
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent development and applications of semipinacol rearrangement reactions
Xiao-Ming Zhang
 a,
Bao-Sheng Li
*b,
Shao-Hua Wang
*a,
Kun Zhang
c,
Fu-Min Zhang
a and
Yong-Qiang Tu
a
a,
Bao-Sheng Li
*b,
Shao-Hua Wang
*a,
Kun Zhang
c,
Fu-Min Zhang
a and
Yong-Qiang Tu
a
aState Key Laboratory of Applied Organic Chemistry and School of Pharmacy, Lanzhou University, Lanzhou, 730000, P. R. China
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400030, P. R. China
cSchool of Biotechnology and Health Sciences, Wuyi University, Jiangmen, Guangdong 529020, P. R. China
First published on 28th June 2021
Abstract
As has been well-recognized, semipinacol rearrangement functions as an exceptionally useful methodology in the synthesis of β-functionalized ketones, creation of quaternary carbon centers, and construction of challenging carbocycles. Due to their versatile utilities in organic synthesis, development of novel rearrangement reactions has been a vibrant topic that continues to shape the research field. Recent breakthroughs in novel electrophiles, tandem processes, and enantioselective catalytic transformations further enrich the toolbox of this chemistry and spur the strategic applications of this methodology in natural product synthesis. These achievements will be discussed in this minireview.
Xiao-Ming Zhang studied chemistry at Lanzhou University where he received his B.S. in 2007. He then joined the research group of Prof. Yong-Qiang Tu and completed his PhD at Lanzhou University in 2013. Currently, he is an associate professor at the Department of Chemistry, Lanzhou University. His research mainly focuses on total synthesis and biomimetic synthesis of natural products. |
Bao-Sheng Li obtained his B.S. at Guizhou University in 2008. He then obtained his PhD degree from Lanzhou University (with Prof. Xiao-Ping Cao) in 2013. After postdoctoral research at Nanyang Technological University, Singapore (with Prof. Yonggui Robin Chi), he was appointed as a Tenure-Track assistant professor at Chongqing University (China) in 2016. His research interest focuses on the construction of nitrogen-containing heterocycles and their application in the synthesis of pharmaceuticals. |
Shao-Hua Wang obtained his PhD in organic chemistry from Lanzhou University with Professor Yong-Qiang Tu in 2006. From 2006 to 2011, he was a postdoctoral associate in Professor Yuan-Ping Pang's group at the Mayo Clinic. He was appointed as an associate professor at the School of Pharmacy Lanzhou University in 2011 and promoted to full professor in 2017. His current research interests involve the development of synthetic methodologies and their application in the syntheses of natural products, and medicinal chemistry. |
Kun Zhang received his B.S. and M.S. from Guangdong polytechnic college in 1985 and 1990, respectively, and obtained his PhD in 1997 from Lanzhou University. He was appointed a lecturer at Guangdong University of Technology in 1985 and promoted to a full professor in 1999. Currently, his research interest includes natural products and Chinese medicines. |
Fu-Min Zhang received his PhD degree from Lanzhou University in 2006 working under the supervision of Prof. Yong-Qiang Tu. Subsequently, he worked as a postdoctoral fellow at the Mayo Clinic with Prof. Yuan-Ping Pang (2007–2008). He returned to Lanzhou University as a lecturer and was then appointed as an associate professor in 2009 and a full professor in 2014. Currently, he is interested in the total synthesis of natural products. |
Yong-Qiang Tu received his B.S. and M.S. from Lanzhou University in 1982 and 1985, respectively. He obtained his PhD in organic chemistry in 1989 from Lanzhou University under the supervision of Prof. Yao-Zu Chen. After spending three years (1993–95) as a postdoctoral fellow in W. Kitching's group at Queensland University, Australia, he was appointed a full professor at Lanzhou University in 1995 and became Director of the State Key Laboratory of Applied Organic Chemistry from 2001 to 2010. In 2009, he was elected an Academician of the Chinese Academy of Sciences. |
Introduction
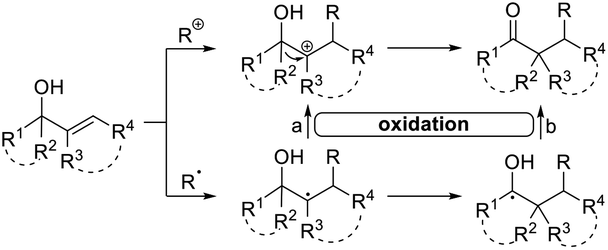
The highly efficient formation of carbon–carbon bonds and construction of corresponding molecular skeletons are two representative key scientific topics in organic synthesis. Therefore, rearrangement reaction related synthetic strategy development, which can simultaneously achieve the above two synthetic goals in a very efficient manner, has been an evergreen topic in the synthetic chemistry community. Among the rearrangement reactions developed so far, semipinacol rearrangement, as a typical 1,2-migration process, has also attracted the broad attention of synthetic chemists. Since the inception of the term “semipinacol” to describe a specific subclass of pinacol rearrangement reactions by Tiffeneau in 1923,1 the definition of this research area has continuously extended with advancement of novel types of reactions.2,3 Generally, despite the diverse reaction patterns semipinacol rearrangement may involve, a typical process often entails the generation of a carbonyl group through a 1,2-C–C or C–H bond migration from an oxygen-containing carbon to a vicinal carbon center, which is archetypally an electrophilic carbon3 though a C-radical center has also been suggested in recent emerging radical rearrangements (Scheme 1).4 A noteworthy aspect of this class of 1,2-migration reactions is that it provides a reliable and efficient approach to access synthetically useful β-functionalized ketones. In addition, the semipinacol rearrangement also serves as a potent and versatile method for the construction of quaternary carbon centers5 that are challenging to create but pervasive in natural molecules and pharmaceuticals.6–13 These remarkable features render this rearrangement reaction a powerful tool in natural product synthesis,3,14,15 and the utilization of this rearrangement to assemble intricate carbocycles from easily accessible ones, probably the most exciting application of such reactions, has become a subject of particular interest.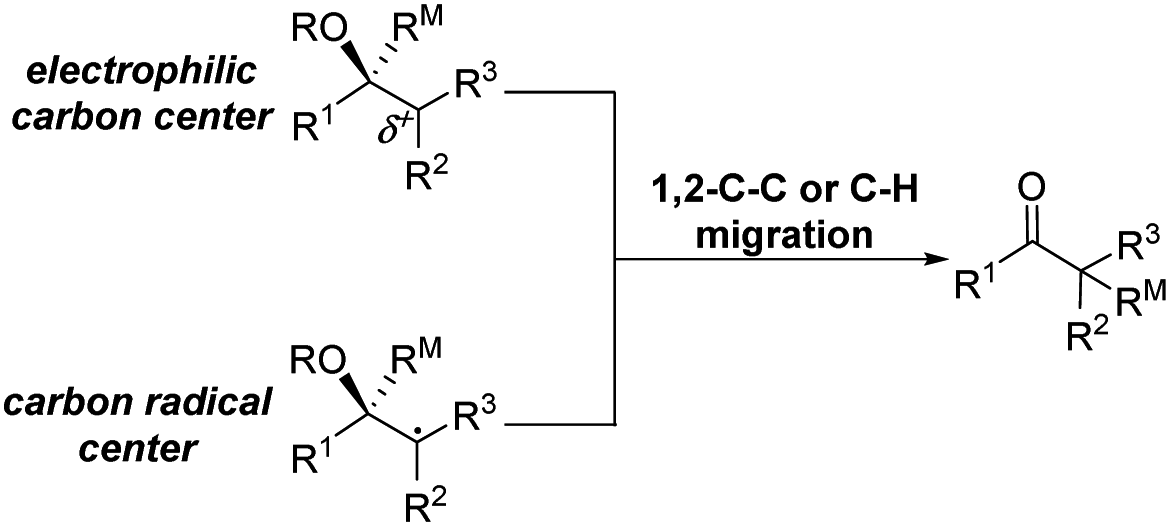 | ||
| Scheme 1 A typical process of semipinacol rearrangement. | ||
As the development of semipinacol rearrangement in the last two decades has gradually become mature, summarization of a certain aspect of the venerable reaction has been documented in several previous reviews.2–5,14–17 Nonetheless, recent advances of semipinacol rearrangement in terms of novel electrophiles, radical processes,4 tandem reactions, and catalytic enantioselective transformations16,17 have further enriched the repertoire of this chemistry. These breakthroughs are accompanied by the prevailing application of the rearrangement reactions as a key step/strategy in the assembly of complex natural products. In some cases, a semipinacolase is even elucidated to be responsible for catalyzing the key rearrangement in the biosynthetic pathway of certain natural molecules.18 Therefore, a summary of these contributions, as presented in this review, would provide not only an up-to-date overview of this research field but also guidance for its future development.
The new patterns and applications of semipinacol rearrangement covered in this review will be classified on the basis of four substrate types—epoxides, allylic alcohols, β-heterosubstituted alcohols, and α-hydroxy ketones and imines. Although some of the basic transformations of each type of substrate have been well-established, recent progress has focused on tandem and asymmetric rearrangement modes. These representative examples along with the extension of radical initiated rearrangement of allylic alcohols will be discussed herein, with an emphasis of the recent synthetic applications in each section.
Epoxides
Epoxides are a classical type of substrate in semipinacol rearrangement. In the presence of an acid, ring-opening of oxirane would result in the formation of an α-hydroxy carbocation that could induce an ensuing 1,2-migration to produce a carbonyl product. Meanwhile, the resulting aldehydes or ketones could also participate in various tandem processes, such as the Tishchenko reaction, allylation, propargylation, and the Schmidt reaction, to produce structurally complex 1,3-diols or amides.5 In these cases, pre-preparation of epoxides from the corresponding alkenes or allylic alcohols is generally required. Thus, the development of a one-pot or tandem epoxidation/semipinacol reaction would be more step-economic since an alkene substrate could be directly used in such a cascade. In this regard, Jiao and coworkers recently developed an oxygenation/semipinacol rearrangement of cyclobutyl-derived allylic carbinol 1 (Scheme 2a).19 Notably, oxygen was used as the epoxidation reagent and Cu(OTf)2 as the catalyst in this reaction, and a catalytic amount of Lewis acid was sufficient to induce the desired rearrangement. Apart from allylic alcohols, a cascade process of ortho-hydroxy styrenes 4 was devised by Sun's group for the synthesis of hydrodibenzofurans 6 (Scheme 2b).20 The formation of an ortho-quinone methide intermediate 5 was suggested after the epoxidation, which would trigger the ring-expansion of the resulting cyclopentanol to generate 6 with two consecutive quaternary stereocenters.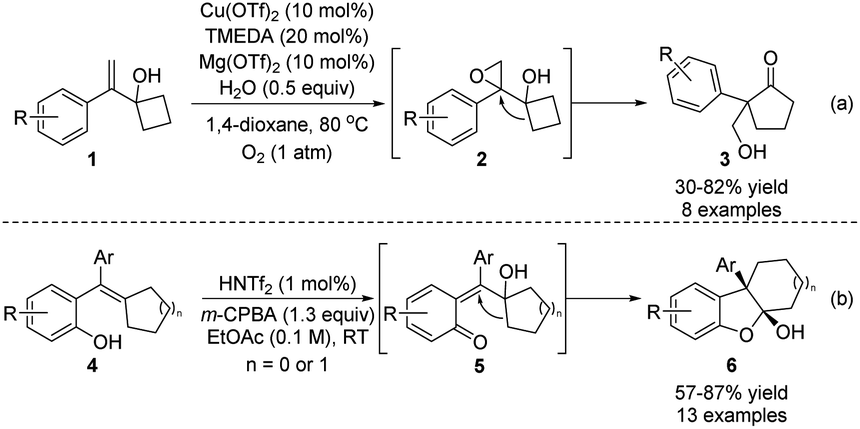 | ||
| Scheme 2 Epoxidation/semipinacol rearrangement reactions. | ||
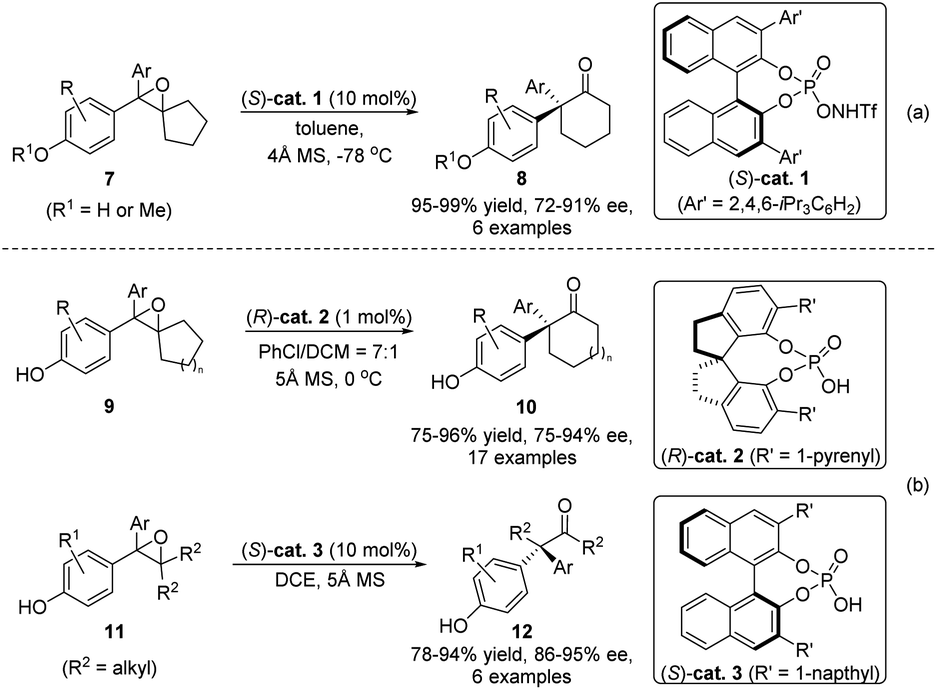
As the 1,2-migration of 2,3-epoxy alcohols often proceeds via a stereospecific process with the migrating group antiperiplanar to the C–O bond of the oxirane, access to enantioenriched β-hydroxy ketone products with these reactions predominantly relies on the initial preparation of enantiopure epoxides. However, for racemic epoxide substrates with only one stereogenic center, regioselective ring-opening of the oxirane would form a prochiral α-hydroxy carbocation intermediate, and thus an ensuing enantioselective migration could potentially take place to produce valuable enantioenriched ketones or aldehydes. Such a catalytic stereoconvergent process has independently been realized by Zhu's and Sun's groups at the same time (Scheme 3).21,22 Under the catalysis of either chiral N-triflyl phosphoramide (S)-cat. 1 or phosphorous acids (R)-cat. 2 or (S)-cat. 3, racemic epoxides 7, 9, or 11 could be converted to enantioenriched α-quaternary ketones 8, 10 or 12, respectively, with high yields and enantioselectivities. Mechanistically, formation of a chiral counteranion of the deprotonated catalyst and the α-hydroxy carbocation was deemed to be crucial for inducing an enantioselective rearrangement.
 | ||
| Scheme 3 Catalytic asymmetric semipinacol rearrangement of racemic epoxides. | ||
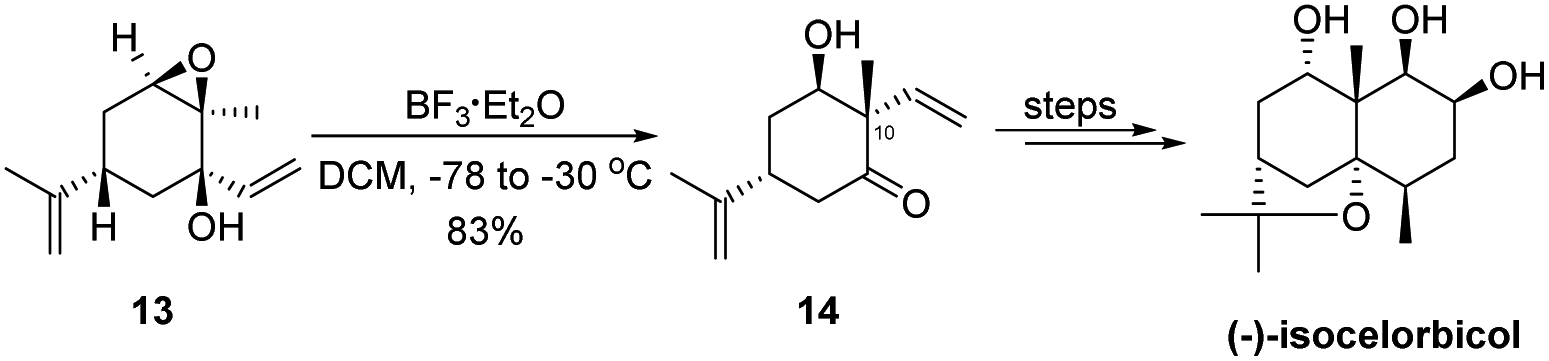
The semipinacol rearrangement of epoxides has found diverse applications in natural product synthesis during the last two decades.3,14,15 The utility of this reaction for quaternary carbon center formation with stereochemical control has been well-recognized and continues to play an essential role in the synthesis of key intermediates of natural molecules. For a recent example, the 1,2-migration of 2,3-epoxy tertiary alcohol 13 would provide α-quaternary-β-hydroxy cyclohexanone 14 that serves as a key building block in the total synthesis of (−)-isocelorbicol by Ogura and coworkers (Scheme 4).23 The correct installation of the C10 quaternary carbon center is attributed to a stereoselective rearrangement of the vinyl group anti to the epoxide.
 | ||
| Scheme 4 Building block preparation in the total synthesis of (−)-isocelorbicol. | ||
In 2020, Chen and Yang's group proposed an elegant total synthesis of (−)-spirochensilide A.24 During their early-stage synthesis, a two-step epoxidation/semipinacol ring-contraction consequence has been developed to convert a trans-octahydronaphthalene system 15 to an octahydroindene carbocycle 17 with vicinal quaternary carbon centers (Scheme 5). Of note, only a single diastereoisomer was obtained in the rearrangement reaction of 16, indicating a highly regio- and diastereochemical control of the ring-contraction process, and the aldehyde functionality introduced through the rearrangement could be further elaborated as a functional handle.
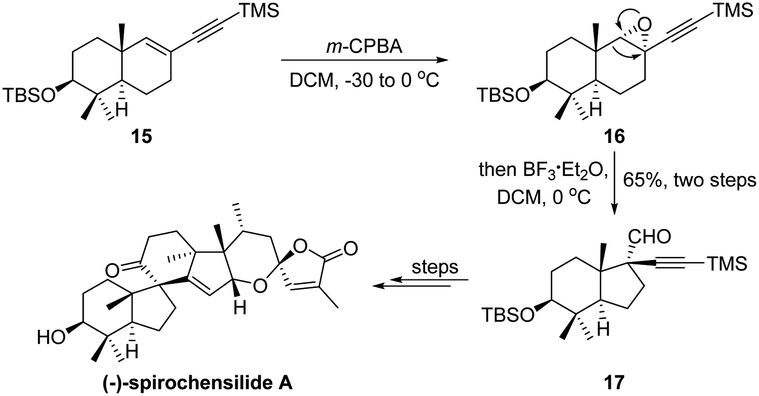 | ||
| Scheme 5 Epoxidation/semipinacol ring-contraction in the total synthesis of (−)-spirochensilide A. | ||
The skeletal 1,2-rearrangement could also be applied as a crucial strategy to guide synthetic planning. For the assembly of highly strained or complex carbocycles, alternation of the cyclic ring system via a retro-1,2-migration might provide an easily accessible precursor from the retrosynthetic viewpoint. As a representative example, such a strategy has been ingeniously applied in the total synthesis of (−)-illisimonin A (Scheme 6).25 As the trans 5–5 system of the molecule presents a well-recognized synthetic challenge, Rychnovsky and coworkers devised a key skeletal rearrangement to access the trans ring-system from its cis counterpart since it is calculated to be less strained (7.03 kcal mol−1 more favorable). Furthermore, this cis-carbocycle 21 could be readily prepared from 2-methylcyclopentane-1,3-dione 18 in three steps via an intramolecular Diels–Alder reaction of α,β-unsaturated cyclopentenone 19. The late-stage semipinacol rearrangement of 2,3-epoxy alcohol 22 derived from ester 21 would provide the tricyclic ketone 23 with the establishment of the strained trans 5–5 ring. Finally, the application of White's acid-directed C–H oxidation of 24 enabled the construction of the bridging lactone and completed the total synthesis.26,27
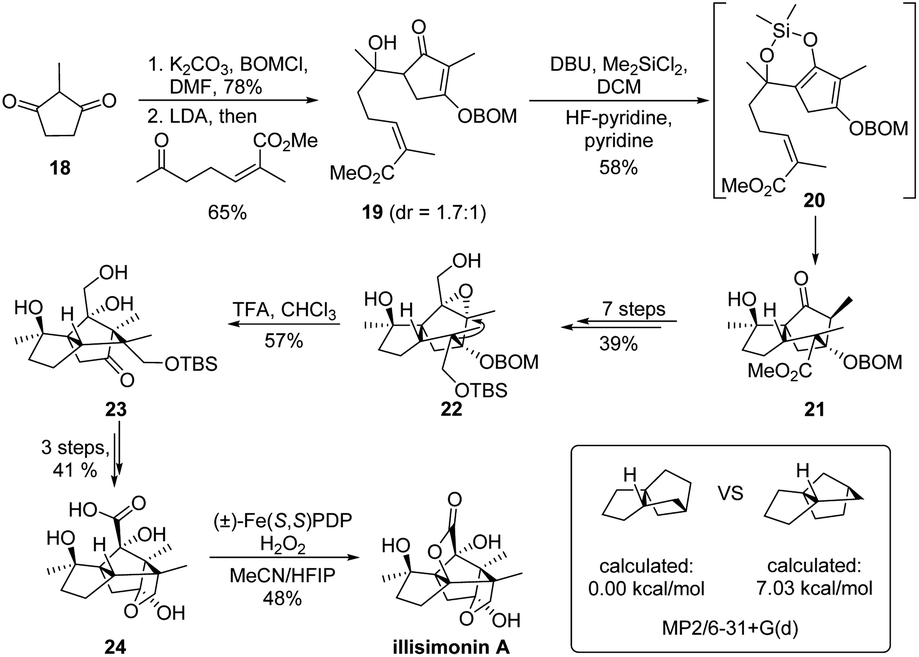 | ||
| Scheme 6 Strategic application of semipinacol rearrangement in the total synthesis of illisimonin A. | ||
Allylic alcohols
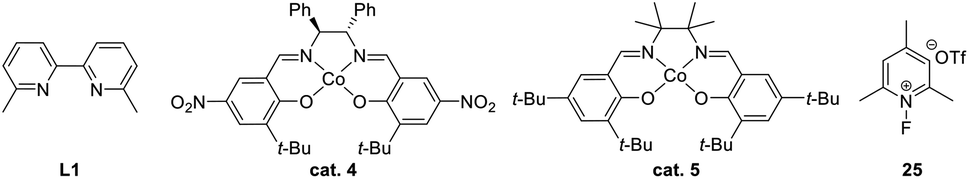
Allylic alcohols are a privileged type of substrate in the development of novel semipinacol rearrangements because of the availability of these compounds from simple chemical feedstocks. In classical reactions, activation of the alkene group by an electrophile would generate an α-hydroxy carbocation intermediate, thus triggering the subsequent 1,2-migration to produce β-functionalized ketones (Table 1). Alternatively, addition of a radical species to the double bond is also feasible. In this case, an intermediary α-hydroxy radical is initially generated, which in most cases is further oxidized to the α-hydroxy carbenium before the rearrangement (Table 1, path a). However, when the migrating group is an arene or heteroarene, a neophyl-type 1,2-radical migration followed by oxidation is usually suggested (Table 1, path b). Although a wide range of electrophiles capable of initiating the rearrangement have been well-documented,3,5 interest in the discovery of novel types of electrophiles and radical initiators4,28 continues. Among these breakthroughs, novel catalytic techniques, such as photocatalysis and electrocatalysis, have also enabled reactions under environmentally benign conditions, thus preventing the use of excess oxidants or toxic reagents.|
|
||||
|---|---|---|---|---|
| Electrophile or radical | Reagent | Reaction conditions | Product | Ref. |
| NO+ | NOBF4 | DTBP (15 mol%), CH3CN, −30 °C | 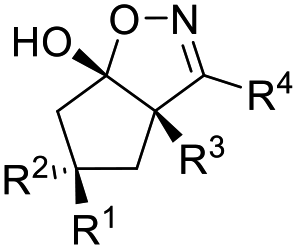 |
29 |
| (SO2Ph)2N˙ | (SO2Ph)2NF | CuCl (10 mol%), L1 (10 mol%), DCE, 70 °C | 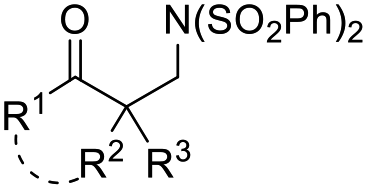 |
30 |
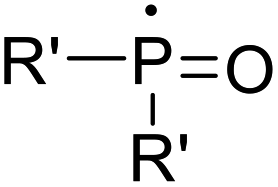 |
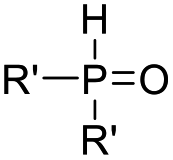 |
Eosin Y (10 mol%), DMA, RT, 12 W blue LEDs | 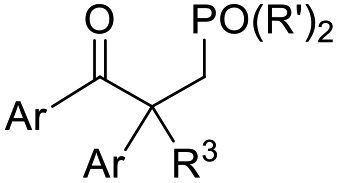 |
31 |
 |
fac-Ir(ppy)3 (1 mol%), dioxane, RT, 5 W blue LEDs | 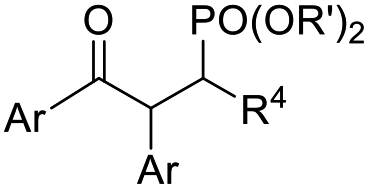 |
32 | |
| ˙SCF3 or +SCF3 | AgSCF3 | K2S2O8 (1.5 equiv.), pyridine, CH3CN, 65 °C | 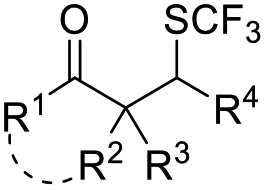 |
33 |
| PhNHSCF3 | CH3CN, AcCl, RT | 34 | ||
| N-Trifluoromethyl thiosaccharin | Et3N (10 mol%), Boc-L-PHE (10 mol%), TMSCl, CH3CN, RT | 35 | ||
| +SCN | (PhSO2)2NSCN | AcCl or TMSCl, DCE, RT | 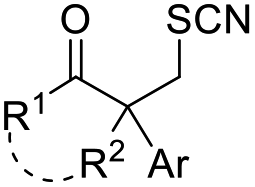 |
36 |
| ˙SO2Ar | ArN2BF4, (DABCO)·(SO2)2 | fac-Ir(ppy)3 (4 mol%), CH3CN, RT, 8 W blue LEDs | 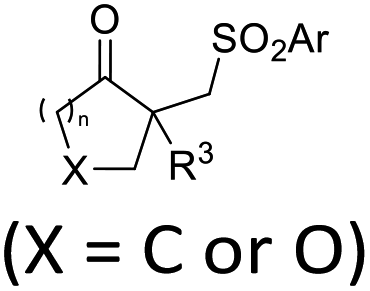 |
39 |
| ArSO2H | fac-Ir(ppy)3 (3 mol%), CH3CN, TBHP, RT, blue LEDs | 40 | ||
| ArSO2Cl | fac-Ir(ppy)3 (3 mol%), CH3CN, RT, blue LEDs | 41 | ||
| ArSO2NH2NH2 | Pt(+)–Pt(−), 5 mA undivided cell, THF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), NaI, RT :1), NaI, RT |
42 | ||
| PhSO2Na | C(+)–Pt(−), 10 mA undivided cell, LiClO4, CH3CN/H2O (1:1), RT |
43 | ||
| +SeR | PhSeBr | DCM, RT | 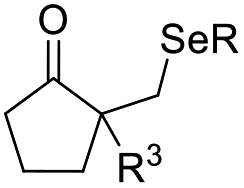 |
37 |
| RSeSeR | I2, EtOH/H2O (1:1), RT |
38 | ||
| +X (X = halogen) | NaX (X = Cl, Br, or I) | PIDA, H2O, 40 °C |  |
44 |
| MgBr2 or MgCl2 | C(+)–Pt(−), 25 mA undivided cell, CH3CN/H2O (6:3), RT |
45 | ||
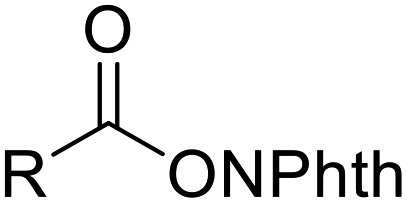
| ˙R (R = alkyl) | RBr | [Au2(dppm)2]Cl2 (5 mol%), Na2CO3, DABCO, CH3CN, UVA LEDs |  |
46 |
| RB(OH)2 | [Ir(ppy)2(dtbbpy)]PF6 (1 mol%), BIOAc, CH3CN, RT, 4 W blue LEDs | 47 | ||
 |
fac-Ir(ppy)3 (3 mol%), DMA, RT, 6 W white LEDs | 48 | ||
| ˙CF3 | 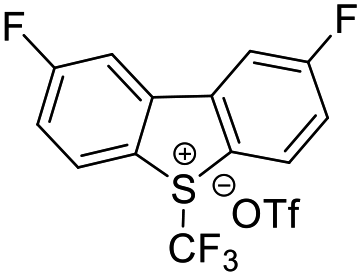 |
DMSO, K2HPO4, RT, 48 W white LEDs | 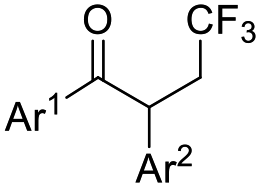 |
49 |
| CF3SO2Na | Pt(+)–Pt(−), 3 mA undivided cell, LiClO4, CH3CN/H2O (1:1), RT |
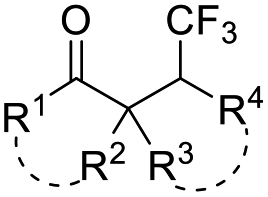 |
50 | |
| CF3SO2Na | C(+)–Pt(−), 15 mA, undivided cell, LiClO4, CH3CN/H2O (2:1), RT |
43 | ||
| ˙H | PhSiH2Me | cat. 4 or cat. 5 (5 mol%), 25, acetone, 0 °C | 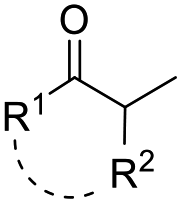 |
51 |
 |
||||
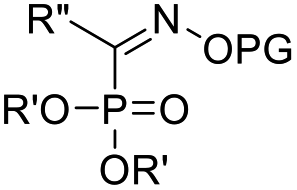
Recent advancements of electrophiles and radicals in the semipinacol rearrangement reactions of allylic alcohols are summarized in Table 1. For a nitrogen group element, nitrosonium (NO+) was proven to be able to promote the rearrangement reaction, producing oximes and oxime ethers with a spirocyclic skeleton.29 This methodology is a rare example of oxime synthesis from non-ketone or hydroxylamine substrates. Besides N-centered electrophiles, an N-radical induced rearrangement has been reported wherein N-fluorobenzenesulfonimide (NFSI) was used as the radical source under the catalysis of Cu(I).30 Additionally, with the assistance of photoredox-catalysis, the phosphinylation/semipinacol rearrangement could be realized either with a diaryl(alkyl)-phosphine oxide31 or oxime phosphonate substrate.32
Chalcogen, specifically sulfur and selenium, related electrophiles and radicals are also viable species in promoting the rearrangement reactions of allylic alcohols. For example, the SCF3 radical could be generated under oxidative conditions,33 while the SCF3, SCN, or SeR electrophile was commonly released from certain electrophilic reagents such as PhNHSCF3,34N-trifluoromethylthiosaccharin,35 (PhSO2)2NSCN,36 PhSeBr,37 or RSeSeR38 in the presence of an acidic promoter. In 2019, a sulfonylation/semipinacol rearrangement has been established independently by several groups. Among these reactions, various types of reagents, such as ArN2BF4 and (DABCO)·(SO2)2,39 ArSO2H,40 and ArSO2Cl,41 were used to generate arylsulfonyl radicals by means of photoredox-catalysis. Of note, electrochemical conditions have also been applied for the generation of arylsulfonyl radicals from either ArSO2NH2NH2 or PhSO2Na,42,43 which provide mild and convenient protocols for the synthesis of β-sulfonated ketones.
The halogenation/semipinacol rearrangement of allylic alcohols has found wide application in the synthesis of β-halogenated ketones. Although electrophilic halogenation reagents have been pervasively used in such transformations, these reagents are generally expensive and toxic. In comparison, the use of inorganic halide salts as halogen sources would be more appealing since they are low-cost and eco-friendly. These approaches have recently been realized taking advantage of either oxidative conditions (PIDA)44 or electrocatalysis.45 In each case, sodium or magnesium halide was used respectively to generate the halogenium in situ to induce the rearrangement.
Apart from heteroatom-centered electrophiles and radicals, carbon-centered radicals are equally competent in inducing migration reactions. The alkyl radicals generated from RBr,46 RB(OH)2,47 or N-acyloxyphthalimides48 under photoredox-catalytic conditions could be utilized for the development of alkylative semipinacol rearrangement reactions. These methodologies provide efficient routes to β-functionalized ketones under mild and environmentally benign conditions. Similarly, trifluoromethylation/rearrangement reactions could also be conducted under photo-49 or electrocatalytic conditions43,50 to produce a wide range of β-trifluoromethyl ketones with high efficiency.
Notably, a catalytic radical-polar cross over reaction has been developed in 2018 in which the generation of a hydrogen radical was responsible for promoting the semipinacol rearrangement of allylic alcohols.51 Compared with the proton induced reactions, this approach features a much broader substrate scope that could be a useful complement to the existing methods.
Advancement of electrophile induced enantioselective semipinacol rearrangement of allylic alcohols has also received considerable attention from the synthetic community with continued development of novel types of catalytic modes.16 In 2019, Gong and coworkers developed a notable catalytic enantioselective tandem allylic alkoxylation/oxidative rearrangement using a chiral iodine catalyst (Scheme 7).52 Oxidation of cat. 6 by selectfluor and TsOH would in situ generate a hypervalent iodine(III) species, which serves as the real catalyst for the enantioselective oxidative rearrangement. Under the optimal conditions, various β-alkoxylated ketones 27 were obtained with good yields and excellent enantioselectivities.
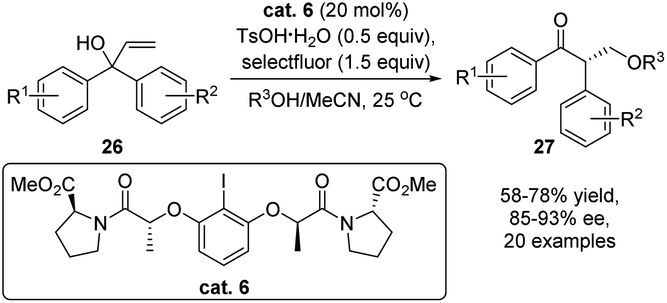 | ||
| Scheme 7 Enantioselective alkoxylation/oxidative rearrangement. | ||
Taking advantage of Lewis base catalysis, Tu's groups accomplished an asymmetric sulfenylation/semipinacol rearrangement that provides a straightforward methodology for the synthesis of a wide range of α-quaternary carbon-containing β-arylthio ketones 30 from allylic alcohols 28 (Scheme 8).53 The best enantioselection was achieved when selenide (R)-cat. 7 was used as the Lewis base catalyst and chiral phosphoric acid (S)-CPA as a cocatalyst. The indispensable role of (S)-CPA for a highly enantioselective transformation was studied by computational methods which indicate a steric repulsion of (R)-cat. 7 and (S)-CPA to be responsible for the facile selection during the sulfenyl group transfer event.
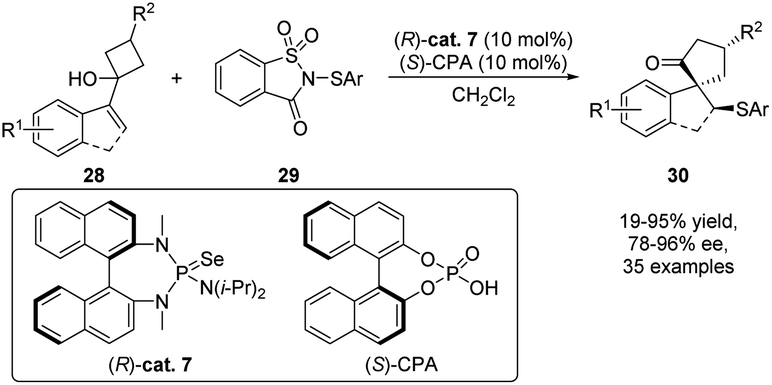 | ||
| Scheme 8 Asymmetric sulfenylation/semipinacol rearrangement. | ||
In the past two decades, significant studies have been devoted to the development of catalytic enantioselective halogenation/rearrangement of allylic alcohols,16 and precedent reactions focusing on the establishment of stereogenic sp3 carbon centers with point chirality were well-established. Recently, Yeung and co-workers further advanced this methodology with an application of the rearrangement for the introduction of axial chirality (Scheme 9).54 As the methyl substituents in the 4,5-dimethylfluorene system (31) are spatially remote, rotation of the diaryl C–C bond is facile, resulting in the racemization of the atropisomers. However, when 4,5-dimethylphenanthrene product 32 was generated via ring expansion of 31, proximity of the methyl substituents in 32 would render a high rotation barrier of the diaryl C–C bond. In this case, two configurationally stable atropisomers could be obtained. With the application of semipinacol rearrangement, the authors developed a dynamic-kinetic resolution strategy to simultaneously assemble the quaternary stereogenic carbon center as well as the axial chirality of 32 from the halogenation/rearrangement of racemic alcohol 31 using (DHQD)2PHAL as the catalyst (cat. 8). The diastereoselectivity of the rearrangement can be rationalized by initial formation of a preorganized intermediate A or B from each atropisomer of 31. The less steric interaction between the catalyst and the substrate in A would favour the formation of 32.
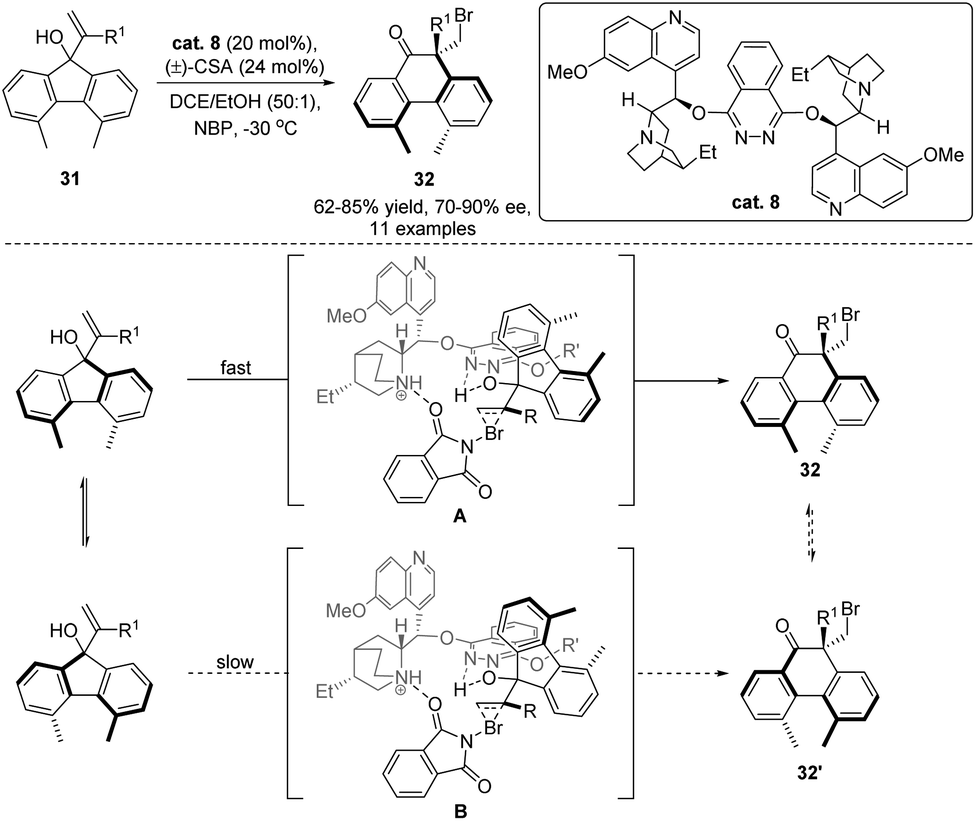 | ||
| Scheme 9 Dynamic-kinetic resolution-semipinacol rearrangement. | ||
Compared with heteroatom-electrophiles, asymmetric semipinacol rearrangement reactions initiated by carbon-electrophiles are rather limited, although they are synthetically more useful in generating complex ketone scaffolds. In 2017, Gaunt and coworkers developed an elegant enantioselective arylation/semipinacol rearrangement of tertiary allylic alcohols (Scheme 10a).55 When Cu(OTf)2 was used as the catalyst in combination with a Box ligand L2, the reactions of 33 or 36 with diaryliodonium salts 34 could proceed smoothly in the presence of 2,6-di-tert-butylpyridine (DTBP) to produce β-aryl spirocyclic ketones 35 or 37, respectively, in high yields and enantiomeric ratios. Notably, the method features a broad substrate scope in terms of diaryliodonium salts and indene or dihydropyran derived allylic alcohols. Ring expansions of cyclobutyl, cyclopentyl, oxetane, and azetidine-containing substrates were all feasible. In the same year, Zhu's group also described their independent development of the tandem reaction (Scheme 10b).56 The CuCl catalyst with another Box ligand L3 was used in this case. Similarly, high yields and enantioselectivities were obtained with mainly indene-derived allylic alcohols.
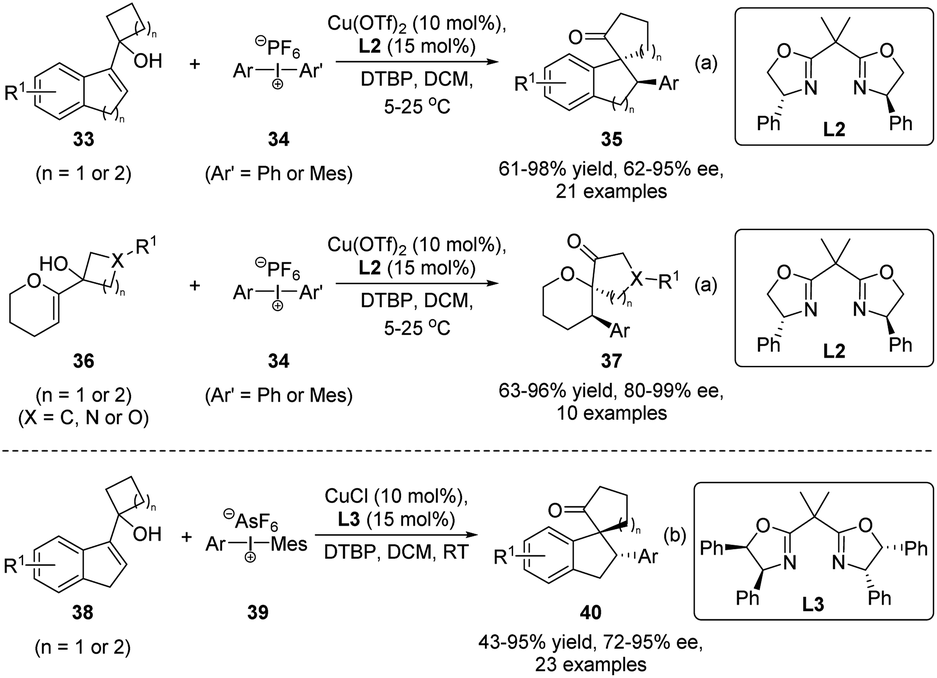 | ||
| Scheme 10 Enantioselective arylation/semipinacol rearrangement. | ||
In addition to cascade reactions initiated by electrophiles and radicals, the semipinacol rearrangement of allylic alcohols could be incorporated into other more complex tandem or one-pot processes. These often involve the combination of other reaction types with the rearrangement for the purpose of generating molecular complexity in an expeditious manner. For example, a tandem hydroacylation/semipinacol rearrangement of alkynyl cyclobutanols with salicylaldehydes has been established by Zhang and co-workers, offering an efficient protocol for the preparation of 2-(2-oxo-2-phenylethyl)cyclopentanones with multiple substitutions (Scheme 11).57 Atom-economical transformation was carried out with a catalytic amount of rhodium(I) catalyst and a PPh3 ligand. While the reactions of terminal alkynes 41 with chelating aldehydes 42 could directly afford 1,4-diketones 43, a step-wise procedure was needed for the transformation of disubstituted alkynes 44.
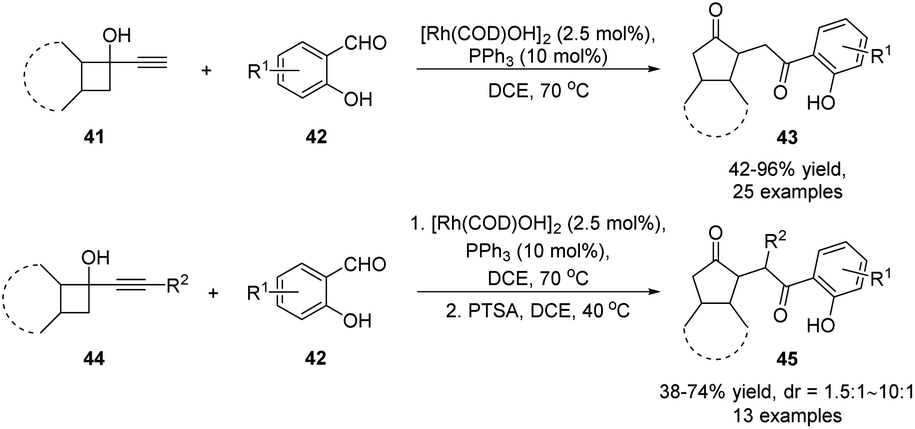 | ||
| Scheme 11 Tandem hydroacylation/semipinacol rearrangement. | ||
A recent report by Zhang and Tu describes a one-pot semipinacol rearrangement/Michael addition/Henry reaction that enables rapid assembly of polyfunctionalized carbocycles from vinylogous α-ketols 46 and nitroalkenes 47 (Scheme 12).58 Initial addition of TMSOTf would trigger the ring-expansion of 46 to form a rearranged enolate intermediate that could then add to the nitroolefin partner upon activation by TiCl4. Once the adduct was formed, an intramolecular Henry cyclization would take place to deliver tricyclic ketones 48 and 48′ as a pair of stereoisomers.
