
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mesoporous ionic solid with 272 AuI6AgI3CuII3 complex cations in a super huge crystal lattice†
Hiroto
Takeda
,
Tatsuhiro
Kojima
 ,
Nobuto
Yoshinari
and
Takumi
Konno
*
,
Nobuto
Yoshinari
and
Takumi
Konno
*
Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka, Japan. E-mail: konno@chem.sci.osaka-u.ac.jp
First published on 16th July 2021
Abstract
Here, we report a unique mesoporous ionic solid (I) generated from a cationic AuI6AgI3CuII3 dodecanuclear complex with D-penicillamine depending on the homochirality and crystallization conditions. I crystallizes in the cubic space group of F4132 with an extremely large cell volume of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 171340 Å3, containing 272 AuI6AgI3CuII3 complex cations in the unit cell. In I, the complex cations are connected to each other through CH⋯π interactions in a zeotype framework, the topology of which is the same as that of the metal–organic framework in MIL-101, with similar but much larger two types of polyhedral pores with internal diameters of 38.2 Å and 49.7 Å, which are occupied by counter-anions and water molecules. Due to the cationic nature of the framework, I undergoes quick, specific exchanges of counter-anions while retaining its single crystallinity. This study realized the creation of a non-covalent mesoporous framework from a single complex salt, providing a conceptual advance in solid chemistry and material science.
171340 Å3, containing 272 AuI6AgI3CuII3 complex cations in the unit cell. In I, the complex cations are connected to each other through CH⋯π interactions in a zeotype framework, the topology of which is the same as that of the metal–organic framework in MIL-101, with similar but much larger two types of polyhedral pores with internal diameters of 38.2 Å and 49.7 Å, which are occupied by counter-anions and water molecules. Due to the cationic nature of the framework, I undergoes quick, specific exchanges of counter-anions while retaining its single crystallinity. This study realized the creation of a non-covalent mesoporous framework from a single complex salt, providing a conceptual advance in solid chemistry and material science.
Introduction
Recent increased interest in porous crystalline materials in chemistry and material science stems from the versatile applications of these materials in, for example, heterogeneous catalysis,1,2 storage and separation of molecules3–6 and ion exchange of ionic species.7–10 The availability of framework pores in crystalline materials as reaction fields to generate chemical species that cannot be obtained via conventional approaches,2,7–10 together with the incorporation and structural determination of molecules that are hardly crystallized,11–13 has also inspired the recent considerable attention to this class of materials. While high porosities and low material densities are needed for the efficient storage and separation of small molecules such as gases and vapors,3,6 other applications also require open channels and pore volumes that are suitable for the sizes of guest molecules, allowing fast diffusion, transfer and specific adsorption in the pores.4,6,14 Thus, the creation of artificial crystalline materials with mesopores with diameters of 2 nm to 50 nm is highly desirable for use in a wide range of applications, such as controlled synthesis of nanoparticles,15 immobilization of biomolecules for catalysts16 and capillary condensation of gaseous molecules,16–18 which are not possible for microporous materials with diameters less than 2 nm and macroporous materials with diameters greater than 50 nm.In 2002, Eddaoudi et al. reported the first crystalline material with three-dimensional (3D) mesopores with a diameter of 2.9 nm, named IRMOF-16, which is a metal–organic framework (MOF) sustained by coordination bonds between ZnII4 nodes and dicarboxylate linkers.19 Since then, a number of mesoporous crystalline materials with MOF structures have been synthesized based on the hierarchical self-assembly of molecular nodes and linkers using a synthetic strategy similar to that applied for IRMOF-16.20–25 One of the well-known examples of this class of materials is MIL-101, whose zeotype framework is composed of CrIII3 nodes and dicarboxylate linkers to form two kinds of cage-type mesopores with diameters of 2.9 nm and 3.4 nm.26 More recently, a unique crystalline MOF with larger mesopores with diameters of 5.0 nm and 6.2 nm, named NU-1301, has also been synthesized via the combination of UO22+ nodes and tricarboxylate linkers.27 Thus, rigid, directional coordination bonds between metal centres and carboxylate ligands have been critical for the creation of mesoporous crystalline compounds, although mesoporous frameworks sustained by covalent bonds and hydrogen bonds have been observed in organic solid materials.28–35
Here, we report the first mesoporous framework sustained by non-covalent CH⋯π interactions in a non-MOF coordination system, the structure of which was established by single-crystal X-ray crystallography. This system consists of a newly prepared AuI6AgI3CuII3 dodecanuclear complex salt, [Au6Ag3Cu3(tdme)2(D-pen)6](TFA)3 (tdme = 1,1,1-tris(diphenylphosphinomethyl)ethane, D-H2pen = D-penicillamine, TFA = trifluoroacetate), which crystallizes in a 3D ionic solid (I) with a very large unit cell containing a total of 272 AuI6AgI3CuII3 complex cations. Notably, the framework of I has the same topology as that of MIL-101, forming two kinds of cage-type mesopores that are larger than those in MIL-101, although the framework is composed of only complex cations linked by intermolecular CH⋯π interactions rather than coordination bonds, with its voids being occupied by counter-anions and water molecules. I is a kinetic product crystallized from aqueous media under ambient conditions and is reproducible and interconvertible with the thermodynamic non-porous solid product (I′) just by changing the crystallization temperature (Fig. 1). Very fast and specific anion exchanges for I, along with the creation of an analogous ionic solid (II) with smaller mesopores by introducing DL-pen instead of D-pen, are also reported.
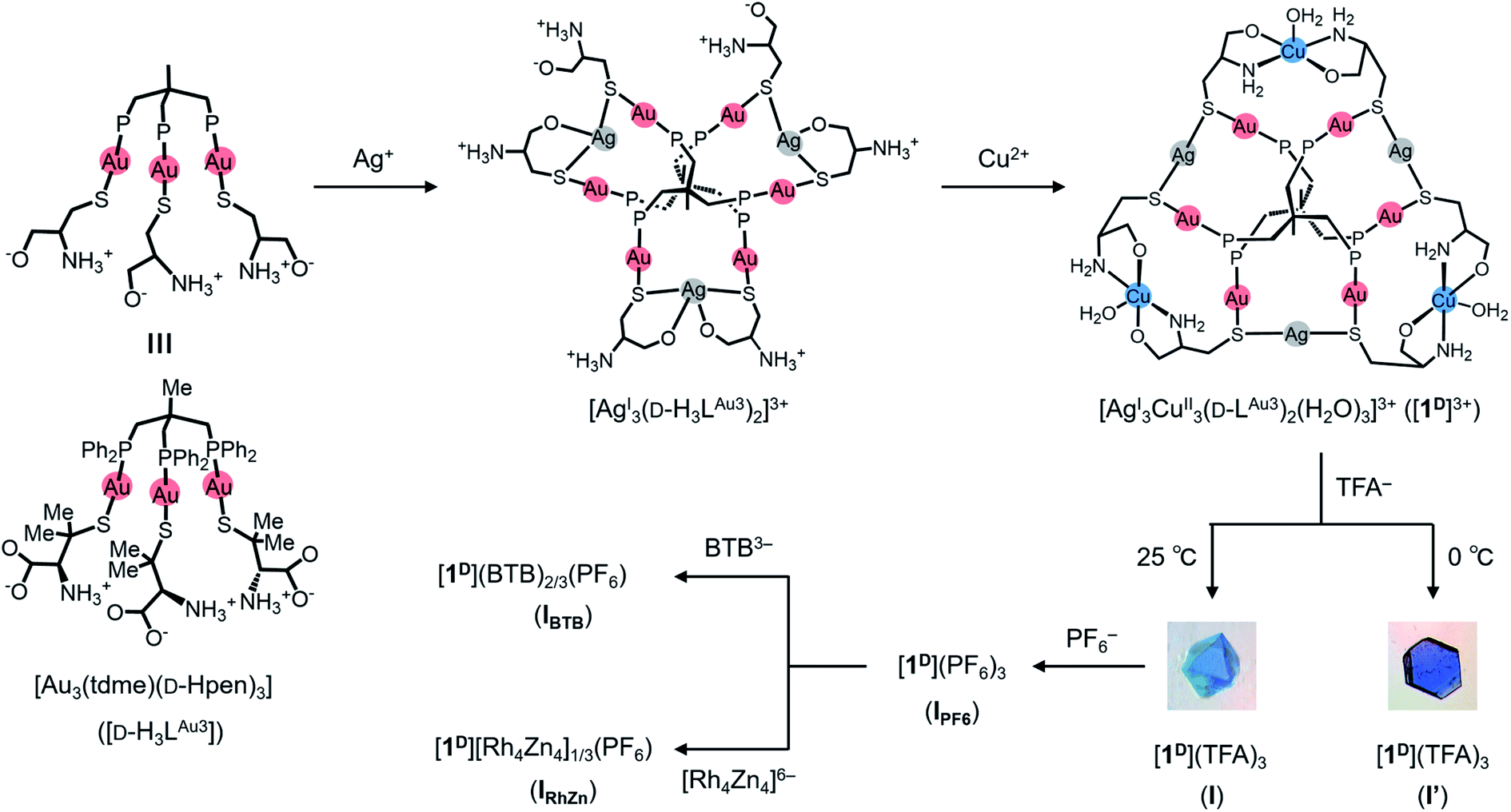 | ||
| Fig. 1 Synthetic routes of I′, I, IPF6, IBTB, and IRhZn. | ||
Results and discussion, experimental
Synthesis, characterization and crystal structure of I′
As a continuation of our recent work on the creation of multinuclear coordination compounds based on the trigold(I) metalloligand with a mixture of hydrophobic 1,1,1-tris(diphenylphosphinomethyl)ethane (tdme) and hydrophilic D-penicillamine (D-H2pen), [Au3(tdme)(D-Hpen)3] (D-H3LAu3),36–38D-H3LAu3 was treated with 1.5 equiv. of silver(I) trifluoroacetate in MeOH/CH3CN at 0 °C. This treatment yielded a colourless solution, from which colourless stick crystals were isolated upon addition of aqueous sodium nitrate. Single-crystal X-ray crystallography (Fig. S1 and Table S1†), together with the X-ray fluorescence (Fig. S2†) and elemental analyses, indicated that the colourless crystals contain the AuI6AgI3 nonanuclear complex with free ammonium groups, [Ag3(D-H3LAu3)2]3+, in which two D-H3LAu3 metalloligands are spanned by three AgI ions through thiolato and carboxylate groups (Fig. S1†).Further treatment of the colourless solution obtained from D-H3LAu3 and silver(I) trifluoroacetate with 1.5 equiv. of aqueous copper(II) acetate at 0 °C yielded a blue solution, from which X-ray-quality blue crystals with a hexagonal plate shape (I′) were obtained by slow evaporation at 0 °C. Based on the IR spectrum (Fig. S3†) and the X-ray fluorescence and elemental analyses, I′ was assigned as an AuI6AgI3CuII3 dodecanuclear complex with the chemical formula [Ag3Cu3(D-LAu3)2](TFA)3·nH2O. The diffuse reflection spectrum of I′ exhibits a broad d–d band at 630 nm due to CuII centers39 and a positive band is observed in the solid-state CD spectrum in this region (Fig. S4†), indicative of the optically active nature of I′.
The structure of I′ was established by single-crystal X-ray crystallography, which revealed the presence of complex cations ([1D]3+ = [Ag3Cu3(D-LAu3)2(H2O)3]3+) and TFA− anions in a 1:3 ratio. As shown in Fig. 2a and b, the complex cation [1D]3+ contains two (D-LAu3)3− metalloligands that are spanned by 3 AgI atoms through thiolato groups and by 3 CuII atoms through amine and carboxylate groups, forming the cage-type AuI6AgI3CuII3 dodecanuclear structure in [AgI3CuII3(D-LAu3)2(H2O)3]3+. The overall structure in [1D]3+ is similar to that in the previously reported [MII3(D-LAu3)2] (M = Co, Cu, Cd), in which each MII centre is octahedrally coordinated by two N,O,S-chelates of D-pen from two (D-LAu3)3− metalloligands.36,37 However, each AgI centre in [1D]3+ adopts an S2 linear geometry coordinated by two thiolato donors, while each CuII centre has a trans-N2O3 square-pyramidal geometry coordinated by two N,O-chelates and a water molecule. In [1D]3+, an S-bridged AuI6AgI3 core is surrounded by 3 hydrophilic CuII centres with amine, carboxylate and aqua donors in the equatorial positions, while its apical positions are covered by 12 hydrophobic phenyl groups. Each [1D]3+ cation is surrounded by 9 TFA− anions so as to construct a 2D layer structure in I′, which is sustained by NH⋯O hydrogen bonds, together with coulombic interactions (Fig. 2c and d). The 2D layers are stacked in an ABAB fashion to form CH⋯π interactions between tdme phenyl groups (Fig. 2e).
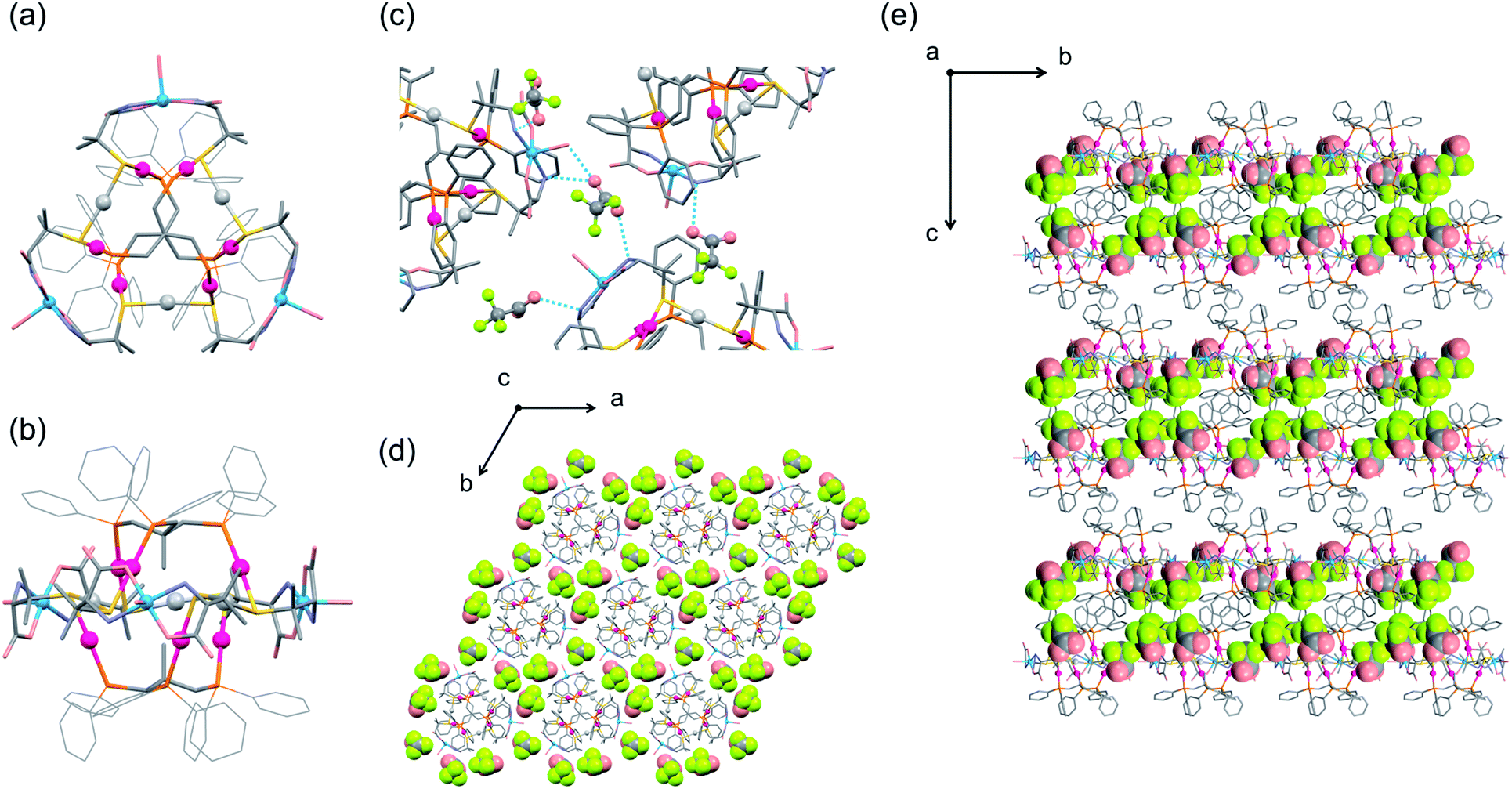 | ||
| Fig. 2 Crystal structure of I′. Top (a) and side (b) views of the structure of the [1D]3+ complex cation. (c) Hydrogen-bonding interactions between TFA− anions and the [1D]3+ complex cations. Top (d) and side (e) views of the packing structure in I′ [colour codes: red, Au; silver, Ag; blue, Cu; orange, P; yellow, S; pink, O; pale blue, N; grey, C; light yellow, F]. | ||
Formation and crystal structure of I
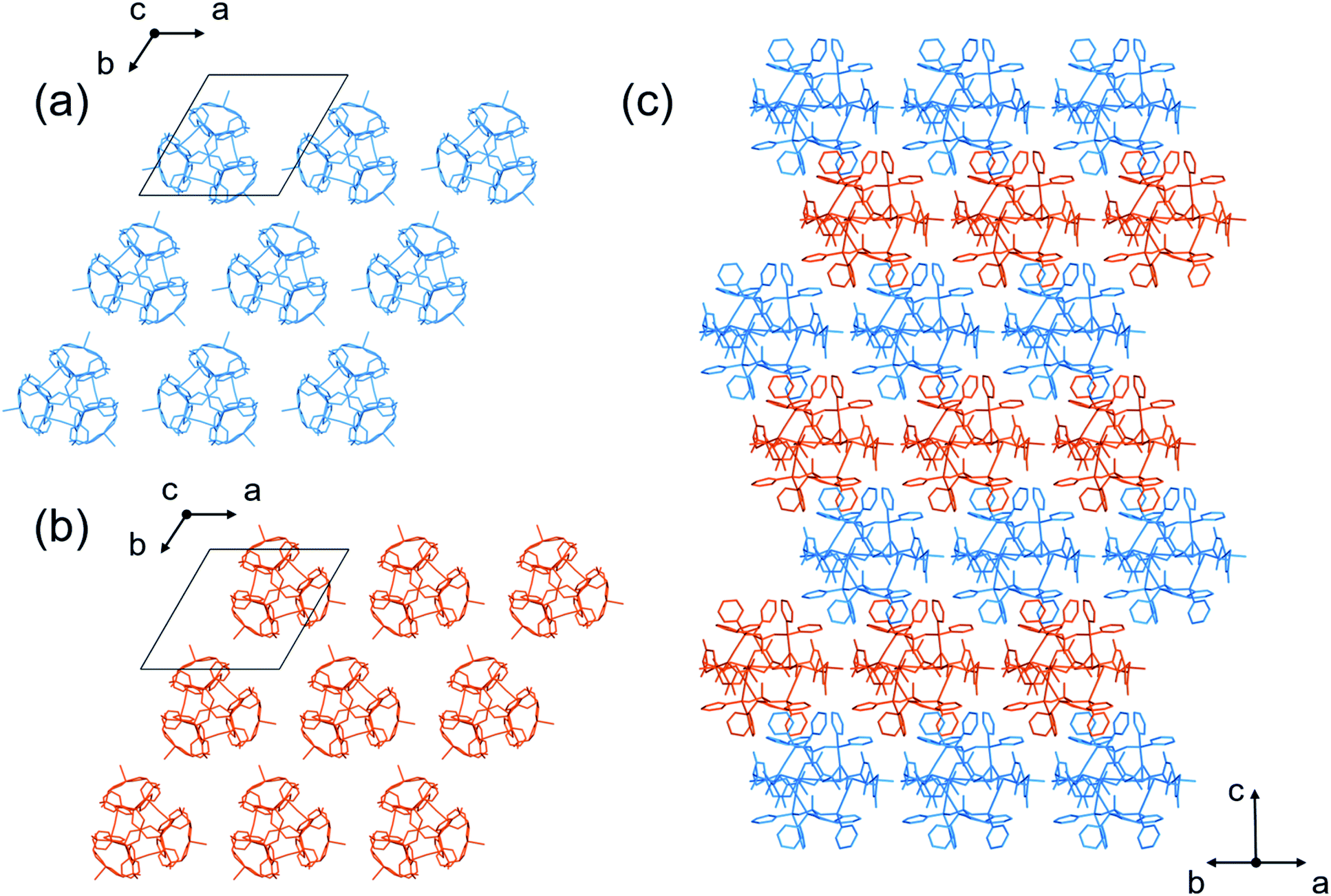
We observed that different blue crystals with octahedral shapes (I) were produced when the blue reaction solution was stored at 25 °C to accelerate evaporation after concentrating it to a small volume at 0 °C. As in the case of I′, I was characterized by IR, diffuse reflection and CD spectra (Fig. S3 and S4†), as well as X-ray fluorescence and elemental analyses, the results of which are all essentially the same as those of I′. Thus, I was also determined to have a formula of [Ag3Cu3(D-LAu3)2](TFA)3·nH2O. However, the powder X-ray diffraction pattern of I differs significantly from that of I′, indicative of the polymorphic relationship between I and I′ (Fig. S5†).Single-crystal X-ray analysis revealed that I crystallized in the cubic space group F4132, which is different from the trigonal space group P321 for I′. Remarkably, I possesses an exceptionally large crystal lattice with a cell volume of 2171340 Å3, which is the largest among non-MOFs and the third largest including MOF crystals reported thus far.27,40 While I afforded very weak X-ray diffraction peaks with a low resolution, comparable to those for protein crystals, we observed electron densities that match well with those of the heavy atoms (Au, Ag, Cu, S and P) in [1D]3+. Finally, the total structure of I was established by fitting the molecular structure of [1D]3+ obtained from the X-ray analysis of I′. The asymmetric unit of I contains one complete [1D]3+ cation, three half cations and a one-third unit of [1D]3+ cations (Fig. S6†). Thus, the total number of [1D]3+ cations in the unit cell was determined to be 272, based on the symmetry operation of the space group F4132. In I, the [1D]3+ cations are linked through CH⋯π interactions between tdme phenyl groups (Fig. S7†), generating a 3D mesoporous framework with a potential porosity of 66% (Fig. 3). Based on the elemental analysis and IR spectrum, the voids of the framework are considered to be occupied by TFA− anions and solvated water molecules. It is assumed that the concentration of the reaction solution at 25 °C results in an aqueous-rich phase due to the faster evaporation of organic solvents, leading to the hydrophobic self-assembly of [1D]3+ cations through CH⋯π interactions to crystallize the kinetic product of I; in contrast, the thermodynamic product of I′ is crystallized by slower evaporation at 0 °C. It should be noted that the recrystallization of I from water at 0 °C afforded I′, while I was generated by recrystallizing I′ at 25 °C.
