
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen bonding in polymer science: towards new smart materials
Robin
Kampes
ab,
Stefan
Zechel
ab,
Martin D.
Hager
 ab and
Ulrich S.
Schubert
*ab
ab and
Ulrich S.
Schubert
*ab
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstraße 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, D-07743 Jena, Germany
First published on 5th July 2021
Abstract
The halogen bond is a special non-covalent interaction, which can represent a powerful tool in supramolecular chemistry. Although the halogen bond offers several advantages compared to the related hydrogen bond, it is currently still underrepresented in polymer science. The structural related hydrogen bonding assumes a leading position in polymer materials containing supramolecular interactions, clearly indicating the high potential of using halogen bonding for the design of polymeric materials. The current developments regarding halogen bonding containing polymers include self-assembly, photo-responsive materials, self-healing materials and others. These aspects are highlighted in the present perspective. Furthermore, a perspective on the future of this rising young research field is provided.
 Robin Kampes | Robin Kampes studied Chemistry at the Friedrich Schiller University Jena and received his master's degree in 2018. In these studies, he worked on halogen bond driven anion sensing. Since then he is working in the group of Prof. Schubert as PhD student focusing on halogen bonding and supramolecular functional materials. |
 Stefan Zechel | Stefan Zechel was born in Blankenburg (Germany) in 1988. He studied chemistry in Jena and performed his PhD under the supervision of Prof. Dr U. S. Schubert. He obtained a stipend from the Verband der Chemischen Industrie (VCI). After completing his PhD, he started his Post-Doc in the group of Prof. Schubert. From 2017 until end of 2019, he was funded by the Carl-Zeiss Foundation. His research interest are supramolecular polymers, self-healing and shape-memory materials as well as digitalization in polymer science. |
 Martin D. Hager | Martin D. Hager studied Chemistry at the Friedrich Schiller University in Jena till 2005. He finished his PhD in 2007. Subsequently, he was PostDoc at the TU Eindhoven. Since 2008 he is a group leader in the group of Prof. Schubert at the FSU Jena. His research interests include in particular organic redox-flow batteries and reversible polymer systems for self-healing materials. |
 Ulrich S. Schubert | Ulrich S. Schubert studied chemistry in Frankfurt and Bayreuth (both Germany) and at Virginia Commonwealth University, Richmond (USA). After PhD studies at the Universities of Bayreuth and South Florida, and postdoctoral training with J.-M. Lehn, he moved to the TU Munich (Germany), where he obtained his Habilitation in 1999. 1999–2000 he was Professor at the University of Munich and 2000–2007 Full Professor at the TU Eindhoven (The Netherlands). Since 2007, he has been a Full Professor at the Friedrich Schiller University Jena (Germany). He is an elected member of the German National Academy of Science and Engineering (acatech) and external scientific member of the Max-Planck-Gesellschaft (MPI for Colloid & Interfaces, Golm). |
Introduction
The combination of supramolecular interactions and polymers gained significant interest during the last decades.1,2 Thus, these non-covalent interactions could be incorporated into polymeric structures enabling many different applications.3 For example, self-assembling materials,4 self-healing materials,5 shape-memory polymers6 and photo-responsive materials/stimuli-responsive materials7 can be based on supramolecular interactions. In particular, main-chain supramolecular polymers have been investigated intensely.8 Herein, the polymerization can be facilitated by the supramolecular interactions. Another rather common approach for functional materials besides the main-chain supramolecular polymers is the functionalization of side-chain polymers.9 The most frequently applied class of supramolecular moieties are hydrogen bond (HB) motifs. Furthermore, metal–ligand coordination,10 ionic interactions11 as well as π–π stacking12 can be utilized to construct polymers featuring supramolecular moieties.In contrast, halogen bonding (XB) is quite uncommon in this field despite its growing impact on several other research areas.13,14 Up to now, only a few examples of polymeric materials with attached XB motives have been reported. However, this interesting field recently gained more and more importance and the incorporation of XB in polymer science accelerates (see Fig. 1). This combination seems to be highly beneficial, since XB feature significant advantages compared to HB and other supramolecular interactions (see Table 1 for comparison). Thus, the perspective will focus on functional materials enabled by the utilization of XB in polymers.
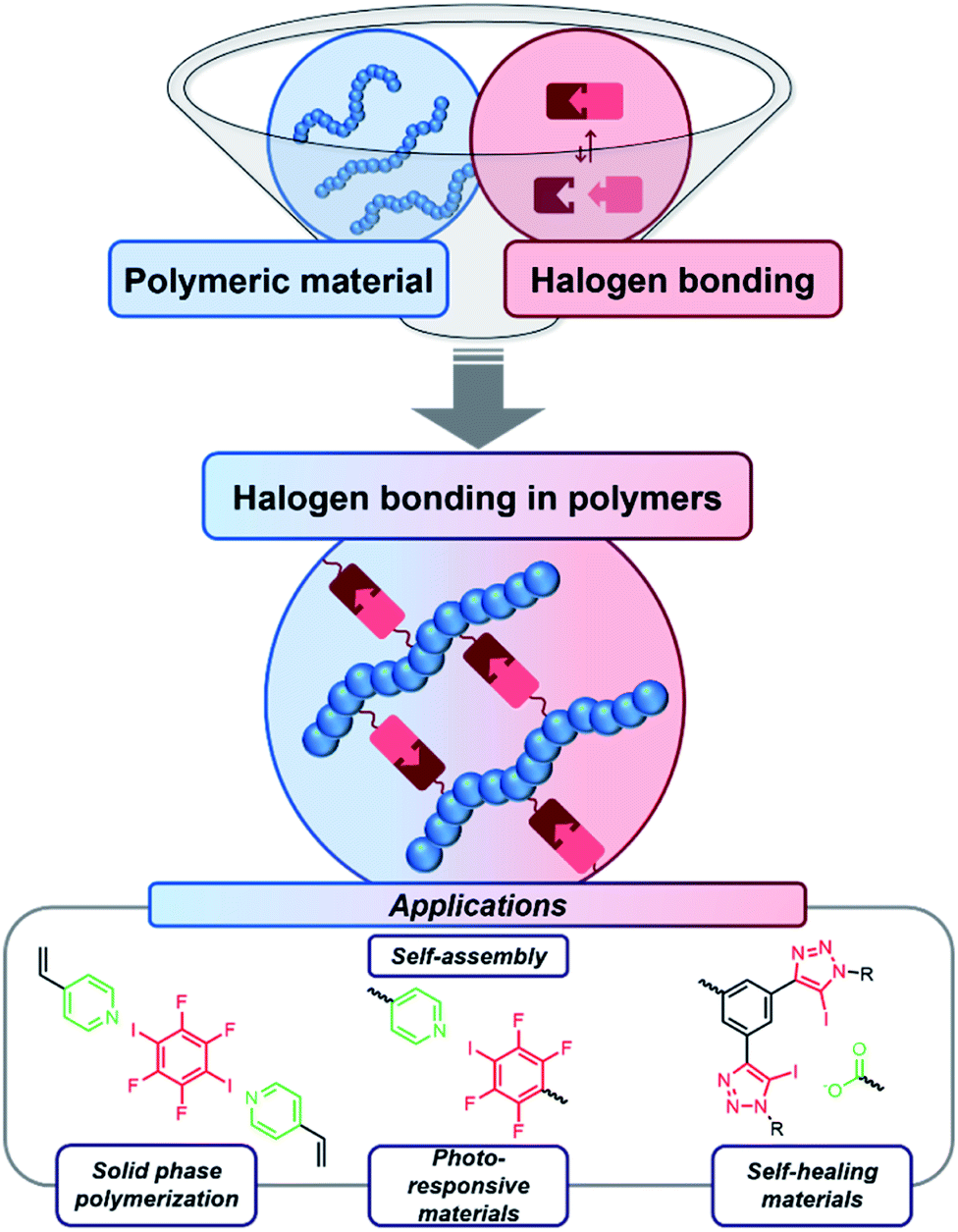 | ||
| Fig. 1 Schematic representation of the concept of halogen bond-driven smart polymer materials. | ||
| XB | HB | Metal to ligand | π–π stacking | |
|---|---|---|---|---|
| Interaction strength | ++ (∼10 to 150 kJ mol−1)13 | ++ (up to ∼155 kJ mol−1)22 | +++ (up to ∼400 kJ mol−1)23 | − (up to ∼50 kJ mol−1)24 |
| Directionality | ++ | + | ++ | + |
| Tunability | + | + | ++ | − |
| Water resistance | ++ | − | ++ | ++ |
| Synthetic effort | − | +/− | +/− | +/− |
| Investigated stimuli | Temperature/pH-change/light | Temperature/pH-change/moisture/mechanical force | Temperature/light/pH-change/redox-reactions/chemicals (other ligands)/mechanical force | Temperature/light |
| Frequency of occurrence | − − | +++ | ++ | +/− |
| Most often used motives | Iodo perfluorobenzene vs. pyridine25 | 2-Ureido-4-pyrimidone26 | Multivalent pyridine vs. Ru(II)10 | Naphthalene diimide vs. pyrene27 |
Halogen bonding