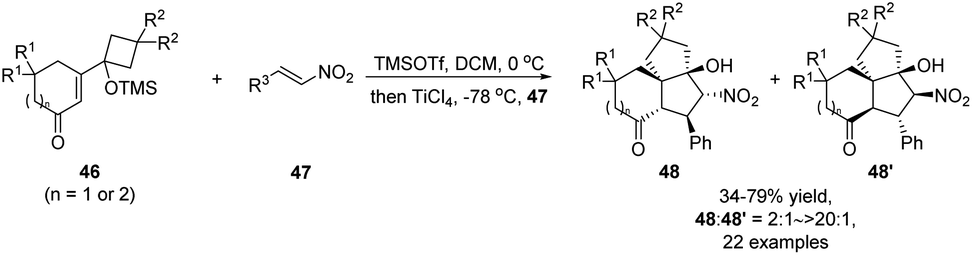 | ||
| Scheme 12 Semipinacol rearrangement/Michael addition/Henry reaction. | ||
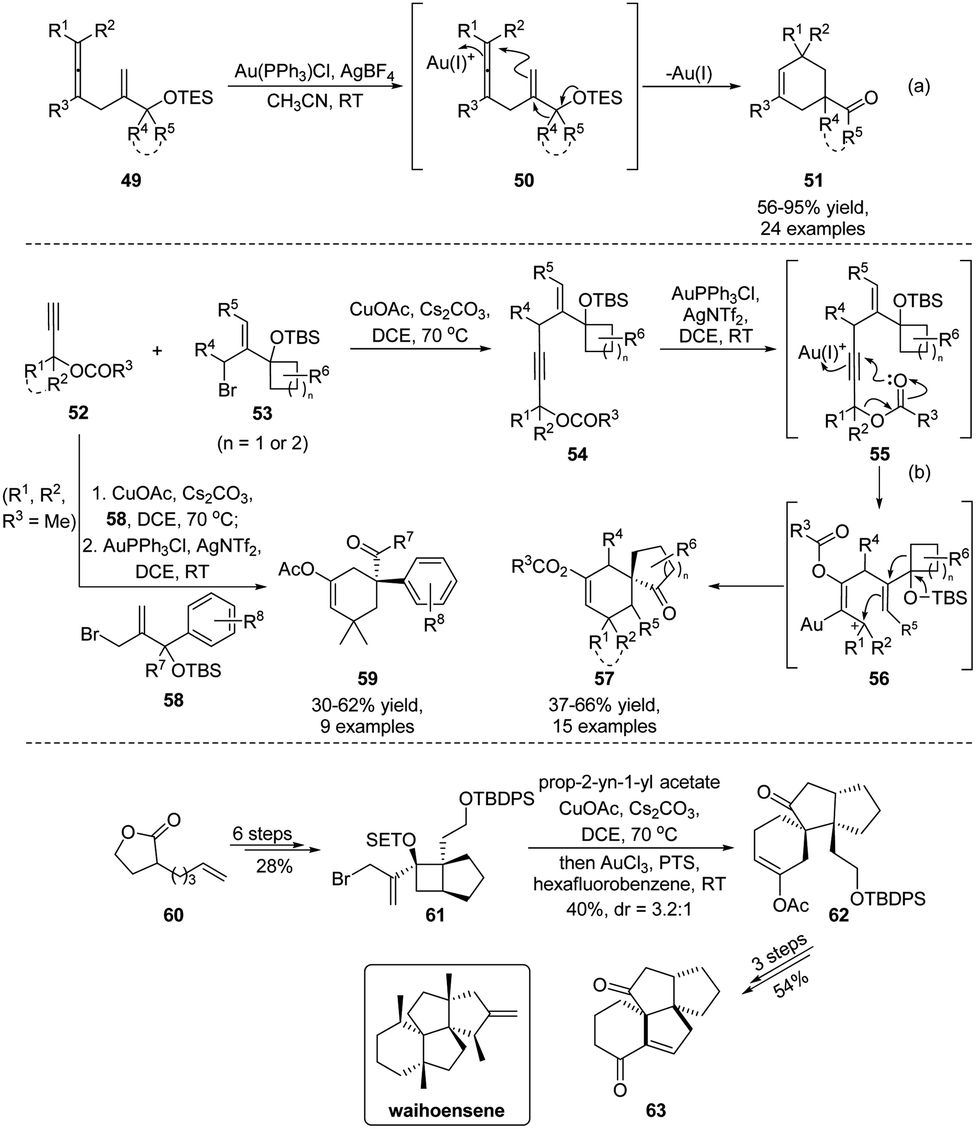
In 2020, Yu and Wang established a Au(I)-catalyzed tandem cyclization/semipinacol rearrangement of allene-substituted alcohols 49 (Scheme 13a),59 which provides a direct synthetic approach for the preparation of quaternary carbon-containing cyclohexenes 51. On the basis of this work, Wang and Tu further developed a two-step Castro–Stephens coupling/tandem acyloxy shift/cyclization/semipinacol rearrangement sequence for the synthesis of such cyclohexenes in a more modular fashion (Scheme 13b).60 The coupling of terminal alkynes 52 with allylic bromides 53 was followed by a Au(I)-catalyzed cascade mechanistically similar to the allene-cyclization/rearrangement process. Specifically, activation of the alkyne group of 54 by Au(I) would trigger an acyloxy shift to generate an allylic carbenium 56 that could induce the cyclization/semipinacol rearrangement, as in the reaction of allene 50, to finally give rise to the spirocyclic ketones 57. Besides the ring-expansion rearrangement, migration of aryl groups was also feasible in this cascade transformation. In these cases, a wide range of cyclohexenes 59 with an aryl substituted quaternary carbon center were obtained. A synthetic application of this highly efficient procedure was also demonstrated in the assembly of the core carbon skeleton of waihoensene, a diterpene compound with a unique tetracyclic ring-system and consecutive quaternary-center arrangement. Cyclobutanol 61 prepared from 60 in 6 steps could be transformed into tricyclic ketone 62 facilely using this methodology. Late-stage functional group manipulations followed by an aldol cyclization finally gave ketone 63 with consistent relative configuration with the natural product.
 | ||
| Scheme 13 Tandem cyclization/semipinacol and Castro–Stephens coupling/tandem acyloxy shift/cyclization/semipinacol rearrangement. | ||
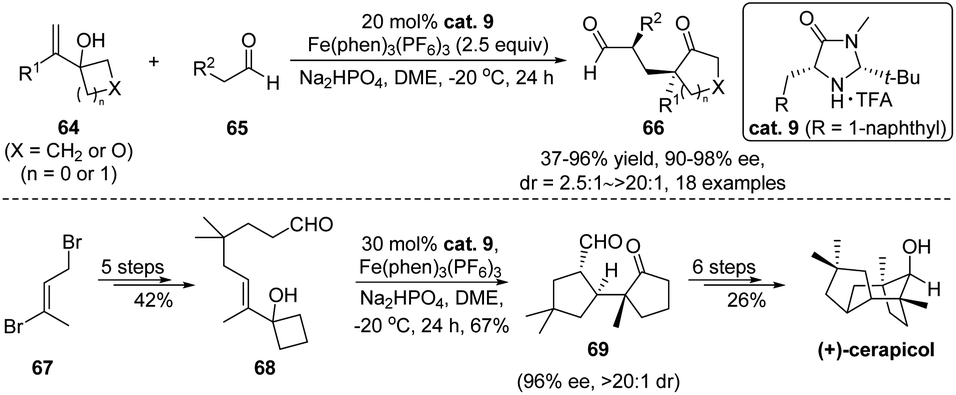
An exciting direction surrounding tandem reactions is their application in natural product synthesis. Accordingly, development of such rearrangement-involving cascades for total synthesis purpose has been an ongoing topic in this research field. Recently, Zhang and Tu developed an enantioselective aldehyde α-alkylation/semipinacol rearrangement taking advantage of organo-SOMO catalysis.61 As depicted in Scheme 14, oxidation of the enamine generated from aldehyde 65 and secondary amine catalyst cat. 9 (ref. 62–64) would form a radical cation that could add to the alkene of allylic alcohol 64, thus promoting the ring-enlargement rearrangement. A broad range of enantioenriched α-quaternary cyclopentanones can be accessed using this method. Notably, a synthetic utility of this approach has been demonstrated in the catalytic asymmetric synthesis of (+)-cerapicol. After preparation of aldehyde 68 from bromide 67 in 5 steps, it was transformed to bicyclic 1,5-dicarbonyl compound 69 through an intramolecular version of the tandem process. Late-stage aldol cyclization and functional group manipulations finally completed the total synthesis.
 | ||
| Scheme 14 Enantioselective aldehyde α-alkylation/semipinacol rearrangement and total synthesis of (+)-cerapicol. | ||
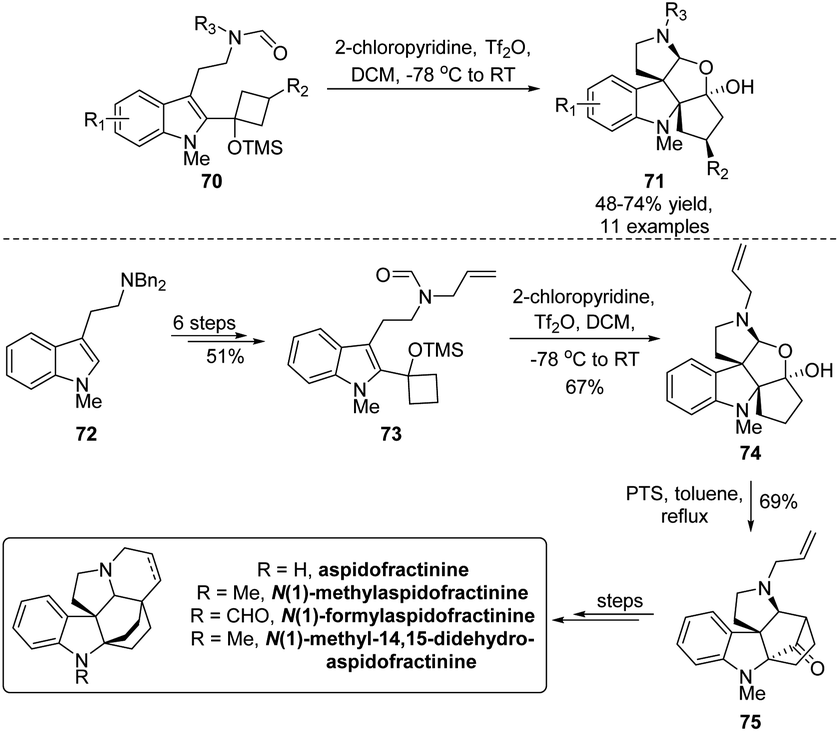
The simultaneous creation of vicinal all-carbon quaternary carbon centers presents a nontrivial task in synthetic chemistry due to their congested chemical environment, although such structural moieties broadly exist in diverse natural molecules, especially indole derived alkaloids. Aiming at assembling a bis(spirocyclic) indole framework with continuous quaternary carbon centers, Tu's group established a Bischler–Napieralski/semipinacol rearrangement that enables the transformation of tryptamine-derived allylic alcohols 70 to tetracyclic indoles 71 in a highly diastereoselective fashion (Scheme 15).65 Upon treatment of bis(spirocyclic) indole 74 under acidic conditions, the following intramolecular Mannich reaction would produce bridge[2.2.1]cyclic ketone 75. This advanced pentacyclic intermediate could be applied in the collective total synthesis of four aspidofractinine alkaloids, further showcasing the synthetic utility of this methodology.
 | ||
| Scheme 15 Bischler–Napieralski/semipinacol rearrangement and collective total synthesis of aspidofractinine alkaloids. | ||
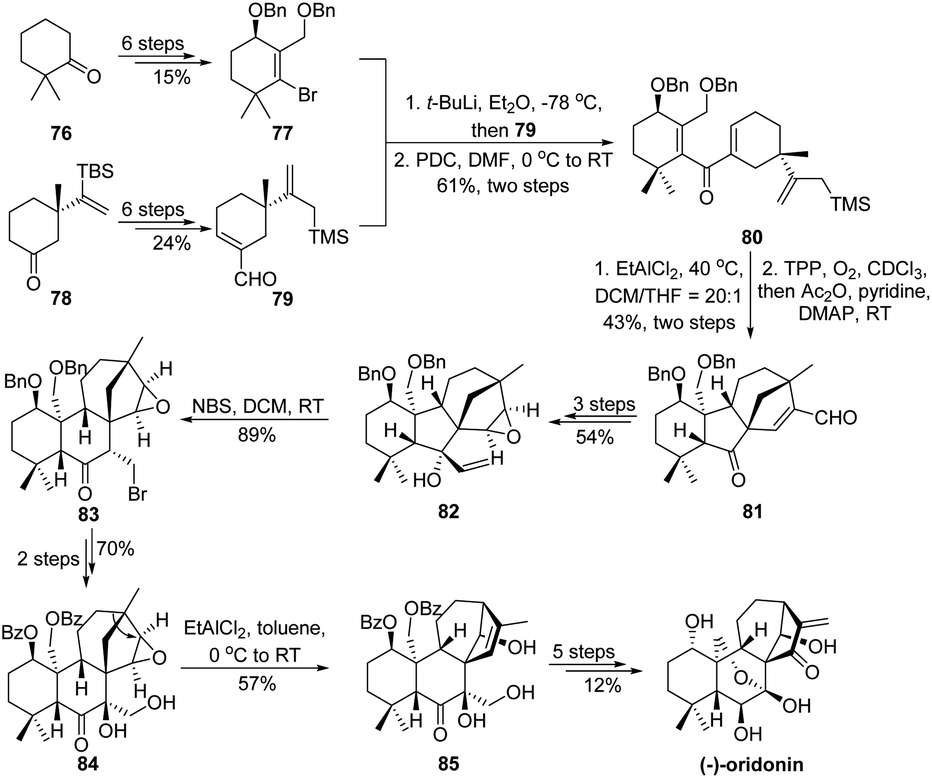
In terpene synthesis, due to the intrinsic complexity of certain carbon skeletons, direct assembly of ring-systems using cyclization methods would sometimes be arduous. In this case, alternation of the carbocycle to an easily accessible one by means of 1,2-carbon migration thereby provides a synthetic solution. Such a strategy has been elegantly performed by Luo and coworkers in the total synthesis of (−)-oridonin (Scheme 16).66 In order to construct the central tetracyclic carbocycles in a rapid fashion, a Nazarov/Hosomi–Sakurai cascade of 80 (prepared from the coupling of segments 77 and 79) followed by a singlet oxygen ene reaction was developed for the synthesis of ketone 81. This highly efficient tandem reaction enables the establishment of a central cyclopentanone and the bicyclo[3.2.1]octene motif as well as the creation of four consecutive stereogenic centers. Although both carbocycles assembled are not consistent with the ones embedded in (−)-oridonin, the authors suggested that later skeletal rearrangement via 1,2-carbon bond migration would adjust the ring-system. Therefore, ketone 81 was transformed into allylic alcohol 82 in a 3-step sequence, which was subjected to a bromination/semipinacol rearrangement to produce β-bromide ketone 83 with alternation of the central cyclopentanone to a cyclohexanone scaffold. Subsequently, after protecting and functional group manipulations, the resulting epoxide 84 could undergo another semipinacol rearrangement in the presence of Lewis acid EtAlCl2 to give 85 with settlement of the functional bridge ring-system. From this advanced intermediate, the total synthesis can be accomplished in another 5-step transformations.
 | ||
| Scheme 16 Total synthesis of (−)-oridonin. | ||
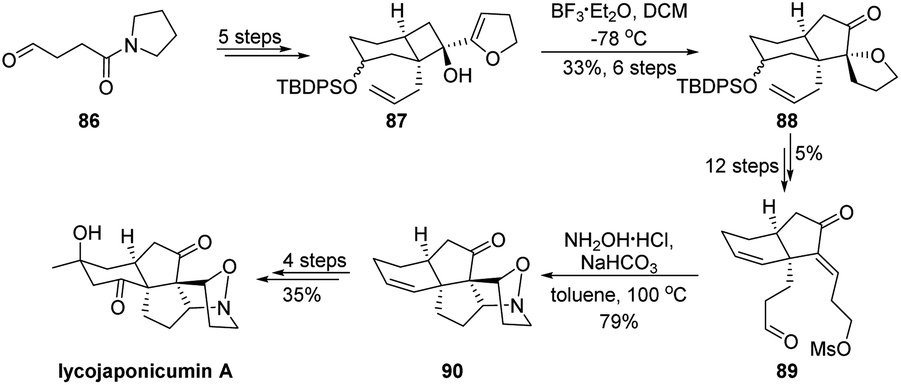 | ||
| Scheme 17 Total synthesis of lycojaponicumin A. | ||
As another strategic application, the ring-expansion semipinacol rearrangement of a dihydrofuran derived cyclobutanol has been used in the total synthesis of natural alkaloid lycojaponicumin A. In Zhang and Tu's studies (Scheme 17),67 a highly functionalized allylic alcohol 87 was prepared from aldehyde 86 in five steps and could be converted to cyclopentanone 88via a highly regio- and diastereoselective ring-enlargement rearrangement. This key transformation established the typical cis-fused[6.5] ring-system of a fawcettimine-type Lycopodium alkaloid. After functional group interconversions, a late-stage transannular 1,3-dipolar cyclization of 89 was developed to produce pentacyclic compound 90 with the construction of the tetrahydroisoxazole ring.68 Finally, another four transformations culminate in the total synthesis. It is worth noting that from advanced intermediate 88 total synthesis of sieboldine A could also be achieved.69
β-Heterosubstituted alcohols
The 1,2-migration of β-heterosubstituted alcohols with loss of a leaving group has found wide application in total synthesis,3 though the development of novel types of such reactions is relatively limited.16 Strategic use of this type of semipinacol rearrangement to transform an easily accessible carbon skeleton into a synthetically complicated one would provide an overall convenient route in natural product synthesis, as represented in the elegant total synthesis of (−)-gardmultimine A by She's group (Scheme 18).70 From known indole ester 91, they developed a synthetic sequence including a C–H borylation to prepare the tetracyclic ketone 92. Next, an oxidative rearrangement of 92 was designed and realized to transform the fused tetrahydro-pyrido indole ring system into a spirooxindole compound 96. From precedent studies,71–73 this cascade reaction may involve the initial formation of β-chlorinated hemiaminal 94 from indole chlorination product 93. Then, an intermediate benzylic carbocation 95 would presumably be formed from 94 and trigger the following ring-contraction semipinacol rearrangement to afford 96 in 75% yield. The total synthesis of (−)-gardmultimine A could then be accomplished in another 11 steps.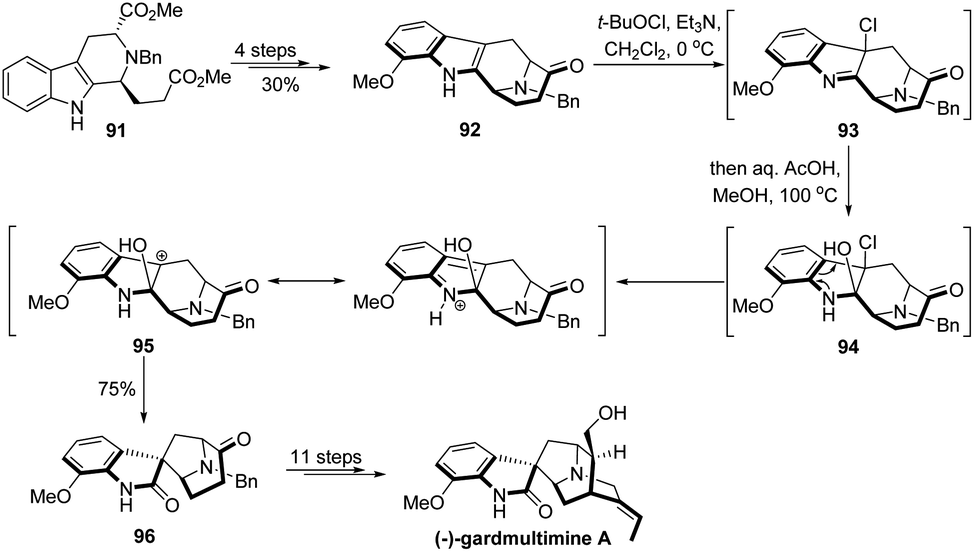 | ||
| Scheme 18 Total synthesis of (−)-gardmultimine A. | ||
In 2020, Ma and coworkers reported the first total synthesis of kopsinitarine E wherein the semipinacol rearrangement of a β-iodinated alcohol was also applied as a crucial strategic step (Scheme 19).74 Their synthesis began with the preparation of tetracyclic bromide 98 from known Boc-protected carbazolone 97 in 9 steps. Then, a highly efficient SmI2-mediated Dieckmann condensation/intramolecular Prins-type cyclization of 98 was developed to assemble the complex 8-membered bridge ring of the natural product and generate a β-hydroxy ketone compound 99. At this stage, switch of the synthetically accessible central N-hetero[3.3.1]bridge cycle to a desired N-hetero[4.2.1]bridge ring system should be achieved. Though challenging, this skeletal reorganization could be successfully accomplished by using the semipinacol rearrangement of β-iodinated alcohol 100 prepared from iodination of 99. Late-stage formation of the hemiaminal and Mannich cyclization finally enabled the total synthesis.
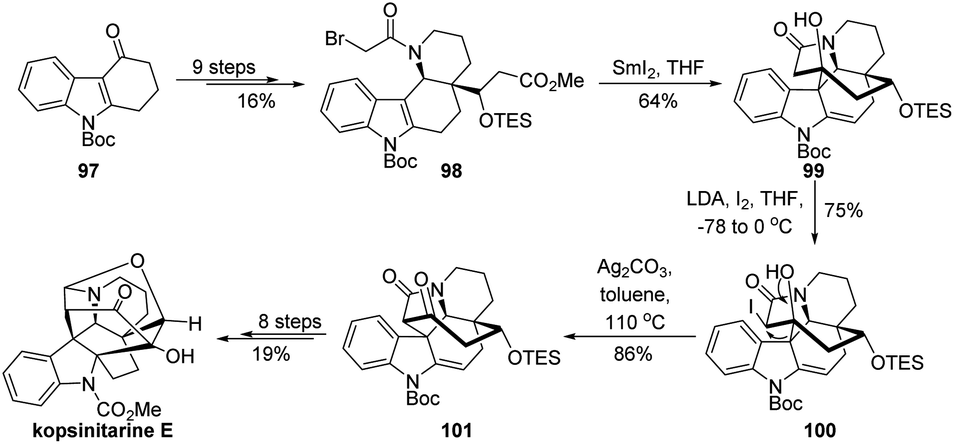 | ||
| Scheme 19 Total synthesis of kopsinitarine E. | ||
Skeletal rearranged steroids pinnigorgiols B and E are appealing targets for chemical synthesis due to their intriguing tricyclic decane framework and potential cytotoxic bioactivities. In 2018, Li's group had developed an effective strategy to construct such core structures in their elegant total synthesis of aplysiasecosterol A,75 though the biosynthetic pathway of the scaffold from a steroid precursor is still yet to be elucidated. Inspired by a proposed biosynthetic α-ketol rearrangement cascade, Gui and coworkers ingeniously accomplished a concise synthesis of pinnigorgiols B and E from an inexpensive steroid.76 As depicted in Scheme 20, dehydroergosterol 102 was converted to mesylate 103 in 5 steps. Subsequent semipinacol rearrangement of 103 under basic conditions would afford ketone 104 with the conversion of the trans-bicyclo[4.4.0]decane to the cis-bicyclo[5.3.0]decane ring system. After cleavage of the alkene and cycloheptanone functionality via a 7-step consequence to give thiolester 105, a highly efficient acyl radical cyclization cascade of 105 was developed that enabled the formation of the key tricyclic decane framework and completed the synthesis of pinnigorgiol E, which was then converted to pinnigorgiol B via deacetylation.
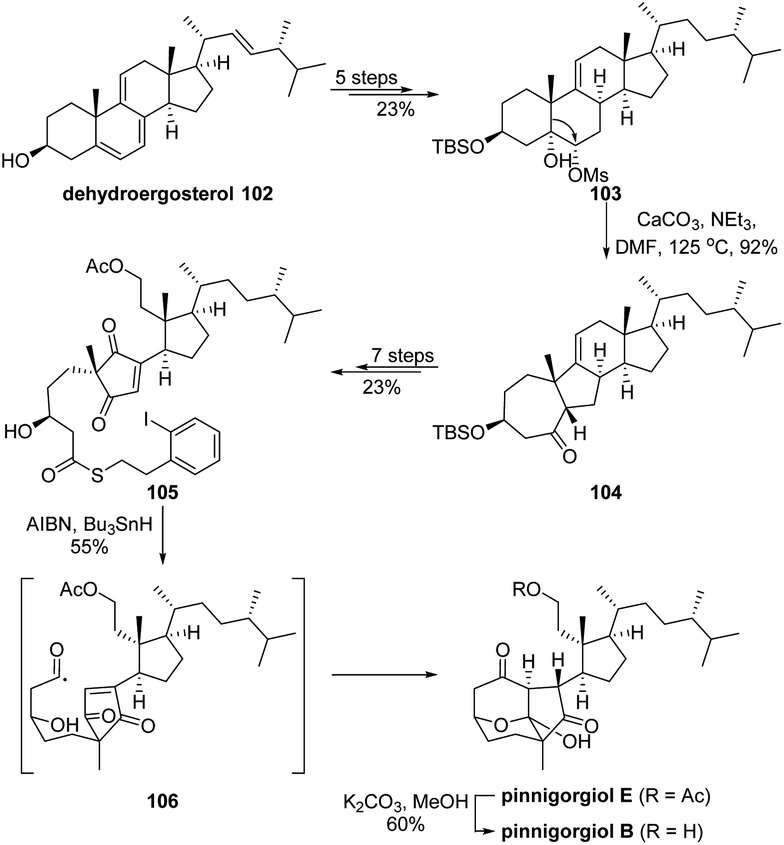 | ||
| Scheme 20 Synthesis of pinnigorgiols B and E. | ||
α-Hydroxy ketones and imines
The semipinacol rearrangement of α-hydroxy carbonyls and imines, also known as acyloin rearrangement, is a rare type of reversible 1,2-migration reaction, which favors the formation of a thermodynamically more stable product from an α-ketol precursor. Because of the reversibility of the rearrangement process, development of such a catalytic enantioselective reaction presents a challenging task in asymmetric catalysis. Breakthroughs from Maruoka's77,78 and Wulff's79 groups have proven chiral Lewis acids to be competent catalysts in these transformations since the complexation of the Lewis acid with the carbonyl would create a chiral environment for subsequent 1,2-migration. This catalytic mode has recently been used for the development of a cyclopropanation/semipinacol rearrangement by Ryu and co-workers (Scheme 21).80 Under the catalysis of a chiral oxazaborolidinium ion cat. 10, asymmetric cyclopropanation of α-silyloxyacrolein 107 with α-phenyldiazoester 108 was followed by a stereospecific semipinacol (acyloin) rearrangement to produce enantioenriched α-hydroxy cyclobutanones 109 in moderate to high yields with good to excellent diastereo- and enantioselectivities. This tandem reaction features a broad substrate scope and provides a convenient protocol for the preparation of cyclobutanones with multiple stereogenic centers.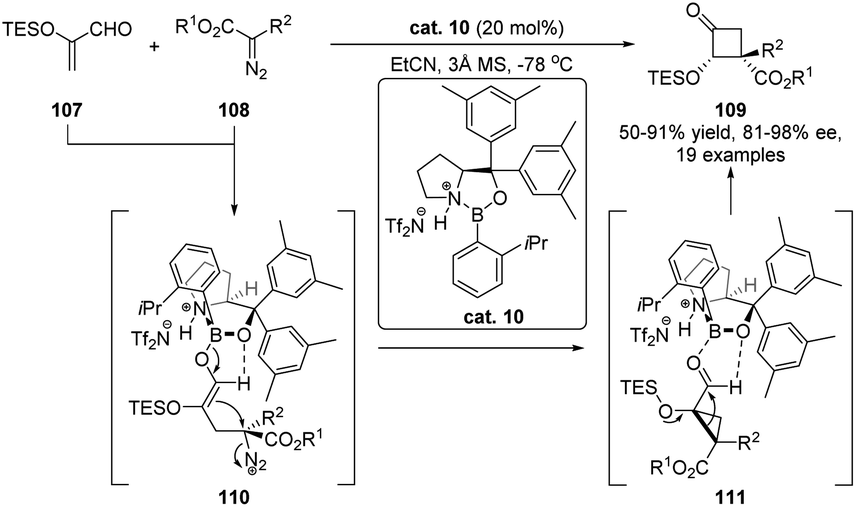 | ||
| Scheme 21 Asymmetric cyclopropanation/semipinacol rearrangement. | ||
Besides Lewis acids, chiral phosphoric acids (CPAs) or their derivatives are also capable of catalyzing enantioselective acyloin rearrangement reactions. A seminal work on organocatalytic semipinacol rearrangement has been reported by Zhu and co-workers in 2017. When α-hydroxy acetals 112 were used as the substrates, their transformations to enantioenriched α-alkoxy ketones 113 performed well under the catalysis of BINOL-derived N-triflyl phosphoramide cat. 1, with high yields and enantioselectivities obtained in most cases (Scheme 22).81 The enantioselection of the 1,2-C–C migration of 112 might be controlled by the formation of an ion pair between the in situ formed oxocarbenium ion and the conjugate anion of the catalyst.
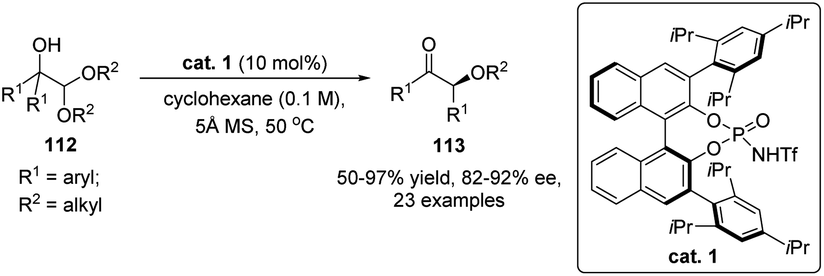 | ||
| Scheme 22 Enantioselective acyloin rearrangement of α-hydroxy acetals. | ||
Compared with the wide substrate patterns of acyloin rearrangements of α-hydroxy carbonyls, studies on the reactions of α-hydroxy imines mainly focus on the rearrangement of indol-3-ols, which represents a powerful methodology to access 2,2-disubstituted indolin-3-ones. Although stepwise preparation of indol-3-ols and semipinacol rearrangement has been well-documented, direct reaction of 2,3-disubstituted indoles in a tandem oxidation/migration process would be more efficient. Such an oxidation/semipinacol rearrangement has been independently investigated by Brasholz's and Lu and Xiao's groups (Scheme 23a and b).82,83 With the aid of photocatalysis, oxygen could be used as an oxidant for the tandem transformation, producing a wide range of 2,2-disubstituted indolin-3-ones (116 or 121) in both catalytic systems. Recently, a catalytic enantioselective variant of the tandem process has been realized by Zhao and Jiang's group (Scheme 23c).84 The SPINOL-derived CPA cat. 11 was found to be an optimal catalyst in this reaction when combined with a dicyanopyrazine-derived chromophore photoredox catalyst. Generally, moderate to high product yields of 2-aryl-2-benzyl substituted indolin-3-ones 123 were obtained with satisfactory enantioselectivities.
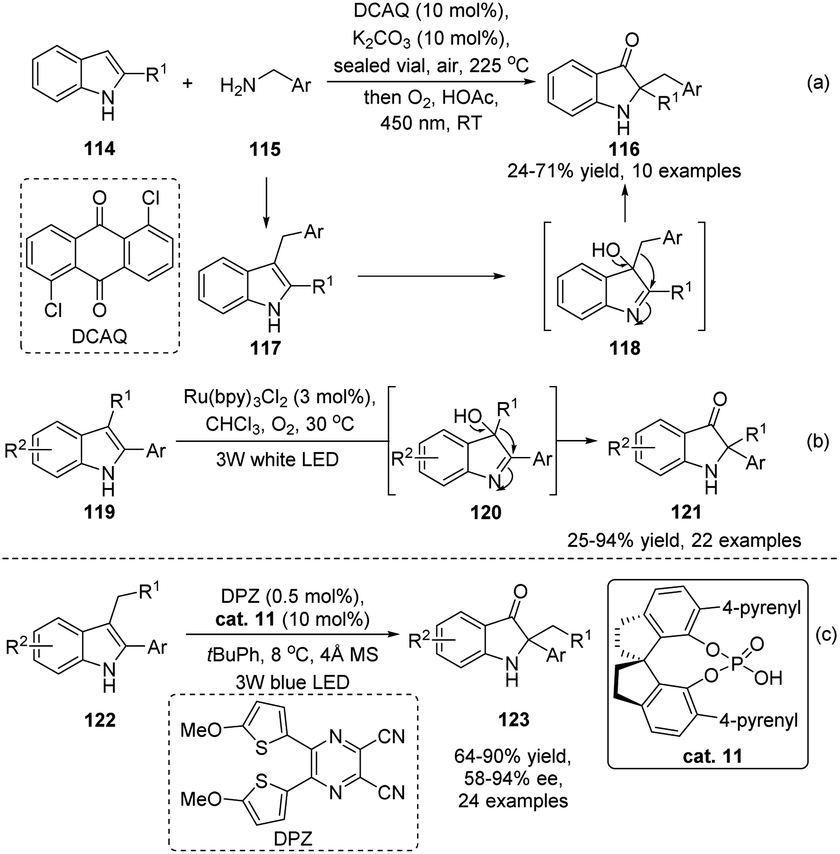 | ||
| Scheme 23 Aerobic oxidation/semipinacol rearrangement of 2,3-disubstituted indoles. | ||
From a biosynthetic viewpoint, the semipinacol rearrangement of indol-3-ols is often suggested to be a crucial step in the formation of indolin-3-one related natural products.3 As a representative example, Lawrence and co-workers showcased a creative application of the rearrangement reaction in the biomimetic total synthesis of (+)-brevianamide A and B (Scheme 24).85 The concise route commenced from the synthesis of (+)-dehydrodeoxybrevianamide E 125, which was surmised to be an authentic biosynthetic precursor of the natural products, from L-tryptophan methyl ester 124 in an efficient 5-step sequence. After oxidation of 125 with m-CPBA to afford two pentacyclic diastereomers 126 and 127, the designed biomimetic cascade retro-5-exo-trig/semipinacol rearrangement/tautomerization/Diels–Alder reaction could take place under basic conditions, delivering both (+)-brevianamide A and B in a combined 63% yield. Likewise, the enantiomers of the two natural products (dr = 93:7) could be derived from 126 with comparable results. It is worth noting that such a highly efficient tautomerization/semipinacol rearrangement/Diels–Alder reaction has also been elucidated to be a biosynthetic pathway and controlled by an isomerase/semipinacolase BvnE, which accounts for the biogenesis of these natural molecules.18
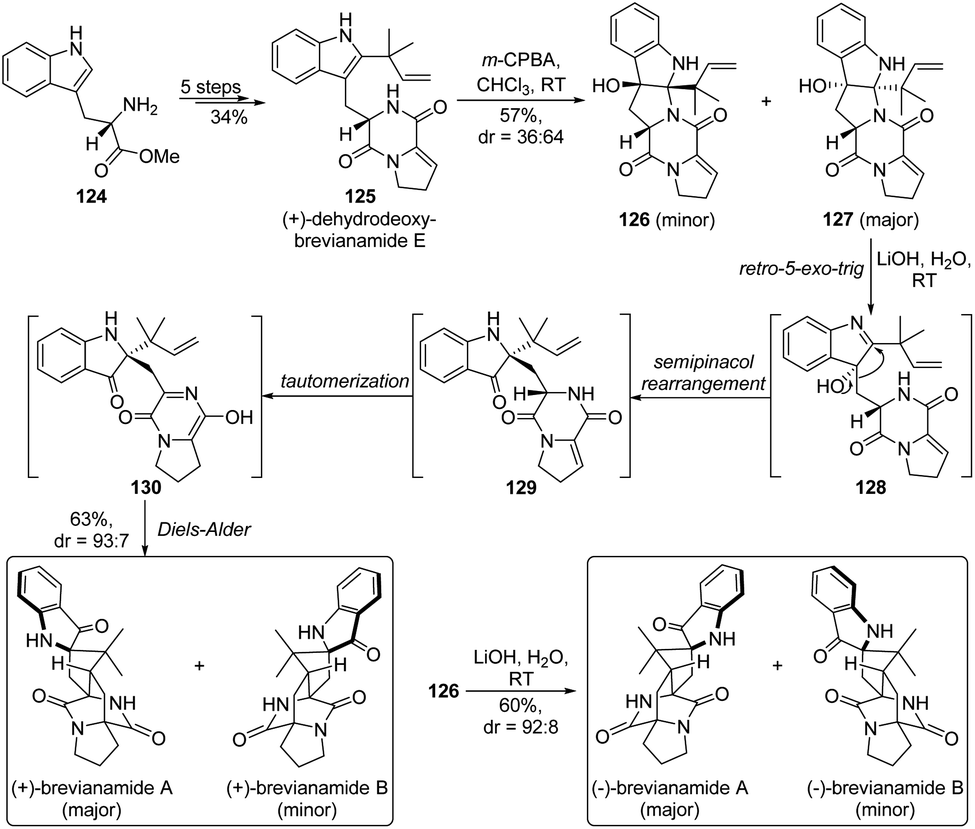 | ||
| Scheme 24 Asymmetric total synthesis of brevianamide A and B. | ||
Conclusion and perspectives
With recent significant advancement in the tandem process and enantioselective catalysis, myriad transformations regarding semipinacol rearrangement have been continuously developed that inarguably demonstrate the vitality of this reaction in chemical synthesis. Given the widely recognized feats of β-functionalized ketone synthesis, quaternary carbon center formation, and skeletal conversion of carbocycles, such a rearrangement reaction has been employed as a guiding strategy in natural product synthesis, inspiring novel disconnection logics in synthesis design. While these impressive contributions have largely boosted the research field, there are still exciting new forefronts to further exploit the reaction potential. For instance, development of carbon-electrophile-initiated enantioselective rearrangements, though in its infancy, is highly appealing because of the molecular complexity that the reaction can generate from simple precursors. Furthermore, most studies focus on the simple use of a 1,2-C–C bond migration reaction, and the application of novel tandem rearrangement reactions in total synthesis is still sparse,15 in particular, the construction of polycyclic rings or systems with multiple stereogenic centers. We hope that our summarization of the existing achievements in this review with an updated outlook of the research field would inspire future innovations on semipinacol rearrangement.Author contributions
All authors conceptualised the review. X.-M. Z., B.-S. L. and S.-H. W. wrote and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China NSFC (No. 21772019, 21772071, 21502080, 21772076, 21871117, 91956203, and 21971095), the Science and Technology Program of Gansu Province (20JR10RA608), the Department of Education of Guangdong Province (No. 2019KZDXM035), the ‘111’ Program of MOE, the Major Project (2018ZX09711001-005-002) of MOST, the STCSM (19JC1430100), and the lzujbky-2020-ct01 for the financial support.Notes and references
- M. Tiffeneau and J. Levy, Comptes rendus, 1923, 176, 312 CAS.
- T. J. Snape, Chem. Soc. Rev., 2007, 36, 1823–1842 RSC.
- Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS.
- W.-Z. Weng and B. Zhang, Chem. - Eur. J., 2018, 24, 10934–10947 CrossRef CAS PubMed.
