
A critical review on the use of DP4+ in the structural elucidation of natural products: the good, the bad and the ugly. A practical guide
Maribel O.
Marcarino
a,
Soledad
Cicetti
a,
María M.
Zanardi
 *b and
Ariel M.
Sarotti
*a
*b and
Ariel M.
Sarotti
*a
aInstituto de Química Rosario (CONICET), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, Rosario 2000, Argentina. E-mail: sarotti@iquir-conicet.gov.ar
bInstituto de Ingeniería Ambiental, Química y Biotecnología Aplicada (INGEBIO), Facultad de Química e Ingeniería del Rosario, Pontificia Universidad Católica Argentina, Av. Pellegrini 3314, Rosario 2000, Argentina. E-mail: zanardi@inv.rosario-conicet.gov.ar
First published on 2nd July 2021
Abstract
Covering: 2015 up to the end of 2020
Even in the golden age of NMR, the number of natural products being incorrectly assigned is becoming larger every day. The use of quantum NMR calculations coupled with sophisticated data analysis provides ideal complementary tools to facilitate the elucidation process in challenging cases. Among the current computational methodologies to perform this task, the DP4+ probability is a popular and widely used method. This updated version of Goodman's DP4 synergistically combines NMR calculations at higher levels of theory with the Bayesian analysis of both scaled and unscaled data. Since its publication in late 2015, the use of DP4+ to solve controversial natural products has substantially grown, with several predictions being confirmed by total synthesis. To date, the structures of more than 200 natural products were determined with the aid of DP4+. However, all that glitters is not gold. Besides its intrinsic limitations, on many occasions it has been improperly used with potentially important consequences on the quality of the assignment. Herein we present a critical revision on how the scientific community has been using DP4+, exploring the strengths of the method and how to obtain optimal results from it. We also analyze the weaknesses of DP4+, and the paths to by-pass them to maximize the confidence in the structural elucidation.
 Maribel O. Marcarino | Maribel Marcarino received her B.S. in chemistry from the National University of Rosario (UNR, Argentina) in 2019. That same year she joined Dr Sarotti's group at IQUIR-UNR as a PhD student, focusing on the development of new computational methods based on NMR calculations. |
 Soledad Cicetti | Soledad Cicetti received her B.S. in chemistry from the National University of Rosario (UNR, Argentina) in 2019. In 2020, she started her PhD studies at IQUIR-UNR in the Sarotti group, working in the ecofriendly synthesis of bioactive compounds and DFT calculations. |
 María M. Zanardi | Maria Marta Zanardi received her PhD from the National University of Rosario (UNR) in 2011 under the supervision of Prof. Alejandra G. Suárez. She was a postdoctoral researcher at the IQUIR-UNR with Dr Ariel Sarotti. Currently she is a researcher of CONICET at the INGEBIO-(UCA). Her research interests include development and optimization of new tools for structural elucidation based on NMR calculation, and the use of artificial intelligence for organic and medicinal chemistry research. |
 Ariel M. Sarotti | Ariel Sarotti received his PhD from the National University of Rosario (UNR, Argentina) in 2007. After postdoctoral training in computational chemistry, he became a researcher of the Argentine National Research Council (CONICET). Currently, he is an Associate Professor of Organic Chemistry at UNR and an Independent Researcher at CONICET. His scientific interests include green chemistry, asymmetric synthesis, computational chemistry, NMR spectroscopy, and medicinal chemistry. |
1 Introduction
The exhaustive description of the molecular architecture of novel compounds, including connectivity, relative and absolute configurations, is of fundamental importance in the discovery of biologically active molecules, as their chemical and biological properties are strongly linked to their 3D structures. Several methods are currently used for structural elucidation, with X-ray crystallography analysis being the most unquestionable technique, though the need to generate diffraction quality crystals limits its scope. In contrast, due to its universality and effectiveness to unravel the structural mysteries of a vast range of organic molecules, NMR spectroscopy has become the leading methodology in the field. Over the last years the advances made in NMR have been noteworthy. Nevertheless, data misinterpretation is not uncommon, leading to a large number of erroneous structures published in the last decades.1–5Gauge-Including Atomic Orbitals (GIAO) NMR calculations at DFT levels emerge as an excellent complement to structural elucidation.6–11 The discipline has experienced a tremendous growth over the last years, and nowadays the results of those calculations are often found as part of the routine in the structural elucidation of natural products. As a general trend, the procedure to determine the most likely structure among several candidates involves the following steps: (1) conformational search at a molecular mechanics level; (2) geometry optimization (in case DFT optimized structures are required); (3) NMR calculations (chemical shifts and/or J couplings); (4) energy calculations (can be done at the same or different levels employed in the previous steps); (5) calculation of the Boltzmann-averaged NMR chemical shifts and/or J couplings; (6) correlation of the calculated data with the experimental values. During the first decade of the XXI century, the agreement between calculated and experimental NMR data was determined with the aid of simple statistical descriptors, such as R2, mean absolute error (MAE) or corrected mean absolute error (CMAE).10 The introduction of CP3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 and DP4,13 both from the Goodman group, catalyzed the emergence of a new series of sophisticated approaches increasing in confidence. Among the Bayesian probability methods that were inspired by DP4 which are worth mentioning, we find DP4.214 and DP4.AI15 (Goodman group), DP4+16 (Sarotti group), J-DP417 (Sarotti and Hernández Daranas groups), and DICE18 (Gonnella group). Among the non-probabilistic approaches, CASE-3D by Gil and Navarro-Vázquez19 and DU8+ by Kutateladze20 stand out. While CASE-3D merges isotropic and anisotropic NMR measurements with DFT NMR calculations and conformational selection, DU8+ is based on fast NMR calculations obtained at low-cost DFT methods coupled with NBO-corrected J calculations.
12 and DP4,13 both from the Goodman group, catalyzed the emergence of a new series of sophisticated approaches increasing in confidence. Among the Bayesian probability methods that were inspired by DP4 which are worth mentioning, we find DP4.214 and DP4.AI15 (Goodman group), DP4+16 (Sarotti group), J-DP417 (Sarotti and Hernández Daranas groups), and DICE18 (Gonnella group). Among the non-probabilistic approaches, CASE-3D by Gil and Navarro-Vázquez19 and DU8+ by Kutateladze20 stand out. While CASE-3D merges isotropic and anisotropic NMR measurements with DFT NMR calculations and conformational selection, DU8+ is based on fast NMR calculations obtained at low-cost DFT methods coupled with NBO-corrected J calculations.
Over the years, the scientific community has embraced computational NMR methods as an alternative and convenient strategy for the structural elucidation. The DP4+ probability is among the most popular and widely used methods today, due to its ease of use and the overall satisfactory and reliable performance. Its usefulness in structural elucidation is evidenced by the large amount of structures of natural products assigned with the aid of DP4+, and the trend suggests an increase of its use in the near future. This highlights the contribution of DP4+ to settle structural issues, preventing misassignments when experimental data is not conclusive, or providing additional support to newly assigned structures. However, all that glitters is not gold. Besides the intrinsic limitations of the method, we noticed that DP4+ was used improperly in many cases, which might affect the quality of the results. Considering the rising popularity of DP4+, we decided to make a critical review of the strengths and weaknesses of it, including a thorough analysis of how it has been applied. This review covers all the papers that cited DP4+ (source: Scopus) up to December 2020, and also includes a final section with descriptions and general recommendations to run DP4+ calculations.
2 The good