XB is a supramolecular interaction between a XB-donor (i.e. Lewis-acid) and an acceptor Y (i.e. Lewis-base) (Fig. 2).15 In detail, XB arises at a donor halogen atom X, which constitutes the XB-donor together with a covalently bound polarizing group R. The polarization of X results in a partial positive polarization on the far side of the R–X axis, which is the called σ-hole.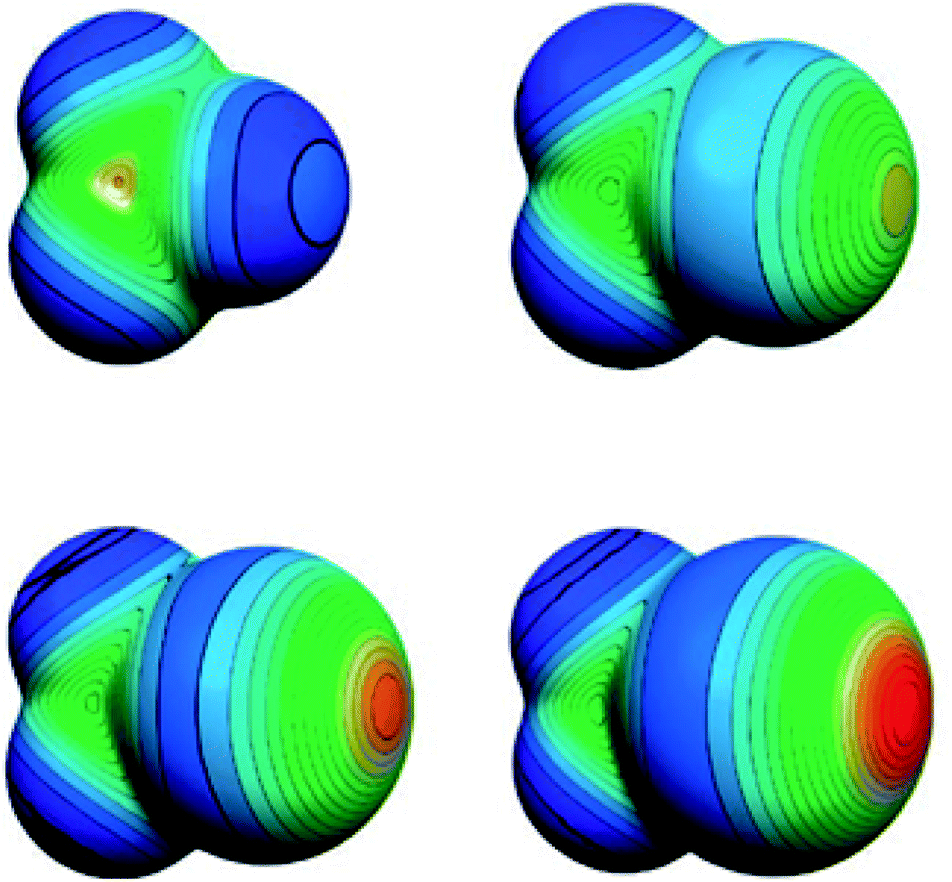 | ||
| Fig. 2 Schematic representation of the surface potential of CF3X (from top left to bottom right X = F, Cl, Br and I) (reprinted with permission from ref. 16). | ||
It allows the halogen atom to act as an electrophile towards a potential XB-acceptor. The σ-hole is surrounded by a belt of negative potential (see Fig. 2).16 Consequently, the XB features high directionality. The well-defined location of the σ-hole leads to a strong tendency towards a linear R–X⋯Y arrangement. Excellent tunability of the XB-donor strength is provided by the choice of X (due to polarizability I > Br > Cl > F, see Fig. 2) and the polarizing strength of R.16 Another benefit of the XB is that common XB-donor functionalities are less hydrophilic compared to the usual HB-donor moieties.13 Consequently, the XB can be an emerging tool in supramolecular chemistry, e.g., in crystal engineering,17 anion recognition18,19 and organocatalysis.20 For detailed information on halogen bonds in non-polymeric surroundings, the interested reader is referred to two excellent review articles.13,14
XB in polymers
In the following chapters, we will introduce several XB-driven materials categorized by possible application field. The currently utilized polymers and the corresponding XB-motives are summarized in Fig. 3. Thus, it could be seen that only very few different motifs are currently applied for the design of polymers featuring supramolecular moieties. The most frequently applied XB-moiety in polymers is based on poly(4-vinylpyridine) and iodoperfluorohydrocarbons. These systems feature the high benefit of an easy synthetic accessibility. However, the interaction strength is rather low and cannot be tuned significantly. Therefore, first approaches focus also on other systems featuring strong supramolecular binding such as phenyl-bis-triazole systems. However, the structural variety is currently limited to only a few different XB-motives.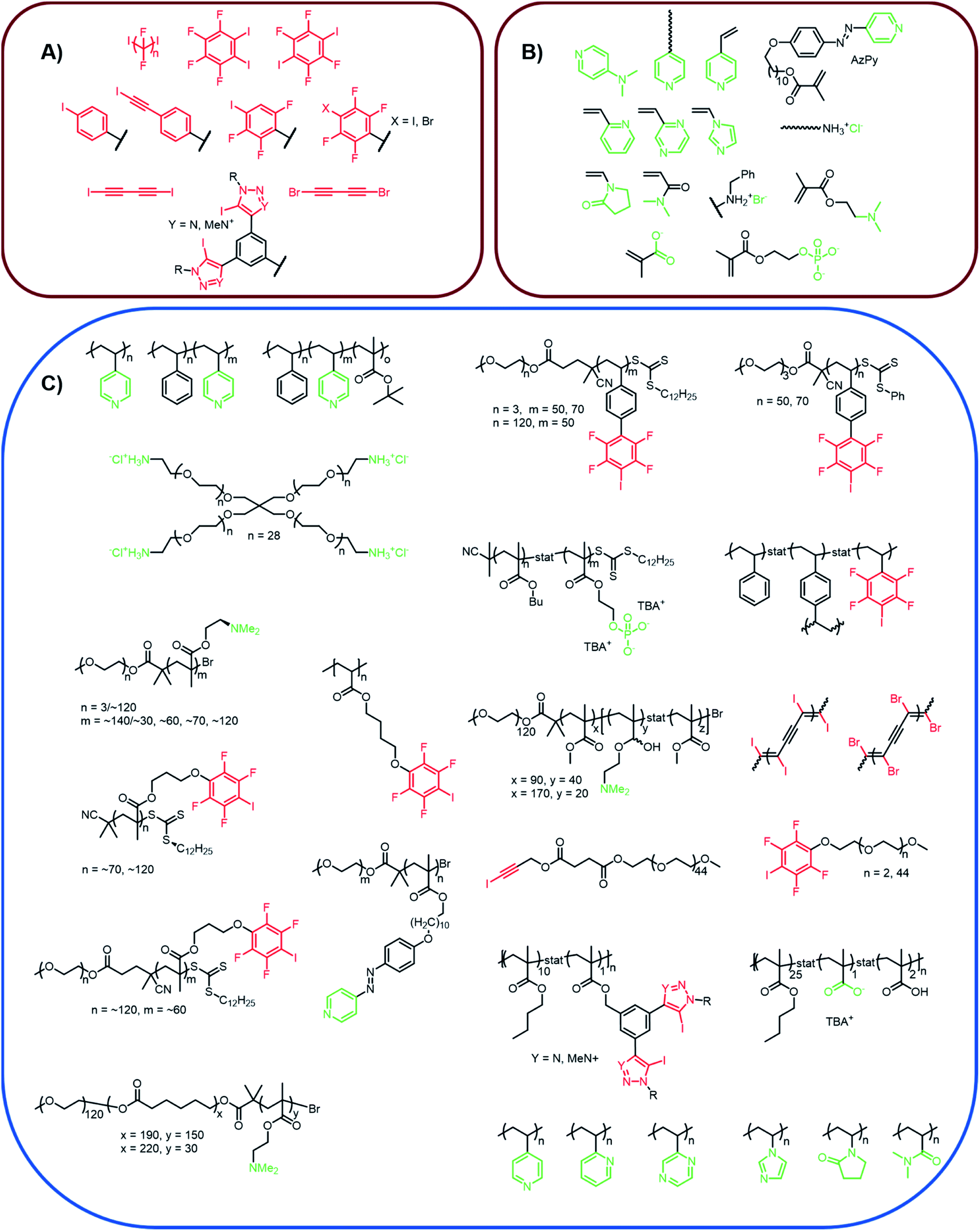 | ||
| Fig. 3 Schematic representation of (A) XB-donors (red) and (B) XB-acceptors (green) introduced into XB-driven polymer materials as well as (C) the realized polymer structures containing XB-donors and acceptors, respectively.25,32–55. | ||
Self-assembly
A major goal in polymer science is the targeted synthesis of polymeric structures featuring an assembly in a defined manner. For this purpose, several different strategies can be applied such as the utilization of block copolymers.28,29 Furthermore, supramolecular interactions can influence the assembly or can even enable or improve the assembly process.30,31One of the first reports involving XB in polymer materials was published in 2002.32 The authors prepared a XB-driven comb-copolymer based on a poly-(4-vinylpyridine) (P4VP) backbone and diiodoperfluorocarbons as XB-donor moieties. Polarized-light optical microscopy revealed that the supramolecular material (i.e. P4VP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :donor 2:1) featured a high tendency to assemble in a macroscopic scale probably due to phase separation of the perfluorinated XB-donors and the P4VP. More recently, this first approach was expanded to the application in block copolymers.33 For this purpose, P4VP-b-polystyrene (P4VP-b-PS) was applied featuring still one XB accessible block (P4VP), which could be utilized for the formation of XB in a polymer by adding 1,8-diiodoperfluorooctane leading to a directed self-assembly. Herein, a lamellae-within-cylinder structure could be obtained. The approach reveals the high benefit of XB-driven self-assembly in polymers since the simple addition of a XB-donor molecule induced the self-assembly behavior of the block copolymer to a rather complex structure. Consequently, this approach was also applied for other materials as well, e.g., by using a combination of P4VP as polymeric XB-acceptor with a polyacrylate bearing XB-donor moieties, i.e. poly(4-(4-iodo-2,3,5,6-tetrafluorophenoxy)-butyl acrylate) (PIPBA).34 Within this study, self-assembled multilayer films were targeted. For this purpose, an amino-functionalized substrate was immersed into solutions of the polymers in THF/chloroform alternately. By this manner, a (PIPBA/P4VP)10 multilayer film could be obtained. To observe the self-assembly behavior of the multilayer films, UV-Vis spectroscopy was applied confirming a linear growth of the film with increasing cycles. Desorption experiments in methanol revealed a worse performance of the purely XB-based film compared to a HB based film prepared out of poly(4-vinylphenole) (PVPh) as HB-donor. In order to further improve the system, both supramolecular moieties were combined to a mixed multilayer of (PIPBA/P4VP/PVPh/P4VP)5. This HB/XB-based multilayer revealed a significantly enhanced stability compared to the pure XB-based multilayer. Furthermore, it is also possible to utilize other XB-acceptor motives for the design of XB-containing polymer films. An example of a very powerful large scale organization by XB-driven self-assembly of star-shaped polyethylene glycol (PEG) and iodoperfluoroalkanes (IPFA) was enabled by using ammonium chloride as XB-acceptor motive.35 The polymer matrix (PEG) featured ammonium chloride end groups, which resulted in a dense packaging with a nanoscale periodicity caused by a clustering of the ionic end groups. However, on the macroscopic scale the pure PEG is isotropic. Small angle X-ray scattering (SAXS) measurements of the supramolecular material revealed a highly ordered lamellar structure based on the complexation of the IPFA. Herein, a combination of two processes caused the strong tendency towards higher order structures: on the one hand, XB occurs between the iodine of the IPFAs and the chloride of the ammonium salt. On the other hand, IPFAs tend to dense lateral packaging to phase separated layers.
:donor 2:1) featured a high tendency to assemble in a macroscopic scale probably due to phase separation of the perfluorinated XB-donors and the P4VP. More recently, this first approach was expanded to the application in block copolymers.33 For this purpose, P4VP-b-polystyrene (P4VP-b-PS) was applied featuring still one XB accessible block (P4VP), which could be utilized for the formation of XB in a polymer by adding 1,8-diiodoperfluorooctane leading to a directed self-assembly. Herein, a lamellae-within-cylinder structure could be obtained. The approach reveals the high benefit of XB-driven self-assembly in polymers since the simple addition of a XB-donor molecule induced the self-assembly behavior of the block copolymer to a rather complex structure. Consequently, this approach was also applied for other materials as well, e.g., by using a combination of P4VP as polymeric XB-acceptor with a polyacrylate bearing XB-donor moieties, i.e. poly(4-(4-iodo-2,3,5,6-tetrafluorophenoxy)-butyl acrylate) (PIPBA).34 Within this study, self-assembled multilayer films were targeted. For this purpose, an amino-functionalized substrate was immersed into solutions of the polymers in THF/chloroform alternately. By this manner, a (PIPBA/P4VP)10 multilayer film could be obtained. To observe the self-assembly behavior of the multilayer films, UV-Vis spectroscopy was applied confirming a linear growth of the film with increasing cycles. Desorption experiments in methanol revealed a worse performance of the purely XB-based film compared to a HB based film prepared out of poly(4-vinylphenole) (PVPh) as HB-donor. In order to further improve the system, both supramolecular moieties were combined to a mixed multilayer of (PIPBA/P4VP/PVPh/P4VP)5. This HB/XB-based multilayer revealed a significantly enhanced stability compared to the pure XB-based multilayer. Furthermore, it is also possible to utilize other XB-acceptor motives for the design of XB-containing polymer films. An example of a very powerful large scale organization by XB-driven self-assembly of star-shaped polyethylene glycol (PEG) and iodoperfluoroalkanes (IPFA) was enabled by using ammonium chloride as XB-acceptor motive.35 The polymer matrix (PEG) featured ammonium chloride end groups, which resulted in a dense packaging with a nanoscale periodicity caused by a clustering of the ionic end groups. However, on the macroscopic scale the pure PEG is isotropic. Small angle X-ray scattering (SAXS) measurements of the supramolecular material revealed a highly ordered lamellar structure based on the complexation of the IPFA. Herein, a combination of two processes caused the strong tendency towards higher order structures: on the one hand, XB occurs between the iodine of the IPFAs and the chloride of the ammonium salt. On the other hand, IPFAs tend to dense lateral packaging to phase separated layers.
Solution phase self-assembly
Beside the solid-state self-assembly process, it is also possible to utilize XB-containing polymers for the assembly in solution. The first example of complementary XB-polymers was reported by the Taylor group in 2015.25 In this study, a XB-donor polymer was obtained by reversible addition–fragmentation chain-transfer (RAFT)-polymerization of a methacrylate monomer containing a tetrafluoro iodobenzene moiety. Complementary, poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) was utilized as XB-acceptor polymer. Both XB-acceptor and -donor polymers were also synthesized as diblock copolymers using a PEG segment as additional block for both polymers. The association constants were investigated for both monomers and polymers applying 19F NMR titration experiments. Herein, higher binding energies could be observed for the polymers indicating the contribution of multiple binding sites to reach stronger association. Moreover, the complementary polymers assembled into higher ordered structures in water and organic solvents. This concept could be expanded resulting in a deeper understanding of the assembly process.36 Therefore, a wide range of morphologies could be obtained using these materials. In addition, the importance of XB for the self-assembly process could be shown in a detailed study. For this purpose, a pentafluorobenzene functionalized polymer, which lacked the XB-donor ability, did not result in any self-assembled morphologies. Furthermore, the addition of iodo pentafluorobenzene, a strong XB-donor, disrupted the self-assembly process. One potential application of self-assembled structures in solution could be drug delivery and one potential aspect could be the formation of multicompartment aggregates. XB-driven self-assembly