- B. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed.
- K. Fuji, Chem. Rev., 1993, 93, 2037–2066 CrossRef CAS.
- E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef.
- C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS.
- B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef CAS.
- P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969–5994 CrossRef CAS.
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed.
- P. Hu, H. M. Chi, K. C. DeBacker, X. Gong, J. H. Keim, I. T. Hsu and S. A. Snyder, Nature, 2019, 569, 703–707 CrossRef CAS PubMed.
- X.-M. Zhang, Y.-Q. Tu, F.-M. Zhang, Z.-H. Chen and S.-H. Wang, Chem. Soc. Rev., 2017, 46, 2272–2305 RSC.
- B. Delayre, Q. Wang and J. Zhu, ACS Cent. Sci., 2021, 7, 559–569 CrossRef CAS PubMed.
- S.-H. Wang, B.-S. Li and Y.-Q. Tu, Chem. Commun., 2014, 50, 2393–2408 RSC.
- H. Wu, Q. Wang and J. Zhu, Eur. J. Org. Chem., 2019, 1964–1980 CrossRef CAS.
- Y. Ye, L. Du, X. Zhang, S. A. Newmister, M. McCauley, J. V. Alegre-Requena, W. Zhang, S. Mu, A. Minami, A. E. Fraley, M. L. Adrover-Castellano, N. A. Carney, V. V. Shende, F. Qi, H. Oikawa, H. Kato, S. Tsukamoto, R. S. Paton, R. M. Williams, D. H. Sherman and S. Li, Nat. Catal., 2020, 3, 497–506 CrossRef CAS PubMed.
- B. Zhu, T. Shen, X. Huang, Y. Zhu, S. Song and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 11028–11032 CrossRef CAS.
- T. H. M. Wong, X. Li, D. Ma and J. Sun, Org. Lett., 2020, 22, 1951–1954 CrossRef CAS PubMed.
- H. Wu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2019, 141, 11372–11377 CrossRef CAS PubMed.
- D. Ma, C.-B. Miao and J. Sun, J. Am. Chem. Soc., 2019, 141, 13783–13787 CrossRef CAS.
- T. Mohri, Y. Takahashi, E. Kwon, S. Kuwahara and Y. Ogura, Org. Lett., 2020, 22, 9234–9238 CrossRef CAS PubMed.
- X.-T. Liang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2020, 142, 8116–8121 CrossRef CAS PubMed.
- A. S. Burns and S. D. Rychnovsky, J. Am. Chem. Soc., 2019, 141, 13295–13300 CrossRef CAS PubMed.
- M. A. Bigi, S. A. Reed and M. C. White, J. Am. Chem. Soc., 2012, 134, 9721–9726 CrossRef CAS PubMed.
- K. Hung, M. L. Condakes, T. Morikawa and T. J. Maimone, J. Am. Chem. Soc., 2016, 138, 16616–16619 CrossRef CAS PubMed.
- Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2015, 44, 5220–5245 RSC.
- J.-W. Dong, T. Ding, S.-Y. Zhang, Z.-M. Chen and Y.-Q. Tu, Angew. Chem., Int. Ed., 2018, 57, 13192–13196 CrossRef CAS PubMed.
- W.-Z. Weng, J.-G. Sun, P. Li and B. Zhang, Chem. – Eur. J., 2017, 23, 9752–9755 CrossRef CAS PubMed.
- Y. Yin, W.-Z. Weng, J.-G. Sun and B. Zhang, Org. Biomol. Chem., 2018, 16, 2356–2361 RSC.
- C. Wang, X. Huang, X. Liu, S. Gao, B. Zhao and S. Yang, Chin. Chem. Lett., 2020, 31, 677–680 CrossRef CAS.
- K. Liu, Q. Jin, S. Chen and P. N. Liu, RSC Adv., 2017, 7, 1546–1552 RSC.
- C.-C. Xi, Z.-M. Chen, S.-Y. Zhang and Y.-Q. Tu, Org. Lett., 2018, 20, 4227–4230 CrossRef CAS PubMed.
- D. Zhu, T.-M. Ding, H.-Y. Luo, H. Ke and Z.-M. Chen, Org. Lett., 2020, 22, 7699–7703 CrossRef CAS PubMed.
- X.-F. Song, A.-H. Ye, Y.-Y. Xie, J.-W. Dong, C. Chen, Y. Zhang and Z.-M. Chen, Org. Lett., 2019, 21, 9550–9554 CrossRef CAS PubMed.
- D. Y. Kim, Synth. Commun., 2019, 49, 2203–2209 CrossRef CAS.
- Y. Kim and D. Y. Kim, Tetrahedron Lett., 2019, 60, 1538–1542 CrossRef CAS.
- F.-S. He, Y. Wu, X. Li, H. Xia and J. Wu, Org. Chem. Front., 2019, 6, 1873–1878 RSC.
- J. W. Park and D. Y. Kim, Bull. Korean Chem. Soc., 2019, 40, 1244–1247 CrossRef CAS.
- J. H. Kim and D. Y. Kim, Synth. Commun., 2020, 50, 207–216 CrossRef CAS.
- Y. J. Kim and D. Y. Kim, Tetrahedron Lett., 2019, 60, 1287–1290 CrossRef CAS.
- J.-C. Kang, Y.-Q. Tu, J.-W. Dong, C. Chen, J. Zhou, T.-M. Ding, J.-T. Zai, Z.-M. Chen and S.-Y. Zhang, Org. Lett., 2019, 21, 2536–2540 CrossRef CAS PubMed.
- Z. Zeng, X. Xun, L. Huang, J. Xu, G. Zhu, G. Li, W. Sun, L. Hong and R. Wang, Green Chem., 2018, 20, 2477–2480 RSC.
- C. Chen, J.-C. Kang, C. Mao, J.-W. Dong, Y.-Y. Xie, T.-M. Ding, Y.-Q. Tu, Z.-M. Chen and S.-Y. Zhang, Green Chem., 2019, 21, 4014–4019 RSC.
- M. Zidan, T. McCallum, L. Thai-Savard and L. Barriault, Org. Chem. Front., 2017, 4, 2092–2096 RSC.
- M. Liu, H. Huang and Y. Chen, Chin. J. Chem., 2018, 36, 1209–1212 CrossRef CAS.
- S. Yao, K. Zhang, Q.-Q. Zhou, Y. Zhao, D.-Q. Shi and W.-J. Xiao, Chem. Commun., 2018, 54, 8096–8099 RSC.
- H. Wang, Q. Xu and S. Yu, Org. Chem. Front., 2018, 5, 2224–2228 RSC.
- H. I. Jung, Y. Kim and D. Y. Kim, Org. Biomol. Chem., 2019, 17, 3319–3323 RSC.
- E. E. Touney, N. J. Foy and S. V. Pronin, J. Am. Chem. Soc., 2018, 140, 16982–16987 CrossRef CAS PubMed.
- D.-Y. Zhang, Y. Zhang, H. Wu and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 7450–7453 CrossRef CAS PubMed.
- Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R.-F. Cao, S.-Y. Zhang and J.-M. Tian, Angew. Chem., Int. Ed., 2019, 58, 12491–12496 CrossRef CAS PubMed.
- Y. Liu, Y.-L. S. Tse, F. Y. Kwong and Y.-Y. Yeung, ACS Catal., 2017, 7, 4435–4440 CrossRef CAS.
- D. H. Lukamto and M. J. Gaunt, J. Am. Chem. Soc., 2017, 139, 9160–9163 CrossRef CAS PubMed.
- H. Wu, Q. Wang and J. Zhu, Chem. – Eur. J., 2017, 23, 13037–13041 CrossRef CAS PubMed.
- R. Guo, X. Mo and G. Zhang, Org. Lett., 2019, 21, 1263–1267 CrossRef CAS PubMed.
- F.-P. Zhu, X. Guo, F.-M. Zhang, X.-M. Zhang, H. Wang and Y.-Q. Tu, Org. Lett., 2020, 22, 2076–2080 CrossRef CAS PubMed.
- T.-L. Zheng, Y. Zhang, A.-L. Gou, F. Cheng, S.-Z. Liu, L. Yu, M.-Y. Cui, X.-T. Xu, K. Zhang and S.-H. Wang, Org. Lett., 2020, 22, 7073–7077 CrossRef CAS PubMed.
- Y. Zhang, T.-L. Zheng, F. Cheng, K.-L. Dai, K. Zhang, A.-J. Ma, F.-M. Zhang, X.-M. Zhang, S.-H. Wang and Y.-Q. Tu, Chem. Sci., 2020, 11, 3878–3884 RSC.
- J. Yang, X.-M. Zhang, F.-M. Zhang, S.-H. Wang, Y.-Q. Tu, Z. Li, X.-C. Wang and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 8471–8475 CrossRef CAS PubMed.
- M. Mečiarová, P. Tisovský and R. Šebesta, New J. Chem., 2016, 40, 4855–4864 RSC.
- R. J. Comito, F. G. Finelli and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 9358–9361 CrossRef CAS PubMed.
- A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Nat. Chem., 2017, 9, 1073–1077 CrossRef CAS PubMed.
- S.-H. Wang, R.-Q. Si, Q.-B. Zhuang, X. Guo, T. Ke, X.-M. Zhang, F.-M. Zhang and Y.-Q. Tu, Angew. Chem., Int. Ed., 2020, 59, 21954–21958 CrossRef CAS PubMed.
- L. Kong, F. Su, H. Yu, Z. Jiang, Y. Lu and T. Luo, J. Am. Chem. Soc., 2019, 141, 20048–20052 CrossRef CAS PubMed.
- H. Shao, K. Fang, Y.-P. Wang, X.-M. Zhang, T.-M. Ding, S.-Y. Zhang, Z.-M. Chen and Y.-Q. Tu, Org. Lett., 2020, 22, 3775–3779 CrossRef CAS PubMed.
- E. H. Krenske, A. Patel and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 17638–17642 CrossRef CAS PubMed.
- H. Shao, Y.-P. Wang, K. Fang, Y.-M. Zhao and Y.-Q. Tu, J. Org. Chem., 2020, 85, 11968–11974 CrossRef CAS PubMed.
- P. Chen, H. Yang, H. Zhang, W. Chen, Z. Zhang, J. Zhang, H. Li, X. Wang, X. Xie and X. She, Org. Lett., 2020, 22, 2022–2025 CrossRef CAS PubMed.
- P. Yu and J. M. Cook, Tetrahedron Lett., 1997, 38, 8799–8802 CrossRef CAS.
- A. C. Peterson and J. M. Cook, J. Org. Chem., 1995, 60, 120–129 CrossRef CAS.
- G. Li, C. Piemontesi, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 2870–2874 CrossRef CAS PubMed.
- K. Nagaraju, D. Ni and D. Ma, Angew. Chem., Int. Ed., 2020, 59, 22039–22042 CrossRef CAS PubMed.
- Z. Lu, X. Zhang, Z. Guo, Y. Chen, T. Mu and A. Li, J. Am. Chem. Soc., 2018, 140, 9211–9218 CrossRef CAS PubMed.
- X. Li, Z. Zhang, H. Fan, Y. Miao, H. Tian, Y. Gu and J. Gui, J. Am. Chem. Soc., 2021, 143, 4886–4890 CrossRef CAS PubMed.
- T. Ooi, A. Saito and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 3220–3221 CrossRef CAS PubMed.
- T. Ooi, K. Ohmatsu and K. Maruoka, J. Am. Chem. Soc., 2007, 129, 2410–2411 CrossRef CAS PubMed.
- X. Zhang, R. J. Staples, A. L. Rheingold and W. D. Wulff, J. Am. Chem. Soc., 2014, 136, 13971–13974 CrossRef CAS PubMed.
- S. Y. Shim, Y. Choi and D. H. Ryu, J. Am. Chem. Soc., 2018, 140, 11184–11188 CrossRef CAS PubMed.
- H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5858–5861 CrossRef CAS PubMed.
- S. Lerch, L.-N. Unkel and M. Brasholz, Angew. Chem., Int. Ed., 2014, 53, 6558–6562 CrossRef CAS PubMed.
- W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Tetrahedron Lett., 2014, 55, 4648–4652 CrossRef CAS.
- L. Bu, J. Li, Y. Yin, B. Qiao, G. Chai, X. Zhao and Z. Jiang, Chem.–Asian J., 2018, 13, 2382–2387 CrossRef CAS PubMed.
- R. C. Godfrey, N. J. Green, G. S. Nichol and A. L. Lawrence, Nat. Chem., 2020, 12, 615–619 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |