
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The switchable phosphorescence and delayed fluorescence of a new rhodamine-like dye through allenylidene formation in a cyclometallated platinum(II) system†‡
Shunan
Zhao
ab,
Yifan
Zhu
b,
Ling
Li
b,
Véronique
Guerchais
 *c,
Julien
Boixel
*c and
Keith Man-Chung
Wong
*b
*c,
Julien
Boixel
*c and
Keith Man-Chung
Wong
*b
aSchool of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 15001, China
bDepartment of Chemistry, Southern University of Science and Technology, 1088 Xueyuan Blvd., Shenzhen, 518055, P. R. China. E-mail: keithwongmc@sustech.edu.cn
cUniv Rennes, CNRS UMR6226, F-3500 Rennes, France. E-mail: veronique.guerchais@univ-rennes1.fr; julien.boixel@univ-rennes1.fr
First published on 7th July 2021
Abstract
A new rhodamine-like alkyne-substituted ligand (Rhodyne) was designed to coordinate a cyclometallated platinum(II) system. The chemo-induced “ON–OFF” switching capabilities on the spirolactone ring of the Rhodyne ligand with an end-capping platinum(II) metal centre can modulate the interesting acetylide–allenylidene resonance. The long-lived 3IL excited state of Rhodyne in its ON state as an optically active opened form was revealed via steady-state and time-resolved spectroscopy studies. Exceptional near-infrared (NIR) phosphorescence and delayed fluorescence based on a rhodamine-like structure were observed at room temperature for the first time. The position of the alkyne communication bridge attached to the platinum(II) unit was found to vary the lead(II)-ion binding mode and also the possible resonance structure for metal-mediated allenylidene formation. The formation of a proposed allenylidene resonance structure was suggested to rationalize these phenomena.
Introduction
Transition metal complexes have attracted much interest due to a variety of potential applications,1–7 associated with the capability of promoting an emissive triplet excited state at room temperature.8,9 Particularly, the controllable population of the triplet excited state by an external stimulus has become an intense research topic of interest, dedicated to the examples of pH- and photo-activatable photodynamic therapy (PDT) reagents,10–12 molecular logic gates13 and data storage,14 or fundamental photochemistry.15,16 The incorporation of switchable organic units into optical-active transition metal complexes is one common strategy to achieve metal-based photo-functional materials with “ON” and “OFF” states in a controllable manner.17–28 The design of multi-chromophoric systems capable of triplet sensitization and emission with a switching unit could lead to remote manipulation of the localization and the lifetime of the triplet emissive state.Rhodamine derivatives are found in equilibrium between two isomers with very different spectroscopic properties,29i.e. colourless and non-fluorescent ring-closed and highly coloured and fluorescent ring-opened structures about the spirocarbon. Because of this impressive attribute, together with their high sensitivity and selectivity, rhodamine derivatives have been widely applied in fluorescent probes.29–32 Although rhodamine was discovered over a century ago and widely investigated,33 most research about rhodamine has focused on its photophysics and photochemistry from the fluorescence of singlet excited states, studies about rhodamine triplet excited states are relatively limited.34,35 The introduction of a heavy atom into a rhodamine system acting as a photosensitizer has been employed to generate its triplet excited state for the application of photodynamic therapy.36–39 The research groups of Zhao40,41 and Eisenberg42 demonstrated that the attachment of a platinum(II) system into the rhodamine unit would facilitate the formation of a rhodamine triplet excited state from its singlet excited state. Our group has recently developed a versatile strategy to generate a rhodamine triplet state as organelle-targeting photosensitizers for efficient PDT through the ligation of rhodamine tethered chelate into transition metal systems.43,44 Such a long-lived triplet excited state of rhodamine is commonly found in a dark state at room temperature which can only be observed by transient absorption spectroscopy.
We have been devoted to the exploration of possible combinations of transition metal complexes with photo-switchable and chemo-switchable units, such as photochromic diarylethenes (DTE)22–24 and rhodamine-based derivatives,43–48 respectively. As an extension of our ongoing interest in controllable luminescence from transition metal systems, our previous results prompted us to design and investigate novel rhodamine-based cyclometallated platinum(II) complexes. A structural modification for a rhodamine-like derivative is rationally designed as a new ligand, in which the triplet excited state of the rhodamine-like ligand could be promoted, while the switching behaviour could remain at the same time. In this study, an ethynyl group on the xanthene unit is introduced to replace the amino or hydroxyl group in rhodamine or Rhodol, respectively, for the coordination of the transition metal centre. Based on the naming of Rhodol with a hydroxyl group, we label this new rhodamine-like ligand as Rhodyne herein (Chart 1).
 | ||
| Chart 1 The molecular structures of rhodamine, Rhodol, and a new rhodamine-like dye with an alkyne substituent on the xanthene unit (named Rhodyne). | ||
On the other hand, diverse examples of transition metal allenylidene complexes have been investigated because of their potential applications in catalysis49 and molecular electronics.50 On the contrary, the luminescence properties of this class of extended unsaturated complexes had been relatively unexplored until recent reports on the room-temperature phosphorescent allenylidene complexes of iridium(III),51 gold(I),52 gold(III)53 platinum(II),53 and palladium(II).54 In these systems, the alkylation or attachment of cationic N-heterocyclic carbene on the alkynyl group is a common strategy to achieve resonance forms of the allenylidene complex. The cationic Rhodyne in its opened form with the alkynyl linker at the 6′ position is anticipated to promote such resonance. As a result, the switching capability from the spirolactone ring on Rhodyne will be able to participate in the allenylidene formation in a controllable manner. Additionally, a higher metal contribution is envisaged to perturb the triplet excited state of the rhodamine-like dye through the delocalization of electron density over the main molecular skeleton, i.e. from the xanthene all the way to the metal centre across the allenylidene bridge, resulting from the extended resonance structure. In view of this, our design and approach in this regard is to tether the newly designed Rhodyne ligand to the cyclometallated platinum(II) luminophore, with the aims of (1) introduction of a convenient and reversible switch into the platinum(II) system to control the photophysical and photochemical properties; (2) exploration of the impact of the platinum(II) centre on the photophysical properties of the rhodamine-like dye and (3) manipulation of the localization and the nature of an emissive triplet excited state in the hybrid from an organic dye and a luminescent transition metal system.
As shown in Chart 2, two cyclometallated platinum(II) complexes, comprising of Rhodyne as a rhodamine-like derivative switch, a luminescent platinum(II) system and an alkynyl bridge, were designed. The ability to form an allenylidene complex, which may impact the luminescent behavior, would be inferred by the different positions of the alkynyl linker on Rhodyne. Herein we report the synthesis, characterization and spectroscopic studies of a new class of cyclometallated platinum(II) complexes, 1 and 2, with the Rhodyne ligand as the switchable regulator (Chart 2). The allenylidene complex formation in the opened-form of 1 with the alkynyl linker at a “suitable position” on Rhodyne is thought to be involved in the unusual rhodamine-like phosphorescence and delayed fluorescence.
 | ||
| Chart 2 The structural design of the cyclometallated platinum(II) complexes 1 and 2 with a switchable function rhodamine-like moiety. | ||
Results and discussion
Synthesis and characterization
In order to construct our designed cyclometallated platinum(II) system, two new Rhodyne ligands with an alkynyl group at different positions, L1 and L2, were firstly designed and synthesized (Scheme S1‡). The coordination of these two ligands into the cyclometallated chloroplatinum(II) complex precursor was achieved in base conditions with the catalysis of CuI, to afford 1 and 2 (Scheme S1‡). Complexes 1 and 2 were fully characterized and their molecular structures determined by X-ray crystallography (Fig. S1–S4‡). As shown in their perspective views in Fig. 1, the platinum(II) metal centre adopts a typical distorted square planar geometry with the cyclometallated N^N^C pincer ligand and the alkynyl linker. The geometry of the quaternary carbon of the spirolactone ring in both 1 and 2 clearly indicates the existence of the ring-closed form in the crystals.29–32 The angles between the xanthene core planes and the platinum(II)-C^N^N framework are 12.12° and 34.31° in 1 and 2, respectively.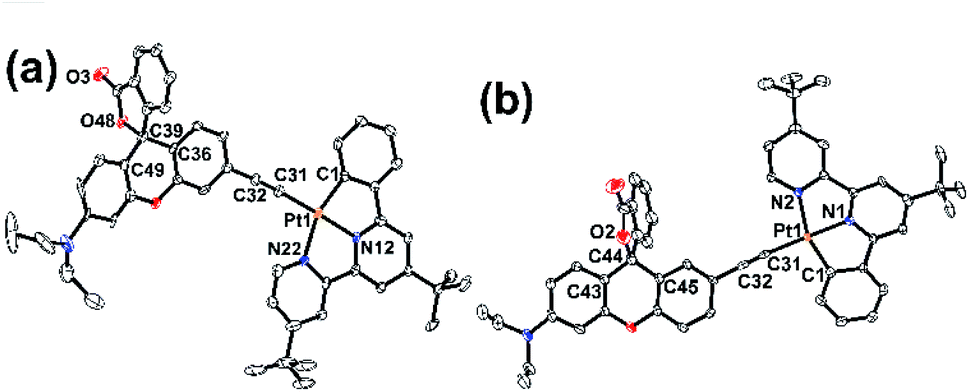 | ||
| Fig. 1 Perspective drawings of (a) 1 and (b) 2. Solvent molecules and hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
Basic photophysical properties in the closed form
Similar to related cyclometallated alkynylplatinum(II) systems,22,23,53,55 low-energy absorption bands at 412 nm and 430 nm (1) and 420 nm and 442 nm (2) were observed in the electronic absorption spectra in acetonitrile at room temperature (Fig. S5a‡), which were assigned to the admixture of [dπ(Pt) → π*(N^N^C)] metal-to-ligand charge transfer (MLCT) and [pπ(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) → π*(N^N^C)] ligand-to-ligand charge transfer (LLCT) transitions. Intense photoluminescence was also found in the acetonitrile solution of 1 (λmax = 572 nm; τ = 215 and 660 ns) and 2 (λmax = 598 nm; τ = 151 ns) (Fig. S5b‡), which originates from the triplet excited state of the 3MLCT/3LLCT character. The red-shift of the low-energy absorption and emission in 2, relative to 1, is ascribed to the stronger influence from the electron-rich alkoxy group at the para-position on the alkynyl ligand, leading to a rise of the HOMO energy level. A deoxygenated acetonitrile solution of 1 (3.1%) was found to show a higher photoluminescence quantum yield (PLQY) than 2 (1.9%). Their nanosecond transient absorption (ns-TA) different spectra (Fig. S6‡) show a positive absorption at around 372 nm which is assigned to the radical anion of N^N^C ligand absorption. In addition, the observation of a broad absorption from 450–800 nm extending beyond the detection range is attributed to the triplet absorption of the 3MLCT/3LLCT excited state.
C) → π*(N^N^C)] ligand-to-ligand charge transfer (LLCT) transitions. Intense photoluminescence was also found in the acetonitrile solution of 1 (λmax = 572 nm; τ = 215 and 660 ns) and 2 (λmax = 598 nm; τ = 151 ns) (Fig. S5b‡), which originates from the triplet excited state of the 3MLCT/3LLCT character. The red-shift of the low-energy absorption and emission in 2, relative to 1, is ascribed to the stronger influence from the electron-rich alkoxy group at the para-position on the alkynyl ligand, leading to a rise of the HOMO energy level. A deoxygenated acetonitrile solution of 1 (3.1%) was found to show a higher photoluminescence quantum yield (PLQY) than 2 (1.9%). Their nanosecond transient absorption (ns-TA) different spectra (Fig. S6‡) show a positive absorption at around 372 nm which is assigned to the radical anion of N^N^C ligand absorption. In addition, the observation of a broad absorption from 450–800 nm extending beyond the detection range is attributed to the triplet absorption of the 3MLCT/3LLCT excited state.
UV-vis absorption titration studies
Upon addition of p-toluene sulfonic acid, new structured absorption bands at 535 nm and 574 nm emerged in 1 as a result of the ring-opening process on Rhodyne, leading to a drastic colour change from yellow to purple in solution (Fig. 2a). In similar conditions, the ring-opened form of 2 obtained by addition of acid into the solution resulted in a colour change to magenta, which is attributed to the appearance of new structured absorption bands at 532 nm and 573 nm and an increase in absorbance at 432 nm (Fig. 2b). A similar amount of acid (1.1–1.2 equivalents of acid) was required to reach the absorbance plateau for the new absorption bands in 1 and 2 (Fig. 2 insets). The observation of a similar absorption response in L1 and L2 (Fig. S7‡) indicates the negligible influence from the alkynyl group at different substitution sites. In contrast, the absorption spectral changes upon ring opening in 1 show essential differences with those in 2. Collectively, the differences in the acid response between them are as follows: (i) the change in the absorbance at around 420 nm in 1 was very tiny while the absorbance at 432 nm was found to increase from 0.16 to 0.50 in 2; (ii) a much higher absorbance (around 6-fold) was generated in 1 for the newly emerged structured absorption band (from 500 to 600 nm) compared to that in 2; (iii) two isosbestic points at 348 nm and 422 nm were observed in 1, which were absent in 2; and (iv) the shapes of the newly emerged absorption bands were different with Huang–Rhys factors of 0.66 (1) and 0.84 (2). | ||
| Fig. 2 The electronic absorption spectral changes of (a) 1 (conc. = 10 μM) and (b) 2 (conc. = 25 μM) upon the addition of various equivalents of protons in acetonitrile at room temperature. The insets show plots of absorbance versus the concentration of protons and the solution colors in the opened forms. | ||
Apart from the acid-promoted ring-opening on the spirolactone ring of Rhodyne, a similar chemo-responsive process could also be triggered by metal ions in 1 and 2. Corresponding absorption titration studies of 1 upon the addition of Pb(II) ion exhibited similar spectral changes with new structured absorption bands, which are ascribed to the opened form of Rhodyne (Fig. 3a). Scrutinizing their spectral changes revealed that the absorption peak at 572 nm upon the addition of 0.5 equivalents of Pb(II) ions was 83% as high as that at its saturation with about 1.2 equivalents of Pb(II) ions (Fig. S8a‡). By monitoring the absorbance at the absorption tail at 620 nm, further addition of Pb(II) ions beyond 0.67 equivalents of Pb(II) ion leads to a decrease in the absorbance, suggesting a two-stage binding process (Fig. S8b‡). Both Job's plot and theoretical binding fitting (Fig. 3a insets) also supported the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb(II) ion = 2:1 binding model, where the initial addition of Pb(II) ion afforded the {[1]2˙Pb2+} species which will be further converted into the {1˙Pb2+} adduct with excess Pb(II) ions. The binding constants of 5.90 ± 0.13 (logK21) and 5.67 ± 0.21 (logK11) as well as the detection limit of 13.9 nM were determined. The normalized absorption spectrum of 1 with excess Pb(II) ions matched very well with that in excess acid (Fig. S8b‡), indicating that the 1:1 bound species mainly exists in the presence of high concentrations of Pb(II) ions. For 2, new structured absorption bands at 520 nm and 567 nm emerged and the absorbance of the peak at ca. 430 nm increased upon the addition of Pb(II) ions (Fig. 3b). In contrast, both the theoretical binding fitting and Job's plot (Fig. 3b insets) supported the 1:1 binding model. The detection limit of 76 nM and the logKs value of 6.32 ± 0.17 for the Pb(II) ion binding to 2 were found. The absence of 2:1 bound species in 2 is possibly due to the unfavourable formation of such a sandwich adduct as a result of stronger mutual repulsion from the bulky cyclometallated platinum(II) moiety at the position in close proximity to the spirolactone group.
:Pb(II) ion = 2:1 binding model, where the initial addition of Pb(II) ion afforded the {[1]2˙Pb2+} species which will be further converted into the {1˙Pb2+} adduct with excess Pb(II) ions. The binding constants of 5.90 ± 0.13 (logK21) and 5.67 ± 0.21 (logK11) as well as the detection limit of 13.9 nM were determined. The normalized absorption spectrum of 1 with excess Pb(II) ions matched very well with that in excess acid (Fig. S8b‡), indicating that the 1:1 bound species mainly exists in the presence of high concentrations of Pb(II) ions. For 2, new structured absorption bands at 520 nm and 567 nm emerged and the absorbance of the peak at ca. 430 nm increased upon the addition of Pb(II) ions (Fig. 3b). In contrast, both the theoretical binding fitting and Job's plot (Fig. 3b insets) supported the 1:1 binding model. The detection limit of 76 nM and the logKs value of 6.32 ± 0.17 for the Pb(II) ion binding to 2 were found. The absence of 2:1 bound species in 2 is possibly due to the unfavourable formation of such a sandwich adduct as a result of stronger mutual repulsion from the bulky cyclometallated platinum(II) moiety at the position in close proximity to the spirolactone group.
 | ||
| Fig. 3 Electronic absorption spectral changes of (a) 1 (conc. = 10 μM) and (b) 2 (conc. = 25 μM) upon the addition of various concentrations of Pb(II) ions in acetonitrile. The insets show plots of absorbance versus the concentration of Pb(II) ions and the theoretical fitting curves; Job's plots; and the solution colours in the opened forms. | ||
Emission titration studies
Emission titration studies of 1 and 2 were carried out in air-saturated acetonitrile and the addition of acid or Pb(II) ion resulted in similar spectral changes for the individual complex (Fig. S9 and S10‡). Upon excitation at the isosbestic point of 348 nm for 1 or at 330 nm for 2 (with minimal changes in absorbance), the original 3MLCT/LLCT emission in their closed forms was found to be diminished with the concentration of acid or Pb(II) ions. Two residual emission band maxima (540 nm and 620 nm for 1; 549 nm and 610 nm for 2) were observed with an excess of acid (Fig. S9‡) or Pb(II) ions (Fig. S10‡). The emission at 540 nm or 549 nm, for those excitation spectra are similar to the corresponding absorption spectra in the closed form, originating from the platinum(II) 3MLCT/LLCT excited state. A related cyclometallated platinum(II) complex with a positively charged alkynyl group was reported to exhibit similar 3MLCT/LLCT emission at 530 nm.53 The binding constants for 1 (logK21, 5.85; logK11, 6.11) and 2 (logK11, 6.62) are in good agreement with those determined by absorption studies, indicating that the emission quenching is attributed to the ring-opening process on Rhodyne.
Since no platinum(II) MLCT/LLCT state could be populated at such low-energy excitation, the ring-opening effect on the Rhodyne excited state is clearly identified. Upon excitation at 540 nm, new emission bands at 620 nm and 610 nm were found to gradually emerge with increasing the concentration of Pb(II) ions in 1 and 2 (Fig. 4). The origin of these emissions is reasonably assigned as the Rhodyne singlet intraligand (1IL) excited state. It is noteworthy that a new near-infrared (NIR) emission band at ca. 750 nm appeared in the solution of 1 with excess Pb(II) ions after removal of the oxygen (Fig. 4a). The excitation spectra of these emissions at 620 nm and 750 nm closely resemble the absorption spectrum of the ring-opened form of 1 featuring the characteristic Rhodyne 1IL absorption (Fig. S11‡), indicating that this emission originates from the opened form of Rhodyne. Given the large stokes shift of 4150 cm−1 (from 572 nm to 750 nm) as well as the nature of the oxygen sensitivity (Fig. 4a), the origin of such NIR emission is assigned as derived from the Rhodyne triplet intraligand (3IL) excited state. Kinetic studies of such NIR emission showing the lifetime in the microsecond regime (vide infra) further support the assignment of triplet parentage. Such phosphorescence of the ring-opened form of 1 was also observed in 1 with excess acid in deoxygenated acetonitrile. Similar to the triplet excited state of organic dyes, the rhodamine-based triplet excited state is commonly considered as a dark state and can be probed by transient absorption spectroscopy.40–44 To the best of our knowledge, this is the first example of direct observation of the rhodamine-like originated phosphorescence in solution state at room temperature. In contrast, such phosphorescence was not found in the deoxygenated solution of 2 in its ring-opened form. On the other hand, the emission responses of L1 and L2 upon ring opening are essentially the same (Fig. S12‡). On the basis of these findings, the position at which the alkynylplatinum(II) unit is attached to the Rhodyne is suggested to be a key factor leading to a significant difference in the Rhodyne 3IL excited state properties in 1 and 2. From consideration of the alkynylplatinum(II) group which is attached to the 6′ position on Rhodyne in 1, the positive charge created at a “suitable position” of the ring-opened form is envisaged to facilitate or contribute to the formation of the allenylidene complex with extended resonance throughout the xanthene core, allenylidene bridge and the platinum(II) metal centre (Chart 3). On the contrary, the alkynylplatinum(II) moiety in 2 is instead tethered at the 7′ position to the xanthene oxygen atom and the generation of the corresponding resonance structure is not allowed. It is reasonably suggested that the Rhodyne phosphorescence in the ring-opened form of 1 is mediated by such an allenylidene complex as a result of the enhanced involvement of the platinum(II) metal centre.
 | ||
| Fig. 4 Emission spectral changes in acetonitrile upon the addition of various concentrations of Pb(II) ions to (a) 1 and (b) 2 at an excitation wavelength of 540 nm; conc. = 10 μM. The insets show plots of the relative emission intensity versus the equivalents of Pb(II) ions. Blue line: deoxygenated solution with an excess of Pb(II) ions. | ||
 | ||
| Chart 3 Resonance structures of 1 and 2 illustrating the possibility of platinum(II) allenylidene formation through the positive charge created on Rhodyne in their ring-opened forms. | ||
Computational studies
In order to elucidate the dependence of the alkynyl position tethered on the Rhodyne for the possibility of allenylidene formation, computational studies were performed. Optimized structures of 1 and 2 (Fig. S13a and b‡) revealed that the spiro-rings are in a closed state, as indicated by their X-ray crystal structures and photophysical behaviours. The opened forms of 1 and 2 were mimicked by replacing the isobenzofuran-1(3H)-one group with a phenyl ring and by adding one more positive charge throughout the whole structure (Fig. S13c and d‡). Compared to its closed form, it is noteworthy that both of the bond distances of Pt–Cα and Cβ–Cγ in the opened form of 1 are shortened by 0.02 Å, while the bond distance of Cα–Cβ is slightly elongated by 0.01 Å (Table S3‡). On the other hand, no such changes in the bond distances of Pt–Cα and Cβ–Cγ between the closed and opened forms of 2 were computed. Given the very similar structures of 1 and 2, their discrepancies from the changes of bond distances are attributed to the variation of the connecting positions between Rhodyne and the alkynylplatinum(II) moiety. For the opened form of 1, the extent of the changes in the bond distances from the theoretical calculations is less significant than those in the iridium(III) system possessing acetylide–allenylidene resonance structure.51 This can be rationalized by the less dominant allenylidene mesomeric structure in the opened form of 1 because the positive charge is delocalized mainly throughout the extended conjugation over the xanthene structure. The computational results reasonably support the formation of the allenylidene complex in the opened form of 1. In sharp contrast, the corresponding allenylidene complex cannot be generated in the opened form of 2 due to the “unmatched” position of the alkynyl group on Rhodyne, as shown in Chart 3.Time-resolved emission and nanosecond transient absorption spectroscopy studies
Nanosecond gated emission measurements of the ring-opened 1 at 532 nm-pulsed laser excitation were performed in order to obtain the emission dynamics from the Rhodyne excited sate, with a minimal influence from the 3MLCT state. The emission spectra in deoxygenated acetonitrile exhibit two emission bands at around 620 nm and 740 nm, while their relative emission intensities were found to be similar going from 0.0 to 14.0 μs after the laser pulse (Fig. 5a). Based on the aforementioned steady-state emission study of the ring-opened form of 1, the emission bands at 620 nm and 740 nm originate from the Rhodyne singlet and triplet excited states, respectively. The emission lifetimes could be estimated by the decay fit of emission intensities at 620 nm and 740 nm (Fig. 5b and c). The long-lived NIR emission at 740 nm (τ = 3.29 μs) suggests the nature of the phosphorescence from the Rhodyne triplet excited state. This is further supported by the observation of long-lived species (τ = 3.39 μs) from the nanosecond transient absorption study (vide infra). It is noteworthy that the lifetime for the Rhodyne-based fluorescence at 620 nm was found to be unprecedentedly long (3.44 μs) and similar to the corresponding phosphorescence within experimental error. The prolonged lifetime suggests that such long-lived emission at 620 nm is attributed to the delayed fluorescence, while the absence of the short-lived Rhodyne prompted fluorescence is reasonably due to the cut-off from time zero setting from the instrument to eliminate the nanosecond laser pulse scattering. Such delayed fluorescence and phosphorescence of the opened form of 1 also emerged in deoxygenated dichloromethane (with 1% acetonitrile) (Fig. S14a‡) with longer lifetimes of τ = 12.88 μs and 13.19 μs (Fig. S14b and c‡), respectively. According to the proposed resonance structure in Chart 3, the d orbitals of the Pt centre can be involved in the IL transition through the linker. As a result, another MLCT (from Pt to Rhodyne) character is mixed with this IL state. This MLCT transition is probably less sensitive to the solvent polarity based on the similar time-resolved emission spectra in acetonitrile and dichloromethane (with 1% acetonitrile). On the other hand, computational studies showed that the contributions to the HOMO and LUMO in their lowest triplet state (T1) from the Pt center and CC group are higher in the opened form of 1 (Table S4 and Fig. S15‡), compared to that of 2, which can further support certain electronic mixing between 3IL and 3MLCT states. In the air-saturated solution of the ring-opened 1 sample, the emission spectra from the pulsed laser excitation also showed the Rhodyne fluorescence and NIR phosphorescence at 620 nm and 740 nm, respectively (Fig. S16a‡). However, the emission lifetime at 740 nm was significantly shortened from 3.54 μs to 420 ns (Fig. S16b‡) in the air-saturated solution, which further confirms the triplet nature of the emission. The observation of the similar lifetime (390 ns) for the emission at 620 nm (Fig. S16c‡) also suggested the assignment of delayed fluorescence, which is essentially generated from the triplet state. For the open form of 2, the main difference is the absence of phosphorescence from the rhodamine triplet excited state as well as the delayed fluorescence, probably due to the forbidden formation of the allenylidene complex.
 | ||
| Fig. 5 (a) Time-resolved emission spectra of 1 in the ring-opened form in deoxygenated acetonitrile at room temperature and the corresponding lifetime decay traces monitored at (b) 620 nm (with the exclusion of the first data point) and (c) 740 nm. Conc. = 15 μM (1) and 45 μM (Pb2+). A pulsed laser at 532 nm was the excitation source. | ||
Three possible mechanisms for delayed fluorescence, (I) triplet–triplet annihilation (TTA) with dominant phosphorescence or dominant delayed fluorescence, (II) recombination of geminately bound electron-hole pairs (GP) and (III) E-type delayed fluorescence or so-called thermally activated delayed fluorescence (TADF), have been reported.56–58 An attempt has been made to investigate the dependence of emission intensity on excitation laser fluence ranging from 1–213 mJ. The relative delayed fluorescence intensity to phosphorescence is similar with a ratio (IDF/IPhos) of about 0.27 (Fig. S17‡). However, no conclusive results could be obtained from the log–log plot of emission intensity versus laser power, probably due to the small changes in the emission intensity in the low laser power regime as well as the saturation effect in the high laser power regime. In spite of this, the aforementioned time-resolved measurements could still prove valuable and give some clues. By virtue of the lifetimes of the phosphorescence at 740 nm and the delayed fluorescence at 620 nm in the opened form of 1 being essentially the same, the assignment of delayed fluorescence from the TTA mechanism is unlikely because the lifetime of the delayed fluorescence from TTA should be half of the phosphorescence in this mechanism.56 In addition, a control experiment was conducted, in which time-resolved emission studies were carried out in viscous solvent, which was anticipated to avoid TTA delayed fluorescence because of the reduced molecular diffusion. The persistent observation of delayed fluorescence with a lifetime of 23.53 μs in glycerol triacetate (Fig. S18‡) was also inconsistent with the TTA mechanism. There was a report on the delayed fluorescence of rhodamine dyes coming from the GP recombination mechanism at 77 K, high energy excitation at 355 nm is required to populate the GP pair.56 In contrast, delayed fluorescence of the ring-closed form of 1 could also be observed from the lower energy of laser pulsed excitations at 532 nm. In addition, both the delayed fluorescence and phosphorescence were found to follow first-order kinetics with no power law relation, suggesting that the GP recombination mechanism is also less possible.58 Therefore, the origin of the delayed fluorescence is tentatively assigned to the TADF. In this mechanism, equal lifetimes of phosphorescence and delayed fluorescence would be suggested because the singlet and triplet state populations are in thermal equilibrium. Variable-temperature emission spectroscopic studies are a common way to verify the TADF mechanism. Upon increasing the temperature, enhanced delayed fluorescence intensity and decreased phosphorescence intensity would be anticipated due to the population of the singlet state from the triplet state through thermally activated process. However, the ring-opened form of 1 will undergo backwards ring-closure conversion in low temperature under −40 °C, the temperature ranging from −5 °C to 60 °C for variable-temperature emission measurement was therefore employed. The phosphorescence intensity at 740 nm was found to drop significantly upon increasing temperature, while the fluorescence intensity at 620 nm only slightly changed (Fig. S19a‡). From the basic knowledge of photophysics, the emission intensity would be weakened as a result of the increase in non-radiative decay with increasing temperature. From the normalized spectra at 740 nm, the relative fluorescence intensity to the phosphorescence is enhanced from −5 °C to 60 °C (Fig. S19b‡). Independent variable-temperature steady-state emission measurements for the opened form of L1 showed that the fluorescence was significantly diminished upon increasing the temperature (Fig. S20‡). In view of this, another opposite effect from TADF is suggested to account for the small change in fluorescence intensity at 620 nm. The energy gap between the singlet and triplet states (ΔS–T) of about 0.39 eV was estimated from the onsets of fluorescence and phosphorescence energies, molecules in comparable ΔS–T values were reported to exhibit TADF.59,60
With reference to other related studies on rhodamine-containing transition metal systems,41–44 transient absorption (TA) spectroscopy is a valuable tool to probe the 3IL of rhodamine. In order to verify the excited state nature, nanosecond transient absorption (TA) spectroscopies of 1 and 2 in their ring-opened forms were investigated with the excitation of 355 nm pulsed laser. The TA spectra of the ring-opened form of 1 in deoxygenated acetonitrile displayed an intense ground state depletion signal at 570 nm, which is characteristic of the negative Rhodyne absorption profile (bleaching) (Fig. 6a). Such a ground state depletion signal at 570 nm with a lifetime of 3.39 μs is assigned as the Rhodyne 3IL state, which is in line with the lifetime of the phosphorescence at 740 nm. For the ring-opened form of 2, a similar strong bleaching signal with a typical negative Rhodyne absorption profile was also found at around 550 nm in the TA spectrum (Fig. 6b), with a prolonged lifetime of 48.15 μs. Both in the opened forms of 1 and 2, the observation of positive TA signals at 350 nm, 410–420 nm and 600–750 nm with similar lifetimes were ascribed to the 3IL absorption of Rhodyne.41,42 On the basis of the shorter lifetime of the Rhodyne 3IL state in 1, a more efficient 3IL → S0 transition arising from the enhanced spin–orbit coupling is anticipated. This can be rationalized by the more substantial involvement or contribution of the platinum(II) metal centre in the Rhodyne 3IL state due to the formation of the allenylidene complex in the opened form of 1. Using a lower-energy wavelength of 532 nm pulsed laser for the excitation of the opened form of 1 also generated the same transient absorption signals in different solvents (Fig. S21‡). The lifetimes for the depletion signal at 570 nm (3.50 μs in MeCN; 12.83 μs in dichloromethane with 1% acetonitrile and 20.75 μs in glycerol triacetate) are in accordance with the phosphorescence lifetimes from the time-resolved emission spectroscopy.
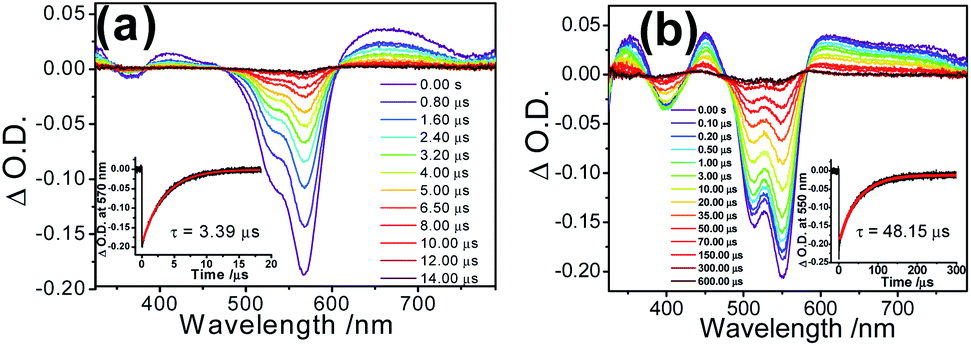 | ||
| Fig. 6 Transient absorption spectral changes of (a) 1 (conc. = 10 μM) and (b) 2 (conc. = 20 μM) in their ring-opened forms with excess amounts of Pb(II) ions in deoxygenated acetonitrile at room temperature. The insets show the transient absorption lifetime fitting of the signals monitored at 570 nm and 550 nm for 1 and 2, respectively. Excitation wavelength = 355 nm. | ||
The presence of the long-lived triplet excited state of Rhodyne is anticipated to interact with molecular oxygen (3O2) to form singlet oxygen (1O2). The ability of the opened forms of 1 and 2 to produce 1O2 was evaluated spectroscopically by the observation of 1O2 emission at about 1270 nm (Fig. S22‡). Moderate 1O2 generation quantum yields (ΦΔ) of 0.38 (1) and 0.35 (2) were determined by using rose Bengal as a reference (ΦΔ in MeCN = 0.45)61 with excitation at 514.5 nm. Alternatively, the singlet oxygen generation abilities of both 1 and 2 in their ring-opened forms were studied using the 1O2 scavenger 1,3-diphenylisobenzofuran (DPBF) indicator. For both compounds, significant absorbance drops were observed at 410 nm (Fig. S23‡), indicating the generation of singlet oxygen.
Based on the steady-state and time-resolved emission, as well as the nanosecond transient absorption spectroscopies in deoxygenated solvents, the simplified energy diagram of the ring-opened form of 1 with various excited states is suggested (Fig. 7). With excitation at around 348 nm (steady state measurements) or 355 nm (time-resolved studies) for the 1MLCT/LLCT transition of the platinum(II) moiety of the ring-opened form of 1, the emission intensity from the original 3MLCT/LLCT state diminished while Rhodyne 1IL fluorescence as well as the 3IL phosphorescence emerged. On the basis of this, together with our recent work on rhodamine-containing transition metal systems,43,44 the higher-lying platinum(II) 3MLCT/LLCT state would be relaxed to the Rhodyne triplet intraligand (3IL) excited state via triplet–triplet energy transfer (TTET). The observation of residual 3MLCT/LLCT emission overlapping with the Rhodyne 1IL fluorescence with such a higher-energy excitation (Fig. S24‡) indicates that the TTET process is partially efficient. However, the possibility of direct population of the 1IL state from the 3MLCT state cannot be completely ruled out. If lower-energy excitation at 540 nm (steady state measurements) or 532 nm (time-resolved studies) was employed, the Rhodyne 1IL state was exclusively excited, which leads to the Rhodyne 1IL fluorescence and 3IL phosphorescence through intersystem crossing (ISC). It is noteworthy that delayed fluorescence, which is derived from the Rhodyne 3IL state through reversed intersystem crossing (RISC), was also observed from different excitation energies. Since the allenylidene complex formation is only possible in complex 1 with the alkynyl linker at a “suitable position” on Rhodyne, unusual rhodamine-like phosphorescence at 740 nm from its triplet state was observed at room temperature. In addition, an interesting delayed fluorescence at 620 nm of this rhodamine-like is also featured. Unlike the reports from Castellano and co-workers on transition metal systems with organic rylenes62–64 which exhibited an equilibrium between the 3MLCT and 3IL states, the delayed fluorescence in this work is derived from an equilibrium between the 1IL and 3IL states of Rhodyne. For the opened form of 2, a higher-energy excitation for the 1MLCT/LLCT transition of a platinum(II) moiety or lower-energy excitation for the Rhodyne 1IL state would also generate a Rhodyne triplet intraligand (3IL) excited state. Such a 3IL excited state is a dark state and could only be probed by nanosecond transient absorption spectroscopy. Unlike the ring-opened form of 1, no phosphorescence was observed in that of 2 from the steady-state and time-resolved emission studies. In addition, the absence of delayed fluorescence from time-resolved emission measurements excludes the occurrence of RISC from the 3IL to 1IL state.
 | ||
| Fig. 7 A simplified energy diagram of the ring-opened form of 1. IL = intraligand excited state of Rhodyne; MLCT/LLCT = metal-to-ligand charge-transfer with mixing of ligand-to-ligand charge transfer character; a superscript of “1” = singlet state; a superscript of “3” = triplet state; ISC = intersystem crossing; RISC = reversed intersystem crossing; TTET = triplet–triplet energy transfer; PF = prompt fluorescence; DF: delayed fluorescence; and Phor. = phosphorescence. The energy levels were estimated based on the onsets of emission energies. | ||
Conclusions
In conclusion, two cyclometallated alkynylplatinum(II) complexes tethered with rhodamine-like (Rhodyne) ligands showing controllable photophysical behaviour were described. Via the judicious choice of the tethering position on Rhodyne, the formation of an allenylidene complex can be generated upon the ring-opening of the spirolactone group. Specifically, NIR phosphorescence originating from Rhodyne 3IL at 740 nm and delayed fluorescence at 620 nm were featured in the solution state at room temperature. Such an observation of NIR phosphorescence and delayed fluorescence originating from a rhodamine-like structure at room temperature occurs for the first time, and it is mediated by allenylidene formation with a resonance structure throughout the xanthene core, an allenylidene bridge, and the platinum(II) metal centre. The present study provides a strategic molecular design for lighting up the dark state of an organic excited state, not only providing new insight into fundamental photophysics but also allowing the exploitation of optical functional materials.Data availability
Data for this paper, including experimental and computational details, characterization, X-ray crystallographic information, other photophysical results are available at https://doi.org/10.1039/d0sc04751a.Author contributions
S. Z. contributed to synthesis and characterization, X-ray diffraction analysis, and optical measurements. Y. Z. contributed to the optical measurements. L. L. contributed to the DFT calculations. V. G. designed the project and performed data analysis. J. B. conducted the project, performed the synthesis and characterization, and prepared the manuscript. K. M. C. W. designed and conducted the project, performed data analysis, and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC–CNRS_PRC Joint Research Projects Program (grant no. 21911530232). K. M. C. W. acknowledges the National Natural Science Foundation of China (grant no. 21771099) and Science, Technology and Innovation Commission of Shenzhen Municipality (grant no. JCYJ20170817110721105 and JCYJ20190809165411528) and the assistance of SUSTech Core Research Facilities. This collaborative work was also financially supported by the CNRS in the framework of a joint PRC program (20181101321). We gratefully acknowledge T. Roisnel and V. Dorcet for X-ray diffraction studies and Ho-Chuen Lam for his initial involvement in ligand synthesis.Notes and references
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762–1785 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- Y. Cakmak, S. Kolemen, S. Duman, Y. Dede, Y. Dolen, B. Kilic, Z. Kostereli, L. T. Yildirim, A. L. Dogan, D. Guc and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 11937–11941 CrossRef CAS PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- J. A. G. Williams, in Photochemistry and Photophysics of Coordination Compounds II, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 205–268 Search PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- J. Zhao, S. Ji, W. Wu, W. Wu, H. Guo, J. Sun, H. Sun, Y. Liu, Q. Li and L. Huang, RSC Adv., 2012, 2, 1712–1728 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- S. O. McDonnell, M. J. Hall, L. T. Allen, A. Byrne, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2005, 127, 16360–16361 CrossRef CAS PubMed.
- B. Li, L. Lin, H. Lin and B. C. Wilson, J. Biophotonics, 2016, 9, 1314–1325 CrossRef CAS PubMed.
- S. Erbas-Cakmak and E. U. Akkaya, Angew. Chem., Int. Ed., 2013, 52, 11364–11368 CrossRef CAS PubMed.
- R. J. Mortimer and N. M. Rowley, Metal Complexes as Dyes for Optical Data Storage and Electrochromic Materials, in Comprehensive Coordination Chemistry II, Pergamon, Oxford, 2003, pp. 581–619 Search PubMed.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W.-W. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- V. W. W. Yam, V. C. Y. Lau and K. K. Cheung, J. Chem. Soc., Chem. Commun., 1995, 259–261 RSC.
- V. W. W. Yam, C. C. Ko, L. X. Wu, K. M. C. Wong and K. K. Cheung, Organometallics, 2000, 19, 1820–1822 CrossRef CAS.
- C. C. Ko, L. X. Wu, K. M. C. Wong, N. Zhu and V. W. W. Yam, Chem.–Eur. J., 2004, 10, 766–776 CrossRef CAS PubMed.
- V. W. W. Yam, C. C. Ko and N. Zhu, J. Am. Chem. Soc., 2004, 126, 12734–12735 CrossRef CAS PubMed.
- C. C. Ko and V. W. W. Yam, Acc. Chem. Res., 2018, 51, 149–159 CrossRef CAS PubMed.
- J. Boixel, V. Guerchais, H. Le Bozec, D. Jacquemin, A. Amar, A. Boucekkine, A. Colombo, C. Dragonetti, D. Marinotto, D. Roberto, S. Righetto and R. De Angelis, J. Am. Chem. Soc., 2014, 136, 5367–5375 CrossRef CAS PubMed.
- J. Boixel, Y. Zhu, H. Le Bozec, M. A. Benmensour, A. Boucekkine, K. M. C. Wong, A. Colombo, D. Roberto, V. Guerchais and D. Jacquemin, Chem. Commun., 2016, 52, 9833–9836 RSC.
- H. Zhao, E. Garoni, R. Roisnel, A. Colombo, C. Dragonetti, D. Marinotto, S. Righetto, D. Roberto, D. Jacquemin, J. Boixel and V. Guerchais, Inorg. Chem., 2018, 57, 7051–7063 CrossRef CAS PubMed.
- H. Tian and J. Zhang, Photochromic Materials: Preparation, Properties and Applications, Wiley-VCH, Weinheim, Germany, 2016 Search PubMed.
- W. Tan, Q. Zhang, J. Zhang and H. Tian, Org. Lett., 2009, 11, 161–164 CrossRef CAS PubMed.
- P. Belser, L. de Cola, F. Hartl, V. Adamo, B. Bozic, Y. Chriqui, V. M. Iyer, R. T. F. Jukes, J. Kühni, M. Querol, S. Roma and N. Salluce, Adv. Funct. Mater., 2006, 16, 195–208 CrossRef CAS.
- M. T. Indelli, S. Carli, M. Ghirotti, C. Chiorboli, M. Ravaglia, M. Garavelli and F. Scandola, J. Am. Chem. Soc., 2008, 130, 7286–7299 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed.
- C. Wang and K. M. C. Wong, Inorg. Chem., 2013, 52, 13432–13441 CrossRef CAS PubMed.
- E. Noelting and K. Dziewonski, Ber. Dtsch. Chem. Ges., 1905, 38, 3516–3527 CrossRef CAS.
- X. Cui, J. Zhao, Z. Lou, S. Li, H. Wu and K. Han, J. Org. Chem., 2015, 80, 568–581 CrossRef CAS PubMed.
- F. Wang, X. Cui, Z. Lou, J. Zhao, M. Bao and X. Li, Chem. Commun., 2014, 50, 15627–15630 RSC.
- B. Calitree, D. J. Donnelly, J. J. Holt, M. K. Gannon, C. L. Nygren, D. K. Sukumaran, J. Autschbach and M. R. Detty, Organometallics, 2007, 26, 6248–6257 CrossRef CAS.
- J. E. Hill, M. K. Linder, K. S. Davies, G. A. Sawada, J. Morgan, T. Y. Ohulchanskyy and M. R. Detty, J. Med. Chem., 2014, 57, 8622–8634 CrossRef CAS PubMed.
- M. R. Detty, P. N. Prasad, D. J. Donnelly, T. Ohulchanskyy, S. L. Gibson and R. Hilf, Bioorg. Med. Chem., 2004, 12, 2537–2544 CrossRef CAS PubMed.
- K. A. Leonard, J. P. Hall, M. I. Nelen, S. R. Davies, S. O. Gollnick, S. Camacho, A. R. Oseroff, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 2000, 43, 4488–4498 CrossRef CAS PubMed.
- L. Huang, L. Zeng, H. Guo, W. Wu, W. Wu, S. Ji and J. Zhao, Eur. J. Inorg. Chem., 2011, 4527–4533 CrossRef CAS.
- P. Majumdar, X. Cui, K. Xu and J. Zhao, Dalton Trans., 2015, 44, 4032–4045 RSC.
- G. Li, M. F. Mark, H. Lv, D. W. McCamant and R. Eisenberg, J. Am. Chem. Soc., 2018, 140, 2575–2586 CrossRef CAS PubMed.
- C. J. Liu, L. H. Zhou, F. F. Wei, L. Li, S. N. Zhao, P. Gong, L. T. Cai and K. M. C. Wong, ACS Appl. Mater. Interfaces, 2019, 11, 8797–8806 CrossRef CAS PubMed.
- L. Zhou, F. Wei, J. Xiang, H. Li, C. Li, P. Zhang, C. Liu, P. Gong, L. Cai and K. M. C. Wong, Chem. Sci., 2020, 11, 12212–12220 RSC.
- C. Y. Wang and K. M. C. Wong, Inorg. Chem., 2011, 50, 5333–5335 CrossRef CAS PubMed.
- K. M. C. Wong, C. Y. Wang, H. C. Lam and N. Y. Zhu, Polyhedron, 2015, 86, 133–140 CrossRef CAS.
- C. Y. Wang, H. C. Lam, N. Y. Zhu and K. M. C. Wong, Dalton Trans., 2015, 44, 15250–15263 RSC.
- Y. K. Cheng, L. Li, F. F. Wei and K. M. C. Wong, Inorg. Chem., 2018, 57, 6439–6446 CrossRef CAS PubMed.
- V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed.
- R. F. Winter and S. Záliš, Coord. Chem. Rev., 2004, 248, 1565–1583 CrossRef CAS.
- F. Kessler, B. F. E. Curchod, I. Tavernelli, U. Rothlisberger, R. Scopelliti, D. Di Censo, M. Gratzel, Md. K. Nazeeruddin and E. Baranoff, Angew. Chem., Int. Ed., 2012, 51, 8030–8033 CrossRef CAS PubMed.
- X.-S. Xiao, C. Zou, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem. Commun., 2016, 52, 4983–4986 RSC.
- X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem.–Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed.
- C. Zou, J. Lin, S. Suo, M. Xie, X. Chang and W. Lu, Chem. Commun., 2018, 54, 5319–5322 RSC.
- W. Lu, B.-X. Mi, M. C. W. Chan, Z. Hui, C.-M. Che, N. Zhu and S.-T. Lee, J. Am. Chem. Soc., 2004, 126, 4958–4971 CrossRef CAS PubMed.
- M. Aydemir, V. Jankus, F. B. Dias and A. Monkman, Chem. Chem. Phys., 2014, 16, 21543–21549 RSC.
- A. Hayer, H. Bässler, B. Falk and S. Schrader, J. Phys. Chem. A, 2002, 106, 11045–11053 CrossRef CAS.
- K. T. Chan, G. S. M. Tong, W. P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352–2364 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- F. Stracke, M. Heupel and E. Thiel, J. Photochem. Photobiol., A, 1999, 126, 51–58 CrossRef CAS.
- J. E. Yarnell, J. C. Deaton, C. E. McCusker and F. N. Castellano, Inorg. Chem., 2011, 50, 7820–7830 CrossRef CAS PubMed.
- F. N. Castellano, Dalton Trans., 2012, 41, 8493–8501 RSC.
- J. E. Yarnell, K. A. Wells, J. R. Palmer, J. M. Breaux and F. N. Castellano, J. Phys. Chem. B, 2019, 123, 7611–7627 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Dr Jean-René Hamon for the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization data, and additional photophysical data. CCDC 1503542 and 1507269. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02787e |
| This journal is © The Royal Society of Chemistry 2021 |