of multicompartment micelles can be achieved on the basis of the Taylor groups polymers.37 For this purpose, the XB-acceptor polymer was extended by a polycaprolactone (PCL) block and a poly(methyl methacrylate) (PMMA) block, respectively. These hydrophobic blocks collapsed during transfer to water resulting in the formation of a secondary compartment.Triblock polymers based on a P4VP, a PS and tert-butyl methacrylate block extended the range of utilized XB-acceptor motives applied for the design of multicompartment micelles.38 The microstructure of triblock copolymer nanoparticles was efficiently manipulated by incorporation of HB and XB. For this purpose, polystyrene-b-poly(4-vinylpyridine)-b-poly(tert-butyl methacrylate) featured a HB- or XB-acceptor in terms of the pyridine nitrogen (see Fig. 4). Furthermore, HB- and XB-donor molecules with different interaction strength were investigated. The findings suggest that not only the interaction strength determines the effect on the microstructure but also the ability for molecular packing of the donor molecules within the matrix. Thus, only the HB-donor cholesteryl hemisuccinate (CHEMS) induced a morphology transition due to its high tendency towards intermolecular packing as well as strong binding properties.
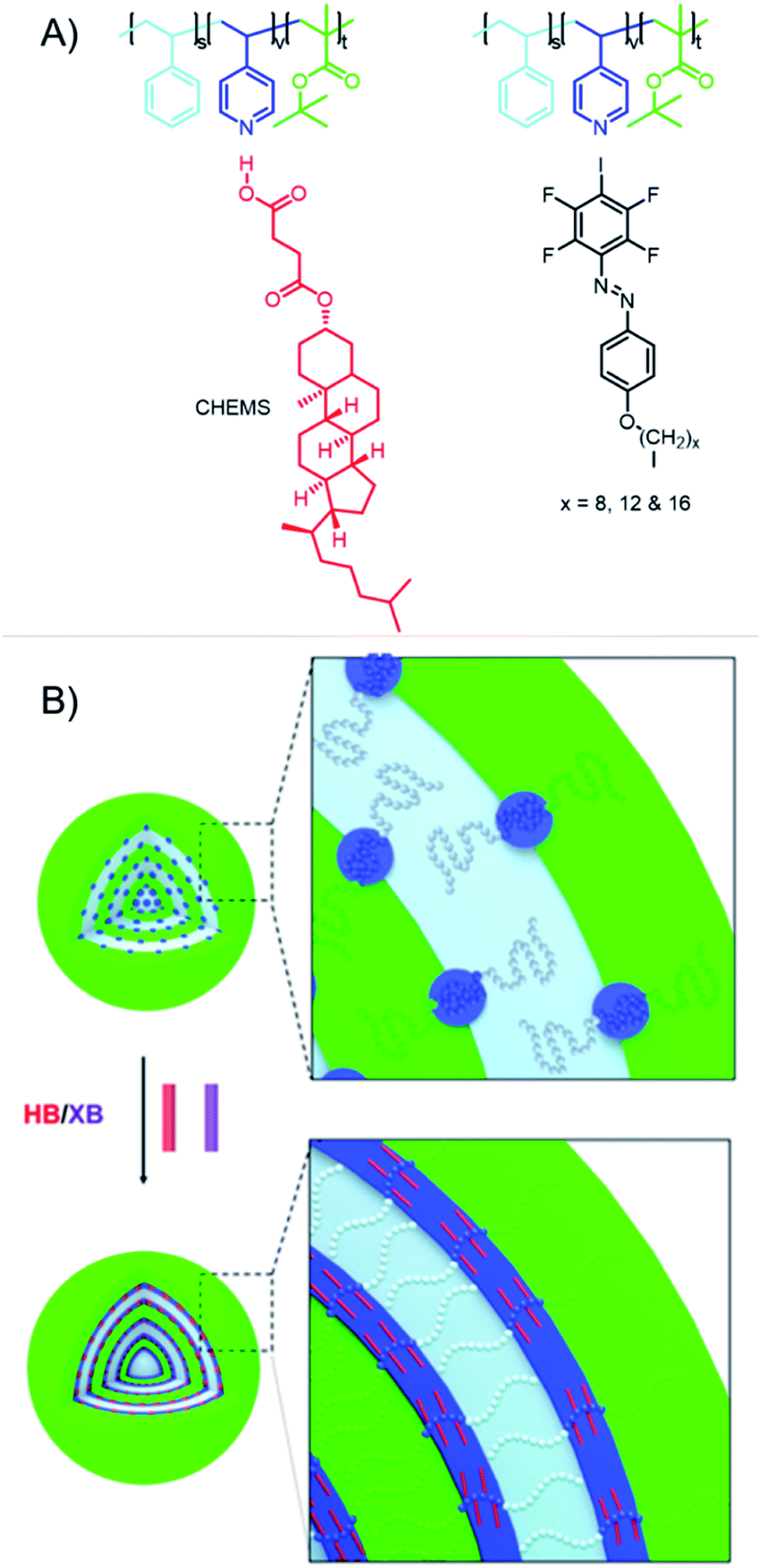 | ||
| Fig. 4 Schematic representation of (A) the acceptor polymers and the HB-donor CHEMS as well as the XB-donors used for self-assembly process and (B) the impact of co-assembly with the HB- and XB-donors on the obtained micelles (reprinted with permission from ref. 38). | ||
A hybrid HB/XB approach on supramolecular graft copolymers for the application via a solution self-assembly process was recently reported exploiting the higher solvent resistance of the XB compared to HB (see Fig. 5).39 The inner component of the assembly process formed by stacking of N1,N3,N5-tris(pyridin-4-ylmethyl)benzene-1,3,5-tricarboxamide (BTA-Py) via HB. Consequently, the formation of supramolecular polymer chains with pyridine motives as XB-acceptor sites pointing outside could be realized. Co-assembly with XB-donor functionalized ω-p-iodo tetrafluorophenyl-PEG (PEG-1) resulted in the formation of spherical micelles, while triethylene glycol functionalized donor (TEG-1) lead to 1D fibers. In contrast to the pure BTA-Py, these supramolecular complexes were soluble in water indicating the complex formation (see Fig. 5C). Due to the basic character of the pyridine moiety, these complexes can be switched simply via pH-changes. During this process, the microstructure returns from spherical micelles to 1D fibers of the pure BTA-Py. Subsequently, the variation of the XB-acceptor towards a pyridyl functionalized naphthalene monoimide (NMI-Py) with one acceptor moiety per molecule led to fibers upon complexation with PEG-1, whereas TEG-1 resulted in vesicles.40
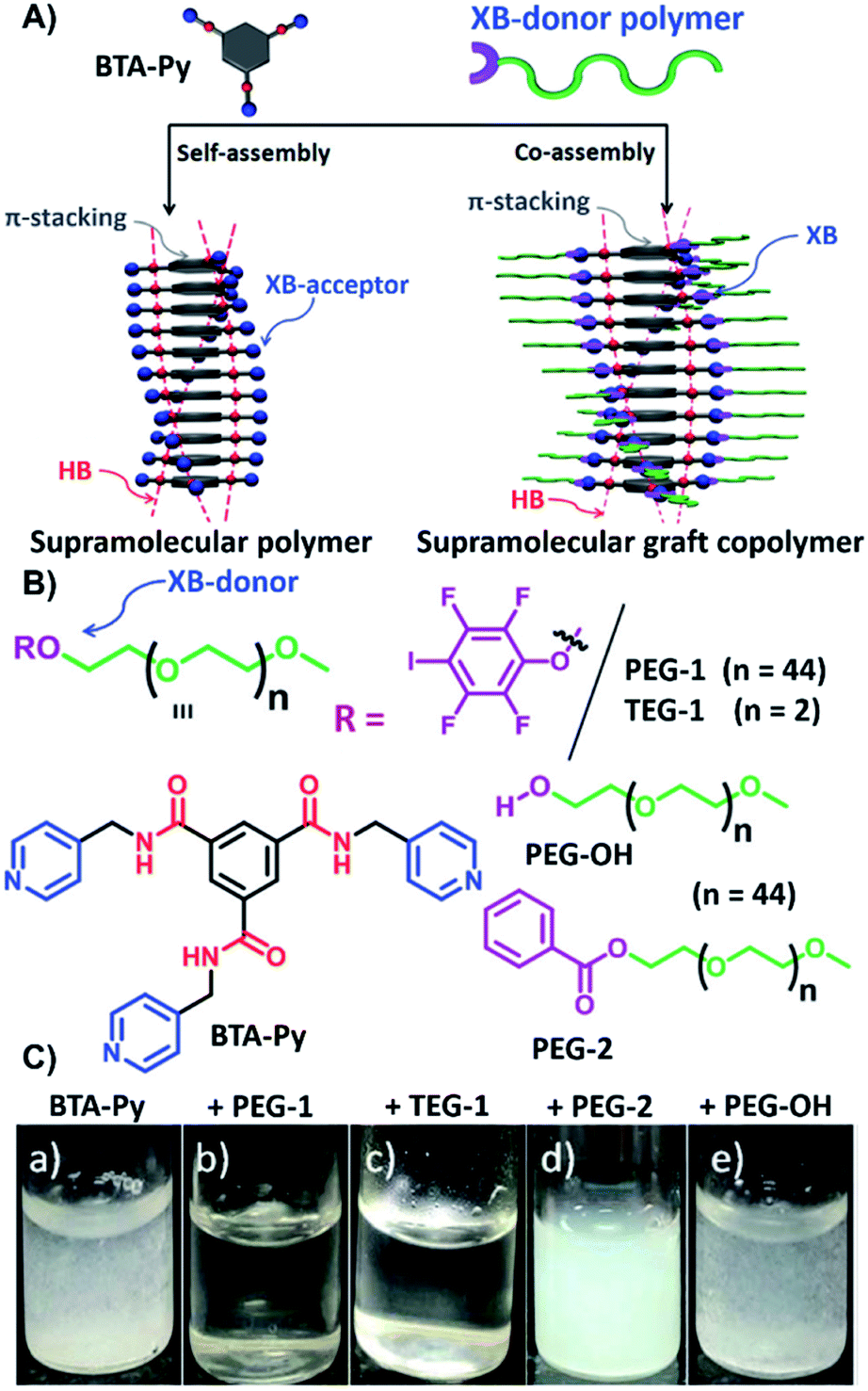 | ||
| Fig. 5 Schematic representation of (A) the self-assembly process of trivalent pyridine derivate and the co-assembly with the XB-donor polymers based on iodotetrafluorobenzene, (B) the different building blocks utilized for self- and co-assembly as well as the control molecules which lack a XB-donor moiety, (C) aqueous solutions of BTA with the different donors (a) acceptor only, (b) with PEG-1 in a ratio of 1:3, (c) with TEG-1 in a ratio of 1:3, (d) with PEG-2 in a ratio of 1:3, and (e) with PEG-OH in a ratio of 1:3 Hereby, the solubility of the BTA supramolecular polymer indicates the co-assembly (reprinted with permission from ref. 39). | ||
Materials for molecular recognition
Non-polymeric XB systems have been intensely utilized in anion recognition chemistry.18 The XB promoted the development of advanced receptor molecules for anion sensing in water, which is a major challenge for anion recognition chemistry.56,57 The ability of anions to act as strong XB-acceptors renders XB very suitable for this approach.18 In addition, other Lewis bases like nitrogen atoms (e.g., in pyridine) can act as XB-acceptors. This behavior was utilized in the first XB-driven imprinted polymer material.46 Imprinted polymers feature specific cavities, which can be created using a template molecule.58 The template forms a complex with the functional monomer during its polymerization into a permanent 3D polymer network.59 For this purpose, tetrafluoro-4-iodostyrene (TFIS) as XB-donor, styrene as a comonomer and divinylbenzene as crosslinker were polymerized in presence of 4-dimethylaminopyridine (DMAP) as template. Subsequent washing of the polymer removed the DMAP yielding a material with imprinted molecular recognition sites. In adsorption tests this polymeric material was able to preferably bind DMAP compared to other aminopyridines.Solid phase polymerization of XB-driven cocrystals
Conjugated polymers have attracted the interest of researchers for instance in fields like photovoltaic or organic light emitting diodes due to their properties such as electric conductivity. A structurally rather simple example of a conjugated polymer is polydiacetylene, which can be obtained by 1,4 polymerization of butadiyne.51,52 However, solution-based synthesis results in unregular polymer structures. To achieve a highly ordered molecular structure, which is required for optical applications, a precise alignment of the monomers in solid phase polymerization (SPP) can be utilized. Aiming for polydiiododiacetylene (PIDA) this topochemical polymerization60 approach was performed based on XB. However, crystals formed form pure diiododiacetylene lack the alignment required for ordered polymerization. To achieve the alignment, the formation of cocrystals with various XB-acceptors was investigated. Within these studies, cocrystals with suitable arrangements between 1 and the hosts 5 to 8 were obtained51,52 (see Fig. 6 and 7). The slow evaporation of a mixture of XB-acceptor 8 and diiododiacetylene in methanol resulted in crystals leading to the formation of PIBA via autopolymerization. For the cocrystals 1 with 5, 6 and 7 the arrangement is less ideal due to the distance between the diacetylene rods exceeding the optimal of 4.9 Å (1 × 5 = 5.11 Å, 1 × 6 = 5.02 Å and 1 × 7 = 5.25 Å). Hence, a polymerization of these crystals could not be achieved. To overcome this challenge the authors utilized pressure induced topochemical polymerizations.53,54 The cocrystals consisting of 1 and 5/6 were exposed to high pressure in a diamond-anvil cell. This process leads to a direct change of the color from colorless to blue to black indicating the successful polymerization process. Polydibromodiacetylene (PBDA) is structurally identical to PIDA. However, it should be significantly more stable rendering it as a potential alternative to PIDA. The major drawback is that the monomer, dibromodiacetylene is explosive at ambient temperature making the polymerization very challenging. To overcome this issue a topochemical polymerization approach analog to PIDA was investigated.55 Unfortunately, dibromodiacetylene is a weaker XB-donor compared to diiododiacetylene making the XB-driven cocrystallization more difficult. Via cocrystallization of 2 with 5 and 9 suitable crystals could be obtained. The cocrystals 2 × 5 were relatively stable at −15 °C. At ambient temperature, polymerization took place immediately. In contrast, the cocrystals 2 × 9 already polymerized at −18 °C indicated by a color change. These findings emphasize the suitability of XB-driven topochemical polymerization also for PIBA synthesis as a conjugated precursor polymer.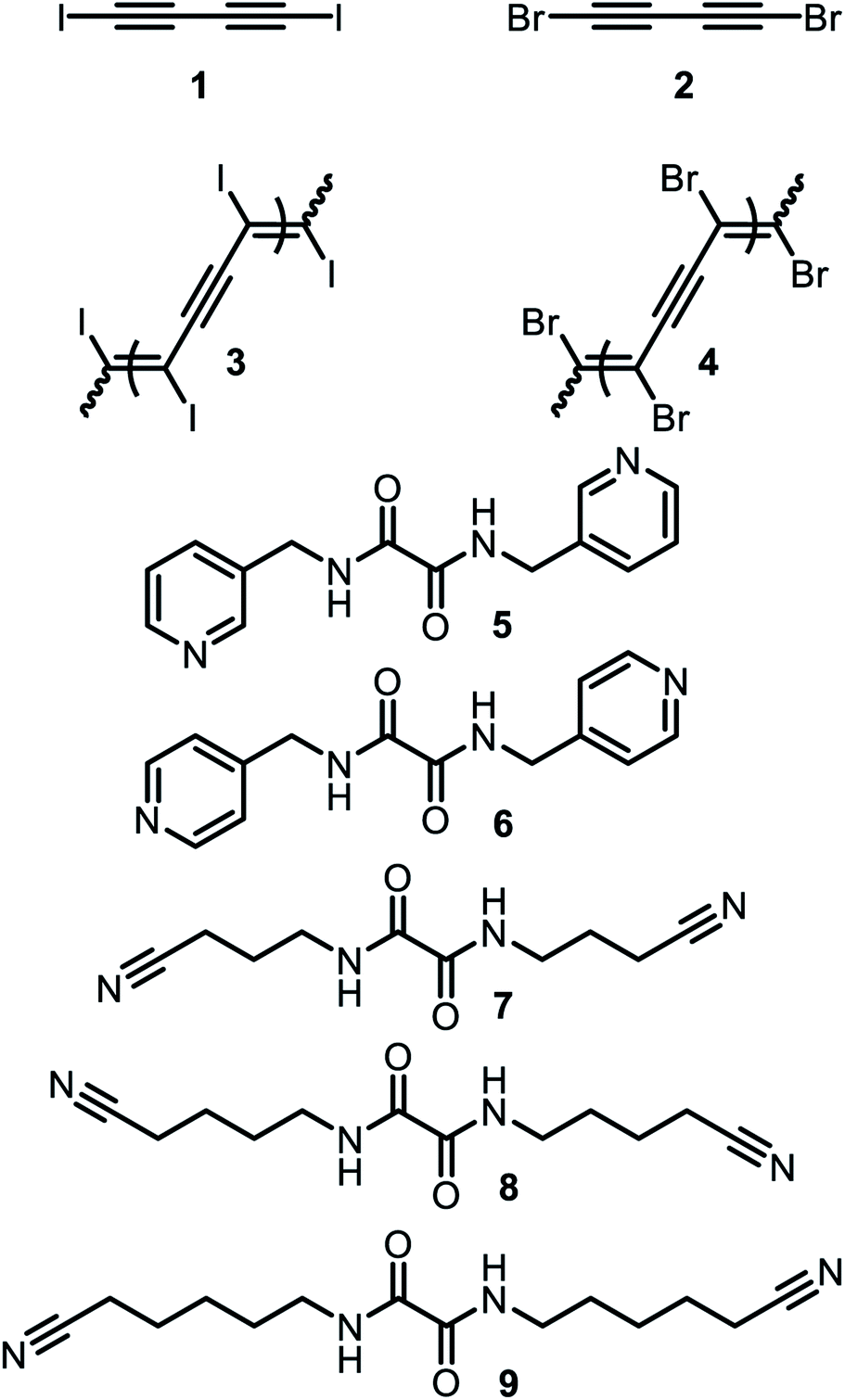 | ||
| Fig. 6 Schematic representation of the XB-donor acetylenes as well as the XB-acceptors utilized for cocrystallization and subsequent topochemical polymerizations by Goroff and coworkers.50–55 | ||
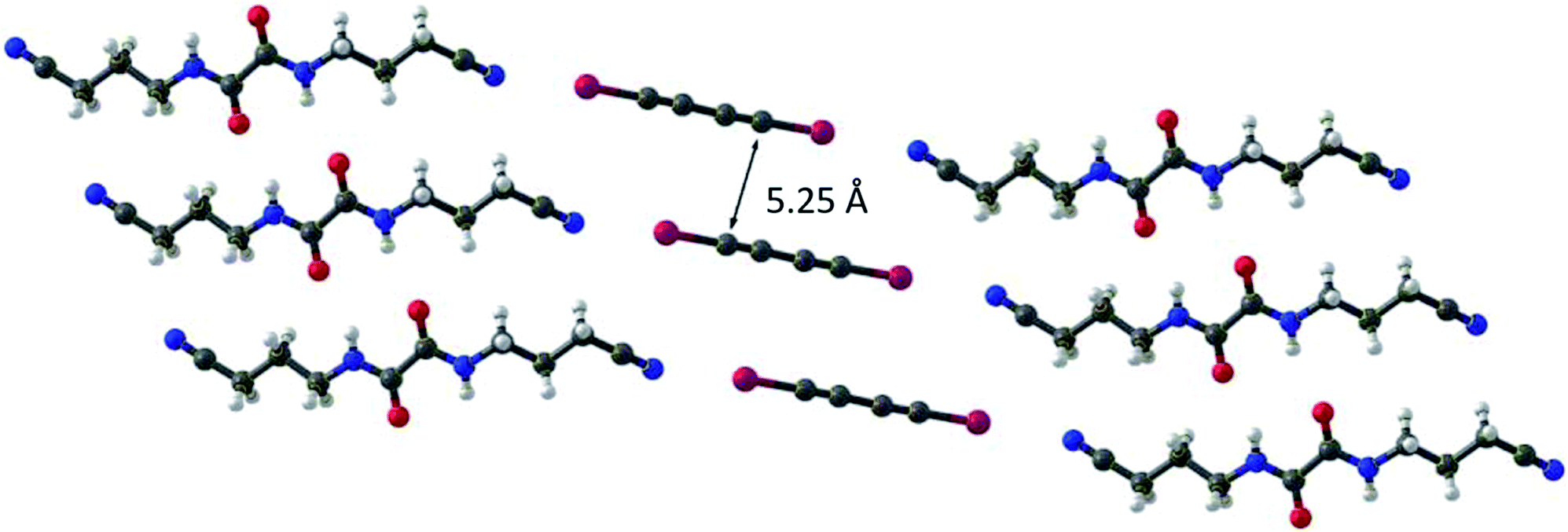 | ||
| Fig. 7 Molecular structure of the cocrystals 1 × 7. The C1 to C4 distance which is necessary for topochemical 1,4-polymerization is displayed (ideally 4.9 Å) (reprinted with permission from ref. 52). | ||
A contrarily approach using a XB-acceptor monomer and a XB donor to enable crystals for topochemical chemistry was introduced by the group of Goto.48 SPP has been utilized for polycondensation reactions to form polyesters and polyamides since several years.61 However, free radical polymerization was also applied in SPP.48 Goto and coworkers utilized nitrogen containing vinyl monomers, which can act as XB-acceptor and 1,4-diiodotetrafluorobenzene (XB-donor) as linker (see Fig. 8). Slowly evaporating solutions of the monomers and the XB linker yielded cocrystals, which were further reacted within the polymerizations. For this purpose, the cocrystals of the former liquid monomers 4-vinylpyridine, 2-vinylpyridine and 1-vinylimidazole were exposed to paraffin oil containing radical initiator and, subsequently, heated to 40 °C for 24 h. Thus, the SPPs resulted in high molar masses with Mn-values up to 7.4 × 105 g mol−1 which were significantly higher in comparison to solution phase polymerization. Furthermore, several monomers yielded narrow molar mass distributions Đ < 2 (down to 1.22). The high benefit of processing the crystals formed by XB of the former liquid monomers is the preparation of pre-shaped structures from liquid monomers (see Fig. 8C and D).
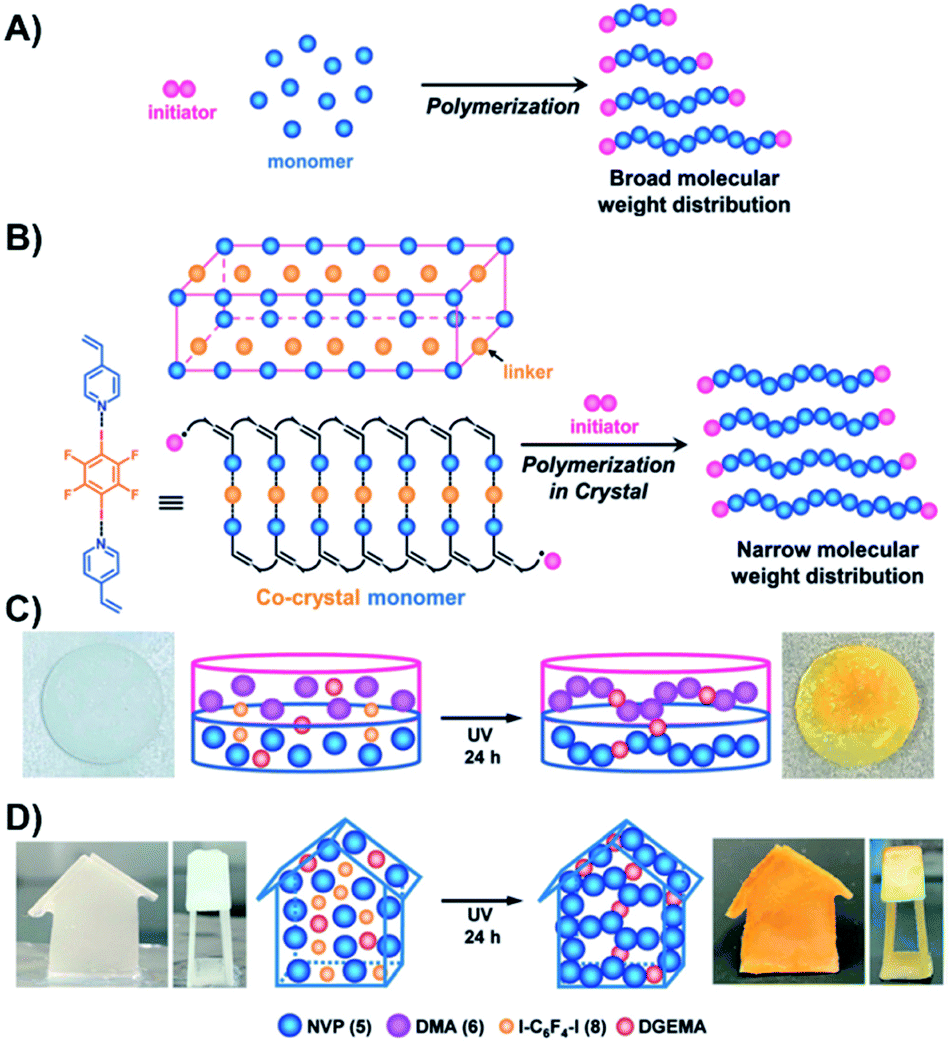 | ||
| Fig. 8 Schematic representation of (A) a radical polymerization in solution, (B) a radical polymerization in the crystalline solid state, (C) a two-layer polymer sheet prepared by photopolymerization of pre-shaped two-layer monomer sheet and (D) a 3D-model obtained by photopolymerization of the 3D pre-shaped monomer crystals (reprinted with permission from ref. 48). | ||
Light responsive materials
Polymers featuring XB have been introduced into the field of light responsive materials in 2012.41 Inspired by studies based on HB62,63 the combination of P4VP as XB-acceptor polymer complexed with photoactive diazo XB-donors led to polymeric films for light induced surface patterning or more exactly surface relief gratings (SRG). In comparison to HB-based films, XB materials revealed advantages in terms of surface patterning efficiency (see Fig. 9). This finding could be attributed to the high directionality of the XB resulting in a more efficient mass transport and the ability to fine-tune the interaction strength. In 2015, XB-donors were further expanded including alkyne based systems.42 This approach enabled an improved understanding of the involved processes in such material, in particular that the SRG performance increases with growing interaction strength. Additionally, the alkyne based XB-donor revealed to be superior to their perfluorinated counterpart due to better photochemical properties. Furthermore, the effect of the complexation degree was investigated for the two most promising azo XB-donors with the highest binding affinity (iodo tetrafluorobenzene and iodo alkyne).44 These investigations revealed that the alkyne donor features faster SRG through the test series from 0.1 to equimolar complexation. Though, the perfluorinated donor offers a larger modulation depth and shows a stronger tendency towards phase separation.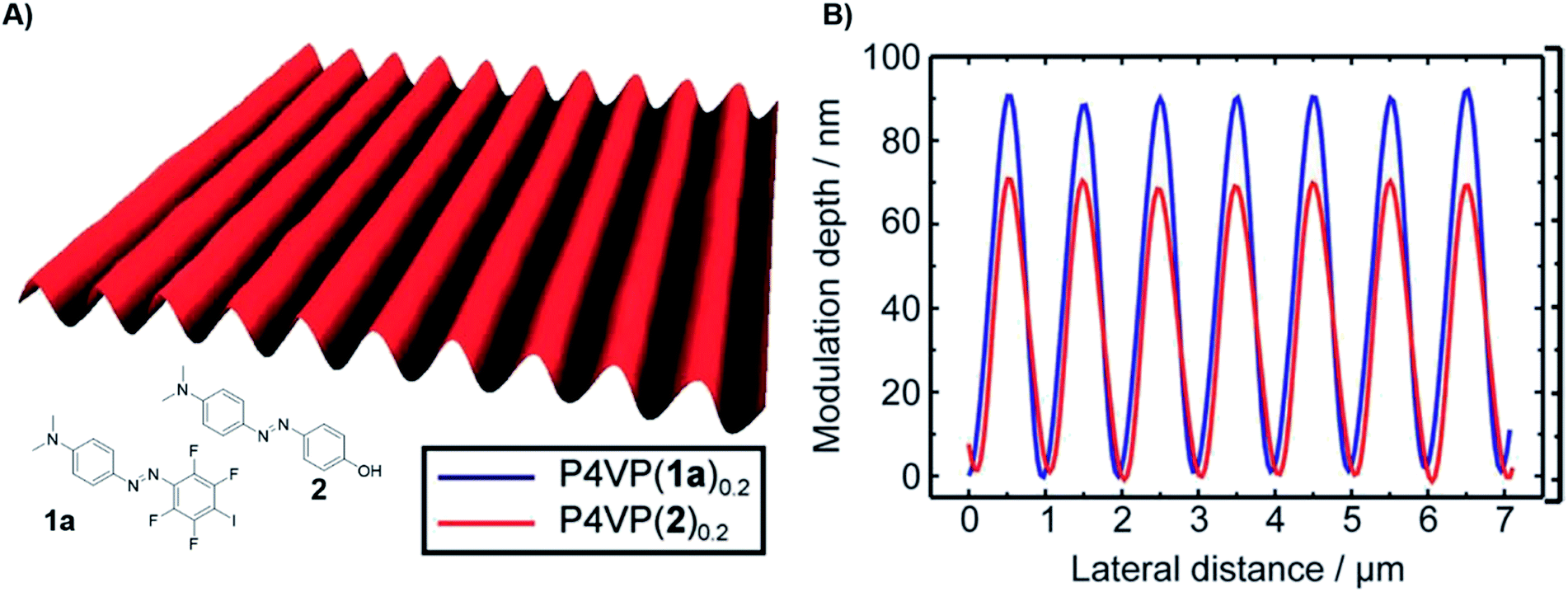 | ||
| Fig. 9 (A) AFM surface relief of the surface relief grating film composed of P4VP and azobenzene containing XB-donor 1a, (B) modulation depth of the HB (2, red) and XB (1a, blue) films (reprinted with permission from ref. 41). | ||
Spin-coated polymer films from P4VP with different azo perfluoro benzene donors (X = I, Br, H) were also investigated for second order nonlinear optical (NLO) response applications using all-optical poling.43 The NLO response (I > Br > H) correlated with the interaction strength while with similar interaction strength (X = Br, H) XB performed better compared to the HB system. In contrast, there was only a small effect when using polystyrene instead of P4VP. Since there is no XB towards the pyridine nitrogen possible in polystyrene, the small effect observed was attributed to weak X⋯π interaction.
The utilization of azo pyridines enables the design of covalently bound photo-switchable materials. For this purpose, a pyridyl azobenzene motive was introduced into a polymeric structure.47 The side chain attached azo pyridine units also provide the pyridine nitrogen as XB-acceptor. Due to complexation with 1,2-diiodo tetrafluorobenzene the material showed a high tendency to form ordered microstructure in polymer films. In addition, these films feature efficient photoalignment and reorientation properties.
Self-healing polymers
Self-healing (SH) polymer materials are in the scope of research since the first discovery in 2001.64 In particular, intrinsic self-healing polymers gained significant attention.65 The principle of intrinsic self-healing materials is based on temporary (cross-) linking, which can be achieved using reversible covalent bonds or supramolecular interactions.66 Among the field of supramolecular interactions HB,67 π–π stacking,68 ionic inter-actions,69 metal–ligand coordination70 and host guest interactions71 have been investigated intensely (for comparison with XB-based self-healing materials see Table 2).72| XB | HB | Metal to ligand | |
|---|---|---|---|
| Publications | 2 | >250 | >25 |
| Applied motives | 2 (one receptor and two donors) | >10 different | Ca. 10 different |
| Healing conditions | 100 °C for 17 h or 3.5 h | Room temperature in a few minutes | Light irradiation (320 to 390 nm; 950 mW cm−2, 30 s) |
| Thermally (60 to 150 °C for several hours) | |||
| Frequently utilized motifs | Phenyl-bis-triazoles and carboxylate and phosphonate | 2-Ureido-4[1H]-pyrimidinone | Terpyridine metal complexes (bis-methylbenzimidazolyl)pyridine metal complexes |
| Ureas, amides | Thiolate metal complexes | ||
| 2,7-Diamido-1,8-naphtyridine | And more | ||
| Hamilton receptor | |||
| Barbituric acid and many more | |||
| Utilized polymers | Poly(butyl methacrylate) networks | Polymer networks of multivalent moieties, polystyrene, polymethacrylates, polyisobutylene and many more | Poly(ethylene-co-butylene) networks, polymethacrylate networks, networks of multivalent moieties |
| Mechanical properties | Hard materials, hardness up to 60 MPa (nanoindentation) | Very soft materials (e.g., E-moduli up to 12 MPa for phase-separating graft-copolymers)75 | Hard materials, indentation modulus up to 1.5 GPa70 |
Even though the successful and often applied utilization of related HB there are so far only two investigations dealing with XB for the design of self-healing polymers. The first reported material was based on the combination of a XB-donor functionalized polymer with a XB-acceptor macromolecule.49 In this study, two HB- as well as two XB-donor polymers were synthesized using triazole and triazolium, respectively. This approach enabled the comparison of HB and XB as well as the investigation of the influence of the interaction strength on the healing behavior. The bidentate receptor design (see Fig. 10) was adapted from earlier anion recognition studies, in which these receptors revealed high potential for anion complexation.73 To obtain the polymeric donors, the monomeric systems were copolymerized with butyl methacrylate via RAFT polymerization. Subsequent quaternization of the triazoles yielded the charged donor polymers P3 and P4 with identical composition. All polymers were intensely investigated regarding their anion complexation behavior via isothermal titration calorimetry (ITC). For the acceptor polymer, methacrylic acid was copolymerized with butyl methacrylate (BMA) and partly deprotonated to obtain the ionic acceptor polymer P6. The XB-crosslinked polymer network was obtained by simply mixing both polymers together. All polymer films revealed self-healing behavior at 100 °C.
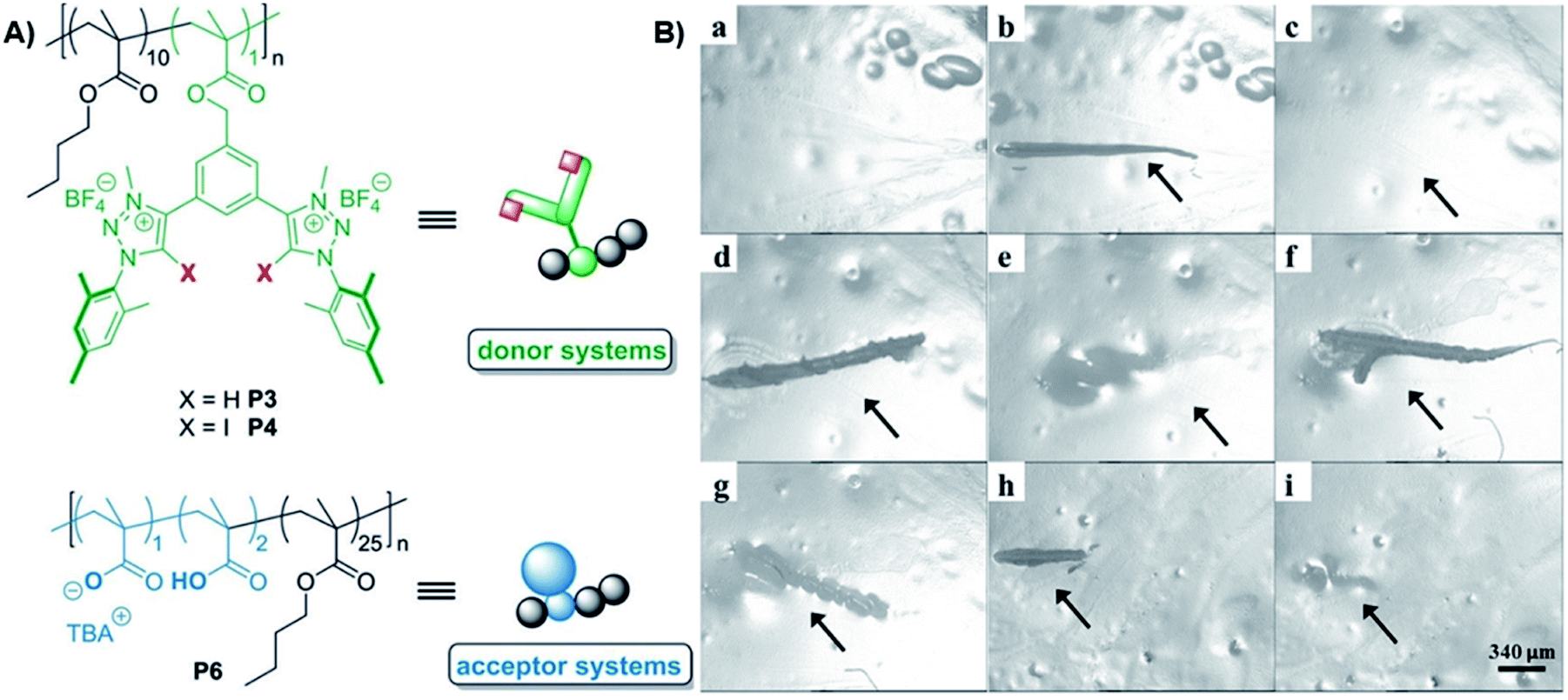 | ||
| Fig. 10 (A) Schematic representation of the polymer XB-donor and -acceptor utilized for the design of a self-healing material, (B) polymer film P46 (combination of P4 and P6) (X = I), (a) initial material, (b) first scratch, (c) healing via 17 h at 100 °C, (d) second scratch, (e) healing via heating for 17 h at 100 °C, (f) third scratch, (g) partial healing via heating for 4 h at 100 °C, (h) fourth scratch, and (i) healing via heating for 69 h at 80 °C (reprinted with permission from ref. 49). | ||
The concept could be expanded by crosslinking of an ionomer via an XB driven crosslinker (see Fig. 11).50 Ionomers usually contain anionic groups, which makes them well suited for crosslinking via XB. For this study a butyl methacrylate copolymer with phosphate side groups was synthesized. The bis-bidentate crosslinker features two of the already utilized receptors linked with C8-spacer. Even the ionomer revealed self-healing behavior, the XB-driven crosslinking increased the hardness of the material by one order of magnitude compared to the pure ionomer while being still able to heal mechanical damage.
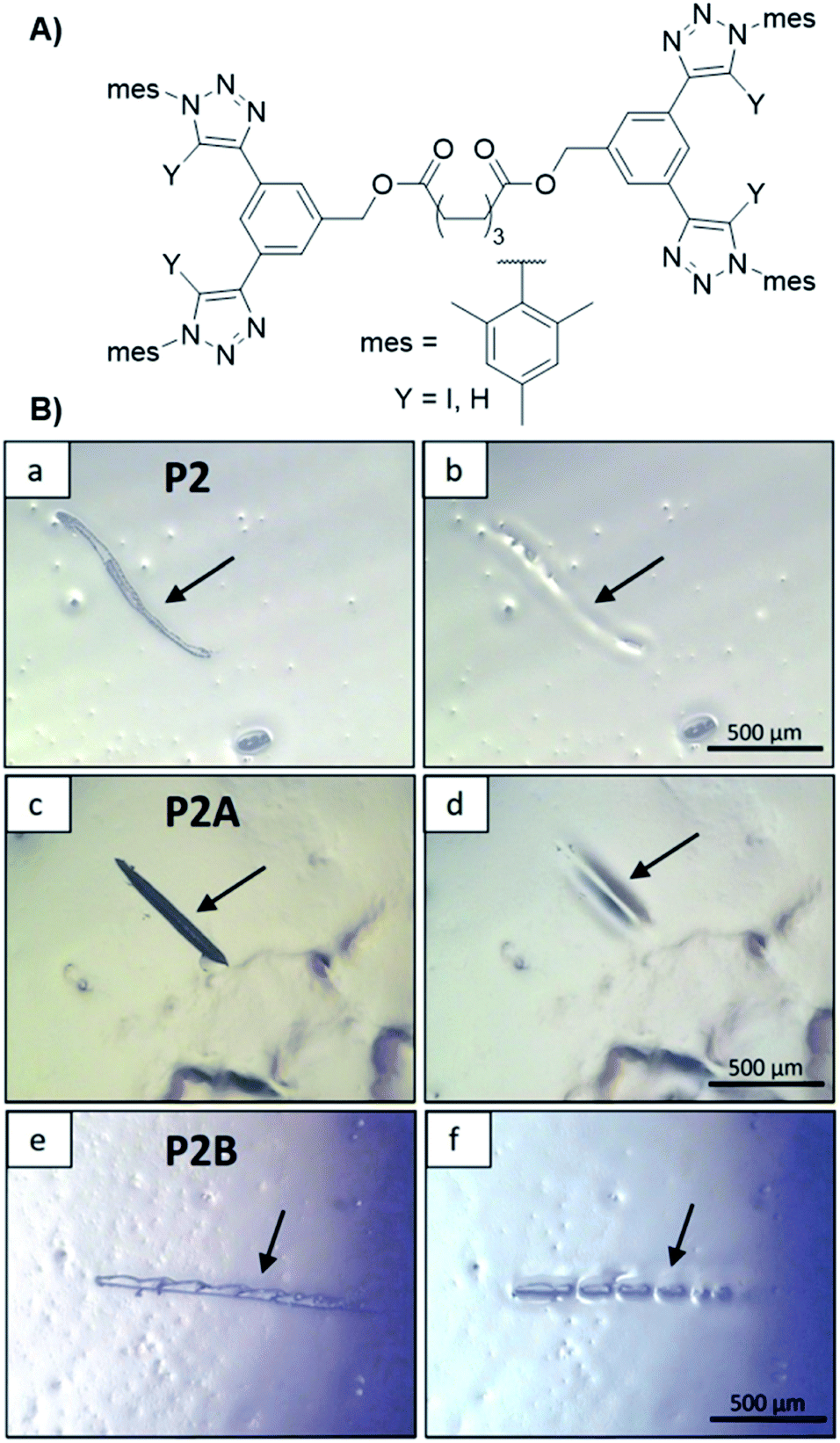 | ||
| Fig. 11 (A) Schematic representation of the XB and HB linker structure utilized for the synthesis of polymer networks capable for self-healing, (B) pictures of the scratch healing at 100 °C (left before, right after healing): (a and b) pure polymer, (c and d) polymer plus XB linker (X = I) and (e and f) polymer plus HB linker (X = H) (reprinted with permission from ref. 50). | ||
Conclusion and outlook
The implementation of XB into polymeric structures enables the development of new functional and tailor-made materials. Despite the still predominant underrepresentation of XB, there are several examples showing the high benefit of using such type of supramolecular interaction for diverse applications, including self-assembly, molecular recognition, photo-responsive and self-healing materials. Hence, the general suitability of XB for functional polymers based on supramolecular interactions could already be demonstrated very successfully. In addition, the obtained results indicate the potential advantageous character of XB compared to other supramolecular moieties, in particular compared to hydrogen bonds (HB). Within several direct comparisons, the XB-driven materials exceeded their HB-driven counterparts in many aspects – for instance in:(1) Water resistance in aqueous self-assembly processes,
(2) Surface relief gratings in photo responsive materials,
(3) Hardness in self-healing films.
Considering the overall small number of publications, there is still a huge potential for further research related to XB driven functional polymeric materials. A significant broadening of the applications analogous to HB is likely. Due to the structural relatedness of both bonds (HB and XB) one might expect that XB can also be utilized for all applications studied so far for HB polymers. One major field of interest will be stimuli-responsive materials. The first examples (i.e. self-healing materials) could already be shown. However, a general understanding of the addressability of the XB in polymers in not obtained up to now. Thus, the investigation of different stimuli and the response of the XB system in the solid will be one focus of research in the future. In particular, the application of other stimuli, beside the already applied temperature and light, will be investigated. For example, mechanoresponsive materials could be one field of interest since many other supramolecular interactions were already studied in this context.76 Furthermore, the defined preparation of XB systems featuring different binding strength can improve the investigation of the stimuli-responsive behavior. This knowledge will later enable more intelligent materials applicable for different proposes including shape-memory, sensors as well as self-healing.