Since its introduction in late 2015, DP4+ has stood out as one of the leading toolboxes in structural elucidation with computational NMR methods and the structures of more than 200 natural and synthetic products were puzzled out with its aid.21–217 This section is devoted to a thorough analysis of those studies highlighting the strengths and advantages of DP4+ during the elucidation stage.Briefly, the DP4+16 probability is an improved version of DP413 in which P(i) is the probability of candidate i (out of m isomers) to be correct, obtained through Bayes's theorem. It is based on the fact that the errors e between experimental, δexp, and calculated chemical shifts, δcalc, (e = δcalc − δexp) for a set of organic molecules obey a t distribution defined by three terms: μ (mean), σ (standard deviation) and ν (degrees of freedom). The original DP4 distributions have μ = 0 due to the scaling procedure to remove systematic errors, which is done according to δs = (δcalc − b)/m, where b and m are the y-intercept and slope of a plot of δcalc against δexp, respectively. Hence, the DP4 method is built with two sets of [σ, ν] values, corresponding to the errors in the carbon and proton series, respectively. However, in DP4+ we also included the errors due to unscaled chemical shifts (δcalc) as we hypothesized that this would improve the stereochemical differentiation among closely related structures. Given the lack of scaling, the unscaled series are no longer centered in zero (μ ≠ 0). In addition, the data should be separated in terms of hybridization to furnish t distributed series (Fig. 1). Hence, six sets of [μ, σ, ν] parameters are needed to build DP4+ (one for scaled data, one for unscaled sp–sp2 data, and one for unscaled sp3 data, both for carbon and proton chemical shifts). The corresponding sixteen parameters (note that the two scaled series have μ = 0) show dependence on the level of theory. In the original publication these were estimated at 24 different levels (combining B3LYP and mPW1PW91 functionals with six Pople's basis sets and two approaches to consider the solvent effect, using B3LYP/6-31G* optimized geometries in all cases). Recently, we reported a new customizable method to be employed with any desired level of theory (vide infra).218 The use of higher levels of theory for the NMR calculation step resulted in another important improvement over the original DP4 protocol (which is based on chemical shifts computed at the fast but far than optimal B3LYP/6-31G**//MMFF level). As shown in Fig. 1, DP4+ was constructed as a function of the corresponding probabilities computed from scaled and unscaled chemical shifts, termed sDP4+ and uDP4+, respectively. Moreover, each DP4+ term can be calculated using only 1H data, 13C data, or both. The great performance improvement due to the introduction of these additional parameters and upgrading the levels of theory is noteworthy. Other advantages of the method are:
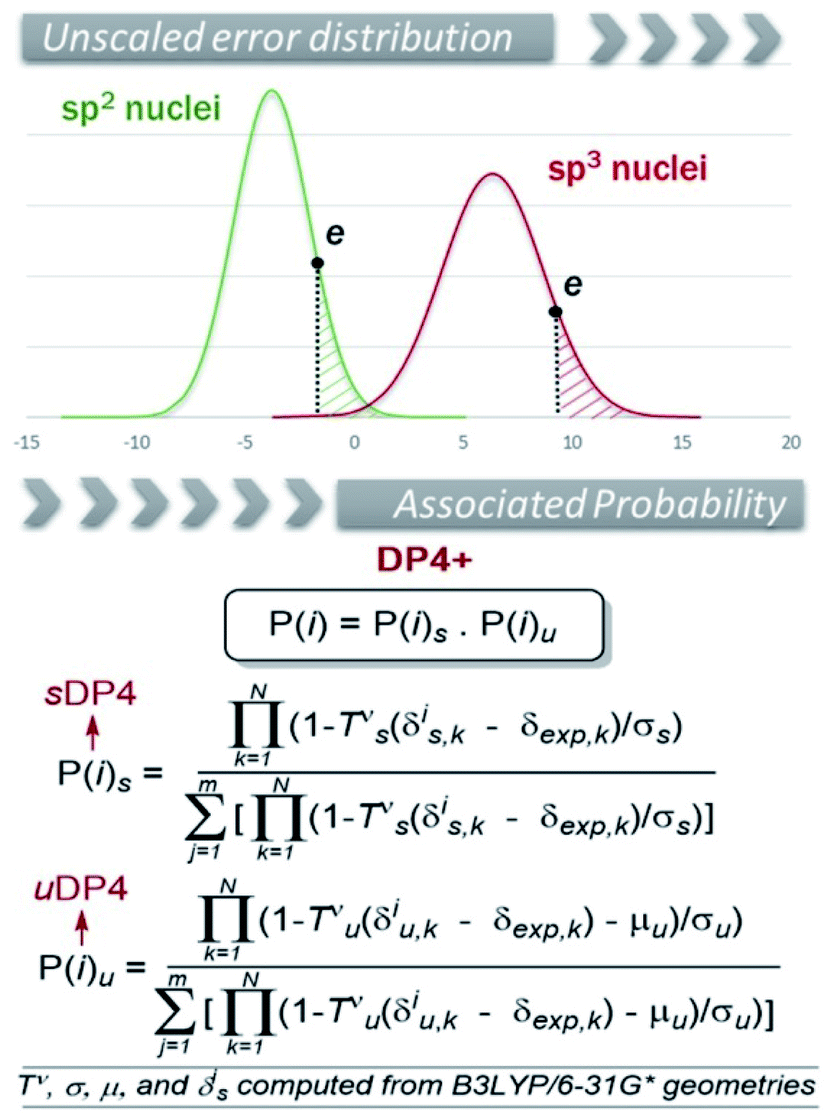 | ||
| Fig. 1 (Up) Error distribution plot of unscaled 13C chemical shifts. (Down) General equation of DP4+, in which P(i) gives the probability that candidate structure i (out of m possible candidates) is the correct one. Tν is the cumulative T distribution function with ν degrees of freedom, whereas μ and σ are the center and standard deviation, respectively, of the error series. | ||
2.1 Simplicity
In our opinion, besides its good overall performance (vide infra), the simplicity of conducting DP4+ calculations is surely one of the main reasons of its popularity. This can be easily done with a ready-to-use excel spreadsheet included by the authors as part of the ESI of the original paper,16 or also available at sarotti-nmr.weebly.com. Following the simple rules provided in the tutorial file, the DP4+ probabilities can be computed at 24 different levels of theory (original version)16 or any desired level of theory (updated version).218 In the last case, the proper estimation of the [μ, ν, σ] terms should be carried out at the desired level. Despite DP4+ can be implemented in Python, C++, Matlab, or any other related platforms without difficulty, its distribution in excel format has certainly contributed to spreading the method among chemists or spectroscopists who are not experts in programming language.2.2 Structural diversity
After a deep literature survey, we observed that the structures analyzed with DP4+ displayed a wide variety of arrangements, featuring diversity of shapes, functional groups and conformational freedom. The overall performance of DP4+ tends to be very good in general, even for challenging structural motifs (such as epoxides and spiroepoxides).219 The range of molecular sizes goes from small structures with a few atoms (less than 20)103 up to molecules with more than a hundred.141,169 The average size of the studied systems was around 60 atoms, with a variable ratio of C, H, O, N as the prevalent atoms. We also found several cases of molecules containing other elements, such as S,29,32,55,92,140,164,212 P,213 Cl,21,26,28,125,147 and Br.29,212 In those cases of carbons attached to bromine (or other atom of the third row or greater) the errors are larger than the average due to the well-known heavy atom effect, consequence of neglecting the spin–orbit contributions from relativistic effects and minor contributions from electron correlation effects.11 In those cases, it would be recommended to exclude those carbons of the DP4+ calculations in order to increase the confidence of the assignment.About 70% of the compounds assigned in silico were further validated by additional studies such as more complex spectroscopic analysis, X-ray crystallography, total synthesis and electronic circular dichroism (ECD) among others, (for instance compounds 1–12, Fig. 2).32,46,57,78,82,103,126,135,138,149,151,173
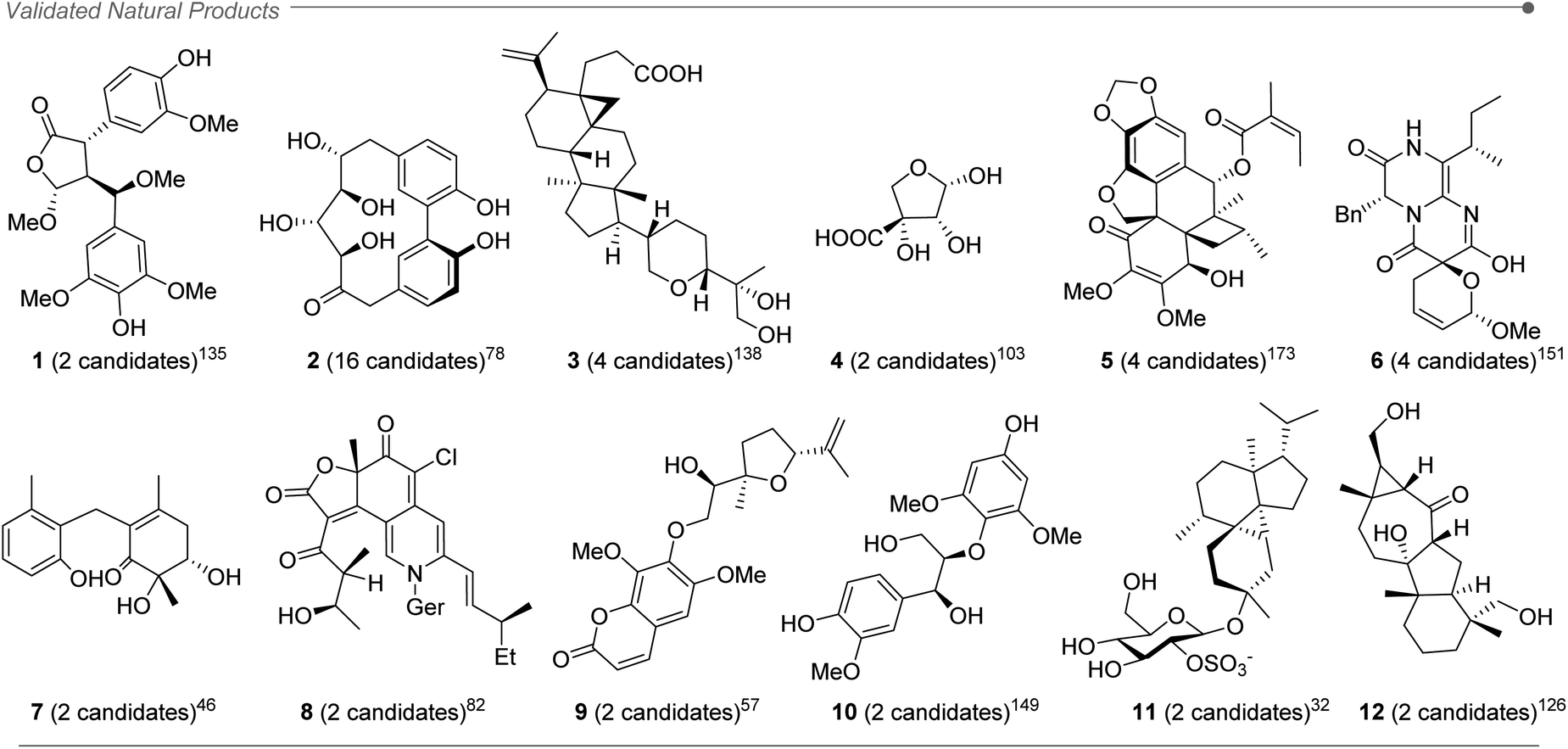 | ||
| Fig. 2 Natural products assigned by DP4+ empirically validated by X-ray crystallography, circular dichroism and total synthesis. The number of compared candidates in each study is between parenthesis. | ||
DP4+ has been mainly employed in the assignment of natural products, but also it has stood out as a valuable tool in the structural elucidation of synthetic compounds (Fig. 3), including reaction intermediates (as compound 13 during Richnovsky's synthesis of kujonins A1 and A2)85 and unexpected impurities (as the case of the demethylated intermediate of (+)-frondosin B which complicated the configuration assignment of the natural product).105 In this case, the impurity of (+)-frondosin B was assigned as (R,R)-14 based on DP4+ calculations. Moreover, despite DP4+ is typically applied to evaluate stable molecules in their ground state, it can provide further insight into the identification of reaction intermediates. For instance, during the mechanistic study of the [Bi(OTf)3] mediated mild electrophilic aromatic formylation with PhSCF2H, three possible intermediates were studied (two conformational isomers 15a and 15b, and their geometric isomer 15c).55
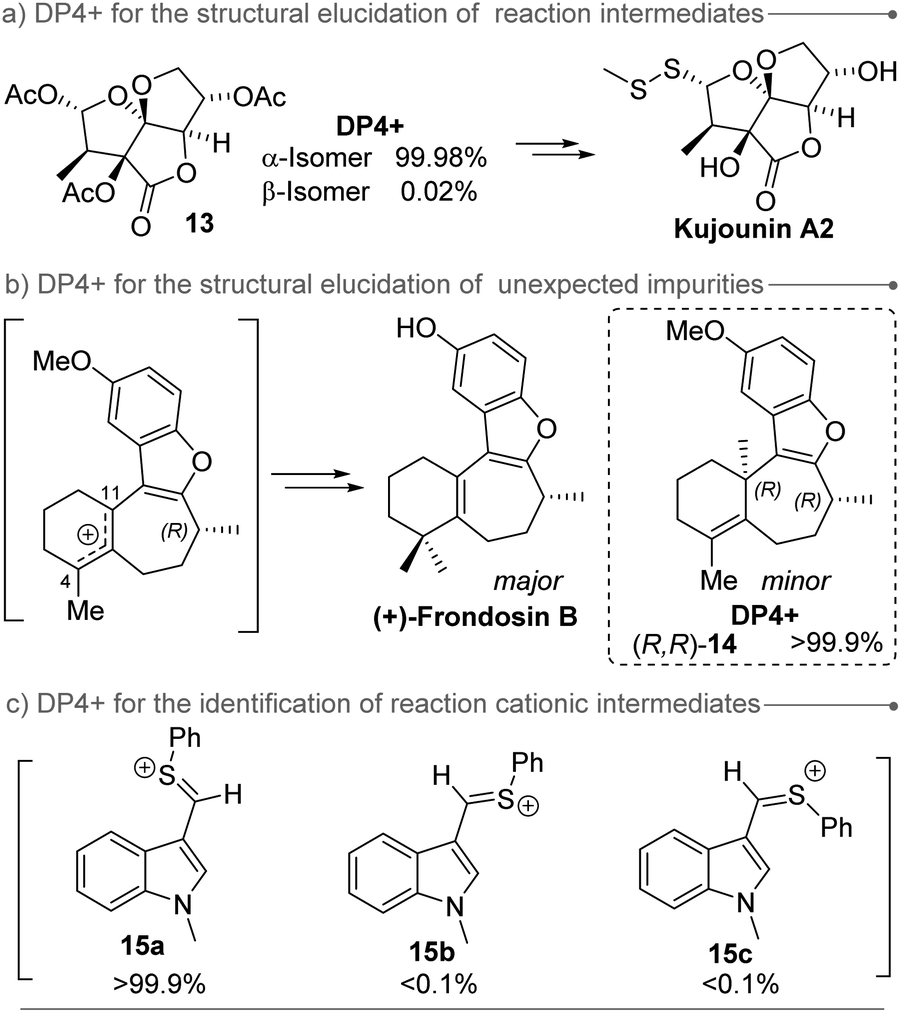 | ||
| Fig. 3 Examples of non-traditional applications of DP4+. | ||
2.3 Isomerism variety
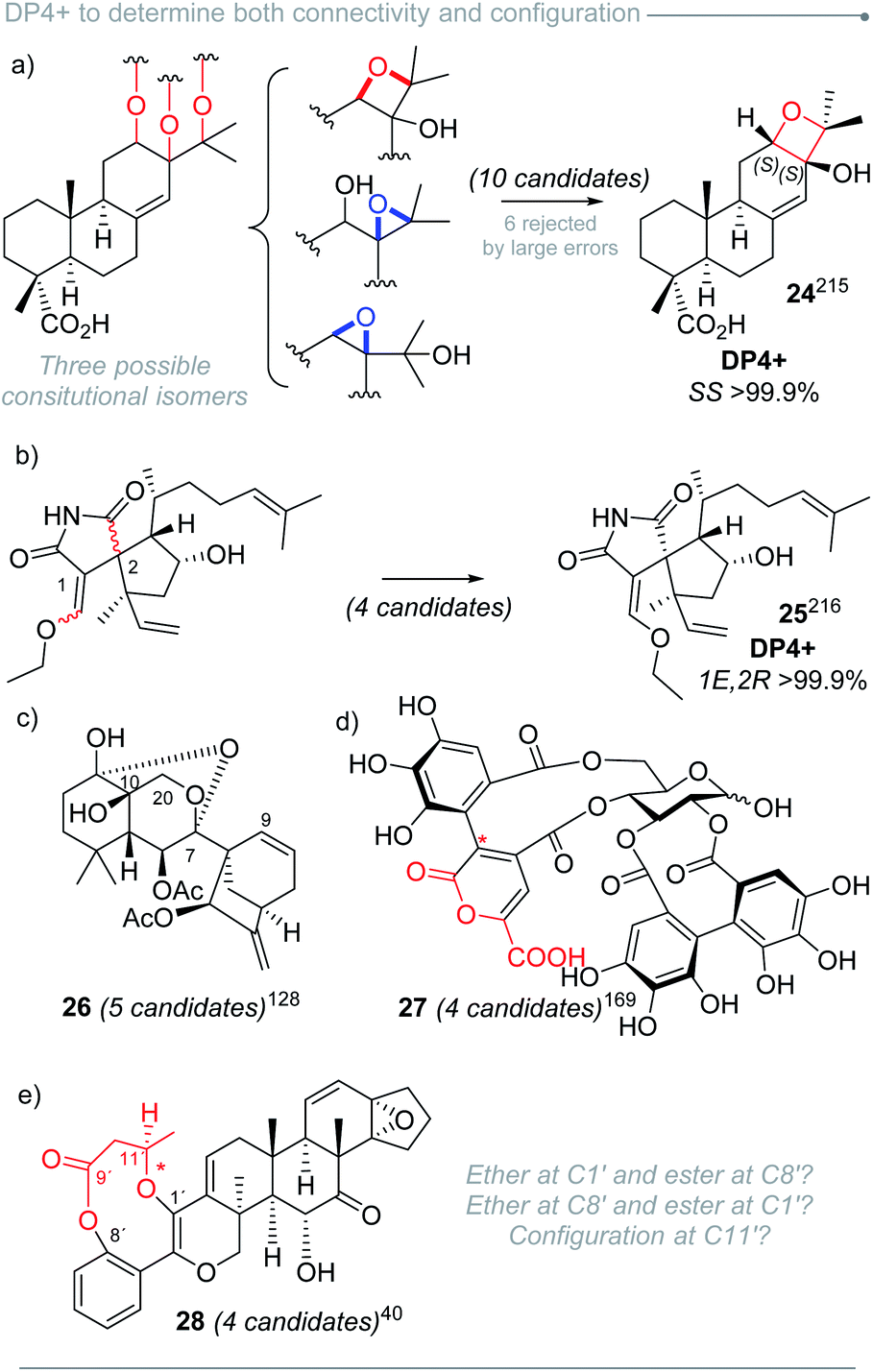
Because of the comparison-based nature of DP4+, it can be used to discriminate among different types of isomerism, including diastereo-, regio- and constitutional isomers. In addition, it can handle both single and combined uncertainty.47 represent additional examples that outline the usefulness of DP4+ to establish molecular connectivity.
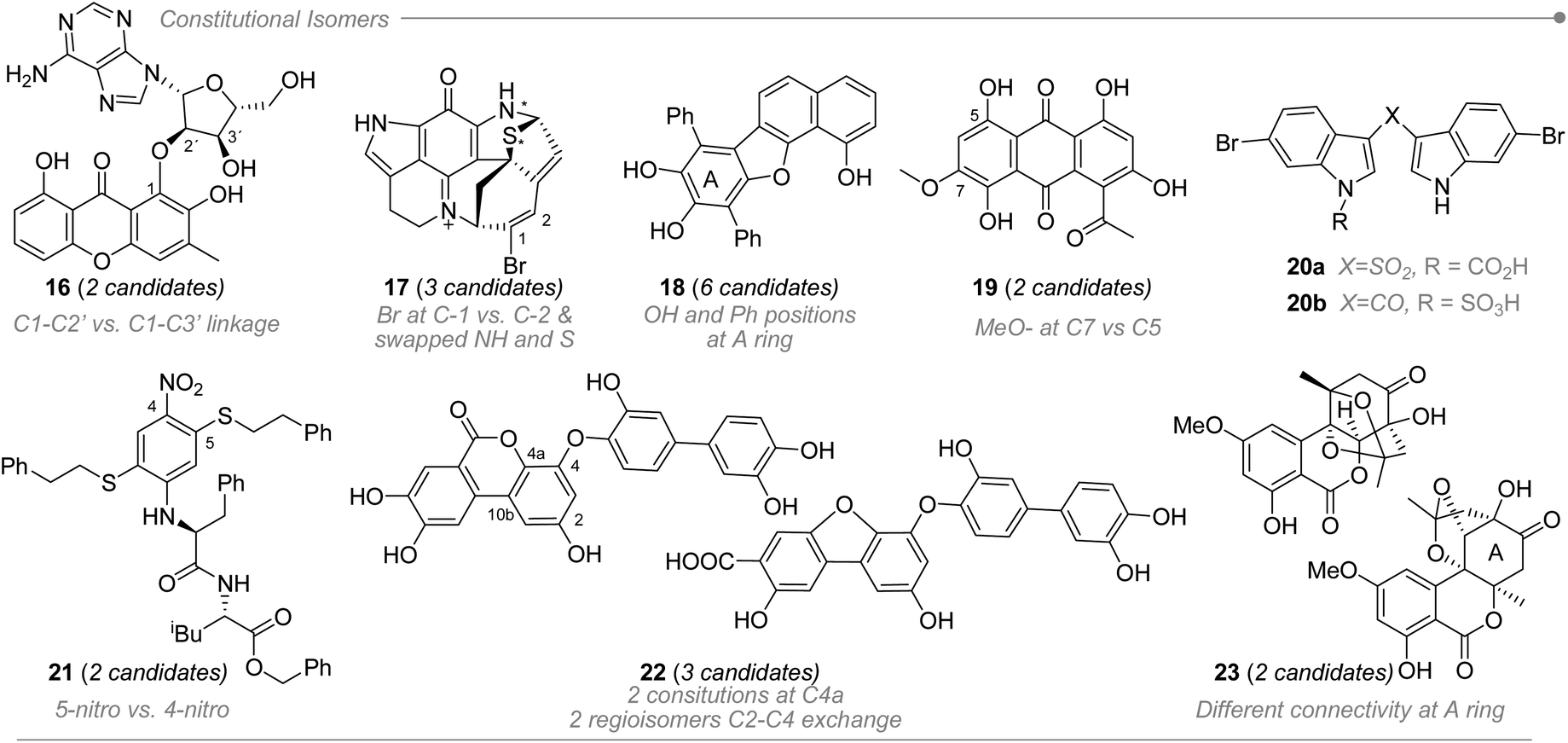 | ||
| Fig. 4 DP4+ in the constitutional assignment for chiral and achiral compounds. | ||
 | ||
| Fig. 5 DP4+ to tackle combined uncertainty problems. | ||
Similarly, we found examples that simultaneously explore geometric and configurational isomers, such as the case of dictyospiromide (25), a diterpenoid with a novel structural scaffold isolated from a marine brown algae (Fig. 5b).216 Its constitution and configuration were proposed after a thorough analysis of NMR and NOESY data, but further studies were needed to provide the full structure of the molecule. GIAO NMR calculations were carried out for four isomers arising from the difference in the configuration at C-2 (R or S) and the exocyclic alkene at C-1 (E or Z). On the basis of RMSD and MAE, isomer 1E,2R was identified as the most probable one, mainly on the basis of carbon data. However, the calculated values showed close similarity to those of the C-2 epimer (1E,2S). The DP4+ calculations revealed that the proton-based probability was ambiguous, but the characteristic compensation of the errors associated to DP4+ allowed the correct assignment by introducing the carbon data. Anisotropic NMR experiments validated the putative structure.
Approximately 20% of the molecules assigned with the aid of DP4+ included stereoclusters separated through flexible spacers (such as ethylenes, non-stereogenic quaternary carbons, alkenes, heteroatoms, etc.). Some examples of this type of arrangement are present in compounds 29–36 (Fig. 6),79,88,89,92,154,164,168,217 and although we demonstrated that DP4+ can be applied to these types of compounds, the challenge of assessing the relative configuration becomes much more complicated than in other cases.16 It is fundamental to make a correct description of the chemical environment in order to avoid a wrong assignment, considering that some of the diastereoisomers might exhibit similar NMR chemical shifts.223 That is the reason for the keen remark in the inclusion of all the data available, especially during the study of this type of systems, to guarantee a confident result when using DP4+.
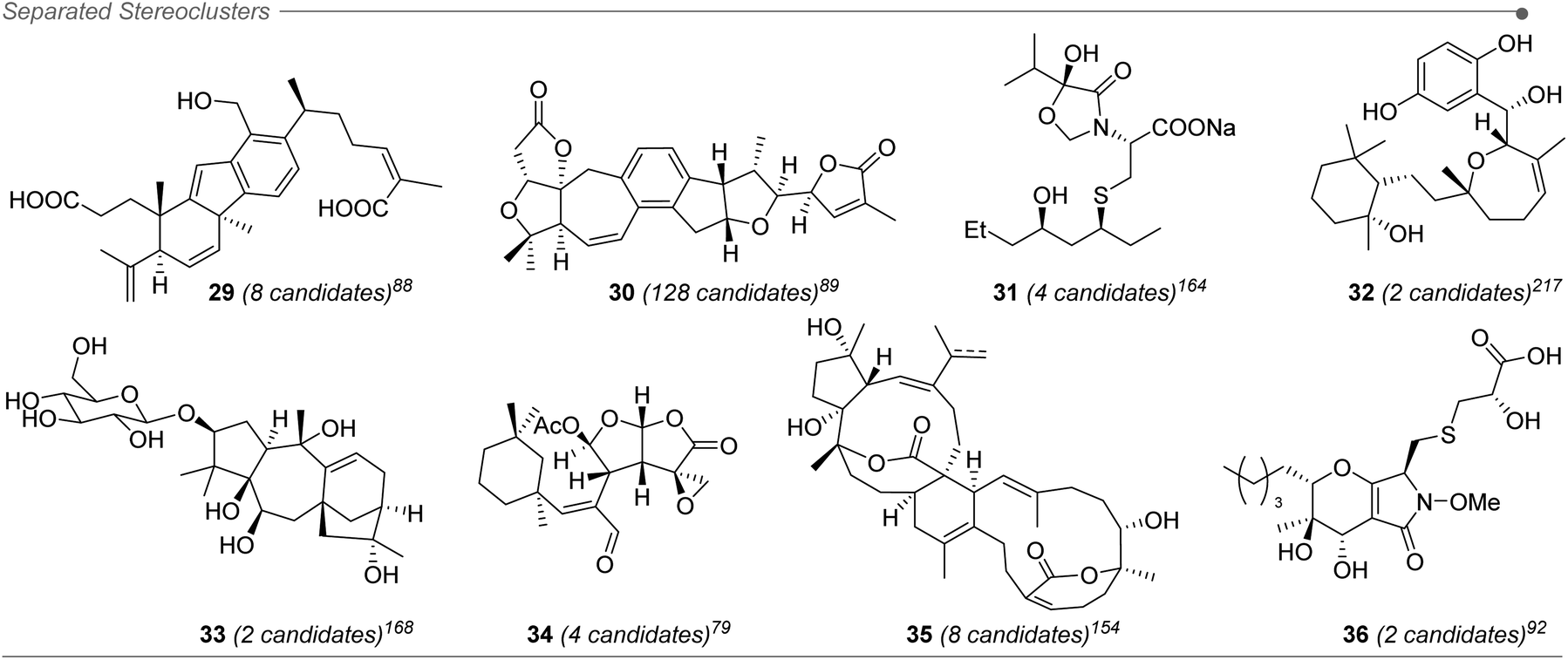 | ||
| Fig. 6 Example of molecules containing separated stereoclusters assigned by DP4+. | ||
The good capacity of DP4+ to connect the relative configuration of separated stereoclusters was exploited in a conceptually novel method to establish absolute configuration (AC) of organic molecules, and it was focused on chiral derivatization agents (CDAs) such as Mosher's reagent or its analogues (Fig. 7).220 Using an ambitious and large set of examples (114 systems) the absolute configuration of 96% of the cases could be determined using only a single derivatization experiment. The best results were achieved for secondary alcohols, along with secondary and primary amines as well. Primary and tertiary alcohols yielded more modest but still exciting predictions. A new DP4+ Integrated Probability (DIP) was then introduced to combine two independent DP4+ predictions into a single descriptor. This new probability proved to be useful in double derivatization approaches with remarkable results.
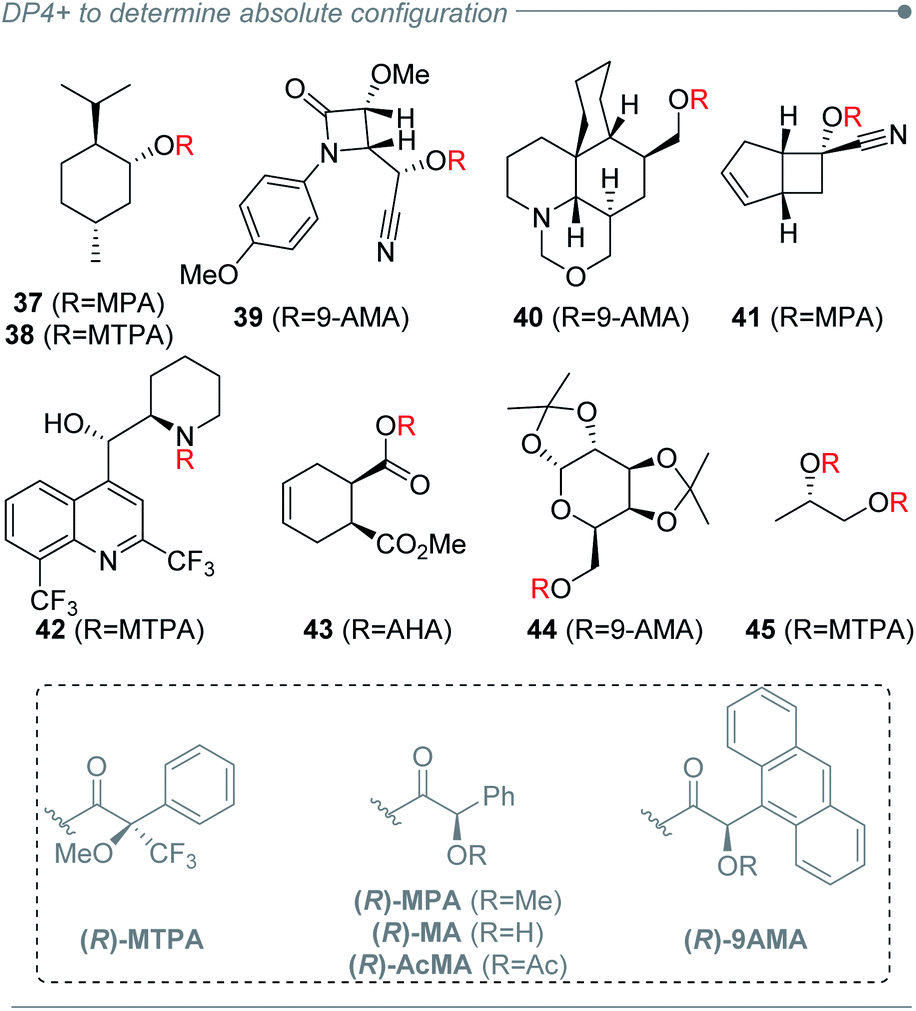 | ||
| Fig. 7 Structural diversity of the compounds assigned by chiral derivatization and DP4+ analysis. | ||
2.4 Variable number of candidate structures
The number of candidates is not a limitation for the method, though the typical applications found in the literature involves the comparison between few isomers. In fact, 70% of the reported DP4+ calculations were carried out considering only two candidates, 25% with four isomers, and the rest comparing more than eight structures. The highest number of isomers studied using DP4+ was 128 in the structure elucidation of pseudorubriflordilactone B (30).89 Only considering a reduced number of isomers is beneficial regarding computational cost, but also because it minimizes the possibility of incorrect isomers fortuitously showing good fits with the experimental data, hence affecting the assignment. This reinforces the importance of conducting a careful analysis of the experimental data, including chemical shifts, J coupling constants and/or NOE interactions, in order to rule out those candidates a priori for further NMR calculations and DP4+ analysis.172.5 Flexibility in the level of theory
In order to provide flexibility when choosing the method to perform the NMR calculations, DP4+ was developed at 24 different levels of theory. These levels were generated combining two functionals (B3LYP and mPW1PW91), six basis sets (6-31G*, 6-31G**, 6-31+G**, 6-311G*, 6-311G**, and 6-311+G**) and two approaches to include the solvent effect (gas phase and PCM) for the GIAO NMR calculations, starting from B3LYP/6-31G* optimized geometries in all cases. Naturally, not all levels provide the same quality in the assignments, which is the reason we recommended PCM/mPW1PW91/6-31+G**//B3LYP/6-31G* for general applications.16,219 We noticed that in about 30% of the publications this recommended level was used,40,55,78–117,214 whereas in 34% of the cases the authors decided to evaluate one of the other 23 levels available for DP4+.24,25,28,32,39,41,43,146–148,150–155,224 Noteworthy, in the remaining 36% papers the analysis was conducted using NMR shifts calculated at other levels for which DP4+ was not parameterized. This indicates a clear inclination of each research group towards conducting the geometry optimizations and GIAO calculations with pre-selected methods well-known to them. In this regard, it should be emphasized that using alternative levels does not represent a mistake by itself, but caution should be taken as the accuracy of the predictions may decrease, mainly when correlating NMR chemical shifts with [μ, σ, ν] values computed at different levels (vide infra). Recently we published the study exploring the sensitivity of the DP4+ method with the probability distribution terms. The results led us to develop a customizable DP4+ methodology, which allows calculations at any desired level of theory. The [μ, σ] terms can be fairly estimated from a small set of 8 rigid molecules for preliminary explorations. However, if more accurate DP4+ results are required, the [μ, σ, ν] parameters should be obtained from a more exhaustive number of molecules, such as those employed in the original publication.2182.6 Flexibility in the NMR data employed
3 The bad
The main limitation of any comparison-based method arises when an incorrect isomer shows a fortuitously better agreement with the experimental data than the right candidate does, and DP4+ is not the exception. Indeed, in the original publication a few molecular systems could not be properly solved by DP4+ (for example, compounds 44–45, Fig. 8),16 even at the recommended level of theory (PCM/mPW1PW91/6-31+G**). In subsequent publications, other unsuccessful examples were provided (for example, compounds 46–49, Fig. 8).220,230,231 The extensive use of DP4+ puts the method constantly under scrutiny, and new cases with inconclusive results arise continuously. This highlights the fact that all current methods may show flaws in some certain scenarios. Understanding the reasons behind such failures, even after proper computational work, provides an excellent opportunity to develop improved methodologies. In this section, we discuss the background that might lead to potential misassignments and the alternatives to overcome those problems.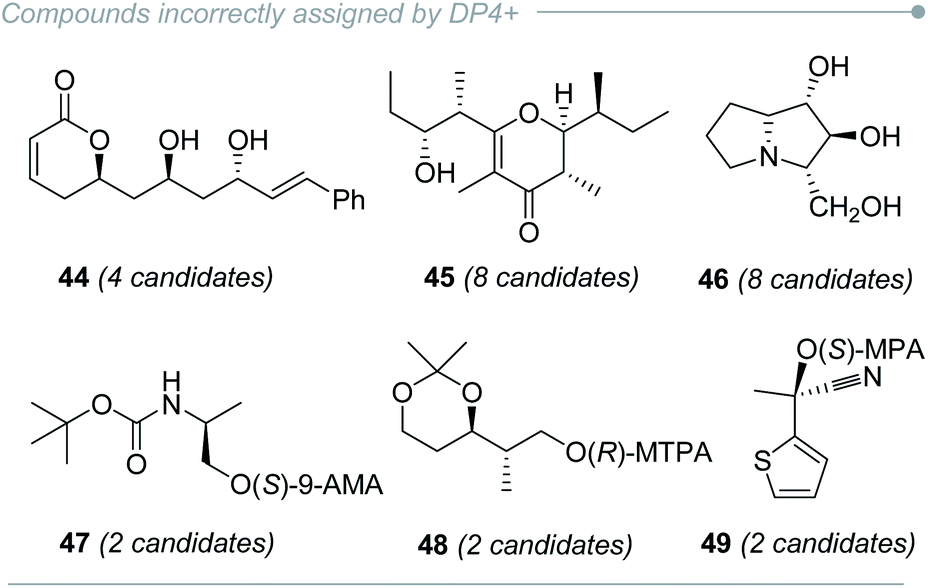 | ||
| Fig. 8 Compounds incorrectly assigned by DP4+. | ||
3.1 Energy miscalculations
Its favorable cost/accuracy ratio makes DFT the preferred alternative for quantum-based NMR calculations. However, this approach has intrinsic simplifications, which affect the quality of the predictions (including the Born–Oppenheimer approximation, the still imperfect nature of the exchange–correlation terms of all functionals, the use of implicit solvation models and the alternatives to tackle the gauge origin problem when using truncated basis sets).11 Hence, error-free calculations should not be expected, not even for the simplest molecules using state-of-the-art computational procedures. Although there are many factors potentially influencing the accuracy of the NMR predictions, they can be summarized in two main categories: tensorial and energetic. The first one is related to the inability to perfectly reproduce the experimental isotropic shielding constants, whereas the second is associated with the errors involved in the energy simulations (only important when dealing with conformationally flexible systems). It would be reasonable to guess that the tensorial source of error would be the most decisive factor, but in our opinion, the energy term could have a greater influence when discriminating among flexible diastereoisomers. This is evidenced by the often excellent results obtained with most methods when applied to rigid molecules with a very limited or null conformational freedom.13,16,19,20 On the contrary, it has been well documented that erroneous energy calculations might affect the Boltzmann-averaging, leading to low quality NMR predictions, with a potentially negative effect in the DP4/DP4+ results.13,16We recently showed that under a low energetic uncertainty (<1 kcal mol−1), DP4+ values are not affected significantly.231 On the other hand, larger errors in the energy calculations might lead to substantial discrepancies between the computed and actual conformational landscape. This becomes manifest in flexible molecules properly functionalized to give rise to intramolecular hydrogen bonding interactions (IHB), mainly when the experimental NMR spectra are collected in polar solvents, such as D2O or CD3OD. The IHB is a long-standing and well-known problem for those in the field, and it can lead to wrong DP4+ results when the most contributing conformations of the right isomer are spurious as a consequence of such interactions. We've thoroughly studied the impact of IHB in the stereoassignment of 40 known compounds of the hyacinthacine family, polyhydroxylated pyrrolizidine alkaloids featuring a rich conformational freedom with the possibility of building multiple IHB arrangements.230,232 Following the standard DP4+ procedure at the recommended level of theory (PCM/mPW1PW91/6-31+G**//B3LYP/6-31G*), we found that 11 isomers were incorrectly assigned as consequence of improper conformational descriptions for the corresponding right candidate, highlighting the importance of IHB to bias the conformational amplitudes. Naturally, not all IHB lead to wrong results, as most of the leading conformations of the 29 compounds correctly assigned also displayed such interactions.
Therefore, when dealing with systems featuring IHB, the main goal is to define if those interactions are harmless or not in terms of DP4+ results, which is obviously impossible without knowing a priori which candidate is the correct isomer of the molecule. There are, however, additional strategies that can be followed to anticipate a possible misassignment as a consequence of IHB. Perhaps the easiest way is to recompute the DP4+ values after removing those featuring suspicious IHB interactions. In a preliminary study on the simplest members of the hyacinthacine family, we proved that these counterintuitive approaches allowed to improve the DP4+ results not only in the isomers where the standard procedure failed, but also keeping unchanged the DP4+ trends of the remaining five examples being correctly assigned.232 In all those cases, removing the biased IHB shapes affected positively the quality of the NMR predictions by lowering the MAE (mean absolute error) values, clearly indicating that the predominance of those structures in the conformational landscape pictured by DFT is wrong. This was evidenced during the assignment of sphaerialactonam (50)91 and peyronetide A (51) (Fig. 9),100 as we noticed large outliers in the calculated 1H and 13C NMR resonances, respectively. In both cases, the conformations with largest amplitudes showed IHB interactions (either between C6–OH and the carbonyl groups at C5′ or C20′, or between C8–OH and ketone oxygen at C2′, respectively). The recomputed NMR shifts after neglecting those conformations significantly improved the match between experimental and calculated values. Although this modification did not change the sense of the assignment (the most likely candidate remained after conformational removal), this procedure allowed to rule out the possibility that DP4+ results could have been biased by a wrong conformational description.
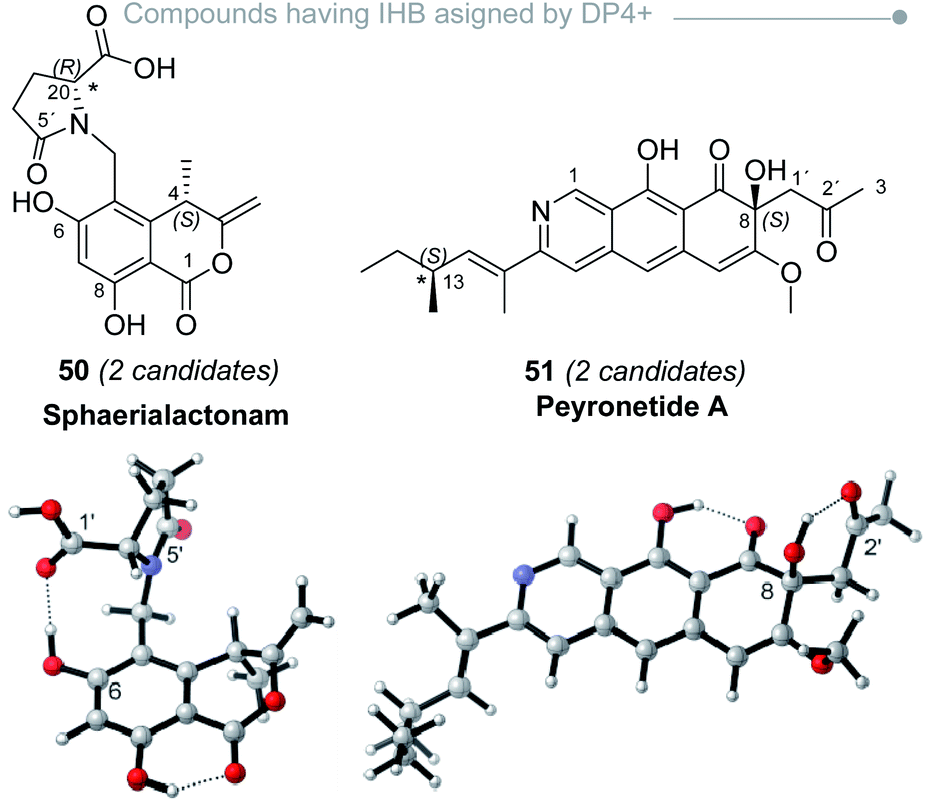 | ||
| Fig. 9 Representative conformations of sphaerialactonam and peyronetide A with spurious IHB responsible for large errors in the DFT NMR calculations. | ||
The removal of conflicting conformations is an easy shortcut to potentially improve the NMR results and to support the assignment made by the standard formalism. There are, however, other alternatives to analyze the impact of spurious conformations in the DP4+ results. One of them is refining the Boltzmann amplitudes using SMD as the solvation model, a version of PCM that decomposes the solvation energy into SCRF bulk electrostatic contributions and the short-range solvent–solute interactions in the first solvation shell.233 In a thorough exploration of the conformational description of the hyacinthacine family, we observed sharped differences between the conformational amplitudes computed with PCM (the recommended for DP4+ for broad applications) and SMD, with the later providing a more realistic description in certain IHB systems.230 Another and more drastic approach involves neglecting all together the relative energies given by DFT calculations, and alternatively creating and evaluating different ensembles generated by removing conformations followed by a random relative energy distribution of the remaining shapes. This is supported by the idea that in a large set of ensembles, the majority of them would point towards the right isomer and reflect the correct final assignment. The averaged overall performance of this approach was excellent with 100% of the compounds belonging to the hycintacine family being correctly classified by DP4/DP4+.230 In comparison with other methods previously developed, this new and exciting approach is very different in its nature. The random ensemble strategy is based on the fact that one should not rely on a single determination to decide whether a putative structure is correct or not (Fig. 10).
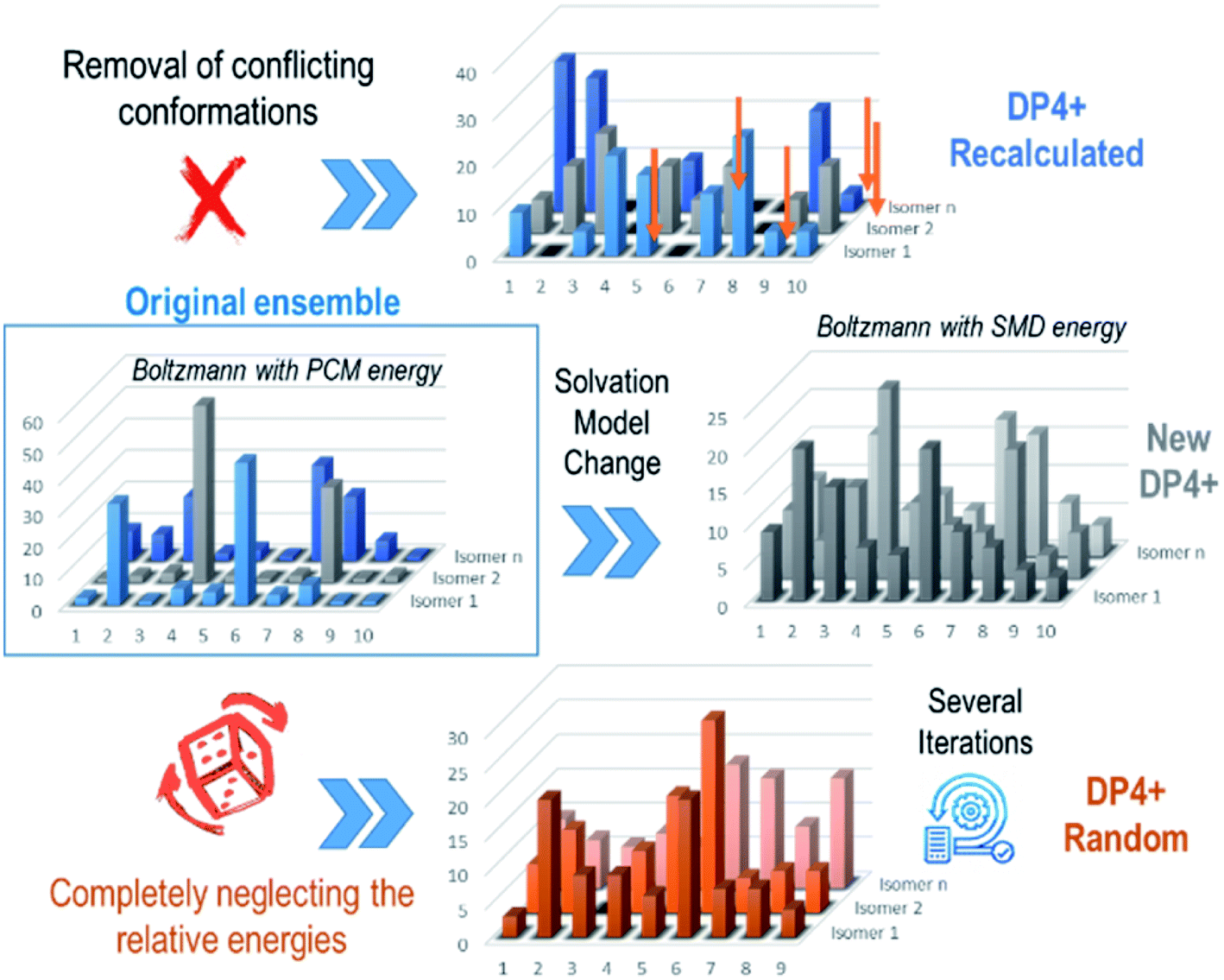 | ||
| Fig. 10 Random strategies employed to avoid the IHB problem. | ||
The inadequate estimation of the conformer populations could be also tackled using advanced hybrid or double-hybrid functionals for the energy calculations, which were shown to provide superior performance than B3LYP.234 For example, in DP4.2 Goodman and co-workers found that better results were obtained by calculating the NMR chemical shifts at the mPW1PW91/6-311G* level and the relative energies for Boltzmann analysis at the M06-2X/6-31G** level.14 In DP4+ the estimation of Boltzmann amplitudes using new levels can be done in different ways. One of them, previously discussed, involves correlating the NMR chemical shifts computed at one of the 24 levels of theory (for example, PCM/mPW1PW91/6-31+G**//B3LYP/6-31G*) with the Boltzmann amplitudes refined at a new level (for example, SMD/M06-2X/6-31+G**).230 The other option is based on running the NMR calculations and/or the geometry optimization steps at new levels, now allowed in the updated version of DP4+. However, it is important to point out that a proper estimation of the [μ, ν, σ] terms should be carried out first to obtain meaningful results.218
In any case, regardless the level of theory used for the geometry and energy calculations; it would be always wise to incorporate as much experimental information as possible to corroborate the DP4+ findings. For example, whenever available, the homo- and heteronuclear 3J coupling constants and interatomic distances (estimated from NOE/ROE correlations) should be used to check consistency of the DP4+ analysis. In case of improper conformational description, the parallel application of these data would allow to detect the failure, hence unleashing a more detailed study (for example, refining the Boltzmann amplitudes with new levels).
3.2 Similar chemical shifts
The DP4+ results are usually robust and correct when the isomers under consideration show acceptable differences in at least a few chemical shifts, as is often the case. However, when two or more isomers show differences in their chemical shifts below the precision limit of the method, the results might become occasionally erratic. In this regard, it is only enough that the incorrect isomer shows a slightly better fit than the correct one to influence the DP4+ results. This could be the situation when dealing with isomers with separated stereoclusters featuring the same relative configuration in some of them but opposite configurations in others. For example, during the first total synthesis and structural elucidation of (+)-cryptoconcatone H93 (Fig. 11) by Pilli and co-workers, DP4+ calculations were carried out over the eight possible diastereoisomeric candidates to validate the putative structure before facing the synthetic effort. The calculations strongly suggested that the putative structure (52a) was erroneous, whereas the two candidates featuring the cis/trans configuration at the tetrahydropyran ring were the most likely structures of the natural product (52b and 52c). The calculated NMR chemical shifts of these two isomers displayed excellent agreement with the experimental data reported for the natural product, with CMAE values of 1.0 and 1.3 ppm (carbon data) and 0.08 and 0.10 ppm (proton data), respectively. The better match observed with 52b, which impacted in the DP4+ probability values (99.9% vs. 0.1%), was mainly due to small errors computed for the conserved regions of the molecule. However, the real structure of the natural product was determined as 52c after total synthesis of the two candidates. Although recomputing the DP4+ by taking into account only the NMR data from the most relevant and differentiating region (in this case, the tetrahydropyran fragment) reduced the preference towards 52b, and this result revealed the potential problem arising whenever two isomers display similar NMR properties. This case study was subsequently analyzed in detail, but we could not find any evident proof of a bad representation of the conformational landscape that might have negatively influenced the NMR predictions of 52c. We concluded that the problem was not related to a poor prediction of the conformational landscape of the correct structure, but merely to a better match with a similar, yet different, shape. In later publications, the potential problems of setting the conformational amplitudes by fitting instead of relying on Boltzmann analysis using the corresponding relative energies computed at DFT levels were also discussed.231,235 Forcing all isomers to have computed chemical shifts as close as possible to the experimental values jeopardizes the assignment as some incorrect isomer could end up having a better agreement, and therefore higher DP4+ values, than the correct structure. | ||
| Fig. 11 Putative structure of (+)-cryptoconcatone H, and most likely structures by DP4+ calculations. | ||
3.3 Neglecting the real structure
Another inherent problem associated with most computational methods for structural elucidation is related to the inability to unequivocally assess the correctness of a given structure.236,237 Instead, the most probable structure is selected among several candidates that are ranked depending on the degree of fit with the experimental values. Hence, if the real structure of the natural product is not included in the set of candidates, the methods will inevitably fail as they were not design to disprove all the provided options (mathematically, the sum of the probabilities of all candidates should be 100%). As stated above, the main application of DP4+ to date has been related to the determination of the relative configuration of a molecule whose planar structure is known. Therefore, the reliability of the final prediction will be linked to that assignment, which if incorrectly conducted would inevitably lead to a DP4+ failure, regardless the probability value computed for the most likely isomer. The elucidation of littordial F represents an interesting case study to exemplify the above. This natural product, isolated in 2019 from the leaves of Psidiumlittorale by Xu and Feng groups (Fig. 12),62 was proposed as a novel meroterpenoid with a unique 6/8/9/4-tetracyclic core structure. In order to establish the configuration at C-10′ the authors evaluated the two possible diastereoisomers with DP4+ using only 13C NMR data, suggesting that the real structure should be the 1R,4S,5R,9S,10′S isomer (53). However, in 2020 George et al. revised the structure to the corresponding 6/6/9/4-tetracyclic core (54) based on biosynthetic considerations, further confirmed by biomimetic total synthesis and NMR studies.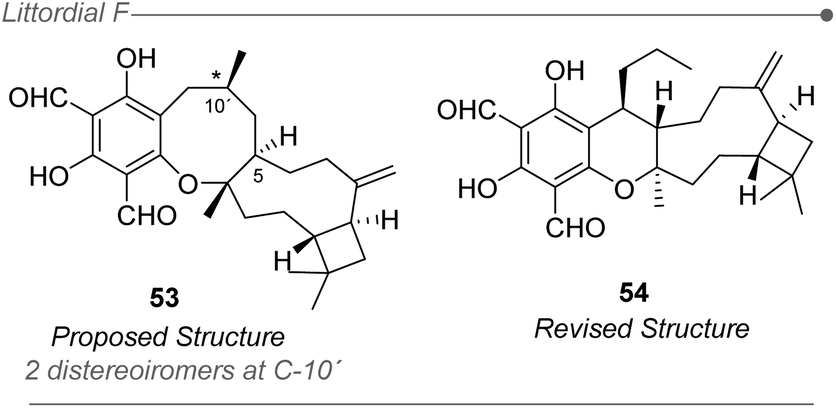 | ||
| Fig. 12 The case of Littordial F reflects the problem of neglecting the real structure. | ||
4 The ugly
DP4+ is a valuable tool for the in silico structural elucidation of natural products, affording overall reliable predictions (see “The good”), though in some challenging and specific cases the performance drops due to the intrinsic limitations of the method (see “The bad”). However, DP4+ results might be affected also by improper computational work and/or misuse of the excel toolbox. From our experience, and after a detailed analysis of the hundreds of cases reporting the use of DP4+, we've detected the most common pitfalls that might end up affecting the results, which will be discussed in this section. To avoid unnecessary speculation about the certainty of the results reported in those few papers where the following errors were detected, we decided not to cite them in this section (though they were cited in this review). After all, this part is dedicated to showing the nature of the errors and how to prevent them in future studies.4.1 Use of inappropriate distributions
DP4+ was built on the basis of the scaled (δs) and unscaled (δu) errors (δcalc − δexp) being random variables that follow t-like distributions. In the case of δu series, the errors depend on the hybridization of the nuclei, leading to two series: one for sp–sp2 carbons (or protons attached to them) and another one for sp3 carbons (or protons attached to them). Since each t distribution is characterized by a set of three parameters: mean (μ), standard deviation (σ) and degrees of freedom (ν), the DP4+ equation requires the knowledge of six [μ, σ, ν] parameter sets (three for 13C data composed of one distribution for scaled shifts and two distributions for unscaled shifts, and the corresponding three distributions for 1H data). Hence, any mistake related to the use of improper distributions might impact on the DP4+ values leading to potentially wrong assignments. The sensitivity of DP4+ to each of the statistical parameters was recently explored.218 This can take place in two different ways: | ||
| Fig. 13 Changes in DP4+ by selecting improper levels in the excel spreadsheet. The isotropic shielding values of 55 and ent-55 were carried out at the PCM/mPW1PW91/6-31+G**//B3LYP/6-31G* level. | ||
A similar and potentially problematic situation arises when the NMR chemical shifts are computed at different levels for which DP4+ has not been parameterized and tested yet. In this case, unless the [μ, σ, ν] values of the new level are known, and are similar to one of those reported 24 levels available in DP4+, any selection made in the excel spreadsheet might end up in a mistaken assignment. This triggers a complex crossroad: without previous knowledge, it is difficult to determine which level to choose in the excel spreadsheet, but the final results will almost certainly depend on that selection. In addition, it is difficult to estimate the exact impact of this mistake, because it largely depends on the system under study and the differences in the [μ, σ, ν] values of the two levels of theory (the employed and the indicated one). Fig. 14 shows how the cumulative probability for a given error (difference between experimental and calculated NMR shifts) changes by choosing improper distributions. To overcome those limitations, we introduced a new customizable DP4+ version, which allows the user to select any level of theory of their preference. The new excel spreadsheet handles the previously explored 24 levels (default settings), and any new level as well (custom settings). If this option is selected, the user must introduce the sixteen [μ, σ, ν] values corresponding to the desired level using a set of known molecules. For preliminary calculations, we showed that these terms can be estimated using a reduced set of 8 molecules with no significant change in the overall DP4+ values. The new excel spreadsheet allows the automatic calculation of the [μ, σ, ν] parameters once the calculated NMR isotropic shielding values for the test set is entered.218
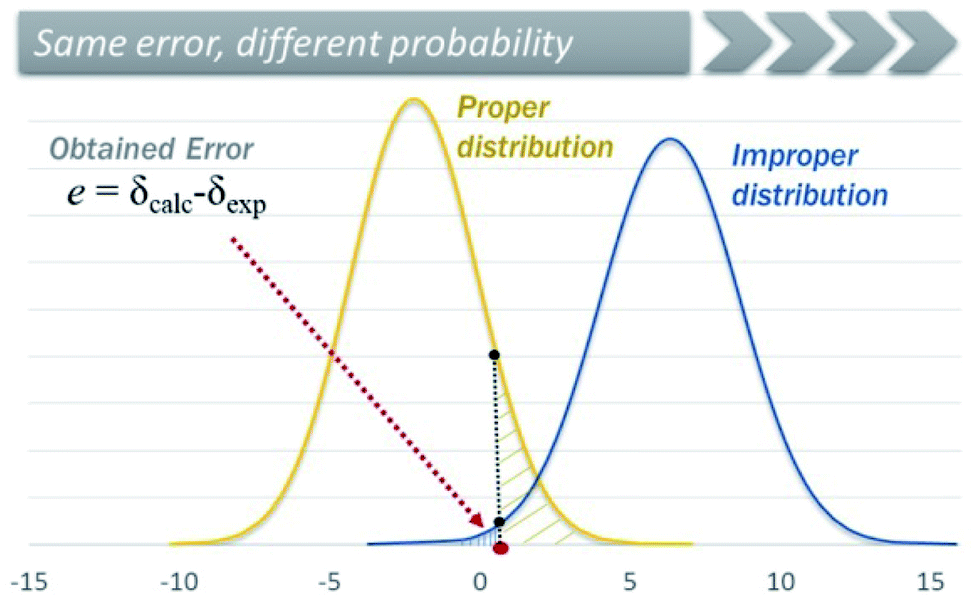 | ||
| Fig. 14 Graphical representation of the change in the cumulative probability for a given error by choosing different distributions. | ||
4.2 Incorrect chemical shifts calculations
The excel toolbox allows three different ways of data entrance: isotropic shielding values, unscaled chemical shifts or scaled chemical shifts. If the first option is selected, the unscaled chemical shifts are computed according to δu = σ0 − σx (where σ0 is the isotropic shielding value of TMS at the level of theory selected in the drop-down menu, and then the scaled chemical shifts are obtained as discussed above. This is the recommended way because it rules out possible errors made by the user during the chemical shift calculation and/or the subsequent scaling procedure. In addition, any other improper computational work will surely affect the results, regardless the way NMR data is inserted in the excel toolbox. The number of situations that might end up in wrong calculations is immense, but the most common ones are briefly discussed below:4.3 Large outliers
Although DP4+ is based on the analysis of the errors that arise from the NMR calculation process, when these errors are too large caution must be taken. The distributions of scaled chemical shifts are centered in zero, with standard deviations in range 1–2 ppm and 0.1–0.2 ppm for carbon and proton shifts, respectively, using DFT optimized geometries. Hence, when alarmingly large scaled errors (>1 ppm for proton data, >10 ppm for carbon data) are observed, this could indicate a potential problem to be further analyzed in detail. The most common situations which may be encountered are described below:4.4 General recommendations
Despite each research group has its own preferences for conducting the NMR calculations, herein we present general recommendations to carry out the DP4+ analysis.(a) When dealing with flexible molecules, a thorough conformational sampling ought to be done using a safe energy cutoff (5 kcal mol−1).
(b) All conformations found should be optimized at the B3LYP/6-31G* level, and duplicates should be removed. A frequency analysis on the most stable structures should be done to verify the nature of the stationary point found.†
(c) The NMR calculations should be done with all significantly populated conformations found in the previous step. It is not recommended to keep only the global minimum for further analysis.
(d) The NMR calculations should be done with the GIAO method, using any of the 24 levels of theory available for DP4+. For general purposes, we recommend PCM/mPW1PW91/6-31+G**.†
(e) When inserting the experimental and calculated data in the excel spreadsheet, it is important to differentiate the sp–sp2 and sp3 nuclei. In addition, the NMR data shall be fully assigned.
(f) The calculated NMR data should be thoroughly revised. Alarmingly large errors (mainly for the most likely candidate) need to be deeply analyzed in search for inconsistencies. If diastereoisomers are scrutinized, very large differences in the calculated NMR values for the same nucleus might be indicative of a mistake.
(g) When dealing with flexible molecules conveniently functionalized with groups that could afford intramolecular H-bonding interactions, whenever the conformational landscape is dominated by shapes featuring IHB interactions, it is recommended to recompute the DP4+ analysis by averaging the isotropic shielding values with SMD-derived Boltzmann amplitudes, or by evaluating the system with the Random DP4+ approach.
(h) It is always recommended to validate the DP4+ results with the experimental NMR information available (such as homo- and heteronuclear coupling values and/or interatomic distances obtained through NOE/ROE experiments).
5 Conclusion
It has been demonstrated that DP4+ is a powerful and easy-to-use toolbox for the structural elucidation of natural products. It can be applied either independently or in combination with other methods. However, to obtain meaningful results the computational work should be done properly, and the data should be manipulated following the suggestions. By understanding the scope and limitations of the method, and how to use it properly, the chances of arriving at a good determination maximize. We consider that with the details and recommendations provided in this review, DP4+ will continue to facilitate the structural determination of new and valuable natural products with high confidence.6 Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
We are grateful to UNR (BIO 316 and BIO 500) and ANPCyT (PICT-2016-0116, PICT-2017-1524 and PICT-2019-4052) for financial support. M. O. M. and S. C. thank CONICET for the award of a fellowship.8 References
- B. K. Chhetri, S. Lavoie, A. M. Sweeney-Jones and J. Kubanek, Nat. Prod. Rep., 2018, 35, 514–531 RSC.
- K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 CrossRef CAS PubMed.
- M. E. Maier, Nat. Prod. Rep., 2009, 26, 1105–1124 RSC.
- T. L. Suyama, W. H. Gerwick and K. L. McPhail, Bioorg. Med. Chem., 2011, 19, 6675–6701 CrossRef CAS.
- H. D. Yoo, S. J. Nam, Y. W. Chin and M. S. Kim, Arch. Pharmacal Res., 2016, 39, 143–153 CrossRef CAS PubMed.
- M. O. Marcarino, M. M. Zanardi, S. Cicetti and A. M. Sarotti, Acc. Chem. Res., 2020, 53, 1922–1932 CrossRef CAS PubMed.
- A. Bagno and G. Saielli, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 228–240 CAS.
- A. G. Kutateladze and T. Holt, J. Org. Chem., 2019, 84, 8297–8299 CrossRef CAS PubMed.
- G. Lauro and G. Bifulco, Eur. J. Org. Chem., 2020, 2020, 3929–3941 CrossRef CAS.
- N. Grimblat and A. M. Sarotti, Chem.–Eur. J., 2016, 22, 12246–12261 CrossRef CAS.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- S. G. Smith and J. M. Goodman, J. Org. Chem., 2009, 74, 4597–4607 CrossRef CAS.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946–12959 CrossRef CAS PubMed.
- K. Ermanis, K. E. B. Parkes, T. Agback and J. M. Goodman, Org. Biomol. Chem., 2019, 17, 5886–5890 RSC.
- A. Howarth, K. Ermanis and J. M. Goodman, Chem. Sci., 2020, 11, 4351–4359 RSC.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS PubMed.
- N. Grimblat, J. A. Gavín, A. Hernández Daranas and A. M. Sarotti, Org. Lett., 2019, 21, 4003–4007 CrossRef CAS PubMed.
- D. Xin, P. J. Jones and N. C. Gonnella, J. Org. Chem., 2018, 83, 5035–5043 CrossRef CAS.
- E. Troche-Pesqueira, C. Anklin, R. R. Gil and A. Navarro-Vázquez, Angew. Chem., Int. Ed., 2017, 56, 3660–3664 CrossRef CAS.
- A. G. Kutateladze and D. S. Reddy, J. Org. Chem., 2017, 82, 3368–3381 CrossRef CAS.
- D. Dardić, G. Lauro, G. Bifulco, P. Laboudie, P. Sakhaii, A. Bauer, A. Vilcinskas, P. E. Hammann and A. Plaza, J. Org. Chem., 2017, 82, 6032–6043 CrossRef.
- J.-Q. Li, H.-W. Zhao and Z.-J. Ma, Tetrahedron Lett., 2020, 151874 CrossRef CAS.
- H. Sun, J. Tan, W. Lv, J. Li, J. Wu, J. Xu, H. Zhu, Z. Yang, W. Wang, Z. Ye, T. Xuan, Z. Zou, Z. Chen and K. Xu, Bioorg. Chem., 2020, 95, 103493 CrossRef CAS PubMed.
- Y. N. Shi, S. Pusch, Y. M. Shi, C. Richter, J. G. Maciá-Vicente, H. Schwalbe, M. Kaiser, T. Opatz and H. B. Bode, J. Org. Chem., 2019, 84, 11203–11209 CrossRef CAS.
- Y. N. Wang, G. Y. Xia, L. Y. Wang, G. B. Ge, H. W. Zhang, J. F. Zhang, Y. Z. Wu and S. Lin, Org. Lett., 2018, 20, 7341–7344 CrossRef CAS.
- W. X. Wang, X. Lei, Y. L. Yang, Z. H. Li, H. L. Ai, J. Li, T. Feng and J. K. Liu, Org. Lett., 2019, 21, 6957–6960 CrossRef CAS.
- M. Ye, W. Xu, D. Q. He, X. Wu, W. F. Lai, X. Q. Zhang, Y. Lin, W. Xu and X. W. Li, J. Nat. Prod., 2020, 83, 362–373 CrossRef CAS.
- J. Bhandari Neupane, R. P. Neupane, Y. Luo, W. Y. Yoshida, R. Sun and P. G. Williams, Org. Lett., 2019, 21, 8449–8453 CrossRef CAS.
- S. Sala, G. L. Nealon, A. N. Sobolev, J. Fromont, O. Gomez and G. R. Flematti, J. Nat. Prod., 2020, 83, 105–110 CrossRef CAS.
- W. X. Wang, G. G. Cheng, Z. H. Li, H. L. Ai, J. He, J. Li, T. Feng and J. K. Liu, Org. Biomol. Chem., 2019, 17, 7985–7994 RSC.
- D. Zhang, W. Yi, H. Ge, Z. Zhang and B. Wu, J. Nat. Prod., 2019, 82, 2800–2808 CrossRef CAS.
- B. Khatri Chhetri, S. Lavoie, A. M. Sweeney-Jones, N. Mojib, V. Raghavan, K. Gagaring, B. Dale, C. W. McNamara, K. Soapi, C. L. Quave, P. L. Polavarapu and J. Kubanek, J. Org. Chem., 2019, 84, 8531–8541 CrossRef CAS PubMed.
- W. X. Wang, Z. H. Li, J. He, T. Feng, J. Li and J. K. Liu, Fitoterapia, 2019, 137, 104278 CrossRef CAS.
- L. H. Martorano, A. L. Valverde, C. M. R. Ribeiro, A. C. F. De Albuquerque, J. W. D. M. Carneiro, R. G. Fiorot and F. M. Dos Santos Junior, New J. Chem., 2020, 44, 8055–8060 RSC.
- H. H. Sun, W. Y. Lv, J. Tan, Y. C. Tang, H. Zhu, J. B. Qu, J. Li, J. P. Wu, X. W. Chang, Z. C. Yang, W. X. Wang, Z. H. Chen and K. P. Xu, Nat. Prod. Res., 2020, 1–7 Search PubMed.
- A. L. Macedo, L. H. Martorano, A. C. F. de Albuquerque, R. G. Fiorot, J. W. M. Carneiro, V. R. Campos, T. R. A. Vasconcelos, A. L. Valverde, D. L. Moreira and F. M. dos Santos, J. Braz. Chem. Soc., 2020, 31, 2030–2037 CAS.
- W. Zhou, F. Kang, L. Huang, J. Li, W. Wang, L. Xiao, Q. Wen, X. Yu, Y. Xu, Z. Zou, H. Zhou, H. Zang, S. Chen and K. Xu, Bioorg. Chem., 2020, 101, 103959 CrossRef CAS PubMed.
- J. Yuan, X. Wen, C. Q. Ke, T. Zhang, L. Lin, S. Yao, J. D. Goodpaster, C. Tang and Y. Ye, Org. Chem. Front., 2020, 7, 1374–1382 RSC.
- R. S. Phansalkar, J. W. Nam, A. A. Leme-Kraus, L. S. Gan, B. Zhou, J. B. McAlpine, S. N. Chen, A. K. Bedran-Russo and G. F. Pauli, J. Nat. Prod., 2019, 82, 2387–2399 CrossRef CAS.
- Y. Y. Fan, L. S. Gan, H. C. Liu, H. Li, C. H. Xu, J. P. Zuo, J. Ding and J. M. Yue, Org. Lett., 2017, 19, 4580–4583 CrossRef CAS.
- D. Pan, X. Zhang, H. Zheng, Z. Zheng, X. Nong, X. Liang, X. Ma and S. Qi, Org. Chem. Front., 2019, 6, 3252–3258 RSC.
- K. S. Salome and C. F. Tormena, J. Org. Chem., 2018, 83, 10501–10504 CrossRef CAS PubMed.
- K. Tanaka, H. Manabe, R. Irie and M. Oikawa, Bull. Chem. Soc. Jpn., 2019, 92, 1314–1323 CrossRef CAS.
- Y. Feng, S. Khokhar and R. A. Davis, Nat. Prod. Rep., 2017, 34, 571–584 RSC.
- X. H. Liu, X. L. Hou, Y. P. Song, B. G. Wang and N. Y. Ji, Fitoterapia, 2020, 141, 104469 CrossRef CAS PubMed.
- Y. F. Liu, Y. H. Zhang, C. L. Shao, F. Cao and C. Y. Wang, J. Nat. Prod., 2020, 83, 1300–1304 CrossRef CAS.
- J. W. Tang, H. C. Xu, W. G. Wang, K. Hu, Y. F. Zhou, R. Chen, X. N. Li, X. Du, H. D. Sun and P. T. Puno, J. Nat. Prod., 2019, 82, 735–740 CrossRef CAS PubMed.
- D. H. Liu, Y. Z. Sun, T. Kurtán, A. Mándi, H. Tang, J. Li, L. Su, C. L. Zhuang, Z. Y. Liu and W. Zhang, J. Nat. Prod., 2019, 82, 1274–1282 CrossRef CAS.
- H. S. Magalhães, A. B. da Silva, N. R. F. Nascimento, L. G. F. de Sousa, M. J. S. da Fonseca, M. I. B. Loiola, N. K. V. Monteiro, F. W. Q. Almeida Neto, K. M. Canuto and O. D. L. Pessoa, Fitoterapia, 2020, 143, 104545 CrossRef.
- A. N. L. Batista, F. M. Dos Santos, A. L. Valverde and J. M. Batista, Org. Biomol. Chem., 2019, 17, 9772–9777 RSC.
- W. P. Ding, K. Hu, M. Liu, X. R. Li, R. Chen, X. N. Li, X. Du, P. T. Puno and H. D. Sun, Fitoterapia, 2018, 127, 193–200 CrossRef CAS.
- Z. hui Huang, X. hua Nong, X. Liang and S. hua Qi, Tetrahedron, 2018, 74, 2620–2626 CrossRef.
- F. Zeng, C. Chen, A. A. Al Chnani, Q. Zhou, Q. Tong, W. Wang, Y. Zang, J. Gong, Z. Wu, J. Liu, J. Wang, H. Zhu and Y. Zhang, Bioorg. Chem., 2019, 86, 176–182 CrossRef CAS PubMed.
- A. Ledoux, A. St-Gelais, E. Cieckiewicz, O. Jansen, A. Bordignon, B. Illien, N. Di Giovanni, A. Marvilliers, F. Hoareau, H. Pendeville, J. Quetin-Leclercq and M. Frédérich, J. Nat. Prod., 2017, 80, 1750–1757 CrossRef CAS.
- N. M. Betterley, S. Kerdphon, S. Chaturonrutsamee, S. Kongsriprapan, P. Surawatanawong, D. Soorukram, M. Pohmakotr, P. G. Andersson, V. Reutrakul and C. Kuhakarn, Asian J. Org. Chem., 2018, 7, 1642–1647 CrossRef CAS.
- S. R. Lee, E. Choi, S. H. Jeon, X. Y. Zhi, J. S. Yu, S. H. Kim, J. Lee, K. M. Park and K. H. Kim, Molecules, 2018, 23, 2732 CrossRef PubMed.
- Z.-Y. Yan, T.-M. Lv, Y.-X. Wang, S.-C. Shi, J.-J. Chen, Bin-Lin, Q.-B. Liu, X.-X. Huang and S.-J. Song, Phytochemistry, 2020, 175, 112361 CrossRef CAS PubMed.
- L. Shao, P. Wu, L. Xu, J. Xue, H. Li and X. Wei, Fitoterapia, 2020, 141, 104465 CrossRef CAS PubMed.
- L. Zhou, F. Y. Han, L. W. Lu, G. D. Yao, Y. Y. Zhang, X. B. Wang, B. Lin, X. X. Huang and S. J. Song, Phytochemistry, 2019, 164, 122–129 CrossRef CAS PubMed.
- T. Huang, S. H. Ying, J. Y. Li, H. W. Chen, Y. Zang, W. X. Wang, J. Li, J. Xiong and J. F. Hu, Phytochemistry, 2020, 169, 112184 CrossRef CAS PubMed.
- C. Huo, X. Lu, Z. Zheng, Y. Li, Y. Xu, H. Zheng and Y. Niu, Phytochemistry, 2020, 170, 112224 CrossRef CAS.
- H. L. Zhu, Y. W. Hu, W. Qu, J. Zhang, E. Y. Guo, X. Y. Jiang, W. Y. Liu, F. Feng and J. Xu, Tetrahedron Lett., 2019, 60, 1868–1870 CrossRef CAS.
- J. Xu, Y. W. Hu, W. Qu, M. H. Chen, L. S. Zhou, Q. R. Bi, J. G. Luo, W. Y. Liu, F. Feng and J. Zhang, Bioorg. Chem., 2019, 90, 103046 CrossRef CAS PubMed.
- Y. P. Song, F. P. Miao, X. H. Liu, X. L. Yin and N. Y. Ji, Mar. Drugs, 2019, 17, 252 CrossRef CAS.
- M. J. Garson, W. Hehre, G. K. Pierens and Suciati, Molecules, 2017, 22, 521 CrossRef.
- X. L. Cheng, H. X. Li, J. Chen, P. Wu, J. H. Xue, Z. Y. Zhou, N. H. Xia and X. Y. Wei, Nat. Prod. Bioprospect., 2021, 11, 63–72 CrossRef CAS PubMed.
- J. Ma, G. Xia, Y. Zang, C. Li, J. Yang, J. Huang, J. Zhang, Y. Su, A. Wang and D. Zhang, Chin. Chem. Lett., 2020, 32, 1173–1176 CrossRef.
- X. Zhen, M. J. Mao, R. Z. Wang, S. S. Chang, T. M. Xiao, Y. X. Wu, L. Y. Yu, Y. L. Song, M. H. Chen and S. Y. Si, J. Asian Nat. Prod. Res., 2020, 1–8 Search PubMed.
- Y. Gao, F. Stuhldreier, L. Schmitt, S. Wesselborg, Z. Guo, K. Zou, A. Mándi, T. Kurtán, Z. Liu and P. Proksch, Front. Microbiol., 2020, 11, 600983 CrossRef PubMed.
- X. Zheng, A. Kadir, G. Zheng, P. Jin, D. Qin, M. Maiwulanjiang, H. A. Aisa and G. Yao, Bioorg. Chem., 2020, 104, 104261 CrossRef CAS PubMed.
- G. Zheng, A. Kadir, X. Zheng, P. Jin, J. Liu, M. Maiwulanjiang, G. Yao and H. A. Aisa, Org. Chem. Front., 2020, 7, 3137–3145 RSC.
- L. Qin, W. Yi, X. Y. Lian, N. Wang and Z. Zhang, Tetrahedron, 2020, 76, 131516 CrossRef CAS.
- A. Mándi, J. Wu and T. Kurtán, RSC Adv., 2020, 10, 32216–32224 RSC.
- W. Li, R. Yan, Y. Yu, Z. Shi, A. Mándi, L. Shen, T. Kurtán and J. Wu, Angew. Chem., 2020, 132, 13128–13136 CrossRef.
- W. Yi, L. Qin, X. Y. Lian and Z. Zhang, Mar. Drugs, 2020, 18, 385 CrossRef CAS PubMed.
- L. J. Zhu, F. Cao, X. X. Su, C. Y. Li, B. Lin, H. F. Wang, X. S. Yao, X. Zhang, J. M. Jia and H. W. Liu, J. Org. Chem., 2020, 85, 8580–8587 CrossRef CAS.
- F. Li, S. Lin, S. Zhang, L. Pan, C. Chai, J. C. Su, B. Yang, J. Liu, J. Wang, Z. Hu and Y. Zhang, J. Nat. Prod., 2020, 83, 1931–1938 CrossRef CAS PubMed.
- A. Cerulli, G. Lauro, M. Masullo, V. Cantone, B. Olas, B. Kontek, F. Nazzaro, G. Bifulco and S. Piacente, J. Nat. Prod., 2017, 80, 1703–1713 CrossRef CAS PubMed.
- L. C. Forster, G. K. Pierens, A. M. White, K. L. Cheney, P. Dewapriya, R. J. Capon and M. J. Garson, ACS Omega, 2017, 2, 2672–2677 CrossRef CAS PubMed.
- P.-E. Campos, E. Pichon, B. Illien, P. Clerc, C. Moriou, N. de Voogd, C. Hellio, R. Trepos, M. Frederich, A. Al-Mourabit and A. Gauvin-Bialecki, Nat. Prod. Chem. Res., 2018, 06, 19–22 Search PubMed.
- Z. Feng, S. Chen, W. Wang, L. Feng, Y. Dong, Y. Zou, C. Ke, C. Tang, S. Yao, H. Zhang, L. Gan, Y. Ye and L. Lin, Fitoterapia, 2019, 139, 104378 CrossRef CAS PubMed.
- W. Wang, J. Yang, Y. Y. Liao, G. Cheng, J. Chen, X. D. Cheng, J. J. Qin and Z. Shao, J. Nat. Prod., 2020, 83, 1157–1166 CrossRef CAS.
- K. Fukaya, D. Urabe, M. Hiraizumi, K. Noguchi, T. Matsumoto and K. Sakurai, J. Org. Chem., 2020, 85, 339–344 CrossRef CAS PubMed.
- W. Li, Y. Q. Tang, J. Sang, R. Z. Fan, G. H. Tang and S. Yin, Org. Lett., 2020, 22, 106–109 CrossRef CAS.
- A. Burtea and S. D. Rychnovsky, Org. Lett., 2018, 20, 5849–5852 CrossRef CAS.
- Q. Y. Zhang, B. C. Yan, K. Hu, X. N. Li, H. D. Sun and P. T. Puno, Fitoterapia, 2020, 142, 104529 CrossRef CAS.
- K. L. Ji, Y. Y. Fan, Z. P. Ge, L. Sheng, Y. K. Xu, L. S. Gan, J. Y. Li and J. M. Yue, J. Org. Chem., 2019, 84, 282–288 CrossRef CAS.
- H. C. Xu, K. Hu, X. H. Shi, J. W. Tang, X. N. Li, H. D. Sun and P. T. Puno, Org. Chem. Front., 2019, 6, 1619–1626 RSC.
- N. Grimblat, T. S. Kaufman and A. M. Sarotti, Org. Lett., 2016, 18, 6420–6423 CrossRef CAS.
- C. Li, A. M. Sarotti, J. Turkson and S. Cao, Tetrahedron Lett., 2017, 58, 2290–2293 CrossRef CAS PubMed.
- P. Huang, C. Li, A. M. Sarotti, J. Turkson and S. Cao, Tetrahedron Lett., 2017, 58, 1330–1333 CrossRef CAS.
- C. S. Li, A. M. Sarotti, P. Huang, U. T. Dang, J. G. Hurdle, T. P. Kondratyuk, J. M. Pezzuto, J. Turkson and S. Cao, Sci. Rep., 2017, 7, 10424 CrossRef PubMed.
- F. Della-Felice, A. M. Sarotti and R. A. Pilli, J. Org. Chem., 2017, 82, 9191–9197 CrossRef CAS.
- Y. S. Cai, A. M. Sarotti, D. Gündisch, T. P. Kondratyuk, J. M. Pezzuto, J. Turkson and S. Cao, Bioorg. Med. Chem. Lett., 2017, 27, 4630–4634 CrossRef CAS.
- A. M. Sarotti, Org. Biomol. Chem., 2018, 16, 944–950 RSC.
- D. A. Heredia, A. M. Durantini, A. M. Sarotti, N. S. Gsponer, D. D. Ferreyra, S. G. Bertolotti, M. E. Milanesio and E. N. Durantini, Chem.–Eur. J., 2018, 24, 5950–5961 CrossRef CAS PubMed.
- Y. H. Tsai, C. M. Borini Etichetti, C. Di Benedetto, J. E. Girardini, F. T. Martins, R. A. Spanevello, A. G. Suárez and A. M. Sarotti, J. Org. Chem., 2018, 83, 3516–3528 CrossRef CAS PubMed.
- Y. S. Cai, A. M. Sarotti, T. L. Zhou, R. Huang, G. Qiu, C. Tian, Z. H. Miao, A. Mándi, T. Kurtán, S. Cao and S. P. Yang, J. Nat. Prod., 2018, 81, 1976–1983 CrossRef CAS PubMed.
- F. Della-Felice, F. F. De Assis, A. M. Sarotti and R. A. Pilli, Synth, 2019, 51, 1545–1560 CrossRef CAS.
- C. Li, A. M. Sarotti, X. Wu, B. Yang, J. Turkson, Y. Chen, Q. Liu and S. Cao, Molecules, 2019, 24, 196 CrossRef.
- F. Della-Felice, A. M. Sarotti, M. J. Krische and R. A. Pilli, J. Am. Chem. Soc., 2019, 141, 13778–13782 CrossRef CAS PubMed.
- J. H. Ke, L. S. Zhang, S. X. Chen, S. N. Shen, T. Zhang, C. X. Zhou, J. X. Mo, L. G. Lin and L. S. Gan, Fitoterapia, 2019, 134, 346–354 CrossRef CAS PubMed.
- V. A. Cosenza, D. A. Navarro and C. A. Stortz, Carbohydr. Polym., 2017, 157, 156–166 CrossRef CAS PubMed.
- M. T. Sibero, T. Zhou, K. Fukaya, D. Urabe, O. K. Karna Radjasa, A. Sabdono, A. Trianto and Y. Igarashi, Beilstein J. Org. Chem., 2019, 15, 2941–2947 CrossRef CAS.
- L. A. Joyce, C. C. Nawrat, E. C. Sherer, M. Biba, A. Brunskill, G. E. Martin, R. D. Cohen and I. W. Davies, Chem. Sci., 2018, 9, 415–424 RSC.
- Y. E. Sim, O. Nwajiobi, S. Mahesh, R. D. Cohen, M. Y. Reibarkh and M. Raj, Chem. Sci., 2020, 11, 53–61 RSC.
- B. Lipp, L. M. Kammer, M. Kücükdisli, A. Luque, J. Kühlborn, S. Pusch, G. Matulevičiūtė, D. Schollmeyer, A. Šačkus and T. Opatz, Chem.–Eur. J., 2019, 25, 8965–8969 CrossRef CAS PubMed.
- H. C. Xu, K. Hu, H. D. Sun and P. T. Puno, Nat. Products Bioprospect., 2019, 9, 165–173 CrossRef CAS PubMed.
- S. M. Isyaka, M. K. Langat, E. Mas-Claret, B. M. Mbala, B. K. Mvingu and D. A. Mulholland, Phytochemistry, 2020, 170, 112217 CrossRef CAS PubMed.
- M. M. Rob, A. Iwasaki, R. Suzuki, K. Suenaga and H. Kato-Noguchi, Plants, 2019, 8, 301 CrossRef CAS.
- P. Klein, P. Johe, A. Wagner, S. Jung, J. Kühlborn, F. Barthels, S. Tenzer, U. Distler, W. Waigel, B. Engels, U. A. Hellmich, T. Opatz and T. Schirmeister, Molecules, 2020, 25, 1451 CrossRef CAS.
- S. Saito, K. Atsumi, T. Zhou, K. Fukaya, D. Urabe, N. Oku, M. R. Ul Karim, H. Komaki and Y. Igarashi, Beilstein J. Org. Chem., 2020, 16, 1100–1110 CrossRef CAS.
- F. R. Jiao, B. Bin Gu, H. R. Zhu, Y. Zhang, K. C. Liu, W. Zhang, H. Han, S. H. Xu and H. W. Lin, J. Org. Chem., 2020 DOI:10.1021/acs.joc.0c02049.
- S. W. Li, C. Cuadrado, X. J. Huan, L. G. Yao, Z. H. Miao, A. Hernandez Daranas and Y. W. Guo, Bioorg. Chem., 2020, 103, 104223 CrossRef CAS.
- M. Alilou, S. Marzocco, D. Hofer, S. F. Rapa, R. Asadpour, S. Schwaiger, J. Troppmair and H. Stuppner, J. Nat. Prod., 2020, 83, 2456–2468 CrossRef CAS.
- S. W. Li, C. Cuadrado, L. G. Yao, A. H. Daranas and Y. W. Guo, Org. Lett., 2020, 22, 4093–4096 CrossRef CAS.
- F. Wang, A. M. Sarotti, G. Jiang, J. C. Huguet-Tapia, S. L. Zheng, X. Wu, C. Li, Y. Ding and S. Cao, Org. Lett., 2020, 22, 4408–4412 CrossRef CAS.
- Y. L. Li, R. X. Zhu, G. Li, N. N. Wang, C. Y. Liu, Z. T. Zhao and H. X. Lou, RSC Adv., 2019, 9, 4140–4149 RSC.
- M. K. Langat, A. Helfenstein, C. Horner, P. Tammela, H. Hokkanen, D. Izotov and D. A. Mulholland, Chem. Biodiversity, 2018, 15, e1800056 CrossRef.
- M. F. Elsebai, H. A. Ghabbour, N. Legrave, F. Fontaine-Vive and M. Mehiri, Med. Chem. Res., 2018, 27, 1885–1892 CrossRef CAS.
- X. C. Guo, L. L. Xu, R. Y. Yang, M. Y. Yang, L. D. Hu, H. J. Zhu and F. Cao, Front. Chem., 2019, 7, 80 CrossRef CAS.
- H. Wakamatsu, S. Tanaka, Y. Matsuo, Y. Saito, K. Nishida and T. Tanaka, Molecules, 2019, 24, 4279 CrossRef CAS.
- Ł. Pecio, M. Alilou, S. Kozachok, I. E. Orhan, G. Eren, F. S. S. Deniz, H. Stuppner and W. Oleszek, Molecules, 2019, 24, 4162 CrossRef PubMed.
- J. Li, C. Li, R. Riccio, G. Lauro, G. Bifulco, T. J. Li, H. Tang, C. L. Zhuang, H. Ma, P. Sun and W. Zhang, Mar. Drugs, 2017, 15, 129 CrossRef.
- R. P. Neupane, S. M. Parrish, J. B. Neupane, W. Y. Yoshida, M. L. Richard Yip, J. Turkson, M. K. Harper, J. D. Head and P. G. Williams, Mar. Drugs, 2019, 17, 423 CrossRef CAS.
- D. B. Pu, B. W. Du, W. Chen, J. B. Gao, K. Hu, N. Shi, Y. M. Li, X. J. Zhang, R. H. Zhang, X. N. Li, H. Bin Zhang, F. Wang and W. L. Xiao, Org. Lett., 2018, 20, 6314–6317 CrossRef CAS.
- D. Pu, X. Li, J. Lin, R. Zhang, T. Luo, Y. Wang, J. Gao, M. A. Zeb, X. Zhang, X. Li, R. Wang and W. Xiao, J. Nat. Prod., 2019, 82, 2067–2077 CrossRef CAS PubMed.
- Q. Yang, K. Hu, B. C. Yan, M. Liu, X. N. Li, H. D. Sun and P. T. Puno, Org. Chem. Front., 2019, 6, 45–53 RSC.
- J. Wang, Q. Ren, Y. Y. Zhang, R. Guo, B. Lin, X. X. Huang and S. J. Song, Fitoterapia, 2019, 138, 104352 CrossRef CAS PubMed.
- C. Ma, C. W. Meng, Q. M. Zhou, C. Peng, F. Liu, J. W. Zhang, F. Zhou and L. Xiong, Fitoterapia, 2019, 138, 104351 CrossRef CAS PubMed.
- P. Zhao, R. Guo, Y. Y. Zhang, H. Zhang, G. D. Yao, B. Lin, X. B. Wang, X. X. Huang and S. J. Song, Bioorg. Chem., 2019, 93, 103354 CrossRef CAS.
- P. Zhou, J. Hu, B. Wen, J. Ding, B. Lou, J. Xiong, G. Yang and J. Hu, Tetrahedron, 2020, 76, 131026 CrossRef CAS.
- H. M. So, J. S. Yu, Z. Khan, L. Subedi, Y. J. Ko, I. K. Lee, W. S. Park, S. J. Chung, M. J. Ahn, S. Y. Kim and K. H. Kim, Bioorg. Chem., 2019, 91, 103145 CrossRef CAS.
- J. Gao, X. Zhang, K. Shang, W. Zhong, R. Zhang, X. Dai, X. Li, Q. Wang, Y. Zou and W. Xiao, Chinese Chem. Lett., 2020, 31, 427–430 CrossRef CAS.
- R. Guo, T. Lv, F. Han, Z. Hou, G. Yao, B. Lin, X. Wang, X. Huang and S. Song, Chinese Chem. Lett., 2020, 31, 1254–1258 CrossRef CAS.
- D. Liu, Q. Yin, Q. Zhang, J. Xiang, C. Ruan, H. Liu, B. Li, W. Zhu, C. ping Yin and J. Fang, Phytochem. Lett., 2019, 34, 91–95 CrossRef CAS.
- T. Zhao, X. H. Nong, B. Zhang, M. M. Tang, D. Y. Huang, J. L. Wang, J. L. Xiao and G. Y. Chen, Phytochem. Lett., 2020, 36, 115–119 CrossRef CAS.
- Y. G. Cao, Y. L. Zhang, M. N. Zeng, M. Qi, Y. J. Ren, Y. L. Liu, X. Zhao, X. K. Zheng and W. S. Feng, J. Nat. Prod., 2020, 83, 1118–1130 CrossRef CAS PubMed.
- H. Cui, Y. Liu, J. Li, X. Huang, T. Yan, W. Cao, H. Liu, Y. Long and Z. She, J. Org. Chem., 2018, 83, 11804–11813 CrossRef CAS PubMed.
- F. Wang, W. Zhao, C. Zhang, S. Chang, R. Shao, J. Xing, M. Chen, Y. Zhang and S. Si, RSC Adv., 2019, 9, 16035–16039 RSC.
- S. G. Li, X. J. Huang, Y. L. Zhong, M. M. Li, Y. L. Li, Y. Wang and W. C. Ye, Chem. Biodiversity, 2019, 16, e1900192 Search PubMed.
- S. X. Yang, W. T. Zhao, H. Y. Chen, L. Zhang, T. K. Liu, H. P. Chen, J. Yang and X. L. Yang, Chem. Biodiversity, 2019, 16, e1900364 CrossRef.
- X. Y. Han, Y. X. Xie, C. Q. Wu, H. L. Ai, X. X. Lei and X. J. Wang, Chem. Biodiversity, 2019, 16, e1900471 CrossRef CAS.
- J. S. Wu, X. H. Shi, Y. H. Zhang, J. Y. Yu, X. M. Fu, X. Li, K. X. Chen, Y. W. Guo, C. L. Shao and C. Y. Wang, Front. Chem., 2019, 7, 763 CrossRef CAS PubMed.
- T. T. Fan, H. H. Zhang, Y. H. Tang, F. Z. Zhang and B. N. Han, Mar. Drugs, 2019, 17, 652 CrossRef CAS.
- D. A. Adpressa, K. J. Stalheim, P. J. Proteau and S. Loesgen, ACS Chem. Biol., 2017, 12, 1842–1847 CrossRef CAS PubMed.
- M. Chen, R. Wang, W. Zhao, L. Yu, C. Zhang, S. Chang, Y. Li, T. Zhang, J. Xing, M. Gan, F. Feng and S. Si, Org. Lett., 2019, 21, 1530–1533 CrossRef CAS PubMed.
- G. Chianese, H. B. Yu, F. Yang, C. Sirignano, P. Luciano, B. N. Han, S. Khan, H. W. Lin and O. Taglialatela-Scafati, J. Org. Chem., 2016, 81, 5135–5143 CrossRef CAS.
- S. R. Lee, H. B. Park and K. H. Kim, Anal. Chem., 2018, 90, 13212–13216 CrossRef CAS.
- J. Li, Y. Hu, X. Hao, J. Tan, F. Li, X. Qiao, S. Chen, C. Xiao, M. Chen, Z. Peng and M. Gan, J. Nat. Prod., 2019, 82, 1391–1395 CrossRef CAS PubMed.
- X. Luo, C. Chen, H. Tao, X. Lin, B. Yang, X. Zhou and Y. Liu, Org. Chem. Front., 2019, 6, 736–740 RSC.
- Y. Nalli, S. Jan, G. Lauro, J. Ur Rasool, W. I. Lone, A. R. Sarkar, J. Banday, G. Bifulco, H. Laatsch, S. H. Syed and A. Ali, Nat. Prod. Res., 2021, 35, 471–480 CrossRef CAS.
- K. J. Park, C. S. Kim, Z. Khan, J. Oh, S. Y. Kim, S. U. Choi and K. R. Lee, J. Nat. Prod., 2019, 82, 1345–1353 CrossRef CAS.
- P. Sun, Q. Yu, J. Li, R. Riccio, G. Lauro, G. Bifulco, T. Kurtán, A. Mándi, H. Tang, T. J. Li, C. L. Zhuang, W. H. Gerwick and W. Zhang, J. Nat. Prod., 2016, 79, 2552–2558 CrossRef CAS.
- Y. Tang, Z. Z. Zhao, K. Hu, T. Feng, Z. H. Li, H. P. Chen and J. K. Liu, J. Org. Chem., 2019, 84, 1845–1852 CrossRef CAS.
- K. Hu, X. R. Li, J. W. Tang, X. N. Li and P. T. Puno, Chin. J. Nat. Med., 2019, 17, 970–981 Search PubMed.
- H. Nguyen Ngoc, M. Alilou, M. Stonig, D. T. Nghiem, L. T. Kim, J. M. Gostner, H. Stuppner and M. Ganzera, J. Nat. Prod., 2019, 82, 2941–2952 CrossRef CAS.
- M. Alilou, D. F. Dibwe, S. Schwaiger, M. Khodami, J. Troppmair, S. Awale and H. Stuppner, J. Nat. Prod., 2020, 83, 1099–1106 CrossRef CAS.
- M. S. Jo, S. Lee, J. S. Yu, S. C. Baek, Y. C. Cho and K. H. Kim, J. Nat. Prod., 2020, 83, 684–692 CrossRef CAS.
- J. Li, M. Chen, X. Hao, S. Li, F. Li, L. Yu, C. Xiao and M. Gan, Org. Lett., 2020, 22, 98–101 CrossRef CAS.
- W. J. Lu, W. J. Xu, Y. Q. Zhang, Y. R. Li, X. Zhou, Q. J. Li, H. Zhang, J. Luo and L. Y. Kong, Org. Chem. Front., 2020, 7, 1070–1076 RSC.
- C. S. Kim, J. Oh, L. Subedi, S. Y. Kim, S. U. Choi and K. R. Lee, Chem. Pharm. Bull., 2018, 66, 839–842 CrossRef CAS.
- F. Cao, T. T. Sun, J. K. Yang, G. Z. Zhao, Q. A. Liu, L. D. Hu, Z. Y. Ma and H. J. Zhu, Nat. Prod. Res., 2019, 33, 2192–2199 CrossRef CAS PubMed.
- T. Seitz, R. E. Millán, D. Lentz, C. Jiménez, J. Rodríguez and M. Christmann, Org. Lett., 2018, 20, 594–597 CrossRef CAS PubMed.
- L. Dong, D. P. Qin, Q. Q. Di, Y. Liu, W. L. Chen, Y. X. Cheng and S. M. Wang, Org. Chem. Front., 2019, 6, 3825–3833 RSC.
- N. Grimblat, T. S. Kaufman and A. M. Sarotti, Org. Lett., 2016, 18, 6420–6423 CrossRef CAS PubMed.
- C. Li, A. M. Sarotti, J. Turkson and S. Cao, Tetrahedron Lett., 2017, 58, 2290–2293 CrossRef CAS PubMed.
- N. Q. Tuan, J. Oh, H. B. Park, D. Ferreira, S. Choe, J. Lee and M. K. Na, Phytochemistry, 2017, 133, 45–50 CrossRef CAS.
- T. Tsujita, Y. Matsuo, Y. Saito and T. Tanaka, Tetrahedron, 2017, 73, 500–507 CrossRef CAS.
- L. L. Gou, K. Hu, Q. Yang, X. N. Li, H. D. Sun, C. L. Xiang and P. T. Puno, Tetrahedron, 2019, 75, 2797–2806 CrossRef CAS.
- L. Chen, J. N. Yao, H. P. Chen, Z. Z. Zhao, Z. H. Li, T. Feng and J. K. Liu, Phytochem. Lett., 2018, 27, 94–100 CrossRef CAS.
- S. Lavoie, A. M. Sweeney-Jones, N. Mojib, B. Dale, K. Gagaring, C. W. McNamara, C. L. Quave, K. Soapi and J. Kubanek, J. Org. Chem., 2019, 84, 5035–5045 CrossRef CAS PubMed.
- X. Wang, J. Liu, P. Pandey, F. R. Fronczek, R. J. Doerksen, J. Chen, X. Qi, P. Zhang, D. Ferreira, F. A. Valeriote, H. Sun, S. Li and M. T. Hamann, Org. Lett., 2018, 20, 5559–5563 CrossRef CAS PubMed.
- S. Zhang, Y. Huang, S. He, H. Chen, Z. Li, B. Wu, J. Zuo, T. Feng and J. Liu, RSC Adv., 2018, 8, 23914–23918 RSC.
- J. J. Orejola, M. Era, Y. Matsuo, Y. Saito and T. Tanaka, Tetrahedron, 2020, 76, 131204 CrossRef CAS.
- I. Park, W. Lee, Y. Yoo, H. Shin, J. Oh, H. Kim, M. A. Kim, J. S. Hwang, J. S. Bae and M. Na, Int. J. Mol. Sci., 2020, 21, 3406 CrossRef CAS.
- S. S. Zhu, J. W. Liu, Y. M. Yan, Y. Liu, Z. Mao and Y. X. Cheng, Org. Lett., 2020, 22, 3428–3432 CrossRef CAS PubMed.
- C. T. Sun, J. P. Wang, Y. Shu, X. Y. Cai, J. T. Hu, S. Q. Zhang, L. Cai and Z. T. Ding, Nat. Prod. Res., 2020 DOI:10.1080/14786419.2020.1806272.
- Y. Zhang, H. Hu and J. Luo, Nat. Prod. Res., 2020 DOI:10.1080/14786419.2020.1830397.
- Y. A. Rincón, G. E. Siless, L. D. Amado, M. V. Dansey, E. Grassi, N. Schenone and G. M. Cabrera, Nat. Prod. Res., 2020 DOI:10.1080/14786419.2020.1752205.
- N. K. T. Pham, T. T. L. Tran, T. H. Duong, N. T. Trung, D. C. T. Phan, D. T. Mai, V. K. Nguyen, B. L. C. Huynh, T. A. T. Nguyen, T. D. Tran, T. N. M. Tran and T. P. Nguyen, Nat. Prod. Res., 2020 DOI:10.1080/14786419.2020.1839456.
- X. Y. Hu, X. M. Li, S. Q. Yang, H. Liu, L. H. Meng and B. G. Wang, Mar. Drugs, 2020, 18, 194 CrossRef CAS.
- T. H. Duong, T. T. Nguyen, C. T. D. Phan, V. D. Nguyen, H. C. Nguyen, T. B. N. Dao, D. T. Mai, N. Niamnont, T. N. M. Tran and J. Sichaem, Nat. Prod. Res., 2020 DOI:10.1080/14786419.2020.1789980.
- K. Calabro, L. K. Jennings, P. Lasserre, R. Doohan, D. Rodrigues, F. Reyes, C. Ramos and O. P. Thomas, J. Org. Chem., 2020, 85, 14026–14041 CrossRef CAS.
- H. Liu, X. Wang, Q. Shi, L. Li, Q. Zhang, Z. L. Wu, X. J. Huang, Q. W. Zhang, W. C. Ye, Y. Wang and L. Shi, ACS Omega, 2020, 5, 10167–10175 CrossRef CAS.
- W. Y. Zhang, Y. Zhong, Y. Yu, D. F. Shi, H. Y. Huang, X. L. Tang, Y. H. Wang, G. D. Chen, H. P. Zhang, C. L. Liu, D. Hu, H. Gao and X. S. Yao, J. Nat. Prod., 2020, 83, 3338–3346 CrossRef CAS.
- R. Kawazoe, Y. Matsuo, Y. Saito and T. Tanaka, J. Nat. Prod., 2020, 83, 3347–3353 CrossRef CAS PubMed.
- K. H. Lee, J. S. Yu, J. H. Choi, S. H. Kim, Y. J. Ko, C. Pang and K. H. Kim, Bioorg. Med. Chem. Lett., 2020, 30, 127641 CrossRef CAS.
- Q. Q. Shi, X. J. Zhang, Y. Zhang, Q. Wang, M. Amin, Q. Li, X. W. Wu, X. L. Li, R. H. Zhang, X. C. Dai and W. L. Xiao, Bioorg. Chem., 2020, 105, 104363 CrossRef CAS PubMed.
- J. S. Yu, C. Li, M. Kwon, T. Oh, T. H. Lee, D. H. Kim, J. S. Ahn, S. K. Ko, C. S. Kim, S. Cao and K. H. Kim, Bioorg. Chem., 2020, 105, 104397 CrossRef CAS PubMed.
- M. K. Langat, E. F. K. Djuidje, B. M. Ndunda, S. M. Isyaka, N. S. Dolan, G. D. Ettridge, H. Whitmore, I. Lopez, A. M. Alqahtani, I. Atiku, J. S. Lobe, E. Mas-Claret, N. R. Crouch, J. O. Midiwo, D. A. Mulholland and A. F. W. Kamdem, Phytochem. Lett., 2020, 40, 148–155 CrossRef CAS.
- N. Nath, J. C. Fuentes-Monteverde, D. Pech-Puch, J. Rodríguez, C. Jiménez, M. Noll, A. Kreiter, M. Reggelin, A. Navarro-Vázquez and C. Griesinger, Nat. Commun., 2020, 11, 4372 CrossRef CAS.
- Z. Hu, Z. Wu, Q. Su, M. Li, S. Wu, R. Meng, W. Ding and C. Li, Bioorg. Chem., 2020, 104, 104300 CrossRef CAS.
- Q. Q. Shi, X. J. Zhang, T. T. Wang, Q. Wang, T. T. Sun, M. Amin, R. H. Zhang, X. L. Li and W. L. Xiao, Org. Lett., 2020, 22, 7820–7824 CrossRef CAS PubMed.
- Y. Shu, J. P. Wang, X. Y. Cai, X. L. Li, J. T. Hu, C. T. Sun, L. Cai and Z. T. Ding, Tetrahedron, 2020, 76, 131520 CrossRef CAS.
- S. G. Li, Y. T. Wang, Q. Zhang, K. B. Wang, J. J. Xue, D. H. Li, Y. K. Jing, B. Lin and H. M. Hua, Org. Lett., 2020, 22, 7522–7525 CrossRef CAS PubMed.
- H. T. Li, R. T. Duan, T. Liu, R. N. Yang, J. P. Wang, S. X. Liu, Y. Bin Yang, H. Zhou and Z. T. Ding, Fitoterapia, 2020, 146, 104711 CrossRef CAS.
- Q. Li, W. Xu, R. Fan, J. Zhang, Y. Li, X. Wang, S. Han, W. Liu, M. Pan and Z. Cheng, J. Nat. Prod., 2020, 83, 2679–2685 CrossRef CAS PubMed.
- J. Wei Tang, K. Hu, X. Zheng Su, X. Nian Li, B. Chao Yan, H. Dong Sun and P. Tenzin Puno, Tetrahedron, 2020, 76, 131475 CrossRef.
- S. Lee, D. Lee, R. Ryoo, J. C. Kim, H. B. Park, K. S. Kang and K. H. Kim, J. Nat. Prod., 2020, 83, 2737–2742 CrossRef CAS.
- Y. Kudo, C. T. Hanifin, Y. Kotaki and M. Yotsu-Yamashita, J. Nat. Prod., 2020, 83, 2706–2717 CrossRef CAS.
- B. Y. Hu, S. X. Wang, Y. M. Yan, J. W. Liu, D. P. Qin and Y. X. Cheng, Org. Chem. Front., 2020, 7, 2710–2718 RSC.
- X. C. Guo, Y. H. Zhang, W. Bin Gao, L. Pan, H. J. Zhu and F. Cao, Mar. Drugs, 2020, 18, 479 CrossRef CAS PubMed.
- Q. Shi, S. Lu, D. Li, J. Lu, L. Zhou and M. Qiu, Fitoterapia, 2020, 145, 104635 CrossRef CAS.
- J. S. Yu, M. Park, C. Pang, L. Rashan, W. H. Jung and K. H. Kim, J. Nat. Prod., 2020, 83, 2261–2268 CrossRef CAS.
- J. P. Wang, Y. Shu, J. T. Hu, R. Liu, X. Y. Cai, C. T. Sun, D. Gan, D. J. Zhou, R. F. Mei, H. Ding, X. R. Zhang, L. Cai and Z. T. Ding, Org. Chem. Front., 2020, 7, 1463–1468 RSC.
- X. R. Peng, Q. Q. Shi, J. Yang, H. G. Su, H. G. Su, L. Zhou, M. H. Qiu and M. H. Qiu, J. Org. Chem., 2020, 85, 7446–7451 CrossRef CAS PubMed.
- A. L. Duddupudi, P. Pandey, H. Vo, C. L. Welsh, R. J. Doerksen and G. D. Cuny, J. Org. Chem., 2020, 85, 7549–7557 CrossRef CAS.
- B. Fan, P. Dewapriya, F. Li, L. Grauso, M. Blümel, A. Mangoni and D. Tasdemir, Mar. Drugs, 2020, 18, 281 CrossRef CAS.
- P. Pel, H. S. Chae, P. Nhoek, Y. M. Kim, P. Khiev, G. J. Kim, J. W. Nam, H. Choi, Y. H. Choi and Y. W. Chin, Bioorg. Chem., 2020, 99, 103869 CrossRef CAS PubMed.
- C. X. Zou, Z. L. Hou, M. Bai, R. Guo, B. Lin, X. B. Wang, X. X. Huang and S. J. Song, Org. Biomol. Chem., 2020, 18, 3908–3916 RSC.
- Y. Zou, X. Wang, J. Sims, B. Wang, P. Pandey, C. L. Welsh, R. P. Stone, M. A. Avery, R. J. Doerksen, D. Ferreira, C. Anklin, F. A. Valeriote, M. Kelly and M. T. Hamann, J. Am. Chem. Soc., 2019, 141, 4338–4344 CrossRef CAS PubMed.
- F. Della-Felice, A. M. Sarotti, M. J. Krische and R. A. Pilli, J. Am. Chem. Soc., 2019, 141, 13778–13782 CrossRef CAS.
- L. Andernach, L. P. Sandjo, J. C. Liermann, R. Schlämann, C. Richter, J. P. Ferner, H. Schwalbe, A. Schüffler, E. Thines and T. Opatz, J. Nat. Prod., 2016, 79, 2718–2725 CrossRef CAS.
- C. S. Kim, J. Oh, L. Subedi, S. Y. Kim, S. U. Choi and K. R. Lee, J. Nat. Prod., 2018, 81, 1795–1802 CrossRef CAS.
- P. Yan, G. Li, C. Wang, J. Wu, Z. Sun, G. E. Martin, X. Wang, M. Reibarkh, J. Saurí and K. R. Gustafson, Org. Lett., 2019, 21, 7577–7581 CrossRef CAS.
- G. Tarazona, G. Benedit, R. Fernández, M. Pérez, J. Rodríguez, C. Jiménez and C. Cuevas, J. Nat. Prod., 2018, 81, 343–348 CrossRef CAS PubMed.
- M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2021, 86, 8544–8548 CrossRef CAS.
- M. M. Zanardi, A. G. Suárez and A. M. Sarotti, J. Org. Chem., 2017, 82, 1873–1879 CrossRef CAS PubMed.
- M. M. Zanardi, F. A. Biglione, M. A. Sortino and A. M. Sarotti, J. Org. Chem., 2018, 83, 11839–11849 CrossRef CAS.
- F. Cen-Pacheco, J. Rodríguez, M. Norte, J. J. Fernández and A. Hernández Daranas, Chem.–Eur. J., 2013, 19, 8525–8532 CrossRef CAS PubMed.
- G. Zuber, M.-R. Goldsmith, T. D. Hopkins, D. N. Beratan and P. Wipf, Org. Lett., 2005, 7, 5269–5272 CrossRef CAS.
- C.-X. Wang, G.-D. Chen, C.-C. Feng, R.-R. He, S.-Y. Qin, D. Hu, H.-R. Chen, X.-Z. Liu, X.-S. Yao and H. Gao, Chem. Commun., 2016, 52, 1250–1253 RSC.
- A. M. White, K. Dao, D. Vrubliauskas, Z. A. Könst, G. K. Pierens, A. Mándi, K. T. Andrews, T. S. Skinner-Adams, M. E. Clarke, P. T. Narbutas, D. C. M. Sim, K. L. Cheney, T. Kurtán, M. J. Garson and C. D. Vanderwal, J. Org. Chem., 2017, 82, 13313–13323 CrossRef CAS.
- M. G. Chini, R. Riccio and G. Bifulco, Eur. J. Org. Chem., 2015, 2015, 1320–1324 CrossRef CAS.
- D. J. Marell, S. J. Emond, A. Kulshrestha and T. R. Hoye, J. Org. Chem., 2014, 79, 752–758 CrossRef CAS.
- W. F. Xu, X. J. Xue, Y. X. Qi, N. N. Wu, C. Y. Wang and C. L. Shao, Nat. Prod. Res., 2021, 35, 490–493 CrossRef CAS.
- C. Jiménez, M. Blanco, C. Cuevas, R. Fernández, J. Rodríguez and G. Tarazona, Org. Lett., 2016, 18, 5832–5835 CrossRef.
- S. C. Baek, K. H. Nam, S. A. Yi, M. S. Jo, K. H. Lee, Y. H. Lee, J. Lee and K. H. Kim, Foods, 2019, 8, 673 CrossRef CAS.
- M. M. Zanardi, M. O. Marcarino and A. M. Sarotti, Org. Lett., 2020, 22, 52–56 CrossRef CAS PubMed.
- M. O. Marcarino, M. M. Zanardi and A. M. Sarotti, Org. Lett., 2020, 22, 3561–3565 CrossRef CAS.
- M. M. Zanardi, M. A. Sortino and A. M. Sarotti, Carbohydr. Res., 2019, 474, 72–79 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- É. Brémond, M. Savarese, N. Q. Su, Á. J. Pérez-Jiménez, X. Xu, J. C. Sancho-García and C. Adamo, J. Chem. Theory Comput., 2016, 12, 459–465 CrossRef PubMed.
- A. M. Sarotti, J. Org. Chem., 2020, 85, 11566–11570 CrossRef CAS.
- M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 9371–9378 CrossRef CAS PubMed.
- A. M. Sarotti, Org. Biomol. Chem., 2013, 11, 4847–4859 RSC.
Footnote |
| † In the case of the new updated version of DP4+, the geometry optimization and NMR calculations can be carried out at any desired level of theory. However, to obtain meaningful analysis a proper estimation of the [μ, ν, σ] set should be carried out.218 |
| This journal is © The Royal Society of Chemistry 2022 |