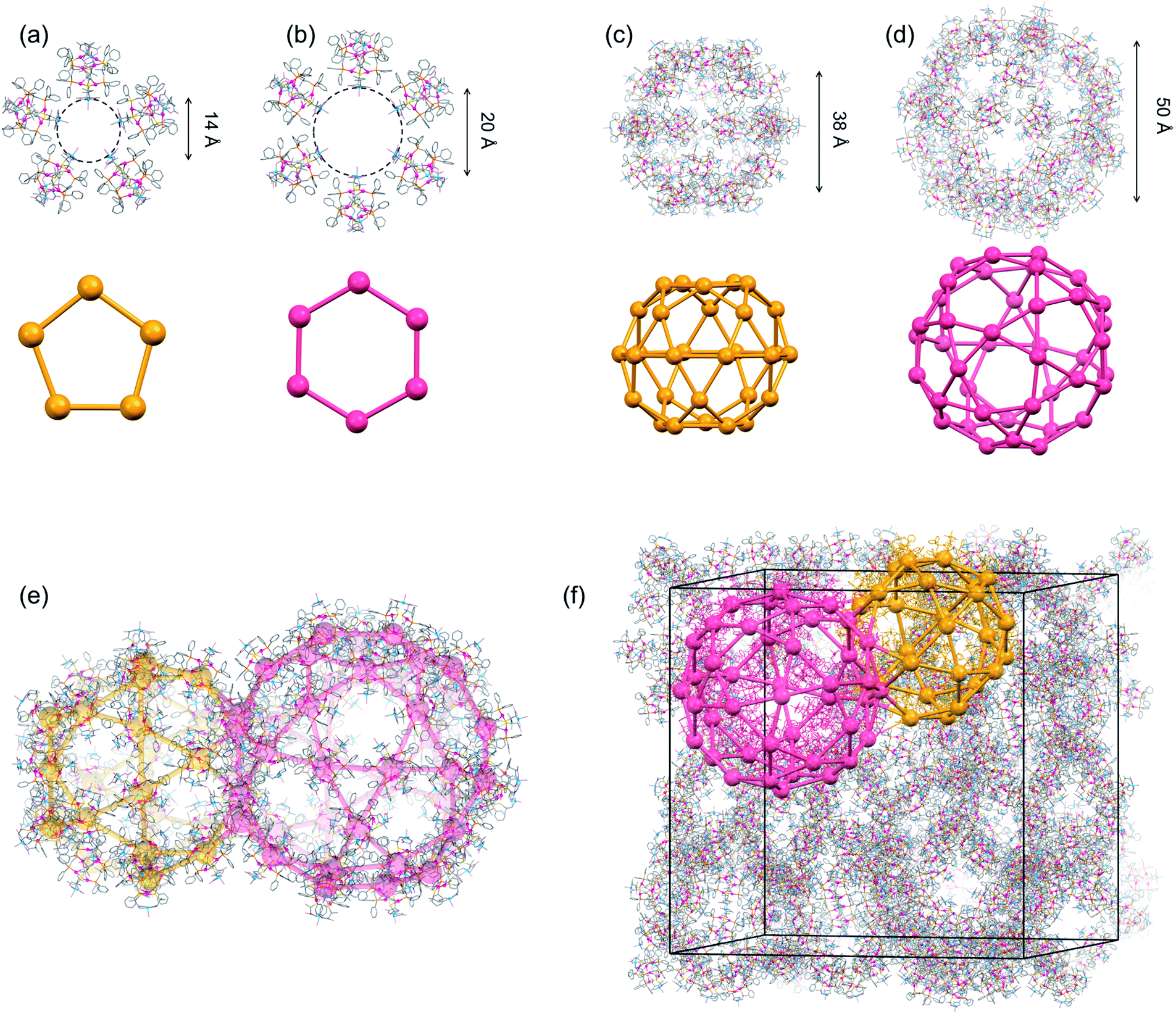 | ||
| Fig. 3 Crystal structure of I.‡ (a) A pentagonal window in cage A and cage B. (b) A hexagonal window in cage A. (c) Cage B. (d) Cage A. (e) Face-sharing cage A and cage B. (f) Schematic representation of the packing structure of a 6-connected mtn-e net (cage A and cage B are represented by red and orange colours, respectively, with [1D]3+ cations shown by balls) [colour codes: red, Au; silver, Ag; blue, Cu; orange, P; yellow, S; pink, O; pale blue, N; grey, C]. | ||
Topological analysis41,42 revealed that the framework in I adopts a 6-connected mtn-e net structure (Fig. 3 and S8†).43 This topology is the same as that of the MOF in MIL-101 with the formula [Cr3F(H2O)2O(1,4-bdc)3]·nH2O (1,4-bdc = 1,4-benzene dicarboxylate),26,43 replacing the trimeric building units of chromium(III) octahedra linked by 1,4-bdc anions through coordination bonds in MIL-101 by dodecanuclear [1D]3+ cations connected by CH⋯π interactions in I. While MIL-101 crystallized in the achiral space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m due to the achiral nature of the trimeric building units and the 1,4-bdc linkers, the homochirality of [1D]3+ cations with D-pen ligands led to crystallization of I in the chiral space group F4132. In addition, the cell volume of I (2171340 Å3) is three times larger than that of MIL-101 (701860 Å3). As in MIL-101, the framework in I contains two types of mesoporous cages (cage A and cage B) in a 1:2 ratio (Fig. 3c and d). Cage A consists of 42 [1D]3+ cations with a [328·512·64] face arrangement,44 bearing 4 hexagonal windows with an internal diameter of 20.2 Å and 12 pentagonal windows with an internal diameter of 14.0 Å (Fig. 3a and b), while cage B consists of 30 [1D]3+ cations with [320·512] face arrangement (icosidodecahedron),44 having 12 pentagonal windows that are shared by cages A. The accessible volumes of cage A and cage B in I, which are occupied by TFA− anions and water molecules, are 64000 Å3 (internal diameter 49.7 Å) and 29000 Å3 (internal diameter of 38.2 Å), respectively. These are much larger than the corresponding cages in MIL-101 (20600 Å3 with an internal diameter of 34 Å and 12700 Å3 with an internal diameter of 29 Å).26
m due to the achiral nature of the trimeric building units and the 1,4-bdc linkers, the homochirality of [1D]3+ cations with D-pen ligands led to crystallization of I in the chiral space group F4132. In addition, the cell volume of I (2171340 Å3) is three times larger than that of MIL-101 (701860 Å3). As in MIL-101, the framework in I contains two types of mesoporous cages (cage A and cage B) in a 1:2 ratio (Fig. 3c and d). Cage A consists of 42 [1D]3+ cations with a [328·512·64] face arrangement,44 bearing 4 hexagonal windows with an internal diameter of 20.2 Å and 12 pentagonal windows with an internal diameter of 14.0 Å (Fig. 3a and b), while cage B consists of 30 [1D]3+ cations with [320·512] face arrangement (icosidodecahedron),44 having 12 pentagonal windows that are shared by cages A. The accessible volumes of cage A and cage B in I, which are occupied by TFA− anions and water molecules, are 64000 Å3 (internal diameter 49.7 Å) and 29000 Å3 (internal diameter of 38.2 Å), respectively. These are much larger than the corresponding cages in MIL-101 (20600 Å3 with an internal diameter of 34 Å and 12700 Å3 with an internal diameter of 29 Å).26
Anion exchange of I
In contrast to the insolubility of MIL-101 in any solvent,26I is soluble in water due to its highly porous 3D framework sustained by CH⋯π interactions.§ While I lost a crystallinity upon desolvation, its crystallinity was retained for at least several weeks without dissolving when soaked in aqueous NaTFA. This was also observed when crystals of I were soaked in other aqueous salts, such as NaX = NaPF6, NaBF4 and NaOTf (OTf = trifluoromethanesulfonate). IR spectroscopy and elemental analysis revealed that the crystals (IX) obtained by soaking crystals of I in aqueous NaX for 1 h contain only X− as counter-anions (Fig. S9†), indicative of the complete exchange of the TFA− anions in I. The retention of the single crystallinity and framework structure in I was confirmed by powder X-ray diffraction studies (Fig. S10†), together with single-crystal X-ray crystallography. Notably, the anion exchange of I was completed just by soaking the samples in aqueous NaPF6 for 1 minute and even by washing the crystals with aqueous NaPF6 on filter paper. To investigate the selective exchange of counter-anions, crystals of I were soaked in a 1:1:1 mixture of aqueous NaPF6, NaBF4 and NaOTf. Again, the single crystallinity was retained and the crystals thus obtained (IPF6/BF4/OTf) contained only PF6− as counter-anions. This implies that PF6− has a higher affinity for the cationic framework than do BF4− and OTf−, presumably due to the effective formation of NH⋯F and Ph⋯F interactions with [1D]3+ cations in the framework. Consistent with these results, IPF6 is much less soluble in water than I, IBF4 and IOTf,§ indicating that the cationic framework is reinforced by introducing PF6− as counter-anions.
Reinforcement of the cationic framework allowed the incorporation of the large organic anion BTB3− (H3BTB = 1,3,5-tris(4′-carboxyphenyl)benzene; Fig. S11†) with 3 carboxylate groups retaining the single crystallinity by soaking crystals of IPF6 in aqueous H3BTB for 1 h;45 the same treatment of crystals of I resulted in the loss of its crystallinity (Fig. S12†). The resulting crystals of IBTB, which are almost insoluble in water,§ are formulated as [1D](BTB)2/3(PF6) based on the IR spectrum (Fig. S13†) and elemental analyses, suggesting the presence of PF6− anions that tightly interact with the cationic framework. Not only the organic BTB3− but also the anionic metal-cluster [Rh4Zn4O(L-cys)12]6− with 12 carboxylate groups (Fig. S11†)46,47 was quickly incorporated into the cationic framework retaining the single crystallinity via the partial exchange of PF6− anions in IPF6; soaking crystals of IPF6 in aqueous K6[Rh4Zn4O(L-cys)12] for 1 h afforded single crystals of IRhZn formulated as [1D][Rh4Zn4O(L-cys)12]1/3(PF6), which are also insoluble in water.§¶
The cationic nature of the mesoporous framework in IPF6 also led to the quick uptake of an anionic dye of resorufin sodium salt from its aqueous solution, which was confirmed by tracing the absorption spectral change of the solution; the characteristic absorption band at 570 nm decreased with time and nearly disappeared on soaking crystals of IPF6 for 6 h (Fig. S16a†). This was also the case for another anionic dye of methyl orange,||** but no significant uptake was observed for a neutral dye of Basic Red 5 and a cationic dye of methylene blue (Fig. S16b–d†). The uptake of an anionic dye in IPF6 is in contrast to the uptake of a cationic dye in NU-1301, which has an anionic MOF structure.27 Note that soaking crystals of IPF6 in an aqueous solution containing methyl orange and methylene blue in a 1:1 ratio led to the disappearance of the absorption due to methyl orange after 6 h (Fig. 4). This result demonstrates that IPF6 has an ability to remove an anionic dye from a mixture of cationic and anionic dyes in solution.
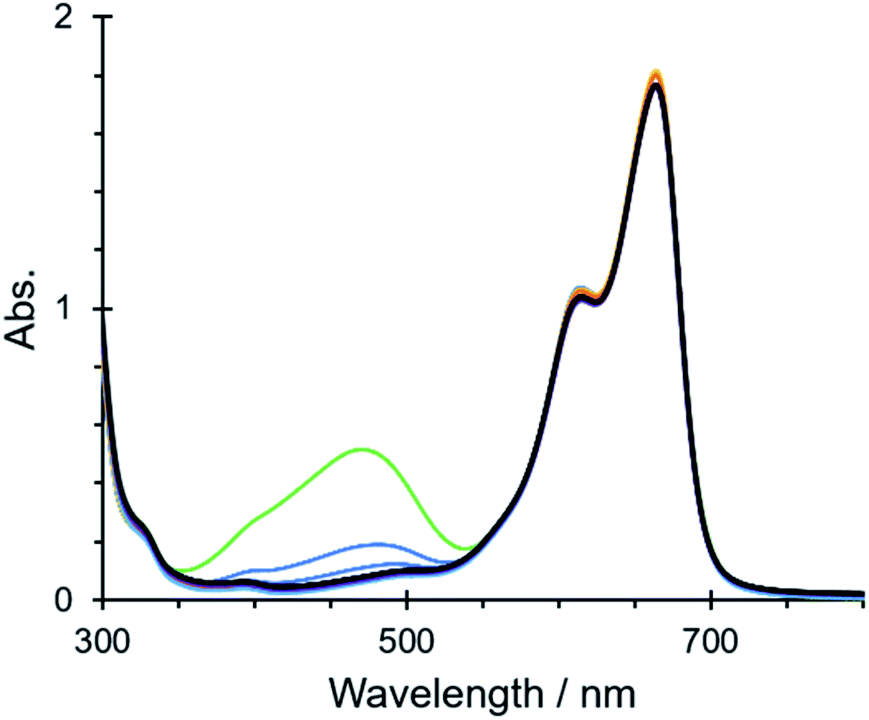 | ||
| Fig. 4 Time-dependent absorption spectral change of a mixture of aqueous methyl orange (4.0 × 10−5 M, 1.5 mL) and aqueous methylene blue (a 4.0 × 10−5 M, 1.5 mL) after soaking crystals of IPF6 (ca. 10 mg) for 0 h (green), 1 h (blue), 2 h (yellow), 4 h (orange), 6 h (light blue), 8 h (purple) and 10 h (black). | ||
Synthesis and crystal structures of II′ and II
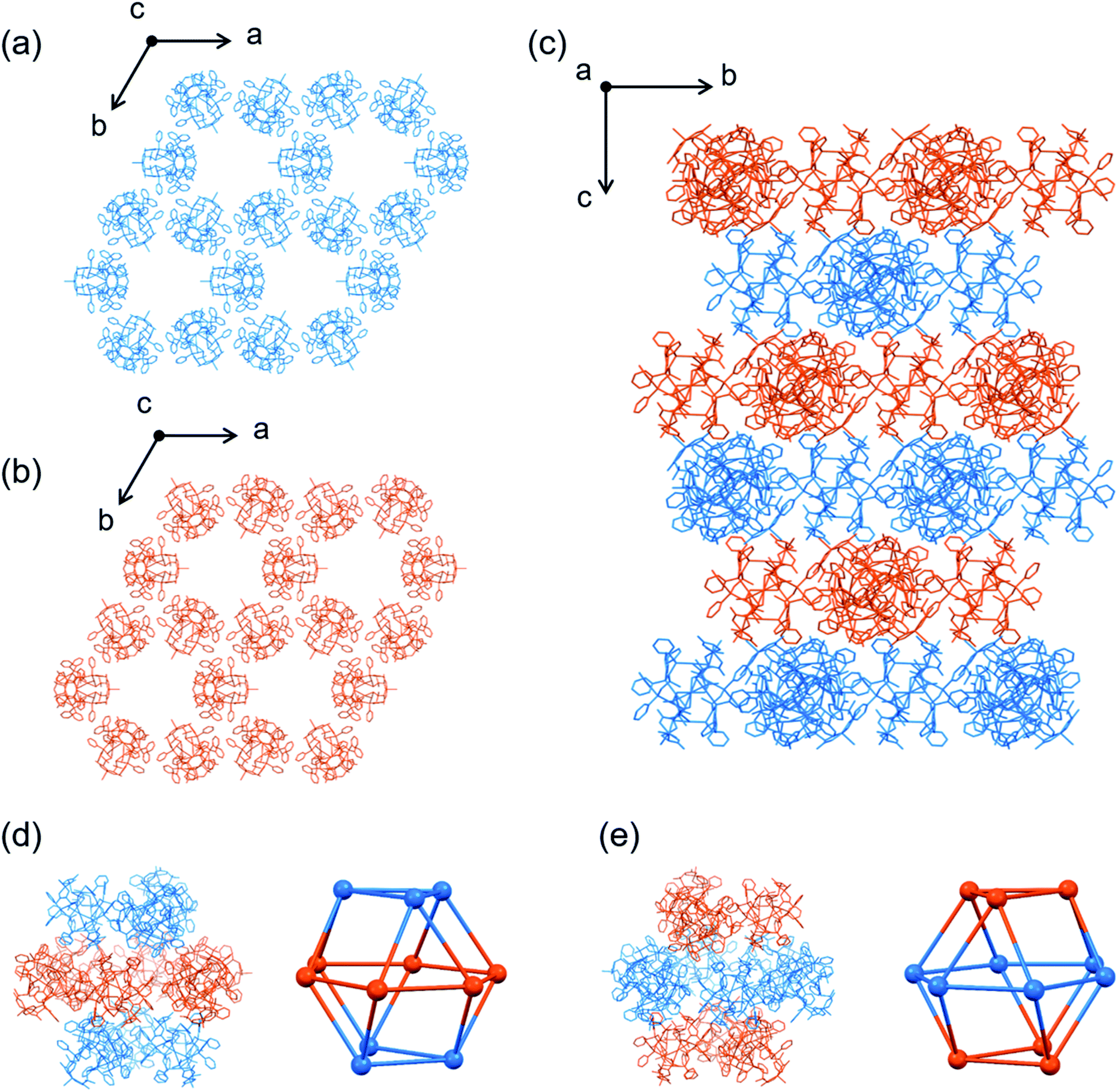
As expected, the use of [Au3(tdme)(L-Hpen)3] instead of [Au3(tdme)(D-Hpen)3] led to the production of hexagonal plate crystals (I′L) and octahedral crystals (IL) of [Ag3Cu3(L-LAu3)2](TFA)3, which correspond to I′ and I, respectively. Single-crystal X-ray crystallography established that the structures of I′L and IL are entirely enantiomeric to those of I′ and I, respectively. Next, we carried out the reactions using a 1:1 mixture of [Au3(tdme)(D-Hpen)3] (D-H3LAu3) and [Au3(tdme)(L-Hpen)3] (L-H3LAu3) to clarify the importance of homochirality in the creation of the highly porous structure in I. From this reaction, blue hexagonal plate crystals (II′) were produced via slow evaporation at 0 °C, together with blue truncated octahedral crystals (II) as a minor product, while fast evaporation at 25 °C dominantly yielded crystals of II. Based on the IR and diffuse reflection spectra (Fig. S3 and S4†), together with the X-ray fluorescence and elemental analyses, II′ and II were both assigned to have the same chemical formula [AgI3CuII3(LAu3)2](TFA)3·nH2O, which corresponds to those of I′ and I. However, II′ and II are CD silent, indicative of the presence of both D-H3LAu3 and L-H3LAu3 in a 1:1 ratio. The powder X-ray diffraction patterns of II′ and II are different from each other and also different from the patterns of I′ and I (Fig. S18†). Single-crystal X-ray analysis indicated that II′ crystallized in the achiral space group of trigonal R3c. Each complex cation in II′ consists of two homochiral metalloligands, (D-LAu3)3− or (L-LAu3)3−, which are spanned by 3 AgI and 3 CuII atoms to give the homochiral AuI6AgI3CuII3 structure in [Ag3Cu3(D- or L-LAu3)2(H2O)3]3+ ([1DorL]3+) (Fig. S19†).
In addition, the homochiral complex cations of [1DorL]3+ were assembled with TFA− ions through hydrogen bonds to form a homochiral 2D layer in [1DorL](TFA)3 (Fig. 5a and b). While the layer structure in II′ is comparable to that in I′, the [1D](TFA)3 and [1L](TFA)3 layers are alternately, closely stacked in an off-set fashion in II′, resulting in an overall heterochiral structure (Fig. 5c).
 | ||
| Fig. 5 Crystal structure of II′. (a) 2D layer structure composed of [1D]3+ cations. (b) 2D layer structure composed of [1L]3+ cations. (c) Side view of the packing structure [color codes: blue, D-[1]3+; orange, L-[1]3+]. | ||
II also crystallized in the achiral but different space group of trigonal Rc. As those in II′, the homochiral [1D]3+ or [1L]3+ cations in II are assembled to form homochiral 2D layers (Fig. 6a and b). In II, however, the complex cations are linked to each other through CH⋯π interactions, forming hexagonal void spaces surrounded by the 6 cations in each layer (Fig. S20†). In addition, the top and bottom of the hexagonal space are each covered by 3 complex cations with opposite chirality through CH⋯π interactions (Fig. 6c), affording a void space surrounded by 6 [1D]3+ and 6 [1L]3+ cations in a cuboctahedral structure, which are occupied by TFA− anions and water molecules (Fig. 6d and e). The pore volume of each cage in II is 16800 Å3 (diameter 15.9 Å) with trigonal and rhomboidal windows (diameters of 8.3 Å and 11.4 Å), which is much smaller than those of the cages in I (64000 Å3 and 29000 Å3). The potential porosity of II (35%) is also less than that of I (66%). Thus, the heterochiral system with a 1:1 mixture of [1D]3+ and [1L]3+ resulted in the creation of a less porous structure in II with much smaller porous cages.
 | ||
| Fig. 6 Crystal structure of II. (a) 2D layer structure composed of [1D]3+ cations. (b) 2D layer structure composed of [1L]3+ cations. (c) Side view of the packing structure.†† (d and e) Enantiomeric nanoporous cages composed of [1D]3+ and [1L]3+ cations [colour codes: blue, D-[1]3+; orange, L-[1]3+]. | ||
Conclusions
We newly prepared the cationic AuI6AgI3CuII3 dodecanuclear complex [AgI3CuII3(D-LAu3)2(H2O)3]3+ ([1D]3+) using the trigold(I) metalloligand D-H3LAu3, by way of the AuI6AgI3 nonanuclear complex [Ag3(D-H3LAu3)2]3+. Cationic [1D]3+ was found to crystallize with TFA− to produce two types of polymorph ionic crystals, I′ and I, depending on the crystallization conditions; slow evaporation at 0 °C yielded the thermodynamic product of I′ with a 2D layer structure by forming hydrogen bonds between [1D]3+ cations and TFA− anions, while fast evaporation at 25 °C produced the kinetic product of I with a 3D porous structure consisting of CH⋯π interactions among [1D]3+ cations. The existence of hydrophobic phenyl groups at the apical positions and hydrophilic CuII moieties at their equatorial positions in [1D]3+ appears to be responsible for this result. Remarkably, I possesses a very large crystal lattice with a cell volume of more than 2 million Å3 containing 272 AuI6AgI3CuII3 complex cations. In addition, I generates a mesoporous framework, the topology of which is identical to that in MIL-101, having similar but much larger two types of mesoporous cages that are occupied by counter-anions and water molecules. While MIL-101 is achiral, I is inherently chiral due to the presence of D-pen in [1D]3+. The importance of the homochiral nature of I in the creation of the mesoporous framework was evidenced by the employment of a racemic mixture of [1D]3+ and [1L]3+, which resulted in the construction of a less porous framework in II. I is highly soluble in water and is easily recrystallized from aqueous media. However, soaking crystals of I in aqueous NaPF6 led to the very quick exchange of TFA− by PF6− to give crystals of IPF6 retaining its single crystallinity, which is ascribed to the presence of mesoporous cages with large solvent-accessible pores. Unlike I, IPF6 is almost insoluble in water, indicative of the reinforcement of the porous framework via non-covalent interactions between [1D]3+ cations and PF6− anions. This allowed not only the incorporation of large organic and metal-cluster anions in the cationic framework to create single crystals that are entirely insoluble in water but also the selective uptake of anionic dyes from aqueous media. The controlled construction of the largest-known non-covalent mesoporous framework from a single complex salt under ambient conditions, together with its chemical modification via simple, very quick, single-crystal-to-single-crystal anion-exchange procedures, is expected to open up a new avenue for innovative development of porous solid materials.Experimental
Preparation of compounds
Blue hexagonal plate crystals of [Ag3Cu3(L-LAu3)2](TFA)3 (I′L) were prepared by the same procedure as that employed for I′, using [Au3(tdme)(L-Hpen)3] instead of [Au3(tdme)(D-Hpen)3]. Anal. calcd for [Au6Ag3Cu3(tdme)2(L-pen)6(H2O)3](TFA)3·12H2O = C118H162Ag3Au6Cu3F9N6O33P6S6: C, 31.94; H, 3.68; N, 1.89%. Found: C, 31.94; H, 3.71; N, 1.99%. IR spectrum (cm−1, ATR): 1615 (νCOO−), 1102 (νP–Ph), 698 (νPh) and 1202 (νTFA−). Crystallographic data for I′L: trigonal P321, a = b = 19.2442(3) Å, c = 26.8194(4) Å, α = β = 90°, γ = 120°, V = 8601.6(3) Å3.
Blue octahedral crystals of [Ag3Cu3(L-LAu3)2](TFA)3 (IL) were prepared by the same procedure as that employed for I, using [Au3(tdme)(L-Hpen)3] instead of [Au3(tdme)(D-Hpen)3]. Anal. calcd for [Au6Ag3Cu3(tdme)2(L-pen)6(H2O)3](TFA)3·20H2O = C118H178Ag3Au6Cu3F9N6O41P6S6: C, 30.93; H, 3.92; N, 1.83%. Found: C, 31.17; H, 4.24; N, 1.93%. IR spectrum (cm−1, ATR): 1626 (νCOO−), 1101 (νP–Ph), 692 (νPh) and 1200 (νTFA−). Crystallographic data for IL: cubic F4132, a = b = c = 128.755(2) Å, α = β = γ = 90°, V = 2134481(119) Å3.
Anal. IPF6: calcd for [Au6Ag3Cu3(tdme)2(D-pen)6(H2O)3](PF6)3·18H2O = C112H174Ag3Au6Cu3F18N6O33P9S6: C, 28.98; H, 3.78; N, 1.81%. Found: C, 29.01; H, 3.72; N, 1.91%. IR spectrum (cm−1, ATR): 1634 (νCOO−), 1102 (νP–Ph), 697 (νPh) and 854 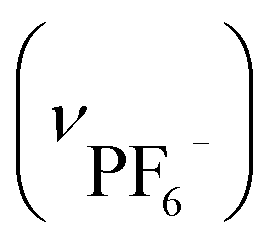 . Crystallographic data for IPF6: cubic F, a = b = c = 129.1080(20) Å, α = β = γ = 90°, V = 2152085(80) Å3.
. Crystallographic data for IPF6: cubic F, a = b = c = 129.1080(20) Å, α = β = γ = 90°, V = 2152085(80) Å3.
Anal. IOTf: calcd for [Au6Ag3Cu3(tdme)2(D-pen)6(H2O)3](OTf)3·18H2O = C115H174Ag3Au6Cu3F9N6O42P6S9: C, 29.68; H, 3.77; N, 1.81%. Found: C, 29.69; H, 3.53; N, 1.87%. IR spectrum (cm−1, ATR): 1634 (νCOO−), 1103 (νP–Ph), 698 (νPh) and 1276, 1170, 1031, 640 (νOTf−). Crystallographic data for IOTf: cubic F, a = b = c = 128.27(6) Å, α = β = γ = 90°, V = 2110298(1600) Å3.
Anal. IBF4: anal. calcd for [Au6Ag3Cu3(tdme)2(D-pen)6(H2O)3](BF4)3·14H2O = C112H166Ag3Au6Cu3F12N6O29P6S6B3: C, 30.61; H, 3.81; N, 1.91%. Found: C, 30.58; H, 3.67; N, 1.89%. IR spectrum (cm−1, ATR): 1634 (νCOO−), 698 (νPh) and 1102 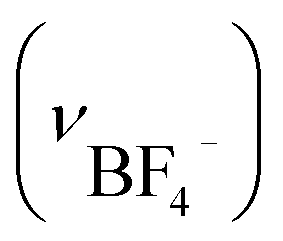 . Crystallographic data for IBF4: cubic F, a = b = c = 128.5(6) Å, α = β = γ = 90°, V = 2121793(19000) Å3.
. Crystallographic data for IBF4: cubic F, a = b = c = 128.5(6) Å, α = β = γ = 90°, V = 2121793(19000) Å3.
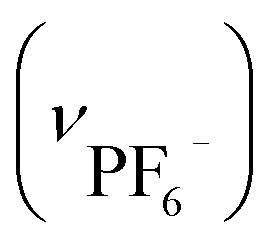 . Crystallographic data for IBTB: cubic F4132, a = b = c = 128.2485(23) Å, α = β = γ = 90°, V = 2109390(66) Å3.
. Crystallographic data for IBTB: cubic F4132, a = b = c = 128.2485(23) Å, α = β = γ = 90°, V = 2109390(66) Å3.
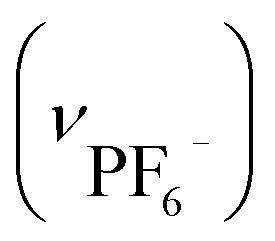 . Crystallographic data for IRhZn: cubic F4132, a = b = c = 128.3373(24) Å, α = β = γ = 90°, V = 2113774(68) Å3.
. Crystallographic data for IRhZn: cubic F4132, a = b = c = 128.3373(24) Å, α = β = γ = 90°, V = 2113774(68) Å3.
Physical measurements
X-ray fluorescence spectrometry was performed on a SHIMADZU EDX-7000 spectrometer. IR spectra were recorded on a JASCO FT/IR-4700 infrared spectrophotometer using the ATR method at room temperature. Solid-state diffuse reflection spectra were measured on a JASCO V-670 UV/Vis/NIR spectrometer and solid-state CD spectra were recorded on a JASCO J-820 spectrometer at room temperature. Elemental analyses (C, H, N) were performed at Osaka University using a Yanaco CHN Corder MT-6. The powder X-ray diffractions were recorded at a controlled temperature in transmission mode [synchrotron radiation λ = 1.0 Å; 2θ range = 2–78°; step width = 0.01°; data collection time = 1 min] on a diffractometer equipped with a MYTHEN microstrip X-ray detector (Dectris Ltd) at the SPring-8 BL02B2 beamline. The crystals were loaded into a glass capillary tube (diameter = 0.3 mm), which was rotated during the measurements.X-ray crystallography
The diffraction data for I, II′, IPF6, IBTB and IRhZn were recorded at 100 K with a PILATUS3 X CdTe 1M with synchrotron radiation (λ = 0.42810 Å) at SPring-8 (BL02B1 beamline). The diffraction images were processed by using RAPID-AUTO. The diffraction data for II and [Ag3(D-H3LAu3)2](NO3)3 were recorded at 100 K with a Rayonix MX225HS CCD area detector with synchrotron radiation (λ = 0.62997 Å for II and 0.6000 Å for [Ag3(D-H3LAu3)2](NO3)3) at the 2D beamline at the Pohang Accelerator Laboratory (PAL). The intensity data were processed using the HKL3000 program and collected by using the ω-scan technique.48 The diffraction data for I′, IOTf and IBF4 were recorded at 100 K with a hybrid pixel array detector with Mo Kα X-ray source (λ = 0.71073 Å). The diffraction images were processed by using CrysAlisPro 1.171.40.75a (Rigaku OD, 2020). The structures were solved by direct methods using SHELXS-2014.49 The structure refinements were carried out using full-matrix least-squares (SHELXL-2018/3).49For [Ag3(D-H3LAu3)2](NO3)3, DFIX, ISOR, DELU and SADI restraints and a FLAT constrain were applied to model nitrate anions. Global ISOR, RIGU and SIMU were applied. All nonhydrogen atoms were refined anisotropically. The contributions of undetermined NO3− ions and solvated water molecules were excluded using the SQUEEZE program in the PLATON package.50 4882 electrons were found in the void with a volume of 11701 Å3 per unit cell by the mask calculation. The number of electrons is not consistent with the presence of 3 NO3− and 34 H2O per formula unit, which gives 3464 electrons per unit cell. The difference in the electron numbers is due to the loss of solvated water molecules in the course of the isolation of the bulk sample.
For I, the positions of Au, Ag, Cu, S and P atoms were solved by direct methods using SHELXS-2014 and refined by using full-matrix least-squares (SHELXL-2018/3).49 The molecular structure of each [1D]3+ complex-cation was fitted with that determined in I′ by using the FRAG command and refined as a rigid body. ISOR, DELU and SIMU restraints and a EADP constrain were applied to coordinated waters. Au and Ag atoms were refined anisotropically and Cu, S, P, O, N and C atoms were refined isotropically. The exclusion of the undetermined TFA− ions and solvated water molecules with SQUEEZE50 was not successful.
For I′, DFIX, ISOR, DELU, SIMU and SADI restraints were applied to model trifluoro acetate anions. All nonhydrogen atoms except for crystal water molecules were refined anisotropically. Six diffractions were omitted to improve the data quality. 669 electrons were found in the void with a volume of 2117 Å3 per unit cell by the mask calculation. The number of electrons is not consistent with the presence of 8 H2O per formula unit, which gives 160 electrons per unit cell. The difference in the electron numbers is due to the loss of solvated water molecules in the course of the isolation of the bulk sample.
For II, ISOR, DELU and SIMU restraints were applied for coordinated water molecules. Global SIMU were applied. All nonhydrogen atoms were refined anisotropically. The contributions of undetermined TFA− ions and solvated water molecules were excluded using the SQUEEZE program in the PLATON package.50 Nineteen diffractions were omitted to improve the data quality. 9168 electrons were found in the void with a volume of 30609 Å3 per unit cell by the mask calculation. The number of electrons is not consistent with the presence of 3 TFA− and 12.5 H2O per formula unit, which gives 5220 electrons per unit cell. The difference in the electron numbers is due to the loss of solvated water molecules in the course of the isolation of the bulk sample.
For II′, DFIX, ISOR, DELU, SIMU, RIGU and SADI restraints were applied to model trifluoro acetate anions and parts of [1D]3+ complex-cation. All nonhydrogen atoms were refined anisotropically. Two diffractions were omitted to improve the data quality. The exclusion of the undetermined TFA− ions and solvated water molecules with SQUEEZE was not successful. 1685 electrons were found in the void with a volume of 6288 Å3 per unit cell by the mask calculation. The number of electrons is compatible with the presence of 2 TFA− and 15 H2O per formula unit, which gives 1560 electrons per unit cell.
Topological analysis
The mtn-e topology was identified using ToposPro (Ver. 5.3.3.3).41,42 An AuI6AgI3CuII3 dodecanuclear complex and CH⋯π contacts between two complexes were simplified as a node and a single edge, respectively.Author contributions
H. T. performed the syntheses and spectroscopic measurements on the compounds; T. Kojima performed single X-ray diffraction analyses. T. Kojima, N. Y. and T. Konno wrote the manuscript; T. Konno conceived and designed the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST CREST (Grant No. JPMJCR13L3) and JSPS KAKENHI (Grant No. 18H05344, 19K05496 and 20K05664). The synchrotron radiation experiments were performed at the BL02B1 and BL02B2 beamlines of SPring-8 with the approval of JASRI (Proposal No. 2018B1481, 2019A1302 and 2019A1350) and at the 2D-SMC beamline of the Pohang Accelerator Laboratory. We would like to thank Mr Kyohei Adachi for a part of the anion-exchange experiments.Notes and references
- J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CrossRef CAS PubMed.
- T. A. Makal, J.-R. Li, W. Lu and H.-C. Zhou, Chem. Soc. Rev., 2012, 41, 7761 RSC.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108 CrossRef CAS.
- L. Gao, C.-Y. V. Li, K.-Y. Chan and Z.-N. Chen, J. Am. Chem. Soc., 2014, 136, 7209 CrossRef CAS PubMed.
- G. Distefano, H. Suzuki, M. Tsujimoto, S. Isoda, S. Bracco, A. Comotti, P. Sozzani, T. Uemura and S. Kitagawa, Nat. Chem., 2013, 5, 335 CrossRef CAS PubMed.
- J. D. Evans, V. Bon, I. Senkovska, H.-C. Lee and S. Kaskel, Nat. Commun., 2020, 11, 2690 CrossRef CAS PubMed.
- L. Liu, Y. Song, H. Chong, S. Yang, J. Xiang, S. Jin, X. Kang, J. Zhang, H. Yua and M. Zhu, Nanoscale, 2016, 8, 1407 RSC.
- Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780 CrossRef CAS PubMed.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed.
- M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139 CrossRef CAS PubMed.
- H.-L. Lu, D.-Q. Lin, M.-M. Zhu and S.-J. Yao, J. Sep. Sci., 2012, 35, 2131 CrossRef CAS PubMed.
- K. Ariga, A. Vinu, Y. Yamauchi, Q. Ji and J. P. Hill, Bull. Chem. Soc. Jpn., 2011, 85, 1 CrossRef.
- Z. Wu and D. Zhao, Chem. Commun., 2011, 47, 3332 RSC.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098 RSC.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS.
- L. Song, J. Zhang, L. Sun, F. Xu, F. Li, H. Zhang, X. Si, C. Jiao, Z. Li, S. Liu, Y. Liu, H. Zhou, D. Sun, Y. Du, Z. Cao and Z. Gabelica, Energy Environ. Sci., 2012, 5, 7508 RSC.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677 RSC.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386 RSC.
- D. Liu, D. Zou, H. Zhu and J. Zhang, Small, 2018, 14, 1801454 CrossRef PubMed.
- H. Y. Guan, R. J. LeBlanc, S. Y. Xie and Y. F. Yue, Coord. Chem. Rev., 2018, 369, 76 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed.
- P. Li, N. A. Vermeulen, C. D. Malliakas, D. A. Gómez-Gualdrón, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624 CrossRef CAS PubMed.
- A. P. Côté, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914 CrossRef PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- Q. Yin, P. Zhao, R.-J. Sa, G.-C. Chen, J. Lü, T.-F. Liu and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 7691 CrossRef CAS PubMed.
- M. J. Białek and R. Klajn, Chem, 2019, 5, 2283 Search PubMed.
- R. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362 RSC.
- Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, Nat. Commun., 2019, 10, 3074 CrossRef PubMed.
- K. Ma, P. Li, J. H. Xin, Y. Chen, Z. Chen, S. Goswami, X. Liu, S. Kato, H. Chen, X. Zhang, J. Bai, M. C. Wasson, R. R. Maldonado, R. Q. Snurr and O. K. Farha, Cell Rep. Phys. Sci., 2020, 1, 100024 CrossRef.
- B. Wang, X.-L. Lv, J. Lv, L. Ma, R.-B. Lin, H. Cui, J. Zhang, Z. Zhang, S. Xiang and B. Chen, Chem. Commun., 2020, 56, 66 RSC.
- Y. Hashimoto, N. Yoshinari, N. Matsushita and T. Konno, Eur. J. Inorg. Chem., 2014, 3474 CrossRef CAS.
- K. Imanishi, B. Wahyudianto, T. Kojima, N. Yoshinari and T. Konno, Chem.–Eur. J., 2020, 26, 1827 CrossRef CAS PubMed.
- N. Yoshinari and T. Konno, Chem. Rec., 2016, 16, 1647 CrossRef CAS PubMed.
- J. Ribas Gispert, Coordination Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed.
- X. Lian, Y.-P. Chen, T.-F. Liu and H.-C. Zhou, Chem. Sci., 2016, 7, 6969 RSC.
- V. A. Blatov, Crystallogr. Rev., 2004, 10, 249 CrossRef CAS.
- V. A. Blatov, IUCr CompComm Newsletter, 2006, 7, 4 Search PubMed.
- D. Kim, X. Liu and M. S. Lah, Inorg. Chem. Front., 2015, 2, 336 RSC.
- V. A. Blatov, D. M. Proserpio and M. O'Keeffe, CrystEngComm, 2010, 12, 44 RSC.
- E. Weber, M. Hecker, E. Koepp, W. Orlia, M. Czugler and I. Csöregh, J. Chem. Soc., Perkin Trans. 2, 1988, 1251 RSC.
- T. Konno, K. Okamoto and J. Hidaka, Inorg. Chem., 1994, 33, 538 CrossRef CAS.
- N. Yoshinari, S. Yamashita, Y. Fukuda, Y. Nakazawa and T. Konno, Chem. Sci., 2019, 10, 587 RSC.
- W. Minor, M. Cymborowski, Z. Otwinowski and M. Chruszcz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 859 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 148 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental detail, PXRD, and XRF, IR, adsorption, diffuse reflectance, and CD spectra, as well as crystal structures and crystallographic data. CCDC 2024986–2024990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02497c |
| ‡ The TFA− anions and solvated water molecules that might be highly disordered in I were not located due to the weak electron densities and the very large cell volume of I. |
| § The solubilities of crystals in water at room temperature were evaluated to be 0.7 mg mL−1, 0.4 mg mL−1, and 0.3 mg mL−1 for IPF6, IRhZn, and IBTB, respectively, based on the absorption spectral measurements. |
| ¶ The presence of the RhIII4ZnII4 cluster anions in IRhZn, together with PF6−, was confirmed by the IR spectrum (Fig. S14†) and the X-ray fluorescence and elemental analyses, together with the solid-state CD spectra (Fig. S15†). |
| || The PXRDs of IPF6 were much broaden after the uptake of methyl orange, which is indicative of the partial collapse of the cationic framework (Fig. S12d†). |
| ** The adsorbed methyl orange was removal from the crystals by soaking in 0.2 M aqueous NaPF6, which was confirmed by the absorption spectral measurements (Fig. S17†). |
| †† The TFA− anions and solvated water molecules were not located due to their positional disorder. |
| This journal is © The Royal Society of Chemistry 2021 |