein samples were separated on a NuPAGE 4–12% bis-tris acrylamide gradient gel (Invitrogen) with MES buffer, transferred to Nitrocellulose membrane (Invitrogen) and probed with primary antibodies for H3K79me2 (Abcam ab3594), H3R2me2a (Abcam ab175007) and total H3 (Abcam ab1791). Following primary antibody incubation, membranes were probed with anti-rabbit HRP secondary antibody (NA934V, GE Healthcare). Visualization was performed on a G:Box imaging system. The test was repeated at least two times for biological replicates.Flow cytometry analysis
Exponentially growing U2OS cells were seeded at 6500 cells per well in 2 mL in a 6-well plate (Costar 3516) and incubated at 37 °C and in 5% CO2 for 2 h. Compound 4 was added to reach a 1 μm or 3.2 μm concentration and cells were incubated for ten days, the control was treated with 0.1% DMSO. Culture media and drugs were replaced every 2–3 days. After incubation, culture media were removed, cells were collected and washed two times with PBS before being fixed with 4% Paraformaldehyde (PFA) for 20 minutes. After fixation, cells were washed one time with PBS and then the aldehyde groups were quenched with 50 mM NH4Cl in PBS for 5 minutes. Cells were then permeabilized with 0.2% Triton X100 in PBS for 10 minutes. Afterwards, cells were washed two times with PBS and blocked with 1% BSA for 1 h at room temperature. For the staining, cells were then incubated overnight at 4 °C with AlexaFluor555 conjugated Histone H3 dimethyl K79 (H3K79me2) antibody (Bioss bsm-33093M-A555) in 1% BSA. Samples were measured using Attune NxT acoustic focusing cytometer (Life Technologies). The voltage for forward scatter (FSC) and side scatter (SSC) were chosen so that the cell population was entirely on scale on an FSC vs. SSC plot. A non fluorescent cell sample was used to appropriately set the YL1 voltage. Individual FSC, SSC, YL1 histograms were checked to ensure that the bell-shaped populations are not cut off on the display. An event rate of ∼1000 events per second was maintained to minimize the chance of coincidence and to improve population resolution. In the FSC vs. SSC plot, a live gate R1 was set around the cell population and a total of 10,000 events inside the gate were measured. All data were analysed by FlowJo Version 9.3.3 analysis software.Mass spectrometry analysis
Exponentially growing U2OS cells were seeded at 6500 cells per well in 2 mL in a 6-well plate (Costar 3516) and incubated at 37 °C and in 5% CO2 for 2 hours. Compound 4 or EPZ5676 was added to reach a 3.2 μm concentration and cells were incubated for 8 days, the control was treated with 0.1% DMSO. Culture media and drugs were replaced every 2–3 days. After incubation, cells were collected and lysed on ice in nuclear extraction buffer (10 mM Tris-HCl, 10 mM MgCl2, 25 mM KCl, 1% Triton X-100, 8.6% Sucrose and Roche protease inhibitor tablet 1836145), followed by centrifugation at 800 × g, 5 minutes at 4 °C. Pellets were collected and histones were extracted for one hour with 0.4 N cold sulfuric acid. Histones were quantified using the BCA protein assay (Pierce 23225). Each sample was repeated five times for biological replicates.50 μg of crude histones purified from cells were run on a precast NuPAGE 4–12% bis–tris acrylamide gradient gel (Invitrogen) with MES buffer and stained with Instant Blue (Sigma). Gel bands corresponding to H2A and H3 histones were cut and destained overnight in 50% acetonitrile and 50 mM NH4HCO3. Gel slices were then treated at 37 °C for 1 h with 100 μL of 30% propionic anhydride in methanol and 40 μL of 50 mM NH4HCO3. Then, samples were washed 2 times 10 minutes in 100 mM NH4HCO3, followed by one wash in 50% acetonitrile and 100 mM NH4HCO3, and then, one wash in acetonitrile.29 The slices were then dried and stored in a 2 mL Eppendorf tube.
Dried gel slices were digested overnight at 37 °C using 0.4 μg of sequencing-grade trypsin (Promega).52 The digests were then acidified in 0.5% trifluoroacetic acid, lyophilized, samples were re-suspended in 40 μL of 50 mM NH4HCO3, and propionylated again in 100 μL of 30% propionic anhydride in methanol at 37 °C for 1 h. Finally, samples were lyophilized and re-suspended in 20 μL of 0.1% of formic acid.29
Tryptic peptides from in-gel digestion were analysed on a Q Exactive Plus instrument (Thermo Fisher Scientific, Bremen) coupled with an EASY nLC 1200 chromatography system (Thermo Fisher Scientific, Bremen). Samples were loaded on an in-house packed 20 cm nano-HPLC column (75 μm inner diameter) with C18 resin (1.9 μm particles, 100 Å pore size, Reprosil-Pur Basic C18-HD resin, Dr Maisch GmbH, Ammerbuch-Entringen, Germany) and equilibrated in 98% solvent A (H2O, 0.1% FA) and 2% solvent B (ACN, 0.1% FA). 70 minutes analysis was carried out at 250 nL min−1 flow rate. Peptides were first eluted using a 6 to 31% gradient of solvent B during 42 min and then a 31 to 62% gradient of solvent B during 10 min. Mass spectrometer was set up in DDA mode (Data Dependent Acquisition). After a survey scan in the Orbitrap (resolution 70 000), the 10 most intense precursor ions were selected for HCD fragmentation (resolution 17 500) with a normalized collision energy set up to 27. Charge state screening was enabled, and precursors with unknown charge state or a charge state of 1 and >7 were excluded. Dynamic exclusion was enabled for 30 seconds.
Data were fully analysed using PEAKS Studio 7.0.53De novo sequencing of peptides was performed with common parameter settings such as trypsin digestion, parent mass tolerance 5 ppm and fragment mass tolerance 0.02![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Da. Carbamidomethylation (C) was add as fixed modification. Multiple variable modifications were also added: Oxidation (M), Methylation (KR), Dimethylation (KR), Trimethylation (KR), Acetylation (K, Nter), Propionylation (K) and Propionyl methyl (K). A customized database was used for the search (H2/H3 histone database, 60 entries) and a label-free quantification (LFQ) was performed using the PEAKS Q quantification module. A cutoff of 1% false discovery rate (FDR) cutoff was applied at the peptide and protein levels. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE54 partner repository with the dataset identifier PXD022873.
Da. Carbamidomethylation (C) was add as fixed modification. Multiple variable modifications were also added: Oxidation (M), Methylation (KR), Dimethylation (KR), Trimethylation (KR), Acetylation (K, Nter), Propionylation (K) and Propionyl methyl (K). A customized database was used for the search (H2/H3 histone database, 60 entries) and a label-free quantification (LFQ) was performed using the PEAKS Q quantification module. A cutoff of 1% false discovery rate (FDR) cutoff was applied at the peptide and protein levels. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE54 partner repository with the dataset identifier PXD022873.
DOT1L enzymatic assay
AlphaLISA assay consists of two steps, the enzyme reaction step and the Alpha signal detection step. In the first step, final concentration of 20 nM DOT1L protein (Reaction Biology # HMT-11-101), 0.2 μm SAM (Sigma-Aldrich ref # A70007), 0.25 ng oligonucleosomes (Reaction Biology # HMT-35-130) and compound 4 or EPZ-5676 with set concentrations were mixed in a Opti-Plate-384 assay plate (white opaque, PerkinElmer) and incubated at room temperature for 90 minutes in the dark, each point is run in triplicate. Then the enzymatic reaction step was stopped by addition of high salt buffer (50 mM Tris–HCl pH 7.4, 1 M NaCl, 0.1% Tween-20, 0.3% poly-L-lysine) and incubated for 15 minutes in the dark. In the Alpha signal detection step, two kinds of beads, anti-Histone H3 Acceptor beads (PerkinElmer # AL147) and biotinylated anti-H3K79me2 antibody (PerkinElmer # AL148), were added according to manufacturer's recommendations and incubated at room temperature for 60 minutes in the dark. Then, streptavidin Donor beads (PerkinElmer # 6760002) were added and incubated for 30 minutes in the dark. DMSO and EPZ-5676 were used as negative or positive compound, respectively. Finally, the signal was read on an EnVision reader (PerkinElmer). Each drug test was repeated at least two times for biological replicates, the data were analysed with Graphpad Prism 8.1.0 software.Co-immunoprecipitation experiments
Exponentially growing U2OS cells were seeded at 6500 cells per well in 2 mL in a 6-well plate (Costar 3516) and incubated at 37 °C and in 5% CO2 for 2 hours. Compound 4 or EPZ5676 was added to reach a 3.2 μm concentration and cells were incubated for 8 days, the control was treated with 0.1% DMSO, each plate contained two repeats of the same concentration for technical repeats. Culture media and drugs were replaced every 2–3 days. After incubation, 5 × 106 cells were collected and washed with pre-cold PBS once and re-suspended in 1 mL cold RIPA buffer (Thermo Scienfic) on ice for 30 minutes. After lysis, cells were centrifuged for 30 minutes at 4 °C with 14000 × g. 500 μg of protein extract were used for immunoprecipitation. Briefly, samples were incubated overnight at 4 °C with the DOT1L antibody (abcam ab72454). Afterwards, mixtures were incubated with protein A/G UltraLink Resin (Thermo Scientific) for 2 h at room temperature, the precipitated fraction was washed with IP buffer, eluted, followed by SDS–PAGE analysis for CARM1 antibody (abcam ab245466) and DOT1L antibody (abcam ab72454). Each experiment was repeated at least two times for biological replicates, western blot analysis was performed following the procedure described above. Vice versa, the same samples were prepared, but incubated overnight at 4 °C with the CARM1 antibody (Abcam ab245466) and SDS–PAGE analysis for DOT1L antibody (Abcam ab72454) and CARM1 antibody (Abcam ab245466).
Histone marks pulldown
Exponentially growing U2OS cells were seeded at 6500 cells per well in 2 mL in a 6-well plate (Costar 3516) and incubated at 37 °C and in 5% CO2 for 2 hours. Compound 4 was added to reach a 3.2 μm concentration and cells were incubated for 8 days, the control was treated with 0.1% DMSO, samples were analysed in duplicate. Culture media and drugs were replaced every 2–3 days. After incubation, cells were collected and lysed on ice in nuclear extraction buffer (10 mM Tris-HCl, 10 mM MgCl2, 25 mM KCl, 1% Triton X-100, 8.6% Sucrose and Roche protease inhibitor tablet 1836145), followed by centrifuge at 800 × g, 5 minutes at 4 °C. Pellets were collected and histones were extracted for one hour with 0.4 N cold sulfuric acid. Histones were quantified using the BCA protein assay (Pierce 23225). 50 μg of total histones were used for the pull down. In brief, samples were incubated in PBS buffer overnight at 4 °C with the corresponding antibody (H3K79me2 (ab3594) or H3R2me2a (ab175007)). After this step, mixtures were incubated with protein A/G UltraLink Resin (Thermo Scientific) for 2 h at room temperature, the precipitated fraction was washed with IP buffer, eluted, followed by SDS–PAGE analysis for H3K79me2 (ab3594), H3R2me2a (ab175007). Each drug test was repeated at least two times for biological replicates, western blot analysis was performed following the procedure described above.Flow cytometric analysis of cell apoptosis
In a 12-well plate, 2 mL exponentially growing MV4-11 cells were plated with a density of 2 × 105 cells per mL. Cells were incubated with a final concentration of EPZ-5676 (1 μm), EPZ-5676/compound 4 (1/1 μm), EPZ-5676/compound 4 (1/3.2 μm), compound 4 (1 μm), compound 4 (3.2 μm) for 15 days, the control was treated with 0.1% DMSO, each plate contained technical duplicates. Culture media and drugs were replaced every 3–4 days and cells were split back to density of 2 × 105 cells per mL at the same time. Cells were harvested on day 15 to allow analysis of Annexin V staining. Apoptosis ratio is detected by an Annexin V-APC/7-AAD double staining assay (SONY 3804610) and cells were prepared according to manufacturer's recommendations. Samples were measured using Attune NxT acoustic focusing cytometer (Life technologies), each test was repeated at least two times for biological replicates, data were analysed by FlowJo Version 9.3.3 analysis software.Analysis of cell proliferation
In a 96-well plate, 200 μL exponentially growing MV4-11 cells were plated with a density of 7 × 104 cells per well. Cells were incubated with a final concentration of EPZ-5676 (1 μm), EPZ-5676/compound 4 (1/1 μm), EPZ-5676/compound 4 (1/3.2 μm), compound 4 (1 μm), compound 4 (3.2 μm) for 15 days, the control was treated with 0.1% DMSO, experiments contained technical duplicates. Viable cell number was determined every 3–4 days for up to 15 days using the trypan blue (Sigma) cell counting by Cellometer Mini (Nexcelom Bioscience) according to the manufacture's protocol. On the days of cell counting, culture media and drugs were replaced and cells were split back to a density of 7 × 104 cells per well at the same time. Each drug test was repeated at least two times for biological replicates, total viable cell number is calculated as split-adjusted viable cells per well as described previously.55Author contributions
Yang Si: conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft. Corentin Bon: investigation; methodology; writing – review & editing the manuscript. Véronique Cadet Daniel/Magalie Duchateau: investigation; methodology. Mariette Matondo/Fabrice Agou: resources; writing – review & editing. Ludovic Halby: conceptualization; supervision; writing – review & editing. Paola B. Arimondo: funding acquisition; conceptualization; project administration; supervision; validation; writing – review & editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors thank Ambre Tafit for the proliferation studies and Alix Boucharlat for the training on the microscope. We thank the “Region Ile de France” for the financing of the PhD of Mr Bon (ArDOC fellowship) and Ligue national contre le Cancer, comité de Paris (grants RS20/75-82 and RS21/75-31 to PBA). This work benefited from a government grant (PPR Antibioresistance) managed by the Agence Nationale de la Recherche – Programme d'Investissements d'Avenir (ANR-20-PAMR-0011).References
- K. E. Stephens, C. A. Miaskowski, J. D. Levine, C. R. Pullinger and B. E. Aouizerat, Biol. Res. Nurs., 2013, 15, 373–381 CrossRef CAS PubMed.
- A. Moosavi and A. M. Ardekani, Iran. Biomed. J., 2016, 20, 246–258 Search PubMed.
- S. Scheer, S. Ackloo, T. S. Medina, M. Schapira, F. Li, J. A. Ward, A. M. Lewis, J. P. Northrop, P. L. Richardson, H. Ü. Kaniskan, Y. Shen, J. Liu, D. Smil, D. McLeod, C. A. Zepeda-Velazquez, M. Luo, J. Jin, D. Barsyte-Lovejoy, K. V. M. Huber, D. D. D. Carvalho, M. Vedadi, C. Zaph, P. J. Brown and C. H. Arrowsmith, Nat. Commun., 2019, 10, 19 CrossRef CAS PubMed.
- S. Sharma, T. K. Kelly and P. A. Jones, Carcinogenesis, 2010, 31, 27–36 CrossRef CAS PubMed.
- D. Bayarsaihan, J. Dent. Res., 2011, 90, 9–17 CrossRef CAS PubMed.
- M. Jakovcevski and S. Akbarian, Nat. Med., 2012, 18, 1194–1204 CrossRef CAS PubMed.
- E. Tzika, T. Dreker and A. Imhof, Front. Genet., 2018, 9, 361, DOI:10.3389/fgene.2018.00361.
- A. Y. Yang, H. Kim, W. Li and A.-N. T. Kong, Curr. Top. Med. Chem., 2016, 16, 697–713 CrossRef CAS PubMed.
- J. E. Audia and R. M. Campbell, Cold Spring Harbor Perspect. Biol., 2016, 8(4), a019521, DOI:10.1101/cshperspect.a019521.
- Z. Wang and D. J. Patel, Q. Rev. Biophys., 2013, 46, 349–373 CrossRef CAS PubMed.
- J. K. Lue and J. E. Amengual, Curr. Hematol. Malig. Rep., 2018, 13, 369–382 CrossRef PubMed.
- Office of the Commissioner, FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer, https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-option-specifically-patients-epithelioid-sarcoma-rare-soft-tissue, (accessed November 24, 2020).
- K. Han, E. E. Jeng, G. T. Hess, D. W. Morgens, A. Li and M. C. Bassik, Nat. Biotechnol., 2017, 35, 463–474 CrossRef CAS PubMed.
- C. Bon, Y. Si and P. B. Arimondo, in Histone Modifications in Therapy, ed. P. Castelo-Branco and C. Jeronimo, Academic Press, 2020, vol. 20, pp. 81–99 Search PubMed.
- X.-C. Cai, K. Kapilashrami and M. Luo, Methods Enzymol., 2016, 574, 245–308 CAS.
- H. Zeng and W. Xu, in Epigenetic Technological Applications, ed. Y. G. Zheng, Academic Press, Boston, 2015, pp. 333–361 Search PubMed.
- A. Dhayalan, E. Dimitrova, P. Rathert and A. Jeltsch, J. Biomol. Screening, 2009, 14, 1129–1133 CrossRef CAS PubMed.
- P. Rathert, X. Cheng and A. Jeltsch, Biotechniques, 2007, 43, 602 CrossRef CAS PubMed.
- J. Wu, N. Xie, Y. Feng and Y. G. Zheng, J. Biomol. Screening, 2012, 17, 237–244 CrossRef CAS PubMed.
- S. Chen, L. Li, Y. Chen, J. Hu, J. Liu, Y.-C. Liu, R. Liu, Y. Zhang, F. Meng, K. Zhu, J. Lu, M. Zheng, K. Chen, J. Zhang, H. Jiang, Z. Yao and C. Luo, J. Chem. Inf. Model., 2016, 56, 527–534 CrossRef CAS PubMed.
- Y. Song, L. Li, Y. Chen, J. Liu, S. Xiao, F. Lian, N. Zhang, H. Ding, Y. Zhang, K. Chen, H. Jiang, C. Zhang, Y.-C. Liu, S. Chen and C. Luo, Bioorg. Med. Chem., 2018, 26, 1751–1758 CrossRef CAS PubMed.
- N. J. Martinez and A. Simeonov, Drug Discovery Today Technol., 2015, 18, 9–17 CrossRef PubMed.
- C. Chen, H. Zhu, F. Stauffer, G. Caravatti, S. Vollmer, R. Machauer, P. Holzer, H. Möbitz, C. Scheufler, M. Klumpp, R. Tiedt, K. S. Beyer, K. Calkins, D. Guthy, M. Kiffe, J. Zhang and C. Gaul, ACS Med. Chem. Lett., 2016, 7, 735–740 CrossRef CAS PubMed.
- J. S. Yi, A. J. Federation, J. Qi, S. Dhe-Paganon, M. Hadler, X. Xu, R. Pierre, A. C. Varca, L. Wu, J. J. Marineau, W. B. Smith, A. Souza, E. J. Chory, S. A. Armstrong and J. E. Bradner, ACS Chem. Biol., 2015, 10, 667–674 CrossRef CAS PubMed.
- B. Zhu, H. Zhang, S. Pan, C. Wang, J. Ge, J.-S. Lee and S. Q. Yao, Chem. – Eur. J., 2016, 22, 7824–7836 CrossRef CAS PubMed.
- S. Kubicek, R. J. O’Sullivan, E. M. August, E. R. Hickey, Q. Zhang, M. L. Teodoro, S. Rea, K. Mechtler, J. A. Kowalski, C. A. Homon, T. A. Kelly and T. Jenuwein, Mol. Cell, 2007, 25, 473–481 CrossRef CAS PubMed.
- Ö. Önder, S. Sidoli, M. Carroll and B. A. Garcia, Expert Rev. Proteomics, 2015, 12, 499–517 CrossRef PubMed.
- H. Ü. Kaniskan, M. L. Martini and J. Jin, Chem. Rev., 2018, 118, 989–1068 CrossRef CAS PubMed.
- P. Robin, L. Fritsch, O. Philipot, F. Svinarchuk and S. Ait-Si-Ali, Genome Biol., 2007, 8, R270 CrossRef PubMed.
- L. Halby, N. Marechal, D. Pechalrieu, V. Cura, D.-M. Franchini, C. Faux, F. Alby, N. Troffer-Charlier, S. Kudithipudi, A. Jeltsch, W. Aouadi, E. Decroly, J.-C. Guillemot, P. Page, C. Ferroud, L. Bonnefond, D. Guianvarc’h, J. Cavarelli and P. B. Arimondo, Philos. Trans. R. Soc., B, 2018, 373, 20170072, DOI:10.1098/rstb.2017.0072.
- The Human Protein Atlas, https://www.proteinatlas.org/.
- F. Liu, D. Barsyte-Lovejoy, F. Li, Y. Xiong, V. Korboukh, X.-P. Huang, A. Allali-Hassani, W. P. Janzen, B. L. Roth, S. V. Frye, C. H. Arrowsmith, P. J. Brown, M. Vedadi and J. Jin, J. Med. Chem., 2013, 56, 8931–8942, DOI:10.1021/jm401480r.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, A. Basavapathruni, L. Jin, P. A. Boriack-Sjodin, C. J. Allain, C. R. Klaus, A. Raimondi, M. P. Scott, N. J. Waters, R. Chesworth, M. P. Moyer, R. A. Copeland, V. M. Richon and R. M. Pollock, Blood, 2013, 122, 1017–1025 CrossRef CAS PubMed.
- V. Kari, S. K. Raul, J. M. Henck, J. Kitz, F. Kramer, R. L. Kosinsky, N. Übelmesser, W. Y. Mansour, J. Eggert, M. Spitzner, Z. Najafova, H. Bastians, M. Grade, J. Gaedcke, F. Wegwitz and S. A. Johnsen, Clin. Epigenet., 2019, 11(1), 4, DOI:10.1186/s13148-018-0601-1.
- D. Pechalrieu, F. Assemat, L. Halby, M. Marcellin, P. Yan, K. Chaoui, S. Sharma, G. Chiosis, O. Burlet-Schiltz, P. B. Arimondo and M. Lopez, ACS Chem. Biol., 2020, 15, 952–961 CrossRef CAS PubMed.
- B. T. Schurter, S. S. Koh, D. Chen, G. J. Bunick, J. M. Harp, B. L. Hanson, A. Henschen-Edman, D. R. Mackay, M. R. Stallcup and D. W. Aswad, Biochemistry, 2001, 40, 5747–5756 CrossRef CAS PubMed.
- E. Shishkova, H. Zeng, F. Liu, N. W. Kwiecien, A. S. Hebert, J. J. Coon and W. Xu, Nat. Commun., 2017, 8, 1–13 CrossRef PubMed.
- D. Cheng, G. Gao, A. Di Lorenzo, S. Jayne, M. O. Hottiger, S. Richard and M. T. Bedford, J. Biol. Chem., 2020, 295, 17060–17070 CrossRef CAS PubMed.
- E. Guccione, C. Bassi, F. Casadio, F. Martinato, M. Cesaroni, H. Schuchlautz, B. Lüscher and B. Amati, Nature, 2007, 449, 933–937 CrossRef CAS PubMed.
- A. D. Lorenzo and M. T. Bedford, FEBS Lett., 2011, 585, 2024–2031 CrossRef PubMed.
- L. Aravind, S. Abhiman and L. M. Iyer, in Progress in Molecular Biology and Translational Science, ed. X. Cheng and R. M. Blumenthal, Academic Press, 2011, vol. 101, pp. 105–176 Search PubMed.
- S. Rakow, S. S. Pullamsetti, U.-M. Bauer and C. Bouchard, Methods, 2020, 175, 53–65 CrossRef CAS PubMed.
- G. S. Baldwin and P. R. Carnegie, Science, 1971, 171, 579–581 CrossRef CAS PubMed.
- M. Sabatino, D. Rotili, A. Patsilinakos, M. Forgione, D. Tomaselli, F. Alby, P. B. Arimondo, A. Mai and R. Ragno, J. Comput.-Aided Mol. Des., 2018, 32, 435–458 CrossRef CAS PubMed.
- D. Steinhilber and R. Marschalek, Biochem. Pharmacol., 2018, 147, 183–190 CrossRef CAS PubMed.
- A. Benedikt, S. Baltruschat, B. Scholz, A. Bursen, T. N. Arrey, B. Meyer, L. Varagnolo, A. M. Müller, M. Karas, T. Dingermann and R. Marschalek, Leukemia, 2011, 25, 135–144 CrossRef CAS PubMed.
- K. Nakayama, M. M. Szewczyk, C. dela Sena, H. Wu, A. Dong, H. Zeng, F. Li, R. F. de Freitas, M. S. Eram, M. Schapira, Y. Baba, M. Kunitomo, D. R. Cary, M. Tawada, A. Ohashi, Y. Imaeda, K. S. Saikatendu, C. E. Grimshaw, M. Vedadi, C. H. Arrowsmith, D. Barsyte-Lovejoy, A. Kiba, D. Tomita and P. J. Brown, Oncotarget, 2018, 9, 18480–18493 CrossRef PubMed.
- H. Hermjakob, L. Montecchi-Palazzi, G. Bader, J. Wojcik, L. Salwinski, A. Ceol, S. Moore, S. Orchard, U. Sarkans, C. von Mering, B. Roechert, S. Poux, E. Jung, H. Mersch, P. Kersey, M. Lappe, Y. Li, R. Zeng, D. Rana, M. Nikolski, H. Husi, C. Brun, K. Shanker, S. G. N. Grant, C. Sander, P. Bork, W. Zhu, A. Pandey, A. Brazma, B. Jacq, M. Vidal, D. Sherman, P. Legrain, G. Cesareni, I. Xenarios, D. Eisenberg, B. Steipe, C. Hogue and R. Apweiler, Nat. Biotechnol., 2004, 22, 177–183 CrossRef CAS PubMed.
- CARM1 – DOT1L Interaction Summary | BioGRID, https://thebiogrid.org/interaction/2538278/carm1-dot1l.html.
- J. Jia, F. Zhu, X. Ma, Z. W. Cao, Y. X. Li and Y. Z. Chen, Nat. Rev. Drug Discovery, 2009, 8, 111–128 CrossRef CAS PubMed.
- J. Wang, S. Yazdani, A. Han and M. Schapira, Drug Discovery Today, 2020, 25, 561–567 CrossRef CAS PubMed.
- C. Addi, A. Presle, S. Frémont, F. Cuvelier, M. Rocancourt, F. Milin, S. Schmutz, J. Chamot-Rooke, T. Douché, M. Duchateau, Q. Giai Gianetto, A. Salles, H. Ménager, M. Matondo, P. Zimmermann, N. Gupta-Rossi and A. Echard, Nat. Commun., 2020, 11, 1941 CrossRef CAS PubMed.
- B. Ma, K. Zhang, C. Hendrie, C. Liang, M. Li, A. Doherty-Kirby and G. Lajoie, Rapid Commun. Mass Spectrom., 2003, 17, 2337–2342 CrossRef CAS PubMed.
- Y. Perez-Riverol, A. Csordas, J. Bai, M. Bernal-Llinares, S. Hewapathirana, D. J. Kundu, A. Inuganti, J. S. Griss, G. Mayer, M. Eisenacher, E. Pérez, J. Uszkoreit, J. Pfeuffer, T. Sachsenberg, S. Yilmaz, S. Tiwary, J. Cox, E. Audain, M. Walzer, A. F. Jarnuczak, T. Ternent, A. Brazma and J. A. Vizcaíno, The PRIDE database and related tools and resources in 2019, Nucleic Acids Res., 2019, 47(D1), D442–D450 CrossRef CAS PubMed.
- S. R. Daigle, E. J. Olhava, C. A. Therkelsen, C. R. Majer, C. J. Sneeringer, J. Song, L. D. Johnston, M. P. Scott, J. J. Smith, Y. Xiao, L. Jin, K. W. Kuntz, R. Chesworth, M. P. Moyer, K. M. Bernt, J.-C. Tseng, A. L. Kung, S. A. Armstrong, R. A. Copeland, V. M. Richon and R. M. Pollock, Cancer Cell, 2011, 20, 53–65 CrossRef CAS PubMed.
- C. Bon, Y. Si, M. Pernak, M. Barbachowska, E. Levi-Acobas, V. Cadet Daniel, C. Jallet, D. Ruzic, N. Djokovic, T. Djikić, K. Nikolic, L. Halby and P. B. Arimondo, Molecules, 2021, 26(17), 5300 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD022873. See DOI: 10.1039/d1cb00095k |
| This journal is © The Royal Society of Chemistry 2022 |