Furthermore, it can be expected that the XB will reveal more advantageous characteristics with increasing applications but also with deeper investigations in the already introduced applications. However, one required precondition will be a broader usable chemistry such as more donor and acceptor moieties for the utilization in polymers. Additionally, a better synthetic availability can enhance the field of XB materials significantly. In particular, the implementation of more complex XB-donor sites as seen in anion recognition chemistry should be considered.
Finally, many different other potential applications were not explored so far. One field of interest could be the utilization in (intelligent) drug delivery systems. Compared to other supramolecular systems, XB bearing polymers can feature many advantages, such as no toxic metal ion or a better water resistance. However, the behavior of XB in biological surroundings was not studied so far.
In conclusion, the XB has great potential to become an important aspect in terms of polymer science in the future, due to its unique characteristics. XB can complement the toolbox of supramolecular interactions besides the well-established HB and metal–ligand interactions. Currently, it is still in its infancy; however, the high future potential was already revealed within the first investigations. It can be assumed that many more interesting examples of XB-based smart materials will follow in the future.
Author contributions
Writing – original draft: R. K. and S. Z. Writing – review & editing: M. D. H. and U. S. S. Supervision: M. D. H. and U. S. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Deutsche Forschungsgemeinschaft (DFG) for financial support (SCHU 1229/24-2 and project number 407426226, SFB/TRR 234 (CataLight, B02)).References
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34–39 CrossRef CAS.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- Y. Yang and M. W. Urban, Adv. Mater. Interfaces, 2018, 5, 1800384 CrossRef.
- Z.-C. Jiang, Y.-Y. Xiao, Y. Kang, M. Pan, B.-J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 20276–20293 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- M. Weck, Polym. Int., 2007, 56, 453–460 CrossRef CAS.
- A. Winter and U. S. Schubert, Chem. Soc. Rev., 2016, 45, 5311–5357 RSC.
- S. L. Craig, Angew. Chem., Int. Ed., 2009, 48, 2645–2647 CrossRef CAS.
- W. Hayes and B. W. Greenland, Adv. Polym. Sci., 2015, 268, 143–166 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- R. Desiraju Gautam, P. S. Ho, L. Kloo, C. Legon Anthony, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 Search PubMed.
- T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS.
- Z. Su, R. Zhang, X.-Y. Yan, Q.-Y. Guo, J. Huang, W. Shan, Y. Liu, T. Liu, M. Huang and S. Z. D. Cheng, Prog. Polym. Sci., 2020, 103, 101230 CrossRef CAS.
- J. Emsley, Chem. Soc. Rev., 1980, 9, 91–124 RSC.
- T. Rossow and S. Seiffert, Adv. Polym. Sci., 2015, 268, 1–46 CrossRef CAS.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS.
- A. Vanderkooy and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 5080–5086 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- H.-A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- J. N. L. Albert and T. H. Epps, Mater. Today, 2010, 13, 24–33 CrossRef CAS.
- J.-F. Gohy, B. G. G. Lohmeijer, S. K. Varshney and U. S. Schubert, Macromolecules, 2002, 35, 7427–7435 CrossRef CAS.
- A. J. Wilson, Soft Matter, 2007, 3, 409–425 RSC.
- R. Bertani, P. Metrangolo, A. Moiana, E. Perez, T. Pilati, G. Resnati, I. Rico-Lattes and A. Sassi, Adv. Mater., 2002, 14, 1197–1201 CrossRef CAS.
- R. Milani, N. Houbenov, F. Fernandez-Palacio, G. Cavallo, A. Luzio, J. Haataja, G. Giancane, M. Saccone, A. Priimagi, P. Metrangolo and O. Ikkala, Chem, 2017, 2, 417–426 CAS.
- F. Wang, N. Ma, Q. Chen, W. Wang and L. Wang, Langmuir, 2007, 23, 9540–9542 CrossRef CAS PubMed.
- N. Houbenov, R. Milani, M. Poutanen, J. Haataja, V. Dichiarante, J. Sainio, J. Ruokolainen, G. Resnati, P. Metrangolo and O. Ikkala, Nat. Commun., 2014, 5, 4043 CrossRef.
- A. Vanderkooy, P. Pfefferkorn and M. S. Taylor, Macromolecules, 2017, 50, 3807–3817 CrossRef CAS.
- A. Vanderkooy and M. S. Taylor, Faraday Discuss., 2017, 203, 285–299 RSC.
- G. Quintieri, M. Saccone, M. Spengler, M. Giese and A. H. Gröschel, Nanomaterials, 2018, 8, 1029 CrossRef PubMed.
- A. Jamadar and A. Das, Polym. Chem., 2020, 11, 385–392 RSC.
- A. Jamadar, C. K. Karan, L. Roy and A. Das, Langmuir, 2020, 36, 3089–3095 CrossRef CAS.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572–2579 CrossRef CAS.
- M. Saccone, V. Dichiarante, A. Forni, A. Goulet-Hanssens, G. Cavallo, J. Vapaavuori, G. Terraneo, C. J. Barrett, G. Resnati, P. Metrangolo and A. Priimagi, J. Mater. Chem. C, 2015, 3, 759–768 RSC.
- M. Virkki, O. Tuominen, A. Forni, M. Saccone, P. Metrangolo, G. Resnati, M. Kauranen and A. Priimagi, J. Mater. Chem. C, 2015, 3, 3003–3006 RSC.
- J. E. Stumpel, M. Saccone, V. Dichiarante, O. Lehtonen, M. Virkki, P. Metrangolo and A. Priimagi, Molecules, 2017, 22, 1844 CrossRef PubMed.
- S. Välimäki, L. Gustavsson, N. K. Beyeh, V. Linko and M. A. Kostiainen, Macromol. Rapid Commun., 2019, 40, 1900158 CrossRef PubMed.
- T. Takeuchi, Y. Minato, M. Takase and H. Shinmori, Tetrahedron Lett., 2005, 46, 9025–9027 CrossRef CAS.
- Y. Chen, S. Huang, T. Wang and H. Yu, Macromolecules, 2020, 53, 1486–1493 CrossRef CAS.
- H. T. Le, C.-G. Wang and A. Goto, Angew. Chem., Int. Ed., 2020, 59, 9360–9364 CrossRef CAS PubMed.
- R. Tepper, S. Bode, R. Geitner, M. Jager, H. Gorls, J. Vitz, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 4047–4051 CrossRef CAS PubMed.
- J. Dahlke, R. Tepper, R. Geitner, S. Zechel, J. Vitz, R. Kampes, J. Popp, M. D. Hager and U. S. Schubert, Polym. Chem., 2018, 9, 2193–2197 RSC.
- N. S. Goroff, S. M. Curtis, J. A. Webb, F. W. Fowler and J. W. Lauher, Org. Lett., 2005, 7, 1891–1893 CrossRef CAS.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030–1034 CrossRef CAS PubMed.
- C. Wilhelm, S. A. Boyd, S. Chawda, F. W. Fowler, N. S. Goroff, G. P. Halada, C. P. Grey, J. W. Lauher, L. Luo, C. D. Martin, J. B. Parise, C. Tarabrella and J. A. Webb, J. Am. Chem. Soc., 2008, 130, 4415–4420 CrossRef CAS PubMed.
- H. Jin, A. M. Plonka, J. B. Parise and N. S. Goroff, CrystEngComm, 2013, 15, 3106–3110 RSC.
- H. Jin, C. N. Young, G. P. Halada, B. L. Phillips and N. S. Goroff, Angew. Chem., Int. Ed., 2015, 54, 14690–14695 CrossRef CAS PubMed.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- A. Borissov, I. Marques, J. Y. C. Lim, V. Felix, M. D. Smith and P. D. Beer, J. Am. Chem. Soc., 2019, 141, 4119–4129 CrossRef CAS PubMed.
- J. J. BelBruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- W. J. Cheong, S. H. Yang and F. Ali, J. Sep. Sci., 2013, 36, 609–628 CrossRef CAS PubMed.
- K. Hema, A. Ravi, C. Raju, J. R. Pathan, R. Rai and K. M. Sureshan, Chem. Soc. Rev., 2021, 50, 4062–4099 RSC.
- S. N. Vouyiouka, E. K. Karakatsani and C. D. Papaspyrides, Prog. Polym. Sci., 2005, 30, 10–37 CrossRef CAS.
- J. Vapaavuori, A. Priimagi and M. Kaivola, J. Mater. Chem., 2010, 20, 5260–5264 RSC.
- A. Priimagi, K. Lindfors, M. Kaivola and P. Rochon, ACS Appl. Mater. Interfaces, 2009, 1, 1183–1189 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- S. J. Garcia, Eur. Polym. J., 2014, 53, 118–125 CrossRef CAS.
- J. Dahlke, S. Zechel, M. D. Hager and U. S. Schubert, Adv. Mater. Interfaces, 2018, 5, 1800051 CrossRef.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251–264 RSC.
- S. J. Kalista, J. R. Pflug and R. J. Varley, Polym. Chem., 2013, 4, 4910–4926 RSC.
- S. Bode, L. Zedler, F. H. Schacher, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2013, 25, 1634–1638 CrossRef CAS PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- M. Enke, D. Döhler, S. Bode, W. H. Binder, M. D. Hager and U. S. Schubert, Adv. Polym. Sci., 2016, 273, 59–112 CrossRef CAS.
- R. Tepper, B. Schulze, M. Jäger, C. Friebe, D. H. Scharf, H. Görls and U. S. Schubert, J. Org. Chem., 2015, 80, 3139–3150 CrossRef CAS PubMed.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2021, 42, 2000573 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |