
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected Markovnikov hydroarylation and hydroalkenylation of alkenes under nickel catalysis†
Zi-Qi
Li
,
Omar
Apolinar
,
Ruohan
Deng
and
Keary M.
Engle
 *
*
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: keary@scripps.edu
First published on 13th July 2021
Abstract
We report a full account of our research on nickel-catalyzed Markovnikov-selective hydroarylation and hydroalkenylation of non-conjugated alkenes, which has yielded a toolkit of methods that proceed under mild conditions with alkenyl sulfonamide, ketone, and amide substrates. Regioselectivity is controlled through catalyst coordination to the native Lewis basic functional groups contained within these substrates. To maximize product yield, reaction conditions were fine-tuned for each substrate class, reflecting the different coordination properties of the directing functionality. Detailed kinetic and computational studies shed light on the mechanism of this family of transformations, pointing to transmetalation as the turnover-limiting step.
Introduction
Catalytic alkene functionalization is an efficient and economical way to build up molecular complexity from readily accessible chemical feedstocks.1 Transition-metal-catalyzed alkene hydroarylation/alkenylation reactions, in particular, represent a straightforward means of constructing C(sp3)–C(sp2) bonds. Various strategies have been developed to control regioselectivity using both conjugated and non-conjugated alkenes, with the latter introducing added complications from alkylmetal chain-walking.2–7 Anti-Markovnikov hydroarylation methods with non-conjugated alkenes have developed rapidly during the past several years.8–12 In these systems selectivity control typically stems from the thermodynamic preference for formation of a primary alkylmetal intermediate. Markovnikov-selective hydroarylation reactions with non-conjugated alkenes, on the other hand, are comparatively rare, with research in this area progressing more slowly (Scheme 1A).13 A notable advance was reported by Shenvi and co-workers 2016, who developed a dual-catalytic Co/Ni metal–hydride H-atom-transfer (MHAT) approach that was effective for the hydroarylation of terminal alkenes with aryl halides, where regioselectivity is controlled by the favorable formation of a secondary alkyl radical via MHAT.13c | ||
| Scheme 1 Background and synopsis of current work. | ||
Pioneered by Zhou and co-workers, nickel(0)-catalyzed redox-neutral hydroarylation enables robust coupling of alkenes and arylboronic acids in alcohol solvents.2i–k Building on foundational work by Zhou using conjugated alkene substrates (i.e., styrenes and 1,3-dienes)2i and later contributions by Zhao using non-conjugated alkenyl carboxamides bearing a bidentate directing auxiliary,7c,d we recently developed a ligand-controlled regiodivergent hydrofunctionalization of simple non-conjugated alkenyl carboxylates. Addition of a carefully tailored Pyrox ligand allowed toggling of regioselectivity, bringing about either anti-Markovnikov or Markovnikov selectivity (Scheme 1B).14 Contemporaneously, Wang and co-workers developed an electron-rich diimine ligand to promote nickel(II)-catalyzed anti-Markovnikov-selective hydroarylation of a range of different non-conjugated terminal alkenes with arylboronic acids.15 Expanding the scope of Markovnikov-selective hydroarylation to other classes of non-conjugated alkene starting materials bearing native functional groups beyond carboxylic acids16 would enhance the preparative utility of this approach. Moreover, understanding the underlying mechanism with greater clarity would support further improvements in scope, selectivity, and efficiency. To this end, in the present study, we report the nickel(0)-catalyzed hydroarylation and -alkenylation of alkenyl sulfonamides,16c ketones,16e and amides16d and investigate the reaction mechanism (Scheme 1C). Across all three substrate classes, high Markovnikov-selectivity arises from substrate directivity without the need for an ancillary ligand.
Results and discussion
To initiate our investigation, we tested various model substrates under the reaction conditions previously optimized to bring about hydroarylation of alkenyl carboxylate substrates.14 However, only moderate to low yields were observed (8–66% yield, Scheme 2, right column). Evaluation of different reaction conditions revealed that each of the individual substrate classes responded differently to changes in key reaction variables. Practically speaking, this observation prompted us to optimize reaction conditions that were tailored for each substrate class, as summarized in Scheme 2. A series of cross-compatibility experiments reveals the extent to which the fine-tuned reaction conditions are substrate-specific. Comparing the optimal conditions for each substrate illustrates common features and important differences that shed light on mechanistic features of this methodology (see below). In all cases the reactions proceed under relatively mild temperatures (rt – 40 °C), in contrast to analogous non-directed reactions that generally require elevated temperatures (≥80 °C).2i,15 Additionally, alcohol solvent was required in all of the protocols, reflecting solvent participation in the key hydronickelation process. Tuning of the steric bulk and pKa for individual substrates presumably serves to control the rate of this step. The optimal inorganic base, both in identity and loading, also varied across substrate class. The base is critically involved in promoting and thus modulating the rate of organoboron transmetalation, but it can also play a deleterious role in mediating alkene isomerization with alkenyl amide and especially alkenyl ketone substrates bearing acidic α-C–H bonds. This latter point required lower base equivalents (5 mol% LiOt-Bu) or weaker base (2 equiv. Cs2CO3), respectively, be used for these two substrate families. It is worth mentioning that even though we screened a wide breadth of different ligands, there was no sign of ligand-based regiodivergence as was reported with alkenyl carboxylates (see ESI†).14 The mechanistic origin of this point remains unclear, though one possible explanation is that the metal is already coordinatively saturated with ligands that cannot be readily displaced in the selectivity determining step (see below).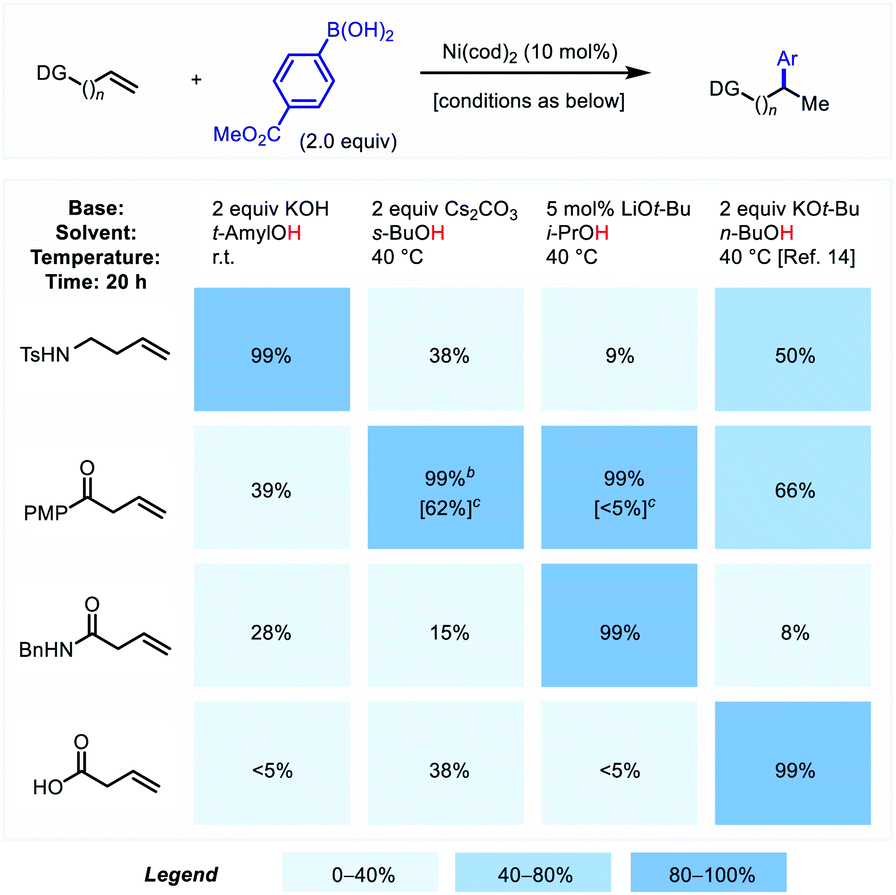 | ||
| Scheme 2 Cross-compatibility of reaction conditions. aAll percentages represent 1H NMR yields of combined regioisomers with CH2Br2 as internal standard. PMP = 4-methoxyphenyl. bReaction time was 2 h instead of 20 h to prevent potential ester exchange with solvent. cValue in brackets represents the reaction outcome using p-TolB(OH)2 as coupling partner. | ||
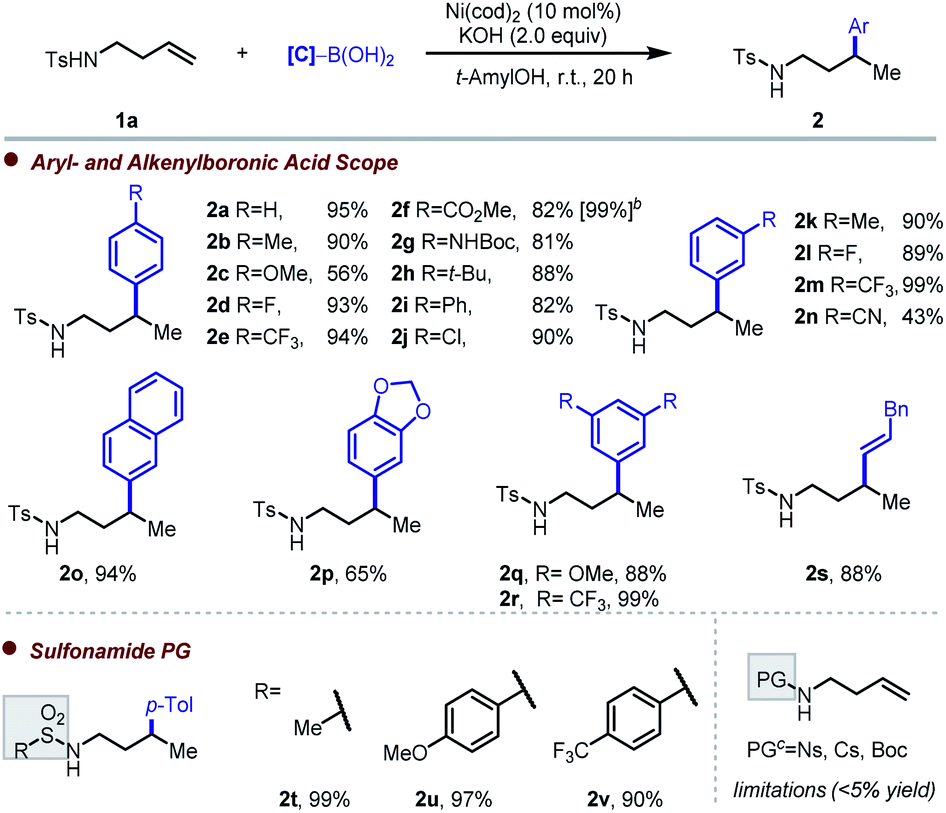
Having identified effective conditions for each family of substrates, we then proceeded to evaluate the scope of each of the three protocols. First, we examined the method for alkenyl sulfonamides,17 where the best conditions were found to be KOH (2 equiv.) as base and t-AmylOH as solvent at room temperature (Table 1). Apart from a moderate yield obtained with electron-rich methoxy substitution at the para-position (2c), electronic variation of arylboronic acid does not affect the yield significantly, and products 2a–2j were prepared in good to excellent yield. When the reaction was performed on 0.6 mmol scale, 2f was obtained in 99% yield. A potentially coordinating meta-CN substituent gave 43% yield (2n). Other substituents on the meta-position were well tolerated (2k–2m). Excellent yield of 2o (94%) was obtained with 2-naphthylboronic acid. With boronic acids bearing more complex substitution patterns, such as a benzodioxazole or 3,5-disubstitution, the reaction proceeded smoothly, giving 2p–2r in good yield. Extension to the analogous hydroalkenylation reaction was successful, with 2s obtained in 88% yield. Other substituents on the sulfonamide group were next tested. With a methanesulfonyl protecting group, quantitative yield was obtained (2t). The electronic influence of the arylsulfonamide was next probed by introducing different groups at the para-position, with 2u (–OMe) and 2v (–CF3) both formed in excellent yield. In terms of limitations, more electron-withdrawing substituents (–CN and –NO2) proved deleterious, with no desired product observed in either case. The lack of product formation in these cases may be due to the oxidizing nature of the arenesulfonyl groups, which could interfere with the nickel(0) catalyst, or alternatively to the attenuated σ-donor strength of the nitrogen atom. The N-sulfonyl group proved to be crucial for reactivity, as replacement with the commonly used tert-butyl carbamate (N-Boc) protecting group yielded only unreacted starting material, potentially reflecting distinct coordination chemistry between sulfonamides and carbamates.16d
We then turned our attention to β,γ-unsaturated ketone substrates, where cesium carbonate (2 equiv.) and s-BuOH were identified as optimal base and solvent, respectively, at a reaction temperature of 40 °C (Table 2). We first evaluated para-substituted arylboronic acid coupling partners with different electronic properties and found that higher yield was obtained with boronic acids bearing an electron-withdrawing substituent (4ac–4ad). A representative example (4ad) was performed on 0.6 mmol scale, and 79% yield was obtained. Although the initial attempt towards 4aa only offered 37% yield, a higher yield could be achieved by using a higher catalyst loading or boronic acid loading. Electronic or steric modifications at the meta-position do not have a significant effect on reaction efficiency, with 4ae–4aj generated in good yields. Potentially reactive electrophilic substituents were well tolerated (4ag and 4ai). When ortho-substituted arylboronic acids were employed, higher yield was observed with electron-deficient aryl groups (4al and 4am), while moderate yield (36%) was obtained with ortho-tolylboronic acid (4ak). High-yielding hydroalkenylation was achieved with both aryl- and alkyl-substituted alkenylboronic acids (4an–4aq). To our delight, heteroaryl boronic acids were tolerated in this reaction, giving products 4ar–4at in moderate to good yield. Subsequently, we examined the scope of alkenyl ketone substrates. Within the aryl allyl ketones series, we found that a variety of aryl substituents were accommodated, leading to moderate to good yields (4ba–4bj). Alkyl-substituted ketones were also tolerated, though in the case of a cyclohexyl group (4bl), a diminished yield of 38% was obtained. To our delight, α-methyl substituted alkenyl ketones gave the corresponding product in 85% yield with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (4bm). When internal alkene was tested, 4bn was obtained in 56% yield. To showcase the synthetic utility of this reaction, the natural product (rac)-turmerone was synthesized in three steps from commercially available starting materials.18
:1 dr (4bm). When internal alkene was tested, 4bn was obtained in 56% yield. To showcase the synthetic utility of this reaction, the natural product (rac)-turmerone was synthesized in three steps from commercially available starting materials.18
| a Reaction conditions: reactions performed on 0.1 mmol scale. Percentages represent isolated yields. PMP = 4-methoxyphenyl, Ar1 = (4-methoxycarbonyl)phenyl. b Initial attempt with 5 mol% catalyst loading led to 37% isolated yield. When 10 mol% catalyst loading was applied, 58% isolated yield was obtained. Yield in parentheses was obtained with 3.0 equiv. of p-TolB(OH)2 and 5 mol% catalyst loading. c Reaction time was 2 h instead of 20 h to prevent potential ester exchange with solvent. Value in brackets represents the isolated yield of a reaction performed on 0.6 mmol scale. d Reaction performed with 10 mol% catalyst loading. |
|---|

|
Having tested sulfonamide and ketone directing groups, our focused then shifted to amide-based substrates. β,γ-Unsaturated amides were found to be prone to isomerization when stoichiometric base was used. Gratifyingly, when catalytic LiOt-Bu (5 mol%) in i-PrOH was employed, both hydroarylation and hydroalkenylation of alkenyl amides proceeded smoothly (Table 3). Generally speaking, compared the analogous ketone-containing substates, alkenyl amides react with lower regioselectivity. With the exception of para-(trifluoromethyl)phenylboronic acid (6a), which delivered only 45% yield, alteration of the electronic properties of the para-substituent did not affect the yield or selectivity in a significant way (6b–6e). When performed on larger scale, 6e was obtained in excellent yield with slightly lower regioselectivity. Electron-donating or -withdrawing groups at the meta-position gave high yield and regioselectivity of approximately 90:10 (6f–6h), whereas a potentially reactive meta-chloro-substituent gave 35% yield (6i). When alkenylboronic acids were used, Markovnikov-selective hydroalkenylation took place with even higher regioselectivity (>95:5, 6j–6l). Attenuated steric hindrance of the alkenylboron coupling partners compared to their aryl counterparts might account for the improved regioselectivity, since this could result in preferential stabilization of the selectivity-determining transmetalation transition state at a five-membered (and more hindered) secondary alkyl nickelacycle (leading to the Markovnikov-selective product) compared to at a six-membered (and less hindered) primary alkyl nickelacycle (leading to the anti-Markovnikov-selective) product (see below). Representative alkenyl amides were then tested to explore the scope and limitations of this method. Both secondary and tertiary amides were tolerated. N-(2,6-Dimethylphenyl)-substituted alkenyl amide gave 82% yield and 90:10 r.r. (6m). N-Alkyl-, N,N-dialkyl-, and N-alkyl-N-aryl-substituted amides gave moderate to good yield (6n–6v). Cyclic tertiary amides exhibited higher regioselectivity (6t, 6v). Notably, when γ,δ-unsaturated amide was tested, 62% combined yield was obtained of a 1:1 mixture of β- (6w) and δ-arylated (6w′) isomers, resulting from carbonyl-directed migratory hydroarylation19 and anti-Markovnikov hydroarylation, respectively (Scheme 3). This observation indicates that the favorable formation of a five-membered nickelacycle provides the driving force for selectivity and that alkylmetal chain walking (or metallacycle contraction) can take place when larger, less stable, metalacycles are formed upon initial hydronickelation. Formation of 6w′ may result from a competitive non-directed pathway.
| a Reactions performed on 0.1 mmol scale. Unless otherwise noted, percentages represent combined isolated yield of the two regioisomers, which were inseparable by silica gel chromatography. Regioisomeric ratio (r.r.) values represent Markovnikov/anti-Markovnikov product ratios, as determined via1H NMR analysis of isolated product mixtures. These values were generally consistent (±5%) with those determined directly from the crude reaction mixture. Ar1 = (4-methoxycarbonyl)phenyl. b Values in brackets represent the isolated yield and r.r. of a reaction performed on 0.6 mmol scale. |
|---|

|
 | ||
| Scheme 3 Reactivity with a representative γ,δ-unsaturated alkene substrate. Reaction performed on 0.1 mmol scale using standard conditions from Table 3. Ar1 = (4-methoxycarbonyl)phenyl. | ||
A detailed mechanistic study was performed to shed more light on the mechanism of the transformation (Scheme 4). First, to exclude a tandem isomerization/1,4-addition mechanism, the α,β-unsaturated amide and ketone that would be formed upon isomerization were tested under the optimal conditions. Only trace amounts (<5%) of the corresponding products were observed, which rules out this alternative pathway. Next, hydroarylation of N-benzyl β,γ-unsaturated amide was chosen as a model reaction for detailed kinetic investigation. In a kinetic isotope effect (KIE) experiment, we found vH/vD = 1.23, suggesting that hydrometallation might not be involved in the turnover-limiting step. In comparison, vH/vD = 2.7 was found in our previous study of Markovnikov-selective hydroarylation of alkenyl carboxylates.14 This distinction indicates that a different mechanism or a different turnover-limiting step is operative in this system.20 Deuterium labeling experiments using EtOD as solvent and boroxine as aryl source were conducted, showing deuterium incorporation mainly on the γ-position with scrambling on the α and β-positions to some extent. Both the deuterium scrambling and the presence of double deuterated product suggests that a reversible hydrometalation step is operative before the selectivity-determining step. To disambiguate between transmetalation and reductive elimination being turnover-limiting step, the experimental rate law was determined using the method of initial rates (see ESI† for detail). We found rate = kobs[5a][ArB(OH)2][Ni]total. This result is consistent with transmetalation being the turnover-limiting step. Altogether, the data are consistent with the following mechanism. First, hydrometalation proceeds through a reversible mechanism. Though a discrete Ni–H intermediate cannot be ruled out at this stage,2i,7c,d a series of related studies have recently pointed to concerted hydronickelation being lower in energy.3c,14,21 Either scenario would result in a common 5-membered alkyl nickelacycle, which rapidly equilibrates between with the corresponding 6-membered species, corresponding to Markovnikov and anti-Markovnikov selectivity, respectively. Next, turnover-limiting and selectivity-determining transmetalation takes place, followed by reductive elimination to furnish the desired product.
 | ||
| Scheme 4 Mechanistic experiments. Percentages represent 1H NMR yields with CH2Br2 as internal standard. (A) Control experiment with α,β-unsaturated ketone/amide as substrate. (B) KIE study of Markovnikov selective hydroarylation of alkenyl amide. (C) Deuterium incorporation study with ethanol-d1 as solvent. (D) Proposed catalytic cycle and experimental rate law, as determined by initial rate measurement and proposed mechanism. | ||
To gain a better understanding of the origin of regioselectivity, we next considered the turnover-limiting transmetalation step and the subsequent reductive elimination step computationally (Scheme 5).22 Despite the formation of a sterically and electronically disfavored secondary alkyl nickel species, the Markovnikov-selective pathway is still favored compared to the anti-Markovnikov-selective pathway by 1.0 kcal mol−1 in the transmetalation step. The same trend was observed when comparing the corresponding intermediates (7_a and 7_m). A structural analysis of these intermediates revealed a shorter bond length between the directing group and the nickel center in 7_m (1.92 Å) compared to 7_a (1.95 Å). This result indicates the formation of a stable five-membered metallacycle with the directing group is the key contributing factor for overcoming the thermodynamic preference of the formation of a primary alkyl nickel species. When alkenyl ketone was used as substrate, a larger energy barrier (ΔΔGsol = 2.4 kcal mol−1) was observed, which explains the higher regioselectivity obtained experimentally (see ESI† for detail). Subsequent C(sp3)–C(sp2) reductive elimination steps were found to have comparatively low barriers of 17.3 and 16.7 kcal mol−1 for TS2_m and TS2_a, respectively, with ethylene as model ligand for the different olefins that could coordinate under the reaction conditions (i.e., COD, substrate, or alkene-containing product). This model stems from previous work demonstrating that π-accepting ligands promote the otherwise high-energy C–C reductive elimination events.16
 | ||
| Scheme 5 (A) Computed energy profile of the hydroarylation of 5a. Calculations were performed at the B3LPY/SDD-6-311+G(d,p), SMD(2-propanol)//B3LYP/SDD-6-31G(d) level of theory. (B) Structural analysis of intermediate 7_a and 7_m. Bond distances are in angstroms. | ||
Conclusions
In summary, we established a series of reliable protocols for Markovnikov-selective hydroarylation/alkenylation of alkenes bearing a sulfonamide, ketone or amide as a directing group. With the support from a detailed mechanistic study, we found transmetalation is likely the turnover-limiting and selectivity-determining step. A computational study revealed that the directing-group-controlled formation of a five-membered alkyl nickel species is the origin of high Markovnikov selectivity.Data availability
All experimental procedures and data related to this study can be found in the ESI. Original NMR data for the products (in MNova format) are included in a master ZIP file.Author contributions
Z.-Q. L. and K. M. E. conceived the project. Z.-Q. L. optimized the reaction conditions and evaluated scope. Z.-Q. L., O. A., and R. D. synthesized the starting materials. Z.-Q. L. performed computational and experimental mechanistic studies. Z.-Q. L. and K. M. E. wrote the manuscript with input from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation (CHE-1800280). We acknowledge the NSF for Graduate Research Fellowships (DGE-1842471 to O. A.). We thank Tanner C. Jankins for careful proofreading and Prof. Peng Liu (University of Pittsburgh) for helpful discussion regarding the DFT studies.Notes and references
- For representative reviews, see: (a) Y. Yamamoto and U. Radhakrishnan, Chem. Soc. Rev., 1999, 28, 199–207 RSC; (b) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed; (c) P. Gandeepan and C. H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (d) E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503–1514 CrossRef CAS PubMed; (e) E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330–2343 CrossRef CAS PubMed; (f) Y. C. Luo, C. Xu and X. Zhang, Chin. J. Chem., 2020, 38, 1371–1394 CrossRef CAS; (g) Z. Liu, Y. Gao, T. Zeng and K. M. Engle, Isr. J. Chem., 2020, 60, 219–229 CrossRef CAS PubMed; (h) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed; (i) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC.
- For representative examples of hydroarylation of styrenes, see: (a) S. Torii, H. Tanaka and K. Morisaki, Chem. Lett., 1985, 14, 1353–1354 CrossRef; (b) G. Bhalla, J. Oxgaard, W. A. Goddard and R. A. Periana, Organometallics, 2005, 24, 3229–3232 CrossRef CAS; (c) Y. Iwai, K. M. Gligorich and M. S. Sigman, Angew. Chem., Int. Ed., 2008, 47, 3219–3222 CrossRef CAS PubMed; (d) Y.-P. Xiao, X.-Y. Liu and C.-M. Che, J. Organomet. Chem., 2009, 694, 494–501 CrossRef CAS; (e) G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258–10261 CrossRef CAS PubMed; (f) S. D. Friis, M. T. Pirnot and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 8372–8375 CrossRef CAS PubMed; (g) K. Semba, K. Ariyama, H. Zheng, R. Kameyama, S. Sakaki and Y. Nakao, Angew. Chem., Int. Ed., 2016, 55, 6275–6279 CrossRef CAS PubMed; (h) L. Jin, J. Qian, N. Sun, B. Hu, Z. Shen and X. Hu, Chem. Commun., 2018, 54, 5752–5755 RSC; (i) L.-J. Xiao, L. Cheng, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461–464 CrossRef CAS PubMed; (j) Y.-G. Chen, B. Shuai, X.-T. Xu, Y.-Q. Li, Q.-L. Yang, H. Qiu, K. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 3395–3399 CrossRef CAS PubMed; (k) X.-Y. Lv, C. Fan, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, CCS Chem., 2019, 1, 328–334 CrossRef CAS; (l) X.-W. Yang, D.-H. Li, A.-X. Song and F.-S. Liu, J. Org. Chem., 2020, 85, 11750–11765 CrossRef CAS PubMed; (m) L. J. Oxtoby, Z.-Q. Li, V. T. Tran, T. G. Erbay, R. Deng, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 8885–8890 CrossRef CAS PubMed.
- For representative examples of hydroarylation of 1,3-dienes, see: (a) L. Liao and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 10209–10211 CrossRef CAS PubMed; (b) J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580–15583 CrossRef CAS PubMed; (c) J. S. Marcum, T. R. Taylor and S. J. Meek, Angew. Chem., Int. Ed., 2020, 59, 14070–14075 CrossRef CAS PubMed; (d) T. Hamaguchi, Y. Takahashi, H. Tsuji and M. Kawatsura, Org. Lett., 2020, 22, 1124–1129 CrossRef CAS PubMed.
- For a review and representative examples of hydroarylation of α,β-unsaturated carbonyl coumpounds, see: (a) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed; (b) A. M. Vasquez, J. A. Gurak Jr, C. L. Joe, E. C. Cherney and K. M. Engle, J. Am. Chem. Soc., 2020, 142, 10477–10484 CrossRef CAS PubMed; (c) S. Cacchi and A. Arcadi, J. Org. Chem., 1983, 48, 4236–4240 CrossRef CAS.
- For representative examples of hydroarylation of heteroatom-substituted alkenes, see: (a) S. Bera and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 13854–13859 CrossRef CAS PubMed; (b) S. Cuesta-Galisteo, J. Schörgenhumer, X. Wei, E. Merino and C. Nevado, Angew. Chem., Int. Ed., 2021, 60, 1605–1609 CrossRef CAS PubMed; (c) Y. He, H. Song, J. Chen and S. Zhu, Nat. Commun., 2021, 12, 638 CrossRef CAS PubMed.
- For representative examples of hydroarylation reactions involving chain-walking, see: (a) Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS PubMed; (b) Y. He, C. Liu, L. Yu and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9186–9191 CrossRef CAS PubMed; (c) Y. Zhang, B. Han and S. Zhu, Angew. Chem., Int. Ed., 2019, 58, 13860–13864 CrossRef CAS PubMed.
- For recent examples involving bidentate directing auxiliaries, see: (a) C. Wang, G. Xiao, T. Guo, Y. Ding, X. Wu and T.-P. Loh, J. Am. Chem. Soc., 2018, 140, 9332–9336 CrossRef CAS PubMed; (b) R. Matsuura, T. C. Jankins, D. E. Hill, K. S. Yang, G. M. Gallego, S. Yang, M. He, F. Wang, R. P. Marsters, I. McAlpine and K. M. Engle, Chem. Sci., 2018, 9, 8363–8368 RSC; (c) H. Lv, L.-J. Xiao, D. Zhao and Q.-L. Zhou, Chem. Sci., 2018, 9, 6839–6843 RSC; (d) H. Lv, H. Kang, B. Zhou, X. Xue, K. M. Engle and D. Zhao, Nat. Commun., 2019, 10, 5025 CrossRef PubMed.
- For reviews of anti-Markovnikov-selective hydroarylation of non-conjugated alkenes with Ar–H coupling partners, see: (a) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS PubMed; (b) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468–3517 CrossRef CAS PubMed; (c) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed. For recent report involving Ni(0) catalysis, see: (d) N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nat. Chem., 2020, 12, 276–283 CrossRef CAS PubMed.
- For recent examples of anti-Markovnikov-selective hydroarylation and alkylation of non-conjugated alkenes involving a silane as the hydride source, see: (a) X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed; (b) J. Nguyen, A. Chong and G. Lalic, Chem. Sci., 2019, 10, 3231–3236 RSC; (c) Z. Wang, H. Yin and G. C. Fu, Nature, 2018, 563, 379–383 CrossRef CAS PubMed.
- For examples of reductive Heck hydroarylation of non-conjugated alkenes, see ref. 2h and 7a, and the following: (a) J. A. Gurak Jr and K. M. Engle, ACS Catal., 2018, 8, 8987–8992 CrossRef PubMed. For a review, see: (b) L. J. Oxtoby, J. A. Gurak Jr, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Trends Chem., 2019, 1, 572–587 CrossRef CAS PubMed.
- S. D. Friis, M. T. Pirnot, L. N. Dupuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 7242–7246 CrossRef CAS PubMed.
- T. Liu, Y. Yang and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 14256–14260 CrossRef CAS PubMed.
- For recent dual catalytic examples involving hydrogen-atom-transfer, see: (a) S. A. Green, J. L. M. Matos, A. Yagi and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 12779–12782 CrossRef CAS PubMed; (b) S. A. Green, S. Vasquez-Cespedes and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 11317–11324 CrossRef CAS PubMed; (c) S. L. Shevick, C. Obradors and R. A. Shenvi, J. Am. Chem. Soc., 2018, 140, 12056–12068 CrossRef CAS PubMed.
- Z.-Q. Li, Y. Fu, R. Deng, V. T. Tran, Y. Gao, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 23306–23312 CrossRef CAS PubMed.
- D.-M. Wang, W. Feng, Y. Wu, T. Liu and P. Wang, Angew. Chem., Int. Ed., 2020, 59, 20399–20404 CrossRef CAS PubMed.
- (a) J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed; (b) V. T. Tran, Z.-Q. Li, T. J. Gallagher, J. Derosa, P. Liu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7029–7034 CrossRef CAS PubMed; (c) J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 1201–1205 CrossRef CAS PubMed; (d) O. Apolinar, V. T. Tran, N. Kim, M. A. Schmidt, J. Derosa and K. M. Engle, ACS Catal., 2020, 10, 14234–14239 CrossRef CAS; (e) R. Kleinmans, O. Apolinar, J. Derosa, M. K. Karunananda, Z.-Q. Li, V. T. Tran, S. R. Wisniewski and K. M. Engle, Org. Lett., 2021, 23, 5311–5316 CrossRef CAS PubMed.
- M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211–5215 CrossRef CAS.
- P. P. Mahulikar and R. B. Mane, J. Chem. Res., 2006, 2006, 15–18 CrossRef.
- Substrate-directed, migratory alkene functionalization via hydronickelation has been previously documented. For oxidative Heck arylation, see ref. 7c. For hydroalkylation, see: (a) X. Chen, W. Rao, T. Yang and M. J. Koh, Nat. Commun., 2020, 11, 5857 CrossRef CAS PubMed. For related nickelacycle contraction processes in 1,3-dicarbofunctionalization, see: (b) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC; (c) P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. KC, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782–7786 CrossRef CAS PubMed.
- In addition, it might also explain our inability to identify effective ligands to promote anti-Markovnikov-selective hydrofunctionalization with these substrates, despite extensive screening (see ESI† for detail).
- (a) F. Wang and Q. Meng, J. Org. Chem., 2020, 85, 13264–13271 CrossRef CAS PubMed; (b) Q. Cheng and Y. Dang, Org. Lett., 2020, 22, 8998–9003 CrossRef CAS PubMed.
- Because the present reaction system does not employ traditional nitrogen- or phosphine-based ancillary ligands, it is difficult to unambiguously establish the preferred coordination environment in the hydronickelation step, which as discussed above also has an unclear mechanism. Due to these uncertainties we were not confident that DFT studies of this step would be conclusive. Additionally, given that hydronickelation was empirically determined not to be involved in the turnover-limiting step, we decided to focus the DFT studies on the transmetalation and subsequent reductive elimination steps.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03121j |
| This journal is © The Royal Society of Chemistry 2021 |