
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a NanoBRET assay to validate inhibitors of Sirt2-mediated lysine deacetylation and defatty-acylation that block prostate cancer cell migration†
A.
Vogelmann
 a,
M.
Schiedel
b,
N.
Wössner
a,
A.
Merz
a,
D.
Herp
a,
S.
Hammelmann
a,
A.
Colcerasa
a,
G.
Komaniecki
c,
JY.
Hong
c,
M.
Sum
d,
E.
Metzger
d,
E.
Neuwirt
efg,
L.
Zhang
h,
O.
Einsle
h,
O.
Groß
efi,
R.
Schüle
df,
H.
Lin
cj,
W.
Sippl
k and
M.
Jung
*af
a,
M.
Schiedel
b,
N.
Wössner
a,
A.
Merz
a,
D.
Herp
a,
S.
Hammelmann
a,
A.
Colcerasa
a,
G.
Komaniecki
c,
JY.
Hong
c,
M.
Sum
d,
E.
Metzger
d,
E.
Neuwirt
efg,
L.
Zhang
h,
O.
Einsle
h,
O.
Groß
efi,
R.
Schüle
df,
H.
Lin
cj,
W.
Sippl
k and
M.
Jung
*af
aInstitute of Pharmaceutical Sciences, University of Freiburg, Albertstraße 25, 79104 Freiburg, Germany. E-mail: manfred.jung@pharmazie.uni-freiburg.de
bDepartment of Chemistry and Pharmacy, Medicinal Chemistry, Friedrich-Alexander-University Erlangen-Nürnberg, Nikolaus-Fiebiger-Straße 10, 91058 Erlangen, Germany
cDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA
dDepartment of Urology and Center for Clinical Research, University of Freiburg Medical Center, Breisacher Strasse 66, 79106 Freiburg, Germany
eInstitute of Neuropathology, Medical Center – University of Freiburg, Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany
fCIBSS – Centre for Integrative Biological Signalling Studies, University of Freiburg, Germany
gFaculty of Biology, University of Freiburg, 79104 Freiburg, Germany
hInstitute of Biochemistry, University of Freiburg, Albertstraße 21, 79104 Freiburg, Germany
iCenter for Basics in NeuroModulation (NeuroModulBasics), Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany
jHoward Hughes Medical Institute; Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA
kDepartment of Medicinal Chemistry, Institute of Pharmacy, University of Halle-Wittenberg, Kurt-Mothes-Str. 3, 06120 Halle, Germany
First published on 1st March 2022
Abstract
Sirtuin2 (Sirt2) with its NAD+-dependent deacetylase and defatty-acylase activities plays a central role in the regulation of specific cellular functions. Dysregulation of Sirt2 activity has been associated with the pathogenesis of many diseases, thus making Sirt2 a promising target for pharmaceutical intervention. Herein, we present new high affinity Sirt2 selective Sirtuin-Rearranging Ligands (SirReals) that inhibit both Sirt2-dependent deacetylation and defatty-acylation in vitro and in cells. We show that simultaneous inhibition of both Sirt2 activities results in strongly reduced levels of the oncoprotein c-Myc and an inhibition of cancer cell migration. Furthermore, we describe the development of a NanoBRET-based assay for Sirt2, thereby providing a method to study cellular target engagement for Sirt2 in a straightforward and accurately quantifiable manner. Applying this assay, we could confirm cellular Sirt2 binding of our new Sirt2 inhibitors and correlate their anticancer effects with their cellular target engagement.
Introduction
Sirtuins are enzymes involved in the regulation of specific biological pathways. Due to deacetylation of lysines in histones, they are classified as class III histone deacylases (HDACs). In contrast to zinc-dependent HDACs, the sirtuin family is characterized by a nicotinamide adenine dinucleotide (NAD+)-dependent mechanism.1 In humans, seven sirtuin isotypes have been identified (Sirt1-7).2 They share the same core domain but differ in their subcellular localization and their substrates. Consequently, sirtuins have a huge variety of cellular functions and contribute to many physiological processes including mitosis, metabolism, cellular stress, DNA damage repair and regulation of gene expression.3Sirt2 is one of the best studied members of the sirtuin family. It is mainly localized in the cytosol but can also shuttle into the nucleus.4,5 Many deacetylation substrates have already been identified, including histones (e.g. H4K16, H3K18)6,7 as well as non-histone proteins like α-tubulin,8 p300,9 NFκB,10 PEPCK1,11 LDH1,12 HIF1α13 and FOXO3.14
In 2013, Sirt2 was discovered to exert defatty-acylase activity as it contains a hydrophobic pocket which allows acyl lysine substrates to bind.15 KRas4a was identified as the first in vivo substrate of Sirt2-catalyzed defatty-acylation. In the following years, two additional deacylase substrates, RalB and ARF6, were discovered and in both cases defatty-acylation regulates their activity and subcellular localization.16,17 All three reported substrates are small GTPases with important cellular functions and their dysregulation has been associated with the pathogenesis of different cancer types. This indicates the importance of Sirt2 as regulator of GTPase activity and implies that Sirt2 defatty-acylation essentially contributes to cellular pathways and functions.18–20
Due to the high number and variety of Sirt2 substrates, Sirt2 is involved in the regulation of many cellular pathways and functions including mitosis, metabolism, aging, inflammation, and gene transcription.21–26 As a consequence, dysregulation of Sirt2 is involved in the pathogenesis of a broad spectrum of diseases, including neurological and metabolic disorders as well as cancer.27–29 Over the years, researchers uncovered that Sirt2 is often dysregulated in cancer and can influence cancer progression by affecting tumour cell cycle and microenvironment. However, whether Sirt2 acts as a tumour suppressor or tumour promotor is not always clear. This is still under discussion as molecular mechanisms of Sirt2 in tumorigenesis are very complex and not fully understood yet.3 Generally, there seems to be a cell type and tissue specific impact, depending on local expression levels and functions of Sirt2 in the respective tissue. For instance, Sirt2 has been reported as a tumour promotor in liver, gastric, breast, colon, and pancreatic cancer, as well as neuroblastoma, whereas there is evidence for a suppressing function in lung cancer, glioma, and renal cell carcinoma.11,12,30–35 For some of the cancer cell lines, conflicting data has been published either suggesting a promoting or suppressing role for Sirt2 in the respective cancer (e.g. breast and lung cancer), which again highlights the complexity of Sirt2 as an anti-cancer target.
The potential of Sirt2 as a target for pharmacological treatment for various diseases has driven researchers to develop Sirt2 inhibitors. In order to develop new selective inhibitors for Sirt2, the knowledge of the unique structural features of this enzyme is essential. The catalytic core of Sirt2 consists of a Rossman fold domain with the NAD+-binding site and a smaller zinc binding domain. A hydrophobic groove separates the two domains and forms the binding site for the acyl-lysine substrate.36 In 2015, we published SirReal2 (1) as a highly selective and potent Sirt2 inhibitor. This compound induces a conformational change upon binding to the enzyme and leads to the formation of a so-called selectivity pocket.37 Optimization of SirReal2 led to a new generation of SirReals with an additional triazole attached to the SirReal scaffold (e.g. compound 2). Due to an additional interaction between the triazole and Arg97 in the acyl lysine channel, these triazole-based SirReals feature an extended binding mode, which results in an improved affinity.38 The family of Sirt2 inhibitors is still growing, with members characterized by different structural scaffolds, binding modes, selectivity and potency profiles. With increasing evidence for the importance of the Sirt2 deacylation function, inhibitor development has been directed towards the inhibition of lysine defatty-acylation besides deacetylation. Fig. 1 highlights Sirt2 inhibitors that have been characterized, at least in vitro, regarding their potential to inhibit Sirt2 deacetylation and deacylation.37,39–46 While some of the compounds such as TM and JH-T4 show inhibition of both reactions, other compounds including SirReal2 and NPD11033 selectively inhibit Sirt2 deacetylation. One of the most recently published inhibitors that blocks both Sirt2 activities in vitro is a peptide-based compound developed by Nielsen and co-workers, which represents the most potent inhibitor for Sirt2 deacetylation (IC50 = 16 nM) to date.42
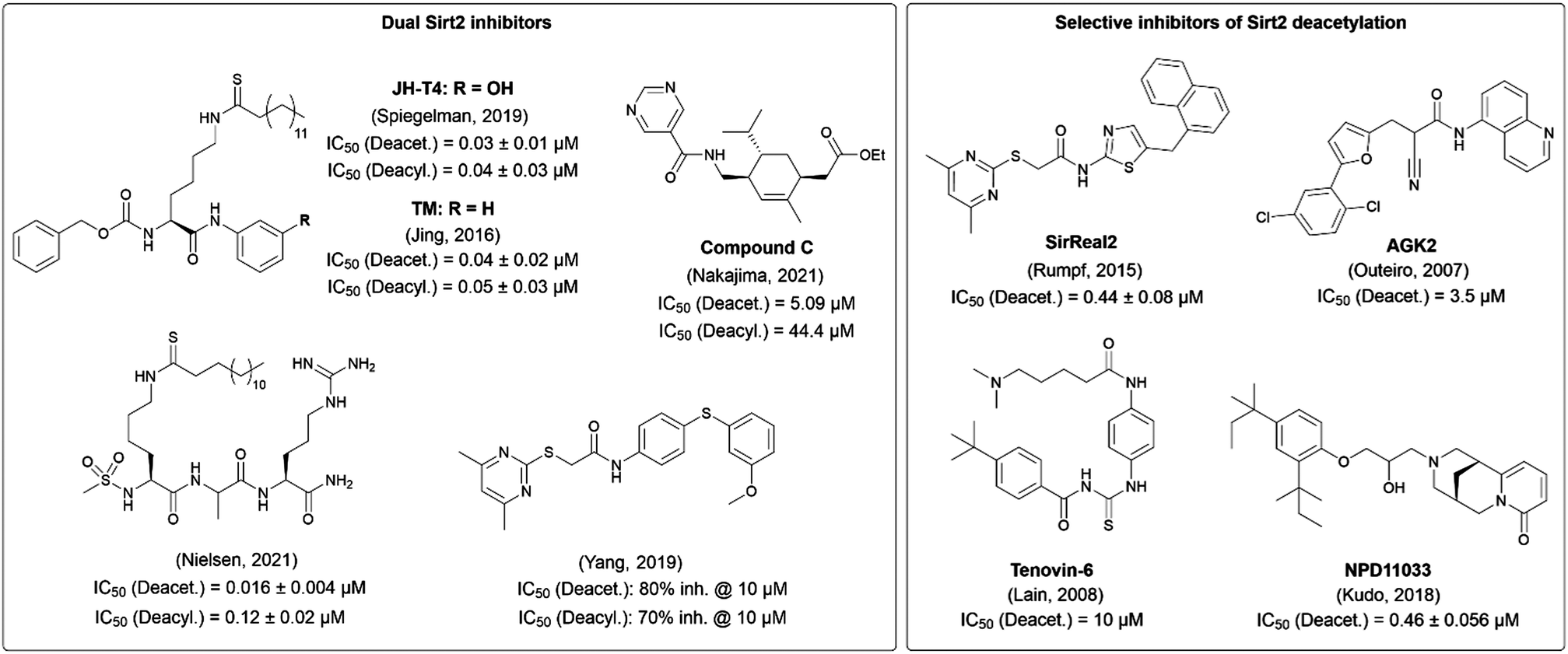 | ||
| Fig. 1 Selected Sirt2 inhibitors characterized regarding their potential to inhibit Sirt2 deacetylation and defatty-acylation activity in vitro.37,39–46 Inhibitors that inhibit both Sirt2 activities are shown on the left, whereas selective inhibitors of Sirt2 deacetylation are presented on the right. | ||
Simultaneous inhibition of Sirt2 activity was reported to result in higher cellular anticancer effects as compared with selective inhibition of Sirt2-mediated deacetylation,47 thereby suggesting the importance of the defatty-acylation activity of Sirt2 for cancer development and progression. This prompted us to develop inhibitors of both Sirt2-catalyzed deacetylation and defatty-acylation reactions based on our highly Sirt2 selective and drug-like SirReal scaffold. In the course of the development of these Sirt2 inhibitors, we put a special focus on methods for studying their cellular target engagement. We present the development of a cellular NanoBRET-based binding assay for Sirt2 as new method to study cellular Sirt2 target engagement in a highly accurate and straightforward manner.
Results and discussion
Design of new SirReal analogues for simultaneous inhibition of Sirt2-catalyzed deacetylation and defatty-acylation
Since additional inhibition of the defatty-acylase activity may play an essential role for anticancer activity of Sirt2 inhibitors, we aimed to develop inhibitors for both Sirt2-catalyzed deacetylation and defatty-acylation, based on our potent, drug-like, and highly Sirt2 selective SirReals.40,47 As SirReal2 (1), our initial lead structure, does not inhibit Sirt2 defatty-acylase activity,48 we wanted to investigate if structural modifications at the SirReal scaffold in the lysine channel towards the entry of the active site would increase potency and Sirt2 affinity and enable the additional inhibition of Sirt2-mediated defatty-acylation. Therefore, we compiled a small library of differently modified SirReals, which are displayed in Fig. 2. In general, the compounds that constitute our small library can be divided into three different groups. For the first group, we chose two already published compounds (3, 4), structurally similar to naphthyl-based SirReal2, which have not been investigated for their effect on deacylation or in a cellular setting yet. These two compounds feature an additional methyl group next to the mercaptopyrimidine and a naphthyl-based aromatic system. The other compounds are based on the core structure of the previously published triazole-based SirReal (2), which features an extended binding mode due to additional polar interactions of the triazole with Arg97 in the acyllysine channel.38 We based the design of these Sirt2 inhibitors on two approaches, reasoning that: (a) targeting the selectivity pocket with lipophilic groups would mimic the binding of fatty acylated substrates and, thus inhibit Sirt2 defatty-acylation activity, (b) extended binding into the lysine channel should block the binding of both acetylated and acylated substrates and lead to simultaneous inhibition of Sirt2 activity.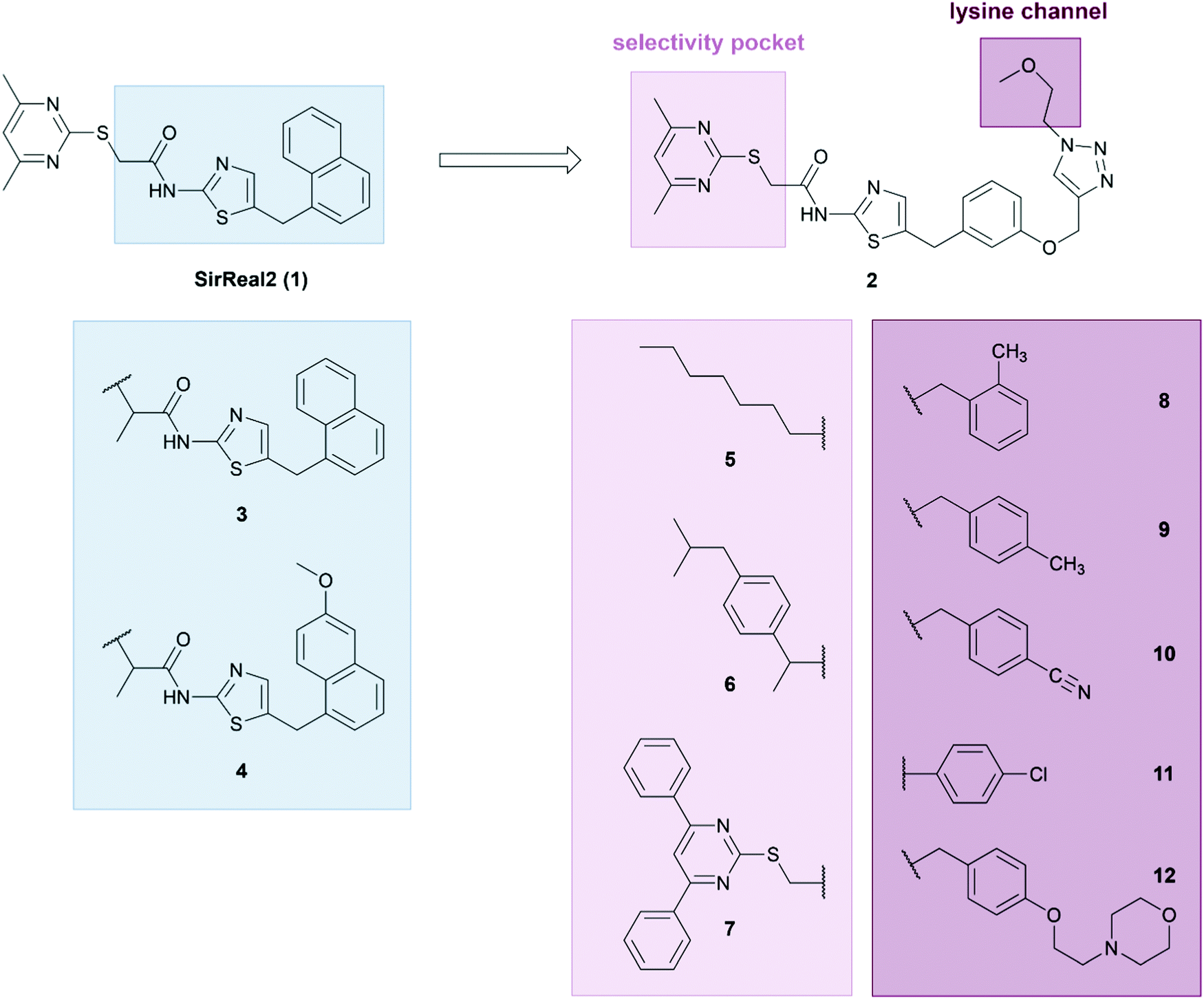 | ||
| Fig. 2 Design of SirReal-based Sirt2 inhibitors for simultaneous inhibition of Sirt2 deacetylase and defatty-acylase activity. | ||
For the first approach, we kept the triazole part of compound 2 unchanged and modified or replaced the pyrimidine part of the molecule, known to be responsible for the formation of the selectivity pocket,37 with a more lipophilic group. As the unique selectivity pocket of Sirt2 also accommodates the long-chain fatty acid of a myristoyl substrate,49 we tried to mimic the binding of fatty acid substrates by replacing the pyrimidine moiety with either a fatty acid (5) or a more bulky hydrophobic group (6). This strategy was further supported by the observation that ligands with bulky or hydrophobic moieties also induce a rearranged conformation of the Sirt2 enzyme.42,46 The hydrophobic group of compound 6 is based on the structure of the cyclooxygenase inhibitor ibuprofen, which is known to act as fatty acid mimic.50 Compound 7 was designed as a negative control, as it was previously shown that an installation of a 4,6-diphenylpyrimidine moiety sterically prevents binding of the respective SirReal analogues to the active site of Sirt2.37 For approach (b), we attached different groups to the triazole ring that extend in the lysine channel towards the enzyme surface to enable further interactions with amino acids forming the substrate channel. The resulting compounds 8–12 contain differently decorated benzyl substituents attached to the triazole. The benzyl substituents are linked to the triazole via a methylene group except for compound 11, where the p-chloro-phenyl group is directly attached to the triazole. For the synthesis of our new SirReal analogues, the reported synthesis route for 238 was adapted to enable late-stage functionalization at different moieties of the SirReal scaffold (Scheme S1, ESI†).
For our set of compounds, we first determined potency and selectivity for Sirt2 inhibition using a biochemical fluorescence-based activity assay.51 SirReal2 (1) served as a positive control together with JH-T4 which was included as a positive control for simultaneous inhibition of both Sirt2 activities. The results are summarized in Table 1. Compound 7 could be confirmed as a negative control, as it showed a more than 1000-fold decreased potency compared to the positive control SirReal2 (1). All our compounds showed selective Sirt2 inhibition over Sirt1 and Sirt3. As we determined IC50 values higher than 100 μM for Sirt1 and Sirt3, the selectivity of our most potent inhibitors (2, 8–12) is at least 1000-fold as compared to the other two class I sirtuins (Sirt1, Sirt3) and HDAC1 as well as HDAC6. Consistent with literature, JH-T4 also inhibited Sirt1 with a ∼4-fold higher IC50 value compared to Sirt2.40 Regarding inhibition of Sirt2-catalyzed deacetylation, compounds 8–12 with the modifications on the triazole ring, resulted in the most potent Sirt2 inhibition, with sub-micromolar IC50 values ranging from 0.11 to 0.17 μM. Compounds 5 and 6, where the pyrimidine moiety was replaced by a hydrophobic motif, showed decreased Sirt2 inhibition compared to SirReal2 (1). However, it should be noted that 5 still exerts a selective inhibition of Sirt2-mediated deacetylation in the sub-micromolar range. This indicates that the fatty acid moiety of 5 is able to bind to the selectivity pocket of Sirt2, thereby leading to a potent and selective Sirt2 inhibition. As previously reported, the naphthyl-based compounds 3 and 4 strongly differ in their effects on Sirt2-catalyzed deacetylation. 3 shows an improved inhibition of Sirt2-mediated deacetylation compared to SirReal2, whereas the activity of 4 is strongly reduced.52 This indicates a high impact of substitutions at the naphthyl moiety on potency. The control compound JH-T4 revealed sub-micromolar inhibition of Sirt2-mediated deacetylation which was slightly weaker than for compounds 8–12 but more potent compared to SirReal2.
| Compound | Sirt1 | Sirt2 | Sirt3 | HDAC1 | HDAC6 | |
|---|---|---|---|---|---|---|
| Deacet. | Deacet. | Demyr. | Deacet. | Deacet. | Deacet. | |
| SirReal2 | >100 | 0.44 ± 0.08 | 30% | >100 | >100 | >100 |
| 2 | >100 | 0.12 ± 0.01 | 2.5 ± 0.2 | >100 | >100 | >100 |
| 3 | >100 | 0.26 ± 0.03 | 16.2% | >100 | >100 | >100 |
| 4 | >100 | 45.7 ± 10.8 | 8.8% | >100 | >100 | >100 |
| 5 | >100 | 0.46 ± 0.21 | 23.2% | >100 | >100 | >100 |
| 6 | >100 | 6.0 ± 3.7 | 16.8% | >100 | >100 | >100 |
| 7 | >100 | 620 ± 72 | >100 | >100 | >100 | >100 |
| 8 | >100 | 0.16 ± 0.02 | 3.9 ± 0.2 | >100 | >100 | >100 |
| 9 | >100 | 0.17 ± 0.02 | 2.0 ± 0.3 | >100 | >100 | >100 |
| 10 | >100 | 0.15 ± 0.01 | 1.1 ± 0.2 | >100 | >100 | >100 |
| 11 | >100 | 0.11 ± 0.005 | 4.2 ± 0.1 | >100 | >100 | >100 |
| 12 | >100 | 0.12 ± 0.01 | 2.4 ± 0.1 | >100 | >100 | >100 |
| JH-T4 | 1.1 ± 0.4 | 0.29 ± 0.01 | 0.80 ± 0.08 | >100 | n.t. | n.t. |
After confirmation of selective Sirt2 inhibition, we used our previously published fluorescence-based assay,53 in order to evaluate inhibition of Sirt2 demyristoylation activity. We were excited to observe additional inhibition of Sirt2-mediated demyristoylation for a subset of our compounds. For our set of compounds, most potent inhibition of Sirt2-catalyzed demyristoylation was evoked by the triazole-based compounds 2 and 8–12 with IC50 values in the low micromolar range. In agreement with previously published data,40,54 SirReal2 only exerted a very weak inhibition of Sirt2-mediated deacylation (30% @20 μM) in our assay, whereas JH-T4 blocked Sirt2-catalyzed demyristoylation with an IC50 value in the sub-micromolar range (IC50 = 0.80 ± 0.08 μM). Inhibition effects of 3, 4, 5 and 6 were weaker than for SirReal2.
These results indicate that (i) the 4,6-dimethylpyrimidine moiety is highly important for the Sirt2 affinity of SirReal analogues by anchoring the inhibitors in Sirt2's selectivity pocket (ii) the modified triazole moiety further improves Sirt2 affinity and leads to simultaneous inhibition of both acetylated and acylated (myristoylated) substrates.
As the simultaneous inhibition of Sirt2 activities went along with increased potency of our inhibitors, a higher Sirt2 affinity induced by additional interactions between the inhibitor and the enzyme might be the key for simultaneous inhibition. We performed kinetic analyses for our acetylated (ZMAL) and myristoylated (ZMML) substrates of the Sirt2 assay. In agreement with literature,40,42,46,54–56 we observed different substrate binding affinities as we obtained KM = 510 ± 95 μM for ZMAL and KM = 6.7 ± 1.0 μM for ZMML. (see Fig. S1 and Table S1, ESI†) Hence, the increased potency of compounds 8–12 presumably allows these inhibitors to better compete with the myristoylated substrate for Sirt2 binding. However, as Sirt2 conformations differ upon binding of the myristoylated or acetylated substrate,56 other reasons cannot be ruled out completely.
Docking study
To rationalize the determined in vitro data, we docked the inhibitors into available X-ray structures of Sirt2 complexed with various SirReals (see Experimental section for details). All inhibitors under study (except the negative control 7) could be docked to the Sirt2 selectivity pocket in agreement with our previous docking studies on Sirt2.38,52 Docking results obtained for PDB ID 5DY538 showed that in case of the SirReal2 (1) derivatives 3 and 4 with an additional methyl group next to the mercaptopyrimidine and a naphthyl-based aromatic system gave a similar binding mode as SirReal2 in its crystal structure (PDB ID 4RMH) but showing weaker interaction energies (Table S2, ESI†). In the case of compound 4, the methoxy substituted naphthyl ring no longer fits well in the binding pocket due to steric effects and is unable to occupy the position of the naphthyl ring of SirReal2 in its crystal structure (Fig. S2, ESI†). The results showed further that the triazole of the potent inhibitors 8–12 forms hydrogen bonds with Arg97 of the cofactor binding loop that is also observed for the co-crystallized triazole probe in PDB ID 5DY5. The N-substituents of the triazole moiety, either aromatic phenyl (11) or benzyl (8–10, 12) rings, interact with an aromatic pocket at the entrance of the acetyl-lysine channel formed by Phe119, His187, and Phe235 (Fig. 3A). In case of 12 the terminal morpholine ring interacts with the backbone of Glu237 located at the surface of Sirt2 (Fig. 3B).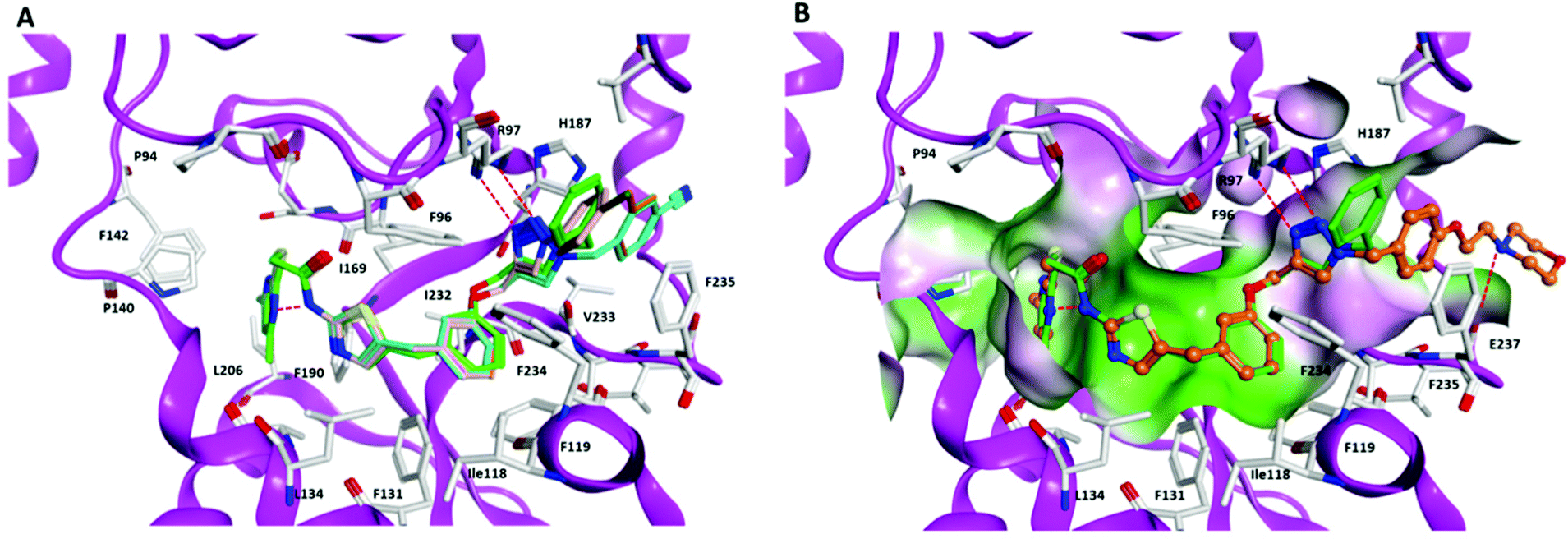 | ||
| Fig. 3 Docking poses of inhibitors 8–12 in Sirt2 (PDB ID 5DY5). (A) Interaction of inhibitors 8–11 at the Sirt2 binding pocket (8 brown, 9 orange, 10 cyan, 11 salmon, triazole–SirReal 5DY5 green). (B) Interaction of inhibitor 12 (colored orange) at the Sirt2 binding pocket (molecular surface colored according to the hydrophobicity: polar regions colored magenta, hydrophobic regions colored green). The co-crystallized triazole–SirReal is colored green. Hydrogen bonds are shown as red dashed lines. | ||
To correlate the docking results with the Sirt2 binding of our compounds, we performed in vitro differential scanning fluorimetry (DSF, also referred to as thermal shift assay (TSA)). In general, the determined thermal shift ΔTM values and the calculated MM-GBSA interaction energies are in good agreement (Table S2, ESI†) indicating that the modelled Sirt2-inhibitor complexes are able to explain the differences in binding.
Effects on cellular viability and proliferation of different cancer cells
After confirming potent and selective Sirt2 inhibition of our set of compounds in vitro, we proceeded with the most promising compounds to cellular experiments and investigated their anticancer effects. SirReal2 with its well-characterized cellular activity37,48 was used as a reference compound for our cellular studies. First, we assessed cytotoxicity by performing MTS assays with human embryonic kidney cells (HEK293T) and different cancer cell lines: human gastric cancer cells (HGC-27), breast cancer cells (MCF-7), acute promyelocytic leukaemia cells (HL-60) and metastatic androgen-independent prostate cancer cells (PC-3M-luc). Due to limited compound solubility, tests were performed at a maximum concentration of 25 μM. For most of the tested compounds, we were therefore unable to obtain complete inhibition curves and, hence, could not determine GI50 values. Thus, we compared inhibition of cell viability at compound concentrations of 10 or 25 μM.As already reported for other Sirt2 inhibitors in different cancer cell lines, the observed effects of our compounds on metabolic activity were rather modest and usually in the micromolar range (Fig. 4A; Fig. S3 and Table S3, ESI†).11,48 For our Sirt2 inhibitors, we could observe distinct differences between the cell lines, which is also in accordance with literature, as Sirt2 is known to have a different impact on different pathways depending on the cell line.3,22 Most pronounced effects for our Sirt2 inhibitors were detected in HGC-27 and HEK293T cells. Only weak or no effects on cell viability were obtained for HL-60, MCF-7 and PC-3M-luc cells. Consistent with our in vitro results, the most potent compounds of our set of Sirt2 inhibitors were 10 and 12 with GI50 values in the low micromolar range for HGC-27 (10: 7.85 ± 0.73 μM and 12: 8.21 ± 0.59 μM) and HEK293T cells (10: 5.87 ± 0.36 μM and 12: 7.70 ± 0.71 μM). A decrease in cell viability in these two cell lines could also be observed for 2, 5, 9 and 11 whereas SirReal2 (1), 6 and 8 exerted only very weak effects on cell viability. Our negative control 7 did not affect cell viability in any of the tested cell lines.
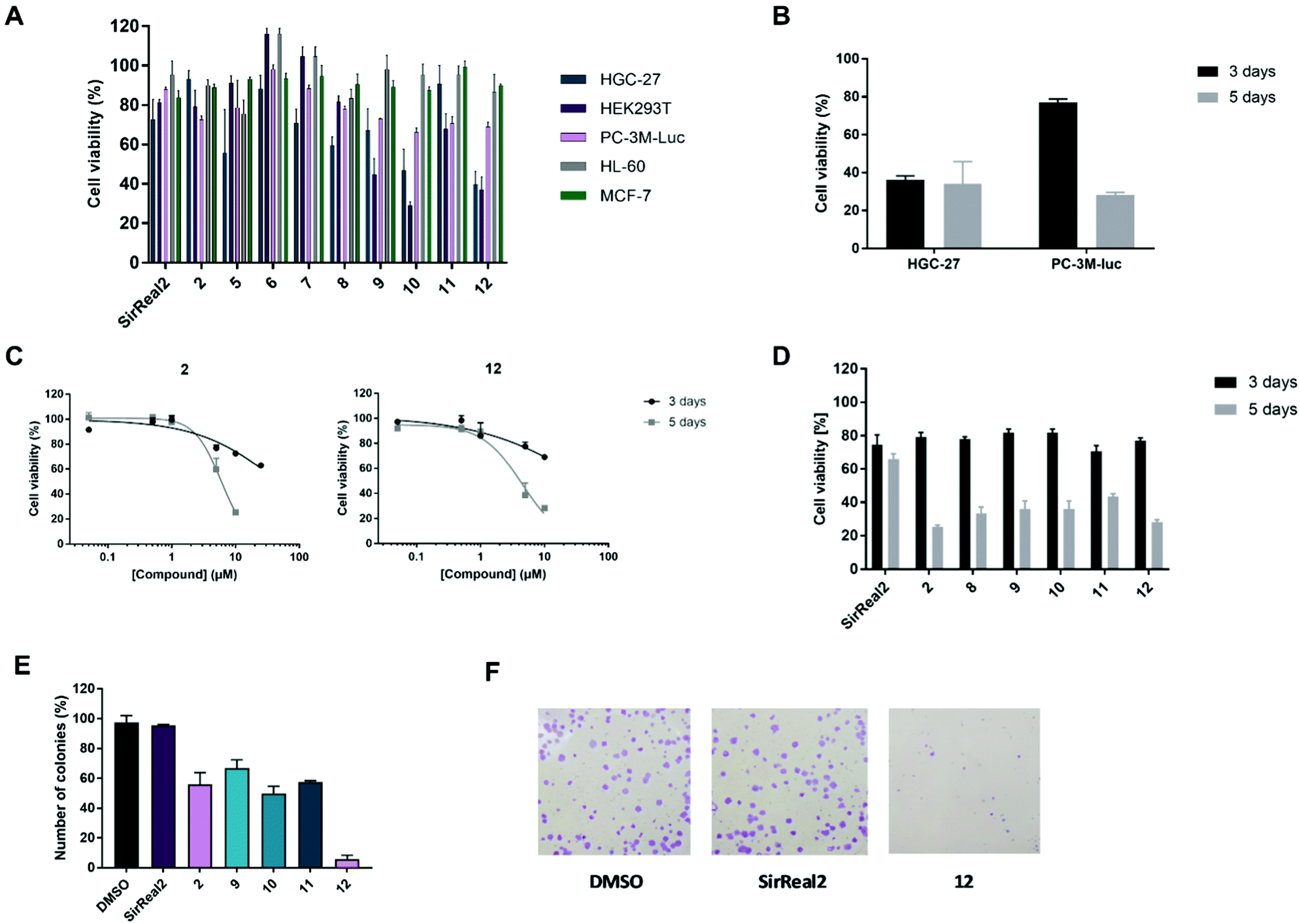 | ||
| Fig. 4 Results of MTS and CFU assays. Values are presented as mean ± SD. MTS data are obtained from two independent experiments performed as triplicates. (A) Results of the MTS assay for different Sirt2 inhibitors (10 μM) in HGC-27, HEK293T, PC-3M-luc, HL-60 and MCF-7 cells. (B) Comparison of cell viability of HGC-27 and PC-3M-luc cells after treatment with 12 (10 μM) and an incubation time of 3 (dark grey) or 5 days (bright grey). (C) Concentration-dependent cell viability curves of PC-3M-luc cells for 2 (left) and 12 (right) after 3 (dark grey) and 5 days (bright grey) of treatment. (D) Comparison of cell viability of PC-3M-luc cells for different Sirt2 inhibitors (10 μM) after 3 (dark grey) and 5 days (bright grey) of treatment. (E) Relative number of colonies of PC-3M-luc cells after treatment with Sirt2 inhibitors (25 μM) or DMSO (n ≥ 2). (F) Pictures of PC-3M-luc colonies after treatment with DMSO (left), SirReal2 (middle) or 12 (right). | ||
Next, we aimed to study if the effects of our Sirt2 inhibitors on cell viability are dependent on the incubation time. Therefore, we prolonged the incubation from 3 days to 5 days. As shown for compound 12 in Fig. 4B, higher effects on the cell viability of PC-3M-luc cells could be observed after prolonged incubation time. In contrast, effects for HGC-27 cells did not depend on the incubation time, thereby indicating that the prostate cancer cells might be able to escape from the effects of the Sirt2 inhibitors for a certain time before cells are affected and cell viability decreases (Fig. S4, ESI†). We could further observe that the time-dependence in PC-3M-luc cells is most pronounced for our potent inhibitors of Sirt2-mediated defatty-acylation (2, 8–12), whereas effects of selective inhibition of Sirt2-catalyzed deacetylation by SirReal2 were weak and similar after 3 and 5 days of treatment (Fig. 4C and D).
Encouraged by the pronounced effects of our Sirt2 inhibitors on cell viability of metastatic prostate cancer cells (PC-3M-luc) and due to the low number of other studies investigating Sirt2 inhibition in prostate cancer, most of the following cellular experiments were focused on the metastatic, androgen-independent prostate cancer cell line PC-3M-luc.
As colony formation of MCF-7 breast cancer cells has been reported to be decreased after treatment with Sirt2 inhibitors,39 we continued with colony formation assays in the prostate cancer cell line PC-3M-luc. Our compounds showed concentration-dependent inhibition of colony formation with the strongest effects for 12 (Fig. 4E; Table S4, ESI†). Colony formation was almost completely prevented by 12 at a concentration of 25 μM. Compounds 2 and 9–11 also reduced colony formation but only to a lower extent compared to 12. SirReal2 did not inhibit colony formation under our test conditions.
After revealing general effects on cell viability and colony formation, we went on with further cellular studies and investigated the effect of our Sirt2 inhibitors on cancer cell migration and the levels of the oncoprotein c-Myc in metastatic androgen-independent prostate cancer.
Inhibition of migration and degradation of c-Myc
Migration is a hallmark of many cancer types and is usually associated with tumour aggressiveness, invasiveness, and poor prognosis. Sirt2 has been shown to contribute to migration in different cancer cells, including breast, gastric, liver and lung cancer and, consequently, treatment with Sirt2 inhibitors resulted in reduced migration.11,30,42,57 We therefore investigated inhibitory effects of our Sirt2 inhibitors on prostate cancer cell migration.As depicted in Fig. 5A, most of our inhibitors showed potent inhibition of migration in the PC-3M-luc cell line. At a compound concentration of 10 μM, 8–12 and 2 showed significant reduction of cell migration and almost completely prevented cells from migrating. Compound 5 revealed 35% of migration inhibition while SirReal2 showed only weak effects. 2, 10 and 12 were selected to determine concentration-dependent effects. The results confirmed a concentration-dependent inhibition of migration for all three compounds (Fig. 5B), which further supported that the effect is Sirt2-dependent.
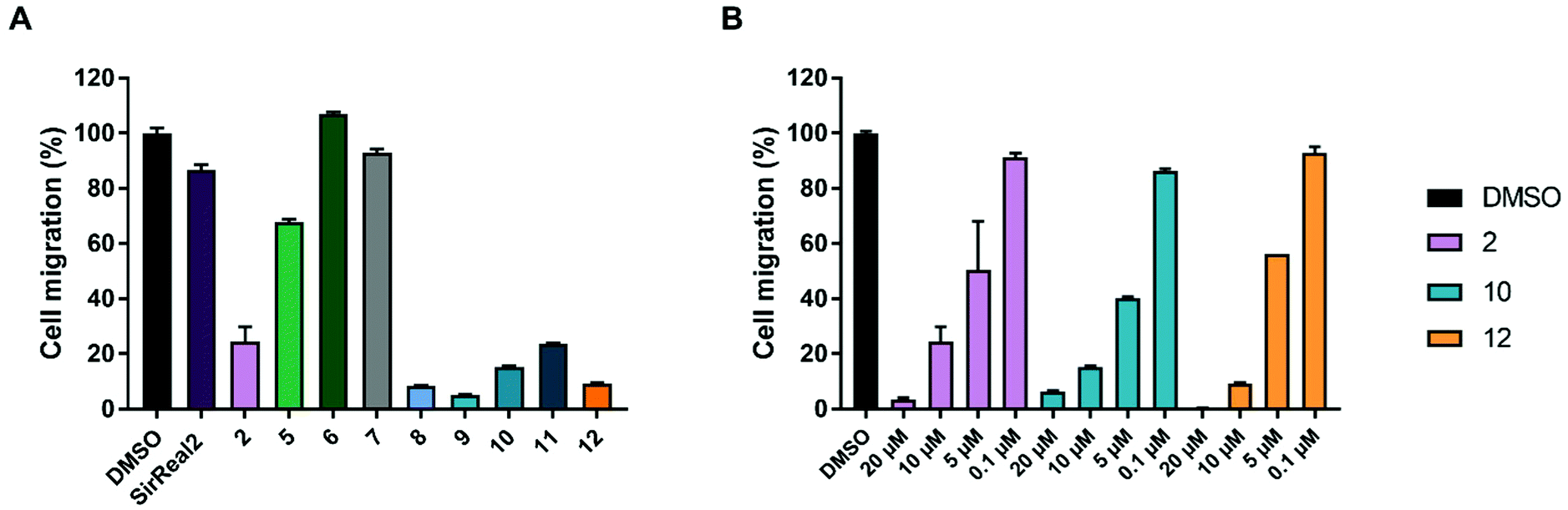 | ||
| Fig. 5 Inhibition of migration in PC-3M-luc cells. Values are presented as mean ± SD. (n = 4) (A) Cell migration after treatment with 10 μM Sirt2 inhibitor or DMSO. (B) Concentration-dependent effect on cell migration after treatment with 2, 10, 12 or DMSO. | ||
We next focused on the oncogene c-Myc, which is a member of the MYC gene family and is involved in the regulation of cell proliferation, cell growth, differentiation, cellular motility, and apoptosis. c-Myc is dysregulated in the majority of human tumours and plays an essential role for tumour pathogenesis.58,59 Previous studies showed that Sirt2 inhibition promotes proteasomal degradation of c-Myc in breast cancer cells.60 As c-Myc has also been reported to be overexpressed and act as a driver of cancer cell proliferation and metastasis in prostate cancer,61–63 we investigated c-Myc levels in the PC-3M-luc cell line upon treatment with our Sirt2 inhibitors.
In metastatic prostate cancer cells, we were able to show a reduction of c-Myc levels upon treatment with our Sirt2 inhibitors. First, we detected c-Myc protein levels after 24 hours of treatment with 10 μM of Sirt2 inhibitor. We detected the strongest effects on c-Myc levels for 2 and 8–12 (Fig. 6A).
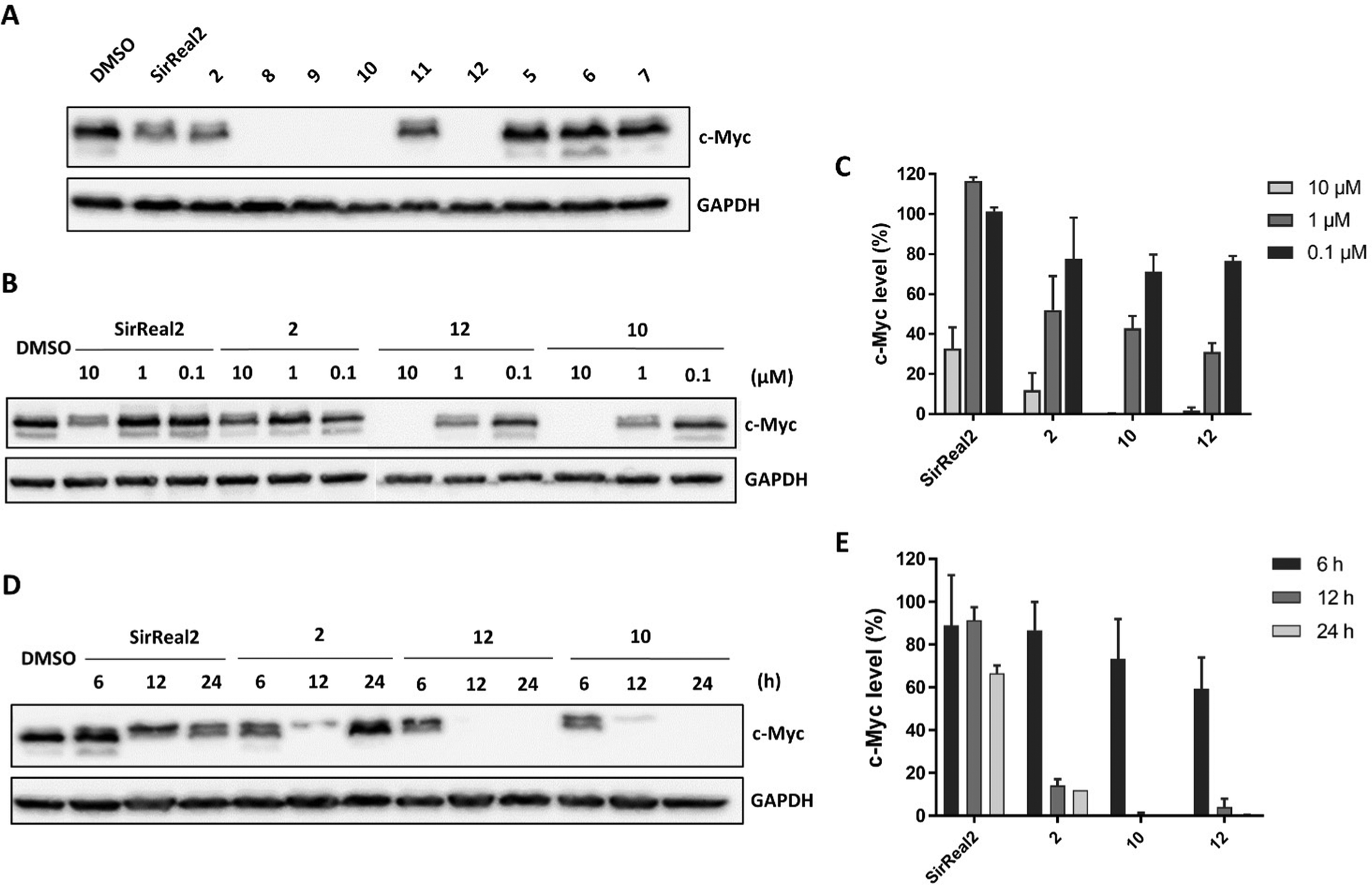 | ||
| Fig. 6 Sirt2 inhibitors induce degradation of c-Myc in PC-3M-luc cells. (A) Immunoblot for c-Myc protein levels after treatment with 10 μM of Sirt2 inhibitors or DMSO for 24 h. (B) Immunoblot for c-Myc protein levels after treatment with different concentrations of Sirt2 inhibitor or DMSO for 24 h. (C) Quantification of concentration-dependent effects on c-Myc protein levels after 24 h of treatment with Sirt2 inhibitor compared to DMSO treated cells. Values are presented as mean ± SD (n = 3). (D) Immunoblot for c-Myc protein levels after treatment with 10 μM Sirt2 inhibitor or DMSO for different periods of time. (E) Quantification of time-dependent effects on c-Myc protein levels of Sirt2 inhibitors compared to DMSO treated cells. Values are presented as mean ± SD (n = 3). | ||
For compounds 2, 10, 12 and SirReal2 we were further able to show that levels of c-Myc were reduced in a concentration-dependent manner (Fig. 6B and C). c-Myc was completely absent after treatment with 12 and 10 at a concentration of 10 μM. SirReal2 and 2 evoked weaker effects on c-Myc protein levels. We continued by investigating the time-dependent effects of our Sirt2 inhibitors on c-Myc degradation. c-Myc protein levels were already significantly reduced after 6 and 12 hours and reached a minimum after 24 hours of treatment for all four inhibitors (Fig. 6D and E). Interestingly, levels increased again after 48 hours (Fig. S5, ESI†). We hypothesize that this might be due increased re-expression of c-Myc. Finally, we investigated whether the degradation was proteasome-based by co-incubating cells with the proteasome inhibitor MG132. Indeed, we could (partially) rescue c-Myc levels by MG132 co-treatment and, thus, confirmed a proteasome-dependent degradation mechanism for c-Myc (Fig. S6, ESI†).
By comparing the results of the c-Myc degradation and the inhibition of migration, we noticed that the compounds inhibiting the cell migration most potently, also led to the lowest levels of c-Myc. Plotting c-Myc levels against the inhibition of migration revealed that both effects are correlating (Fig. S7, ESI†). This indicates an important role of c-Myc for the migration of PC-3M-luc cells and that the degradation of c-Myc, induced by Sirt2 inhibitors might, at least in part, contribute to the inhibiting effects on cell migration in metastatic, androgen-independent prostate cancer cells.
Inhibition of Sirt2 deacetylation and defatty-acylation in cells
After observing the different anticancer effects of our Sirt2 inhibitors, we were curious whether we could correlate them with cellular inhibition of Sirt2. Therefore, we continued our cellular studies on a more mechanistic level by evaluating inhibition of Sirt2 deacetylation and defatty-acylation activity in cells.α-Tubulin is a well-known substrate of Sirt2-mediated deacetylation8 and tubulin hyperacetylation has widely been used as functional readout to study inhibition of Sirt2 deacetylation in cells. Based on our aforementioned promising results with the androgen-independent metastatic prostate cancer cell line, we went on with determining the effects of our new Sirt2 inhibitors on tubulin hyperacetylation using this cell line. PC-3M-luc cells were treated for 5 hours with 20 μM of the respective Sirt2 inhibitor, before immunostaining and assay readout by immunofluorescence microscopy. Acetylation levels of α-tubulin were elevated after the treatment with 2, 10 and 12 (Fig. 7; Fig. S8, ESI†). Only weak effects could be observed for SirReal2 and our negative control 7. The observed changes in cellular tubulin acetylation are consistent with the in vitro data for inhibition of Sirt2-mediated deacetylation (Table 1).
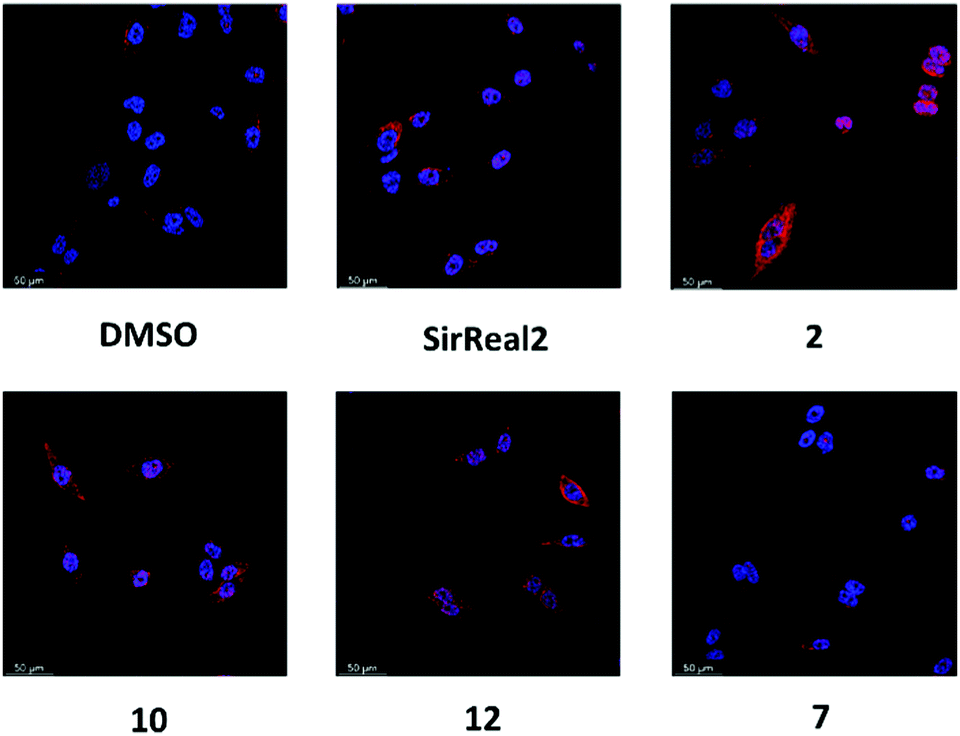 | ||
| Fig. 7 Cellular target engagement studies for Sirt2 inhibitors by determining the α-tubulin acetylation levels. PC-3M-luc cells were treated with 20 μM of Sirt2 inhibitor or DMSO (vehicle) for 5 hours before imaging. (n = 3) The images show acetylation levels of α-tubulin in red and the DAPI-stained nuclei in blue. | ||
After confirming the inhibition of Sirt2 deacetylation, we aimed to investigate inhibition of Sirt2 defatty-acylation activity in cells as well. For these studies, we selected our most potent Sirt2 inhibitor 12, which showed simultaneous inhibition of Sirt2-catalyzed deacetylation and deacylation (demyristoylation) in vitro (Table 1) and revealed the most potent effects in our cellular studies. We chose SirReal2 as negative control as it showed only weak inhibition of Sirt2 deacylation activity in vitro. The Sirt2 inhibitor JH-T4 was used as positive control as it was previously described as an inhibitor of Sirt2-mediated defatty-acylation with cellular activity.40 According to a previously published procedure,40 we investigated changes of the acylation level of the small GTPase KRas4a, a known target of Sirt2 defatty-acylation, after the treatment of HEK293T cells with the different Sirt2 inhibitors. As shown in Fig. 8, compound 12 led to increased acylation levels of KRas4a compared to DMSO treated cells, which confirmed its ability to inhibit Sirt2 defatty-acylation in cells. Effects were as strong as for the positive control JH-T4. As expected, SirReal2 did not show an effect.
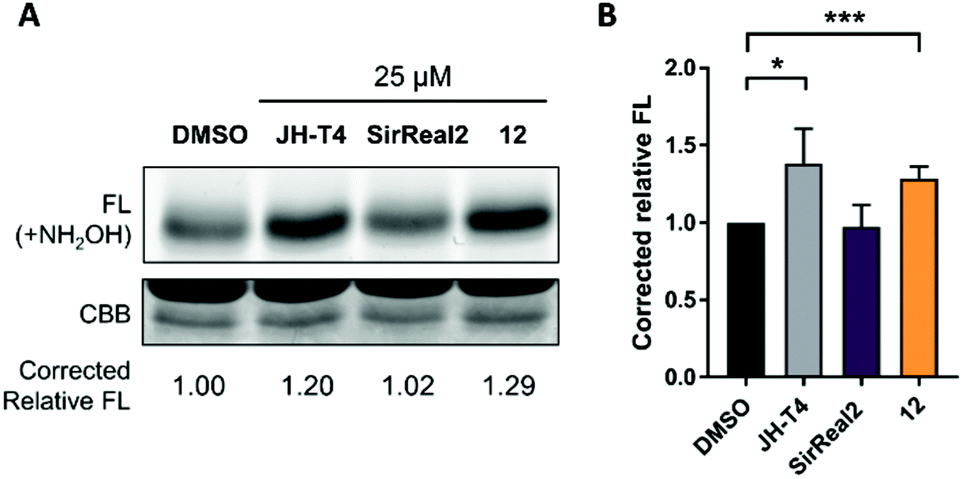 | ||
| Fig. 8 Investigation of the inhibition of Sirt2 defatty-acylation activity in cells. (A) Detection of KRas4a fatty acylation levels after treatment with Sirt2 inhibitors (25 μM) or DMSO (n = 4). FL: Fluorescence, CBB: Coomassie blue. (B) Quantified relative fluorescence signals of the Sirt2 inhibitors and the DMSO control. Error bars represent mean ± SD. Statistical evaluation was performed using a Student's t test. *p < 0.05, ***p < 0.001. | ||
After confirming the simultaneous inhibition of Sirt2 activity in cells by our Sirt2 inhibitor 12, we decided to round up our cellular experiments by investigating the cellular target engagement of our Sirt2 inhibitors.
Investigation of cellular target engagement
As described above, α-tubulin hyperacetylation is used to confirm cellular inhibition of Sirt2 deacetylation and, hence, is often considered as an indirect readout of cellular Sirt2 target engagement. However, as acetylation levels of α-tubulin are essential for cell morphology and cellular functions, its regulation must be adaptive and underlies several different mechanisms. Hence, α-tubulin acetylation is not only influenced by Sirt2 activity but also by other factors such as HDAC6 activity, oxidative stress or high glucose levels.64,65 For this reason, the effects of certain compounds on tubulin acetylation must not necessarily be a consequence of cellular Sirt2 inhibition. Additionally, the overall effect of selective Sirt2 inhibition on tubulin acetylation is not highly pronounced, as upregulation of tubulin acetylation via Sirt2 inhibition can be partially counteracted by HDAC6-mediated tubulin deacetylation. In order to provide a method that allows a more accurate determination of Sirt2 target engagement in a cellular environment, we decided to design and develop a cellular NanoBRET assay for Sirt2, which was not available so far.The NanoBRET technology is proximity-based and relies on bioluminescence resonance energy transfer (BRET) from a donor (Nanoluciferase (Nluc)-labelled fusion protein) to an acceptor (e.g. fluorescently labelled ligand). The energy transfer is enabled by the overlap of the excitation spectrum of the acceptor with the bioluminescence spectrum of the donor Nluc (see Fig. S9, ESI†). In a displacement setup, binding of unlabelled small molecule ligands to the targeted binding site can be detected via the displacement of the fluorescent ligand (tracer), which results in a reduced BRET signal.66 In contrast to the other methods that have already been applied to study cellular Sirt2 target engagement, NanoBRET assays can be performed in a microtiter plate format following a straightforward homogeneous assay protocol, which does not require any antibodies or washing steps. The assay readout can be performed with a plate reader in a highly accurate and high-throughput manner. Design and principle of our Sirt2 NanoBRET assay are illustrated in Fig. 9C. To obtain a BRET donor, we fused the small Nanoluciferase (Nluc) to our target protein Sirt2. As a tracer we used a cell-permeable TAMRA-labelled SirReal2 (13, Fig. 9B), which has already been published by our group as a tool compound for the development of an in vitro Sirt2 binding assay based on fluorescence polarization and had shown cellular permeability.67
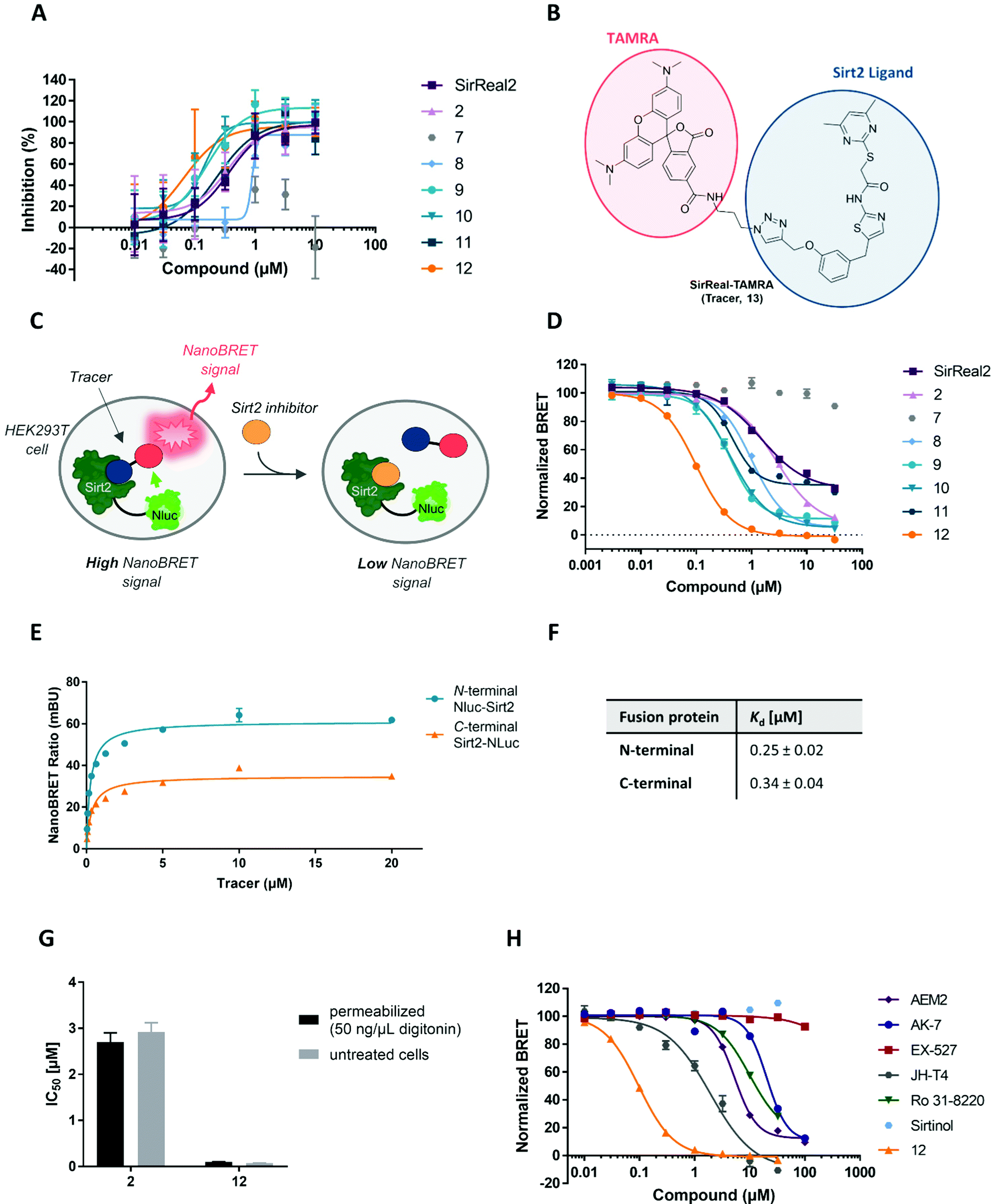 | ||
| Fig. 9 Development of a NanoBRET assay for Sirt2 as a new method to study cellular target engagement for Sirt2. (A) Representative IC50 curves from in vitro fluorescence polarization assay (FP assay). (B) Structure of the SirReal-based tracer (13) consisting of a Sirt2 binding ligand (blue) and a fluorophore (TAMRA, red) connected via a linker. (C) Schematic illustration of the design of our NanoBRET assay for Sirt2. A Nanoluciferase (Nluc)-tagged Sirt2 fusion protein is expressed in HEK293T cells. Upon tracer binding to Sirt2, resonance energy is transferred from Nluc to the tracer fluorophore, which results in a fluorescence emission. The addition of a Sirt2 inhibitor prevents tracer binding to Sirt2 and leads to a decreased fluorescence signal. (D) NanoBRET assay curves displaying the relative affinity of our Sirt2 inhibitors in HEK293T cells. Two independent experiments were performed as triplicates. (E) Saturation binding curves of the tracer for the N-terminal (blue) and the C-terminal (orange) fusion protein. (F) Kd values (mean ± SD) obtained for the N- and C-terminal fusion protein. (G) Comparison of IC50 values (mean ± SD) determined for 2 and 12 in permeabilized (digitonin treatment) and untreated cells. (H) NanoBRET assay curves displaying the relative Sirt2 affinity of different published Sirt2 inhibitors and 12. | ||
Before we started with the assay development in cells, we performed an in vitro fluorescence polarization assay (FP assay) to investigate and compare Sirt2 binding to the selectivity pocket and confirm the suitability of the tracer to determine Sirt2 binding of our set of inhibitors. As depicted in Fig. 9A the Sirt2 inhibitors differed in their Sirt2 binding. IC50 values are presented in Table S5 of the ESI.† Compound 12 showed most potent binding (IC50 = 0.07 ± 0.02 μM). For the compounds 8–11 we also obtained lower IC50 values compared to SirReal2. 5, 6 and 7 as well as 3 and 4 showed weaker binding. As already published by our group, 2 showed similar binding in the FP assay compared to SirReal2.67
Next, we went on to develop the NanoBRET assay in cells. First, we transiently transfected HEK293T cells with a vector either encoding for an N- or C-terminally Nluc-tagged Sirt2. This allowed us to study which of the fusion proteins is more suitable for the development of cell-based Sirt2 target engagement assays. In saturation binding experiments, both fusion proteins led to a tracer-dependent increase of the NanoBRET signal (Fig. 9E). The corresponding Kd values were both in the sub-micromolar range with a Kd = 0.25 ± 0.02 μM for the N-terminally and Kd = 0.34 ± 0.04 μM for the C-terminally labelled fusion proteins (Fig. 9F). This is in good agreement with a previously reported Kd value of the fluorescent tracer (Kd = 0.16 μM) that was determined by means of the in vitro fluorescence polarization assay.67 Furthermore, the obtained results from our NanoBRET assay indicate that neither N-terminal nor C-terminal fusion of Nluc to Sirt2 had a significant impact on ligand binding properties and protein folding of Sirt2. Since the N-terminally labelled Nluc-Sirt2 fusion protein showed higher BRET signals and a lower Kd value, compared to the C-terminally labelled Nluc-Sirt2, we chose the N-terminally labelled construct for further investigations. Next, we evaluated cellular permeability of the tracer by treating the cells with the non-ionic detergent digitonin (50 μg mL−1). Digitonin only disrupts the plasma membrane, while mitochondrial and nuclear membranes remain intact.68 As we detected highly similar binding curves for digitonin permeabilized and non-permeabilized cells, we confirmed the good cellular permeability of our tracer (Fig. S10, ESI†).
After having shown that the binding of our fluorescent tracer (13) to N-terminally labelled Nluc-Sirt2 can be monitored via NanoBRET, we were curious, if we could use our method in order to study cellular target engagement of Sirt2 inhibitors. For our displacement setup, HEK293T cells were transiently transfected with the N-terminal fusion protein and treated with the fluorescent tracer 13 (2 μM) in the presence of varying concentrations of the unlabelled competitors. The results of the cellular target engagement studies are presented in Fig. 9D, H and Table 2. Under the applied conditions, 12 showed the highest binding affinity (IC50 = 0.098 ± 0.004 μM) followed by 9 (IC50 = 0.40 ± 0.03 μM) and 10 (IC50 = 0.41 ± 0.03 μM). Compound 2 and SirReal2 revealed similar target engagement in the low micromolar range. For the compounds 5 and 7, we did not detect cellular Sirt2 binding under the applied conditions. Additionally, we calculated the corresponding Ki values of our compounds according to the Cheng–Prusoff equation (Table 2).69 In contrast to IC50 values that are highly dependent on the used assay setup, Ki values are independent of applied assay conditions and can therefore be compared between different assay systems. For our most potent compound and novel lead structure 12, we also performed a linearized Cheng–Prusoff analysis to confirm the low Ki value and further explore the opportunities of our NanoBRET assay. Dose-titration experiments with varying concentrations of 12 in the presence of different concentrations of the fluorescent tracer (13) yielded a Ki value of 0.005 ± 0.003 μM, which again confirmed the very high Sirt2 affinity of 12. (Fig. S11, ESI†) Furthermore, we permeabilized cells with digitonin for 2 and 12, to investigate if membrane permeability affects cellular target engagement of our inhibitors. None of the tested compounds revealed significant differences in the NanoBRET signals between permeabilized (digitonin) and untreated cells (Fig. 9G; Fig. S10, ESI†). The obtained results suggest that for these compounds not cellular permeability, but, indeed, Sirt2 affinity is the driver for cellular target engagement.
| Compound | In vitro Sirt2 activity assay | NanoBRET assay | Compound | In vitro Sirt2 activity assay | NanoBRET assay | ||
|---|---|---|---|---|---|---|---|
| IC50 [μM] | IC50 [μM] | K i value [μM] | IC50 [μM] (ref) | IC50 [μM] | K i value [μM] | ||
| a For the calculation we used the tracer concentration in the medium as this is a common procedure in literature.74–76 | |||||||
| SirReal2 | 0.44 ± 0.08 | 43%@10 μM | — | AEM2 | 3.8 (70) | 5.2 ± 0.2 | 0.58 ± 0.02 |
| 2 | 0.12 ± 0.01 | 2.7 ± 0.1 | 0.30 ± 0.01 | AK-7 | 15.5 (71) | 20 ± 1.6 | 2.3 ± 0.2 |
| 7 | 624 ± 71.9 | n.i. | — | EX-527 | 32.6 (72) | n.i. | — |
| 8 | 0.16 ± 0.02 | 1.0 ± 0.05 | 0.11 ± 0.005 | JH-T4 | 0.29 ± 0.01 | 1.9 ± 0.2 | 0.28 ± 0.06 |
| 9 | 0.17 ± 0.02 | 0.40 ± 0.02 | 0.04 ± 0.002 | Ro 31-8220 | 0.8 (73) | 9.9 ± 0.6 | 1.1 ± 0.07 |
| 10 | 0.15 ± 0.01 | 0.41 ± 0.02 | 0.05 ± 0.002 | Sirtinol | 38–58 (72) | n.i. | — |
| 11 | 0.11 ± 0.005 | 37%@10 μM | — | ||||
| 12 | 0.12 ± 0.01 | 0.098 ± 0.004 | 0.01 ± 0.005 | ||||
Finally, we investigated the applicability of our new NanoBRET assay for Sirt2 inhibitors that are structurally not based on SirReals. Therefore, we tested a set of published Sirt2 inhibitors. As shown in Fig. 9H, the inhibitors showed different relative Sirt2 affinities in cells, and all revealed a weaker binding compared to our most potent compound 12. The most potent compound of this series was JH-T4 with a submicromolar IC50 value (IC50 = 0.29 ± 0.01 μM). For the Sirt2-selective inhibitors AEM2 and AK-7, we obtained IC50 and Ki values in the (sub)micromolar range for cellular Sirt2 binding. This agrees with their reported in vitro Sirt2 inhibition and elevated acetylation levels of cellular Sirt2 substrates (e.g. p53 and α-tubulin) after treatment with these two compounds.70,77 The Sirt2 inhibitor Ro 31-8220, originally identified as inhibitor of the protein kinase C (PKC),78 also showed cellular Sirt2 affinity in the micromolar range. Due to its additional activity as kinase inhibitor, cellular effects of Ro 31-8220, including α-tubulin hyperacetylation, could also be a consequence of kinase inhibition. Even though we still cannot completely rule out potential off-target effects, our results suggest that Ro 31-8220 acts as Sirt2 inhibitor in cells and the compound-induced tubulin hyperacetylation can be related, at least in part, to inhibition of Sirt2 in cells. Finally, we did not observe cellular Sirt2 binding for EX-527 and Sirtinol. For compound EX-527, this confirms its selectivity for Sirt1 in cells and is consistent with published data where EX-527 does not lead to elevated acetylation levels of α-tubulin.72 In contrast, the dual Sirt1/Sirt2 inhibitor Sirtinol has been reported to induce hyperacetylation of α-tubulin, besides many other cellular effects. However, the hypothesis that Sirt2 inhibition is responsible for the observed cellular effects of Sirtinol has already been questioned by others,79 as Sirtinol might also inhibit other enzymes by aggregation, precipitation or its function as iron chelator.80 The absence of cellular Sirt2 binding of Sirtinol in our assay suggests that off-target effects are responsible for the α-tubulin hyperacetylation after Sirtinol treatment and not Sirt2 inhibition.
With these results, we could confirm the suitability of our NanoBRET assay to study cellular target engagement of Sirt2 inhibitors with different scaffolds. Furthermore, the results highlight the fact that the levels of α-tubulin hyperacetylation are not necessarily a reliable indicator for the potency of cellular Sirt2 inhibition.
Conclusion
Here, we present a set of new SirReal-based, potent and selective inhibitors for Sirt2 with increased Sirt2 affinity compared to SirReal2. Besides inhibiting Sirt2-mediated deacetylation, a subset of our compounds also showed an inhibition of defatty-acylation (demyristoylation) activity in vitro. Effects on cell viability were cell type-dependent with pronounced and time-dependent effects in the metastatic androgen-independent prostate cancer cell line PC-3M-luc. Furthermore, only Sirt2 inhibitors that blocked both deacetylation and long-chain defatty-acylation induced downregulation of the oncoprotein c-Myc and led to an inhibition of cell migration of PC-3M-luc cells. Strongest effects in cells regarding cell viability, downregulation of c-Myc, inhibition of migration and cellular target engagement were evoked by compound 12. The proposed interaction of its basic center with Glu237 at the protein surface might explain the strongest effects in thermal stabilization and cellular target engagement assays. Thus, we have potentially discovered a new key interaction for the development of further potent Sirt2 inhibitors with high cellular efficacy. We chose 12 as our best Sirt2 inhibitor and could show simultaneous inhibition of Sirt2-mediated deacetylation and defatty-acylation in cells, as treatment with 12 led to increased acetylation levels of α-tubulin and increased fatty-acylation levels of KRas4a. In addition to the TM-based PROTACs and the unselective Sirt2 inhibitor JH-T4,40,47 this is the first time that for a potent and selective SirReal-based Sirt2 inhibitor a simultaneous inhibition has been shown in cells. This allows us to conclude that the observed increase in anticancer activity is indeed a consequence of simultaneous inhibition of Sirt2 activity and not caused by any off-target inhibition of other sirtuins isotypes (e.g. Sirt1, Sirt3). Hence, our data strongly suggests that inhibition of both Sirt2-mediated deacetylation and defatty-acylation indeed leads to increased anticancer effects compared to sole inhibition of Sirt2-mediated deacetylation. To further elucidate the role of Sirt2-mediated defatty-acylation, selective inhibitors for the latter would be essential molecular tools, however such compounds have not been published to date.In order to provide a straightforward method for directly studying cellular target engagement for Sirt2, we developed a NanoBRET assay based on a cell permeable fluorescently labelled SirReal-based probe rather than to rely on hyperacetylation of α-tubulin as indirect method. To our best knowledge, this is the first NanoBRET assay reported for NAD+-dependent lysine deacetylases (sirtuins). In contrast to existing methods, our NanoBRET assay is not antibody-based and can accurately be quantified with a plate reader in a high-throughput manner. Specifically, we provide additional evidence of the low on-target specificity of the broadly used Sirt2 probe Sirtinol, which has a great impact on sirtuin chemical biology. Thus, our new Sirt2 NanoBRET assay represents a major advance for the field of Sirt2 inhibitor development, as it allows to put anticancer effects in context with cellular Sirt2 binding.
Experimental section
General experimental information, chemical synthesis, compound characterization data, additional methods, and NMR data of novel compounds are provided in the electronic supplementary information (ESI†).Protein expression and purification
Recombinant human Sirt1134-747 and Sirt256-356 were expressed as described previously.81 Chemically competent E. coli BL21 Star (DE3) cells were transformed with the expression vectors pET30S-hSirt1134-747 or pET30S-hSirt256-356. Cells were grown to an OD600 of 0.6 at 37 °C in 2× YT medium (supplemented with 50 μg mL−1 kanamycin). Overexpression of Sirt1134-747 and Sirt256-356 was induced by IPTG (final concentration 1 mM) and after further cultivation at 20 °C for 12 h cells were harvested by centrifugation (15 min, 5000 g). Cells were resuspended in lysis buffer (100 mM Tris/HCl buffer at pH 8.0, 150 mM NaCl and 10% (v/v) glycerol) and cell lysis was achieved by ultrasonication (Branson Digital Sonifier 250) at 70% amplitude for 10 min (3 s working, 10 s pause). After centrifugation with 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 1 h the supernatant was loaded on a Strep-Tactin Superflow cartridge (5 mL bed volume, IBA Lifescience, Germany). After elution with lysis buffer containing D-desthiobiotin (5 mM, IBA Lifescience, Germany) the proteins were further purified by size-exclusion chromatography (Superdex S200 26/60, GE Healthcare, IL, USA) equilibrated with Tris/HCl buffer (20 mM, 150 mM NaCl, pH 8.0) and concentrated. Purity and identity of the target proteins were verified by SDS–PAGE and protein concentration was determined by BCA-assay using BSA as a standard.
000 g for 1 h the supernatant was loaded on a Strep-Tactin Superflow cartridge (5 mL bed volume, IBA Lifescience, Germany). After elution with lysis buffer containing D-desthiobiotin (5 mM, IBA Lifescience, Germany) the proteins were further purified by size-exclusion chromatography (Superdex S200 26/60, GE Healthcare, IL, USA) equilibrated with Tris/HCl buffer (20 mM, 150 mM NaCl, pH 8.0) and concentrated. Purity and identity of the target proteins were verified by SDS–PAGE and protein concentration was determined by BCA-assay using BSA as a standard.
Sirtuin deacetylation activity assays
Inhibition of Sirt1,2 and 3 activities was determined using a trypsin-based fluorescence assay, previously described by Heltweg et al.82 In black 96-well plates (OptiPlateTM -96F, black, 96 well, Pinch bar design, PerkinElmer, USA) the respective sirtuin (Sirt1134-747, Sirt256-356 or Sirt3118-395) was mixed with 5 μL ZMAL (12.6 mM stock solution in DMSO, 10.5 μM final assay concentration), 3 μL inhibitor in DMSO in varying concentrations or DMSO as a control (final DMSO concentration 5% (v/v)) and filled up to 55 μL with assay buffer (50 mM Tris/HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0). Substrate conversion was adjusted to 15–30% for the DMSO control and a blank control with no enzyme and a 100% conversion control without enzyme but with AMC (12.6 mM stock solution in DMSO, 10.5 μM final assay concentration) instead of ZMAL were performed as well. The enzymatic reaction was started by addition of 5 μL NAD+ (6 mM, final assay concentration 500 μM) and plates were incubated at 37 °C and 140 rpm for 4 h. Addition of 60 μL of a trypsin containing stop solution (50 mM Tris, 100 mM NaCl, 6.7% (v/v) DMSO, 5.5 U μL−1 trypsin, 8 μM nicotinamide, pH 8.0) stopped the catalytic reaction. Further incubation at 37 °C and 140 rpm for 20 min led to release of the fluorophore AMC. Fluorescence intensity was measured using a microplate reader (λEx = 390 nm, λEm = 460 nm, BMG POLARstar Optima, BMG Labtech, Germany). Inhibition was calculated in % in relation to the DMSO control after blank subtraction. IC50 values were determined using OriginPro 9G (OriginLab, USA) or GraphPad 7.0 by a non-linear regression to fit the dose response curve. All experiments were performed at least in duplicates.Sirt2 demyristoylation activity assay
The assay was performed as described for sirtuin deacetylation activity but instead of ZMAL, ZMML, a myristoylated substrate, was used and the Tris buffer was exchanged for a HEPES buffer (25 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.015% Triton X-100, pH 8.0). AMC was used again as 100% conversion control. OriginPro 9.0 and GraphPad 7.0 were used for the analysis of results.HDAC1 and HDAC6 activity assays
The assays were performed according to a previously published procedure.82 OptiPlate-96 black microplates (PerkinElmer) were used. The total assay volume was 60 μL. 52 μL of human recombinant HDAC1 (BPS Bioscience, catalogue no. 50051) or human recombinant HDAC6 (BPS Bioscience, catalogue no. 50006) in incubation buffer (50 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mg mL−1 BSA) were incubated with 3 μL of different concentrations of inhibitors in DMSO and 5 μL of the fluorogenic substrate ZMAL (Z-(Ac)Lys-AMC) (126 μM) for 90 min at 37 °C. After the incubation time, 60 μL of the stop solution, comprising 33 μM trichostatin A (TSA) and 6 mg mL−1 trypsin in trypsin buffer (Tris-HCl 50 mM, pH 8.0, NaCl 100 mM), was added. The plate was incubated again at 37 °C for 30 min, and fluorescence was measured on a BMG LABTECH POLARstar OPTIMA plate reader (BMG Labtechnologies, Germany) with an excitation wavelength of 390 nm and an emission wavelength of 460 nm.Thermal shift assays
Human Sirt256–356 (0.2mgml−1 final concentration) was mixed with inhibitor (25 μM), NAD+ (5 mM) and Sypro Orange (1:4000) in assay buffer (25mM Tris/HCl, 150mM NaCl, 1 mM DTT, 5% (v/v) DMSO, pH 8.0). Fluorescence was monitored during a temperature ramp from 25–95°C (1°Cmin−1) using a Bio-Rad iCycler iQ5 (4titude, FrameStar 96-well plates, 4ti-0771, 4titude qPCR Seal, 4ti-0560). Melting temperatures were determined according to published procedures83 using Graphpad Prism software.
Fluorescence Polarization Assay
A 1.43× concentrated solution containing 286 nM Sirt256–356, (200 nM final concentration) and a 4× concentrated solution containing 160 nM of the fluorescence-labelled probe (13, 40 nM final concentration) were prepared in assay buffer. For negative controls a 20× concentrated solution containing 200 μM of the unlabelled SirReal (12, 10 μM final concentration) was prepared. The assay was performed with a DMSO concentration of 5%. Test compounds were prepared as a 20× dilution series. Blank controls were performed with DMSO instead of the test compound and negative controls (Pneg, complete prevented binding of the fluorescent probe) were performed with an excess of the unlabelled SirReal (12, 10 μM final concentration). Positive controls (Ppos, complete binding of the fluorescent probe) were performed in presence of the fluorescence-labelled probe and the enzyme. Test samples containing the potential ligand (PI) were prepared as follows. 14 μL of the 286 nM solution of Sirt2 (200 nM final concentration) were added to 1 μL of the 20× concentrated solution of the potential ligand and incubated for 10 min at 37 °C. Then, 5 μL of the 160 nM solution of fluorescently labelled probe were added and incubated at 37 °C for another 30 min. Inhibition values (I) were calculated using the following equation: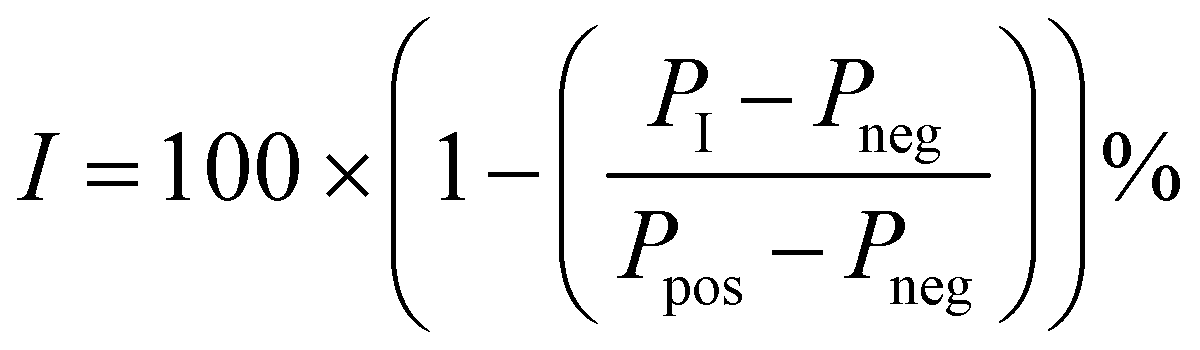
Cell culture
All cell culture media contained 10% (v/v) heat-activated fetal bovine serum (FBS; PAN Biotech GmbH, Aidenbach, Germany), 2 mM glutamine (PAN Biotech GmbH) and 1% penicillin–streptomycin (PAN Biotech GmbH) unless otherwise specified. Human MCF-7, HL-60 and PC-3M-luc cells were grown in RPMI 1640 media (PAN Biotech GmbH). Human HEK293T cells were grown in DMEM media (PAN Biotech GmbH). HGC-27 cells were maintained in DMEM:Ham's 12 medium (1:1) (CLS Cell Lines Service GmbH) supplemented with 5% fetal calf serum (FCS, PanBiotech).
Cell viability assay (MTS Assay)
Cell viability was determined by using the Celltiter 96 AQueous nonradioactive Proliferation Assay (Promega). Cells were seeded in sterile 96-well plates at a density of 2000 cells per well and incubated for 72 h or 120 h at 37 °C and 5% CO2. Compound and vehicle were added to a final concentration of 0.5% DMSO. After 72 or 120 h of incubation time, 20 μL of a mixture (20:1) consisting of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl-2H-tetrazolium) and PMS (phenazine methosulfate) were added to each well. Absorbance was measured after another 2–4 h with a BMG LABTECH POLARstar OPTIMA plate reader (BMG Labtechnologies, Germany). Experiments were performed in triplicates and GI50 values were calculated using the Graphpad Prism software. GI50 was defined as the concentration that led to 50% viable cells.
Colony Forming Unit (CFU) assay
Cells were seeded into six-well plates at 250 cells per well. After 2–3 h at 37 °C and 5% CO2, compound and vehicle were added to a final concentration of 0.5% DMSO. The cells were incubated for 7 days at 37 °C and 5% CO2. Afterwards, cells were washed twice with PBS and fixed with 4% paraformaldehyde (PFA) diluted in demineralized water. After 20 minutes of incubation at RT, wells were washed twice with water and a solution of 0.01% crystal violet was added. After another 30 minutes, cells were washed twice and left drying before counting the number of colonies.Migration assay
PC-3M-luc cells were starved overnight in RPMI-1640 Medium without FCS. Cell migration was monitored using the xCelligence system (Roche, Basel, Switzerland) that measures electrode impedance upon cell attachment to the surface of CIM-plate chambers. PC-3M-luc cells were seeded with a number of 5 × 104 cells into the transwell chamber containing 0% FCS RPMI 1640 in the upper chamber and 10% FCS RPMI 1640 in the lower chamber. Before starting the experiment, inhibitors were added at different concentrations to the cells in the upper chamber. The negative control was performed with 0% FCS RPMI 1640 in both chambers. Cell indices were automatically recorded every 15 minutes for 72 h by the xCelligence system software (Roche). Relative velocities represent the change of the cell index over time.Western Blot analysis
PC-3M-luc cells were seeded into 6-well plates at around 80% confluency. After incubation overnight at 37 °C and 5% CO2, different concentrations of compounds or respective volume of DMSO were added and cells treated for 6, 12, 24, or 48 hours. Next, the cells were washed twice with PBS and lysed with 80 μL lysis buffer (RIPA buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% Sodiumdeoxycholate) supplemented with Protease inhibitor cOmplete (Roche; cat. #4693132001), 2 mM PMSF and PhosSTOP (Roche; cat. #4906845001). Cells were incubated at 4 °C for 30 min on a shaker, collected by scraping and treated with sonication (UP200St with Vial Tweeter, Hielscher Ultrasonics GmbH, Teltow, Germany) for 1 min. Cells were shortly vortexed and pelleted by centrifugation (4 °C, 20 min, 20000 rcf). The supernatant was transferred into a new Eppendorf and BCA assay (Pierce BCA Protein Assay Kit, Thermo Fisher, cat. #23225) performed to determine the protein concentration.
The lysates were resolved by SDS–PAGE in 12.5% Polyacrylamid gels with Tris running buffer (0.25 M Tris, 1.92 M Glycin, 0.5% (m/v) SDS, pH 8.3) and proteins were transferred on a nitrocellulose membrane using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked in 5% milk in TBS-T (TBS + 0.1% Tween-20) for 1 hour at RT. Then, membranes were washed three times with TBS-T (3 × 5 min) and incubated with primary antibody in 3% milk in TBS-T (1:1000) overnight at 4 °C. The membranes were washed three times with TBS-T before addition of horseradish peroxidase (HRP) conjugated secondary antibody diluted in 3% milk in TBS-T (1:5000) for 1 hour at RT. After another three cycles of washing, the proteins were detected in Fusion Xpress using enhanced chemiluminescent reagents (Clarity Western ECL Substrate, Bio-Rad, cat. #1705060). Blots were further analysed with the FusionCapt Advance Software and ImageJ.
Detection of acetylation levels of α-tubulin by immunofluorescence
PC-3M-luc cells (20000 cells per well) were plated in ibidi 8-well slides (Ibidi, cat. #80826) and incubated overnight at 37 °C, 5% CO2. Next, cells were treated with 20 μM of inhibitor. After 5 h, the medium was removed, the cells washed with PBS and fixed with 4% PFA for 8–10 min at RT. Cells were rinsed three times with PBS and lysed with extraction buffer (PBS, 0.1% Triton X-100) for 3–5 min at RT. After another washing step with PBS, blocking buffer (PBS, 0.1% Triton, 5% FCS) was added for at least 10 min before incubating with monoclonal acetylated α-tubulin antibody (1:500, Sigma-Aldrich, cat. #T6793) in blocking buffer overnight at 4 °C. The cells were rinsed three times with blocking buffer and incubated with goat anti-mouse IgG H&L Alexa Fluor 647 (Abcam, #ab150115), diluted 1:2000 in blocking buffer, for 30 min in the dark. The cells were rinsed two times with blocking buffer and once with PBS and DAPI in mounting medium (VECTASHIELD HardSet Antifade Mounting Medium with DAPI, #H-1500-10) diluted 1:50 in PBS was added and it was incubated for 10 min in the dark. Confocal microscopy was performed with a Leica SP8 confocal microscope equipped with a 40×/1.40 oil objective (Leica Microsystems) keeping the laser settings of the images constant to allow direct comparison of signal intensities between images.
In cell inhibition of SIRT2 defatty-acylation of KRas-4a
HEK293T cells stably overexpressing Flag-tagged K-Ras4a were treated with 25 μM SirReal2, 12, JH-T4 or equal volume DMSO (vehicle control) for 14 hours before alkyne treatment. Cell culture media was then changed to media including 50 μM Alk14 and 25 μM of the indicated inhibitors or DMSO and incubated for 6 hours. The cells were washed twice with cold phosphate-buffered saline (PBS), scraped, and collected at 1000 × g for 5min. Cells were then lysed in NP-40 lysis buffer (5mM Tris-HCl pH 7.4, 150mM NaCl, 10% glycerol, and 1% Nonidet P-40) with protease inhibitor cocktail for 30min at 4 °C with rocking. Lysates were centrifuged for 20 min at 4 °C and transferred to a fresh tube. Cleared lysates were incubated with anti-FLAG affinity beads (Sigma) at 4°C for 2h with rocking. The affinity beads were then washed three times with IP wash buffer (25mM Tris-HCl pH 7.4, 150mM NaCl, and 0.2% Nonidet P-40) and then re-suspended in 20μL of IP washing buffer. The click chemistry reaction was performed by adding the following reagents: TAMRA azide (1μL of 2mM solution in DMSO), TBTA (1μL of 10mM solution in DMF), CuSO4 (1μL of 40mM solution in H2O), and TCEP (1μL of 40mM solution in H2O). The reaction was allowed to proceed at room temperature for 30 min. Protein loading dye was added to 2× final concentration and the beads were heated at 95°C for 5min. After centrifugation at 17000 × g for 2min, 5 M hydroxylamine was added to a final concentration of 300 mM and samples were tapped to mix and heated at 95°C for an additional 5min. Samples were run on SDS–PAGE gels and in-gel fluorescence was detected with ChemiDoc MP (BioRad). Protein loading was analysed by staining the gel with Coomassie blue. The quantifications were measured by ImageJ.
Plasmid construction
Vectors from the pNLF1 family of Promega were used according to the manufacturer's protocol to generate fusion proteins for Sirt2 with Nanoluciferase (Nluc) either on the N-terminus or the C-terminus. For the N-terminal fusion, we used the pNLF1-N vector (#N1351) and designed a flexible Gly–Ser–Ser–Gly linker between NLuc and Sirt2. For the C-terminal fusion protein we used the pNLF1-C vector (#N1361) encoding the same linker between Sirt2 and Nluc as for the N-terminal fusion. Instead of the full Sirt2 enzyme, we used a truncated Sirt250-356 for cloning in both vectors.Cell Transfections and BRET measurements
NanoBRET experiments were performed in HEK293T cells. Cells were plated in 6-well plates (Sarstedt, cat. # 83.1839.300) at a density of 8 × 105 cells per well and incubated 2–4 h at 37 °C and 5% CO2 before transfection. The fusion protein plasmids were transfected using Fugene HD Transfection reagent (Promega) according to the manufacturer's protocol. First, 2 μg fusion protein DNA were dissolved in 100 μl medium without serum and phenol red to obtain a concentration of 0.02 μg DNA per μL. Next, Fugene reagent was added to form DNA:Fugene complexes in a ratio of 1:3 and the mix was shortly vortexed and incubated for 15 min at RT. The mix was added dropwise to the HEK293T cells followed by incubation for 20–24 h at 37 °C and 5% CO2. Cells were trypsinized, resuspended in medium without serum and phenol red and adjusted to a concentration of 2 × 105 cells per mL. All compounds were prepared as concentrated stock solutions dissolved in DMSO. For saturation binding experiments, serially diluted tracer was added to the cells in the presence or absence of unlabelled ligand (10 μM 12). Plates were incubated at 37 °C and 5% CO2 for 2 h before BRET measurements. To determine affinities of the inhibitors, a final tracer concentration of 2 μM was used. Serially diluted inhibitors and tracer were added to the cell suspension and 100 μL were seeded in 96-well white, sterile nonbinding surface plates (Greiner Bio-One, cat. #655083). Plates were incubated at 37 °C and 5% CO2 for 2 h. For BRET measurements, NanoBRET NanoGlo Substrate (Promega cat. #N1571) was added to the wells according to the manufacturer's protocol and incubated for 2–3 min at RT. For all measurements, the 2102 EnVisionTM Multilabel reader (PerkinElmer) was used, equipped with 460 nm filter (donor) and 615 nm (acceptor) filter. Data analysis was performed with GraphPad 7.0. Milli-BRET units (mBU) are the BRET values multiplied with 1000. Tracer affinities were calculated using the following equation (eqn (1)):| Y = Bmax × X/(Kd + X) | (1) |
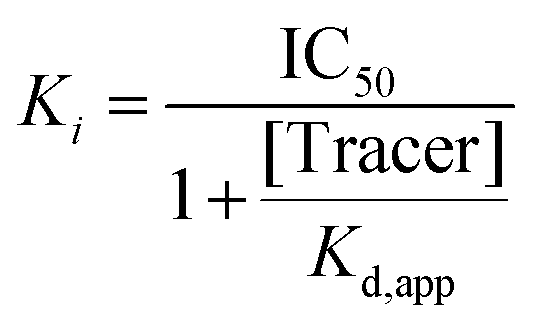 | (2) |
Author contributions
M. S., N. W., D. H., S. H. performed the chemical synthesis; A. V. and A. C. performed the biochemical characterization; docking studies were conducted by W. S.; A. C., E. N., J. H., G. K., M. S. performed cell-based assays, A. V., M. S. and M. J. wrote the original draft of the manuscript; all authors have given approval to the final manuscript. The study was supervised by O. G., O. E., E. M., R. S., H. L., W. S. and M. J.Abbreviations
| AMC | 7-Amino-4-methylcoumarin |
| BRET | Bioluminescence resonance energy transfer |
| BSA | Bovine serum albumin |
| CFU | Colony forming unit |
| DAPI | 4′,6′-Diamidino-2-phenylindole |
| DMSO | Dimethylsulfoxide |
| FBS | Fetal bovine serum |
| HDAC | Histone deacetylase |
| HEK293T | Human embryonic kidney 293 cells |
| HEPES | (4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid) |
| HRP | Horseradish peroxidase |
| GI50 | Half growth inhibitory concentration |
| IC50 | Half maximal inhibitory concentration |
| MCF-7 | Michigan cancer foundation - 7 |
| NAD | Nicotinamide adenine dinucleotide |
| NAM | Nicotinamide |
| PBS | Phosphate-buffered saline |
| SIRT | Sirtuin |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through SFB 992 (Project ID 192904750), Ju295/14-1, Si868/15-1, GRK1976 (Project ID 235777276), SFB 1160 (Project ID 256073931), SFB/TRR 167 (Project ID 259373024), SFB 1425 (Project ID 422681845), SFB 1479 (Project ID 441891347), GRK 2606 (Project ID 423813989). Furthermore, this study was funded by the DFG under Germany's Excellence Strategy (CIBSS - EXC-2189 - Project ID 390939984), as well as the European Research Council (ERC) through Starting Grant 337689 and Proof-of-Concept Grant 966687 (to O.Gr.). M. S. (Li 204/04) is supported by the Verband der Chemischen Industrie (VCI).Notes and references
- A. J. M. de Ruijter, A. H. van Gennip, H. N. Caron, S. Kemp and A. B. P. van Kuilenburg, Biochem. J., 2003, 370, 737–749 CrossRef CAS PubMed.
- S. Michan and D. Sinclair, Biochem. J., 2007, 404, 1–13 CrossRef CAS PubMed.
- G. Chen, P. Huang and C. Hu, Int. J. Cancer, 2020, 147, 3297–3304 CrossRef CAS PubMed.
- B. J. North and E. Verdin, PLoS One, 2007, 2, e784 CrossRef PubMed.
- T. Inoue, M. Hiratsuka, M. Osaki, H. Yamada, I. Kishimoto, S. Yamaguchi, S. Nakano, M. Katoh, H. Ito and M. Oshimura, Oncogene, 2007, 26, 945–957 CrossRef CAS PubMed.
- H. A. Eskandarian, F. Impens, M.-A. Nahori, G. Soubigou, J.-Y. Coppée, P. Cossart and M. A. Hamon, Science, 2013, 341, 1238858 CrossRef PubMed.
- A. Vaquero, R. Sternglanz and D. Reinberg, Oncogene, 2007, 26, 5505–5520 CrossRef CAS PubMed.
- B. J. North, B. L. Marshall, M. T. Borra, J. M. Denu and E. Verdin, Mol. Cell, 2003, 11, 437–444 CrossRef CAS PubMed.
- J. C. Black, A. Mosley, T. Kitada, M. Washburn and M. Carey, Mol. Cell, 2008, 32, 449–455 CrossRef CAS PubMed.
- K. M. Rothgiesser, S. Erener, S. Waibel, B. Lüscher and M. O. Hottiger, J. Cell Sci., 2010, 123, 4251–4258 CrossRef CAS PubMed.
- Y. Li, M. Zhang, R. G. Dorfman, Y. Pan, D. Tang, L. Xu, Z. Zhao, Q. Zhou, L. Zhou, Y. Wang, Y. Yin, S. Shen, B. Kong, H. Friess, S. Zhao, L. Wang and X. Zou, Neoplasia, 2018, 20, 745–756 CrossRef CAS PubMed.
- D. Zhao, S.-W. Zou, Y. Liu, X. Zhou, Y. Mo, P. Wang, Y.-H. Xu, B. Dong, Y. Xiong, Q.-Y. Lei and K.-L. Guan, Cancer Cell, 2013, 23, 464–476 CrossRef CAS PubMed.
- K.-S. Seo, J.-H. Park, J.-Y. Heo, K. Jing, J. Han, K.-N. Min, C. Kim, G. Y. Koh, K. Lim, G.-Y. Kang, J. Uee Lee, Y.-H. Yim, M. Shong, T.-H. Kwak and G. R. Kweon, Oncogene, 2015, 34, 1354–1362 CrossRef CAS PubMed.
- F. Wang, C.-H. Chan, K. Chen, X. Guan, H.-K. Lin and Q. Tong, Oncogene, 2012, 31, 1546–1557 CrossRef CAS PubMed.
- J. L. Feldman, J. Baeza and J. M. Denu, J. Biol. Chem., 2013, 288, 31350–31356 CrossRef CAS PubMed.
- T. Kosciuk, I. R. Price, X. Zhang, C. Zhu, K. N. Johnson, S. Zhang, S. L. Halaby, G. P. Komaniecki, M. Yang, C. J. DeHart, P. M. Thomas, N. L. Kelleher, J. C. Fromme and H. Lin, Nat. Commun., 2020, 11, 1067 CrossRef CAS PubMed.
- N. A. Spiegelman, X. Zhang, H. Jing, J. Cao, I. B. Kotliar, P. Aramsangtienchai, M. Wang, Z. Tong, K. M. Rosch and H. Lin, ACS Chem. Biol., 2019, 14, 2014–2023 CrossRef CAS PubMed.
- K. Suda, K. Tomizawa and T. Mitsudomi, Cancer Metastasis Rev., 2010, 29, 49–60 CrossRef CAS PubMed.
- R. Li, C. Peng, X. Zhang, Y. Wu, S. Pan and Y. Xiao, Life Sci., 2017, 182, 80–84 CrossRef CAS PubMed.
- H. Khawaja, A. Campbell, J. Z. Roberts, A. Javadi, P. O'Reilly, D. McArt, W. L. Allen, J. Majkut, M. Rehm, A. Bardelli, F. Di Nicolantonio, C. J. Scott, R. Kennedy, N. Vitale, T. Harrison, O. J. Sansom, D. B. Longley, E. Evergren and S. van Schaeybroeck, Cell Death Dis., 2020, 11, 930 CrossRef CAS PubMed.
- S. C. Dryden, F. A. Nahhas, J. E. Nowak, A.-S. Goustin and M. A. Tainsky, Mol. Cell. Biol., 2003, 23, 3173–3185 CrossRef CAS PubMed.
- Y. Wang, J. Yang, T. Hong, X. Chen and L. Cui, Ageing Res. Rev., 2019, 55, 100961 CrossRef CAS PubMed.
- H.-S. Kim, A. Vassilopoulos, R.-H. Wang, T. Lahusen, Z. Xiao, X. Xu, C. Li, T. D. Veenstra, B. Li, H. Yu, J. Ji, X. W. Wang, S.-H. Park, Y. I. Cha, D. Gius and C.-X. Deng, Cancer Cell, 2011, 20, 487–499 CrossRef CAS PubMed.
- E. Jing, S. Gesta and C. R. Kahn, Cell Metab., 2007, 6, 105–114 CrossRef CAS PubMed.
- Y. Cha, M.-J. Han, H.-J. Cha, J. Zoldan, A. Burkart, J. H. Jung, Y. Jang, C.-H. Kim, H.-C. Jeong, B.-G. Kim, R. Langer, C. R. Kahn, L. Guarente and K.-S. Kim, Nat. Cell Biol., 2017, 19, 445–456 CrossRef CAS PubMed.
- H. Luo, W.-C. Mu, R. Karki, H.-H. Chiang, M. Mohrin, J. J. Shin, R. Ohkubo, K. Ito, T.-D. Kanneganti and D. Chen, Cell Rep., 2019, 26, 945–954.e4 CrossRef CAS PubMed.
- M. Erburu, I. Muñoz-Cobo, T. Diaz-Perdigon, P. Mellini, T. Suzuki, E. Puerta and R. M. Tordera, Neuropharmacology, 2017, 117, 195–208 CrossRef CAS PubMed.
- R. M. de Oliveira, H. Vicente Miranda, L. Francelle, R. Pinho, É. M. Szegö, R. Martinho, F. Munari, D. F. Lázaro, S. Moniot, P. Guerreiro, L. Fonseca-Ornelas, Z. Marijanovic, P. Antas, E. Gerhardt, F. J. Enguita, B. Fauvet, D. Penque, T. F. Pais, Q. Tong, S. Becker, S. Kügler, H. A. Lashuel, C. Steegborn, M. Zweckstetter and T. F. Outeiro, PLoS Biol., 2017, 15, e2000374 CrossRef PubMed.
- H. Watanabe, Y. Inaba, K. Kimura, M. Matsumoto, S. Kaneko, M. Kasuga and H. Inoue, Nat. Commun., 2018, 9, 30 CrossRef PubMed.
- J. Chen, A. W. H. Chan, K.-F. To, W. Chen, Z. Zhang, J. Ren, C. Song, Y.-S. Cheung, P. B. S. Lai, S.-H. Cheng, M. H. L. Ng, A. Huang and B. C. B. Ko, Hepatology, 2013, 57, 2287–2298 CrossRef CAS PubMed.
- W. Zhou, T. K. Ni, A. Wronski, B. Glass, A. Skibinski, A. Beck and C. Kuperwasser, Cell Rep., 2016, 17, 1302–1317 CrossRef CAS PubMed.
- P. Y. Liu, N. Xu, A. Malyukova, C. J. Scarlett, Y. T. Sun, X. D. Zhang, D. Ling, S.-P. Su, C. Nelson, D. K. Chang, J. Koach, A. E. Tee, M. Haber, M. D. Norris, C. Toon, I. Rooman, C. Xue, B. B. Cheung, S. Kumar, G. M. Marshall, A. V. Biankin and T. Liu, Cell Death Differ., 2013, 20, 503–514 CrossRef CAS PubMed.
- Z. Li, J. Huang, H. Yuan, Z. Chen, Q. Luo and S. Lu, Oncotarget, 2016, 7, 18927–18939 CrossRef PubMed.
- M. G. Cheon, W. Kim, M. Choi and J.-E. Kim, Cancer Lett., 2015, 356, 637–645 CrossRef CAS PubMed.
- Y. Li, D. Dai, Q. Lu, M. Fei, M. Li and X. Wu, Biochem. Biophys. Res. Commun., 2013, 441, 661–667 CrossRef CAS PubMed.
- H. Yuan and R. Marmorstein, J. Biol. Chem., 2012, 287, 42428–42435 CrossRef CAS PubMed.
- T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Oláh, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovádi, M. Schutkowski, W. Sippl, O. Einsle and M. Jung, Nat. Commun., 2015, 6, 6263 CrossRef CAS PubMed.
- M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, S. Gerhardt, J. Ovádi, W. Sippl, O. Einsle and M. Jung, Angew. Chem., Int. Ed., 2016, 55, 2252–2256 CrossRef CAS PubMed.
- H. Jing, J. Hu, B. He, Y. L. Negrón Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y.-L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS PubMed.
- N. A. Spiegelman, J. Y. Hong, J. Hu, H. Jing, M. Wang, I. R. Price, J. Cao, M. Yang, X. Zhang and H. Lin, ChemMedChem, 2019, 14, 744–748 CrossRef CAS PubMed.
- Y. Nakajima, M. Kawaguchi, N. Ieda and H. Nakagawa, ACS Med. Chem. Lett., 2021, 42, 9249 Search PubMed.
- A. L. Nielsen, N. Rajabi, N. Kudo, K. Lundø, C. Moreno-Yruela, M. Bæk, M. Fontenas, A. Lucidi, A. S. Madsen, M. Yoshida and C. A. Olsen, RSC Chem. Biol., 2021, 85, 405 Search PubMed.
- L.-L. Yang, W. Xu, J. Yan, H.-L. Su, C. Yuan, C. Li, X. Zhang, Z.-J. Yu, Y.-H. Yan, Y. Yu, Q. Chen, Z. Wang, L. Li, S. Qian and G.-B. Li, MedChemComm, 2019, 10, 164–168 RSC.
- T. F. Outeiro, E. Kontopoulos, S. M. Altmann, I. Kufareva, K. E. Strathearn, A. M. Amore, C. B. Volk, M. M. Maxwell, J.-C. Rochet, P. J. McLean, A. B. Young, R. Abagyan, M. B. Feany, B. T. Hyman and A. G. Kazantsev, Science, 2007, 317, 516–519 CrossRef CAS PubMed.
- S. Lain, J. J. Hollick, J. Campbell, O. D. Staples, M. Higgins, M. Aoubala, A. McCarthy, V. Appleyard, K. E. Murray, L. Baker, A. Thompson, J. Mathers, S. J. Holland, M. J. R. Stark, G. Pass, J. Woods, D. P. Lane and N. J. Westwood, Cancer Cell, 2008, 13, 454–463 CrossRef CAS PubMed.
- N. Kudo, A. Ito, M. Arata, A. Nakata and M. Yoshida, Philos. Trans. R. Soc. London, Ser. B, 2018, 373, 20170070 CrossRef PubMed.
- J. Y. Hong, H. Jing, I. R. Price, J. Cao, J. J. Bai and H. Lin, ACS Med. Chem. Lett., 2020, 11, 2305–2311 CrossRef CAS PubMed.
- N. A. Spiegelman, I. R. Price, H. Jing, M. Wang, M. Yang, J. Cao, J. Y. Hong, X. Zhang, P. Aramsangtienchai, S. Sadhukhan and H. Lin, ChemMedChem, 2018, 13, 1890–1894 CrossRef CAS PubMed.
- J. L. Feldman, K. E. Dittenhafer-Reed, N. Kudo, J. N. Thelen, A. Ito, M. Yoshida and J. M. Denu, Biochemistry, 2015, 54, 3037–3050 CrossRef CAS PubMed.
- C. J. Fowler, G. Tiger and A. Stenström, J. Pharmacol. Exp. Ther., 1997, 283, 729–734 CAS.
- B. Heltweg and M. Jung, J. Biomol. Screening, 2003, 8, 89–95 CrossRef CAS PubMed.
- M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, J. Oláh, S. Gerhardt, J. Ovádi, W. Sippl, O. Einsle and M. Jung, J. Med. Chem., 2016, 59, 1599–1612 CrossRef CAS PubMed.
- D. Monaldi, D. Rotili, J. Lancelot, M. Marek, N. Wössner, A. Lucidi, D. Tomaselli, E. Ramos-Morales, C. Romier, R. J. Pierce, A. Mai and M. Jung, J. Med. Chem., 2019, 62, 8733–8759 CrossRef CAS PubMed.
- I. Galleano, M. Schiedel, M. Jung, A. S. Madsen and C. A. Olsen, J. Med. Chem., 2016, 59, 2847 CrossRef CAS PubMed.
- Y.-L. Chiang and H. Lin, Org. Biomol. Chem., 2016, 14, 2186–2190 RSC.
- Y.-B. Teng, H. Jing, P. Aramsangtienchai, B. He, S. Khan, J. Hu, H. Lin and Q. Hao, Sci. Rep., 2015, 5, 8529 CrossRef CAS PubMed.
- N. A. Spiegelman, X. Zhang, H. Jing, J. Cao, I. B. Kotliar, P. Aramsangtienchai, M. Wang, Z. Tong, K. M. Rosch and H. Lin, ACS Chem. Biol., 2019, 14, 2014–2023 CrossRef CAS PubMed.
- M. Vita and M. Henriksson, Semin. Cancer Biol., 2006, 16, 318–330 CrossRef CAS PubMed.
- D. M. Miller, S. D. Thomas, A. Islam, D. Muench and K. Sedoris, Clin. Cancer Res., 2012, 18, 5546–5553 CrossRef CAS PubMed.
- H. Jing, J. Hu, B. He, Y. L. Negrón Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y.-L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS PubMed.
- D. Hawksworth, L. Ravindranath, Y. Chen, B. Furusato, I. A. Sesterhenn, D. G. McLeod, S. Srivastava and G. Petrovics, Prostate Cancer Prostatic Dis., 2010, 13, 311–315 CrossRef CAS PubMed.
- C. M. Koh, C. J. Bieberich, C. V. Dang, W. G. Nelson, S. Yegnasubramanian and A. M. de Marzo, Genes Cancer, 2010, 1, 617–628 CrossRef CAS PubMed.
- J. Y. Kim, T. Valencia, S. Abu-Baker, J. Linares, S. J. Lee, T. Yajima, J. Chen, A. Eroshkin, E. A. Castilla, L. M. Brill, M. Medvedovic, M. Leitges, J. Moscat and M. T. Diaz-Meco, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6418–6423 CrossRef CAS PubMed.
- A. Matsuyama, T. Shimazu, Y. Sumida, A. Saito, Y. Yoshimatsu, D. Seigneurin-Berny, H. Osada, Y. Komatsu, N. Nishino, S. Khochbin, S. Horinouchi and M. Yoshida, EMBO J., 2002, 21, 6820–6831 CrossRef CAS PubMed.
- L. Li, S. Jayabal, M. Ghorbani, L.-M. Legault, S. McGraw, A. J. Watt and X.-J. Yang, Cell. Mol. Life Sci., 2019, 76, 3621–3640 CrossRef CAS PubMed.
- N. C. Dale, E. K. M. Johnstone, C. W. White and K. D. G. Pfleger, Front. Bioeng. Biotechnol., 2019, 7, 56 CrossRef PubMed.
- S. Swyter, M. Schiedel, D. Monaldi, S. Szunyogh, A. Lehotzky, T. Rumpf, J. Ovádi, W. Sippl and M. Jung, Philos. Trans. R. Soc. London, Ser. B, 2018, 373, 20170083 CrossRef PubMed.
- Y. Nonnenmacher, R. Palorini, A. F. d'Herouël, L. Krämer, M. Neumann-Schaal, F. Chiaradonna, A. Skupin, A. Wegner and K. Hiller, Metab. Eng., 2017, 43, 147–155 CrossRef CAS PubMed.
- C. Yung-Chi and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef.
- G. Hoffmann, F. Breitenbücher, M. Schuler and A. E. Ehrenhofer-Murray, J. Biol. Chem., 2014, 289, 5208–5216 CrossRef CAS PubMed.
- D. M. Taylor, U. Balabadra, Z. Xiang, B. Woodman, S. Meade, A. Amore, M. M. Maxwell, S. Reeves, G. P. Bates, R. Luthi-Carter, P. A. S. Lowden and A. G. Kazantsev, ACS Chem. Biol., 2011, 6, 540–546 CrossRef CAS PubMed.
- B. Peck, C.-Y. Chen, K.-K. Ho, P. Di Fruscia, S. S. Myatt, R. C. Coombes, M. J. Fuchter, C.-D. Hsiao and E. W.-F. Lam, Mol. Cancer Ther., 2010, 9, 844–855 CrossRef CAS PubMed.
- J. Trapp, A. Jochum, R. Meier, L. Saunders, B. Marshall, C. Kunick, E. Verdin, P. Goekjian, W. Sippl and M. Jung, J. Med. Chem., 2006, 49, 7307–7316 CrossRef CAS PubMed.
- M. B. Robers, J. D. Vasta, C. R. Corona, R. F. Ohana, R. Hurst, M. A. Jhala, K. M. Comess and K. V. Wood, Methods Mol. Biol., 2019, 1888, 45–71 CrossRef CAS PubMed.
- L. L. Ong, J. D. Vasta, L. Monereau, G. Locke, H. Ribeiro, M. A. Pattoli, S. Skala, J. R. Burke, S. H. Watterson, J. A. Tino, P. L. Meisenheimer, B. Arey, J. Lippy, L. Zhang, M. B. Robers, A. Tebben and C. Chaudhry, SLAS Discovery, 2020, 25, 176–185 CrossRef CAS PubMed.
- L. Grätz, K. Tropmann, M. Bresinsky, C. Müller, G. Bernhardt and S. Pockes, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- X. Chen, P. Wales, L. Quinti, F. Zuo, S. Moniot, F. Herisson, N. A. Rauf, H. Wang, R. B. Silverman, C. Ayata, M. M. Maxwell, C. Steegborn, M. A. Schwarzschild, T. F. Outeiro and A. G. Kazantsev, PLoS One, 2015, 10, e0116919 CrossRef PubMed.
- P. D. Davis, C. H. Hill, G. Lawton, J. S. Nixon, S. E. Wilkinson, S. A. Hurst, E. Keech and S. E. Turner, J. Med. Chem., 1992, 35, 177–184 CrossRef CAS PubMed.
- T. T. Y. Wang, N. W. Schoene, E.-K. Kim and Y. S. Kim, Mol. Carcinog., 2013, 52, 676–685 CrossRef CAS PubMed.
- M. Schiedel, D. Robaa, T. Rumpf, W. Sippl and M. Jung, Med. Res. Rev., 2018, 38, 147–200 CrossRef CAS PubMed.
- N. Wössner, Z. Alhalabi, J. González, S. Swyter, J. Gan, K. Schmidtkunz, L. Zhang, A. Vaquero, H. Ovaa, O. Einsle, W. Sippl and M. Jung, Front. Oncol., 2020, 10, 657 CrossRef PubMed.
- B. Heltweg, J. Trapp and M. Jung, Methods, 2005, 36, 332–337 CrossRef CAS PubMed.
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212–2221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00244a |
| This journal is © The Royal Society of Chemistry 2022 |