
Anti-mycobacterial natural products and mechanisms of action†
Jianying
Han
 a,
Xueting
Liu
b,
Lixin
Zhang
b,
Ronald J.
Quinn
*a and
Yunjiang
Feng
*a
a,
Xueting
Liu
b,
Lixin
Zhang
b,
Ronald J.
Quinn
*a and
Yunjiang
Feng
*a
aGriffith Institute for Drug Discovery, Griffith University, Brisbane, QLD 4111, Australia. E-mail: y.feng@griffith.edu.au; r.quinn@griffith.edu.au
bState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 6th July 2021
Abstract
Covering: up to June, 2020
Tuberculosis (TB) continues to be a major disease with high mortality and morbidity globally. Drug resistance and long duration of treatment make antituberculosis drug discovery more challenging. In this review, we summarize recent advances on anti-TB natural products (NPs) and their potential molecular targets in cell wall synthesis, protein production, energy generation, nucleic acid synthesis and other emerging areas. We highlight compounds with activity against drug-resistant TB, and reveal several novel targets including Mtb biotin synthase, ATP synthase, 1,4-dihydroxy-2-naphthoate prenyltransferase and biofilms. These anti-TB NPs and their targets could facilitate target-based screening and accelerate TB drug discovery.
1. Introduction
Tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis (Mtb), is one of the top 10 causes of death worldwide.1 Approximately 1.7 billion people are latently infected with Mtb and they are at risk of developing TB.2 The emergence of drug resistant strains of Mtb has further complicated the situation, resulting in an urgent medical need for new drugs with novel mechanisms of action.3 The standard chemotherapy regimen for drug-sensitive TB involves treatment with rifampicin, isoniazid, ethambutol and pyrazinamide for 2 months, followed by treatment with rifampicin and isoniazid for an additional 4 months. Whereas treatment of drug-resistant TB often requires more than 20 months.4 The long treatment regimens for multiple and extensively drug-resistant (MDR/XDR) TB are associated with compliance issues, making new anti-TB drug discovery imperative. Since the discovery of streptomycin, a series of antitubercular drugs have been developed.5–7 However, few New Chemical Entities (NCEs) have been discovered in recent decades to treat emerging drug resistance and to shorten the duration of TB treatment. Recently, collective global efforts have been made to eradicate TB.8,9 An intensive search for novel and more effective agents is now under way, generating a number of lead molecules with varying potential for progression into optimization, preclinical development, and clinical trials (Table S1 and Fig. S1†).6Natural products (NPs) are important sources for anti-TB drug discovery and have provided a number of novel molecular scaffolds.10–13 Most of these active compounds were identified through phenotypic screening against different mycobacteria strains. Several NPs have advanced to market, including streptomycin, cycloserin, kanamycin, rifampicin, capreomycin, and amikacin (kanamycin derivative) (Fig. 1). However, no new NPs have emerged since the 1970s, mainly due to the shifting focus of pharmaceutical programs away from NPs, constant re-discovery of known NP scaffolds, and the lack of understanding of their drug targets.14,15
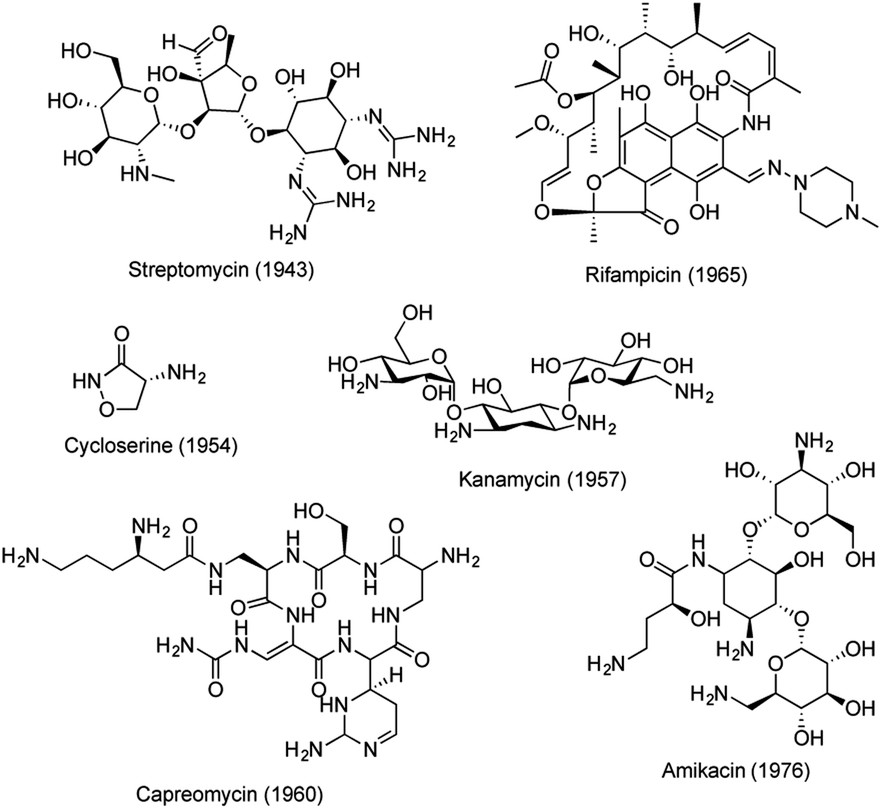 | ||
| Fig. 1 Anti-TB drugs derived from NPs. | ||
Target-based approaches have been a dominant paradigm to discover new classes of drugs with novel modes of action.16,17 Research has shown there are almost 4000 proteins in the Mtb pathogen, consisting of more than 500 essential proteins.18 To date, 576 proteins from M. tuberculosis H37Rv have been selected as potential druggable targets and have proceeded to the structure determination pipeline (https://www.ssgcid.org/) (Table S2†). These proteins are involved in different metabolic pathways, such as dehydrogenases, PE-PGRS family proteins, oxidoreductases, transporters, lipases, kinases, PPE family proteins, proteases, hydrolases, hydratases, RNA polymerases, ligases and others (Fig. 2). Unfortunately, anti-TB drug discovery has only focused on a few clinically validated targets. The drugs in clinical use mainly include fatty acid biosynthesis inhibitors isoniazid (target KatG, InhA, and AhpC), pyrazinamide (target PncA), ethionamide (target InhA), prothionamide (target InhA), and thioacetazone (target CmaA2); inhibitors of nucleic acid synthesis rifampicin (target RpoB), ofloxacin (target GyrA and GyrB), levofloxacin (target GyrA and GyrB), moxifloxacin (target GyrA and GyrB), and ciprofloxacin (target GyrA and GyrB); inhibitors of protein synthesis streptomycin (target RpsL, Rrs, and GidB), kanamycin (target Rrs, and Eis), amikacin (target Rrs), capreomycin (target Rrs and TylA), and linezolid (target RplC). Their mechanisms of action have been previously reviewed by Tiwari and Ryoo.6,19 However, a large number of putative targets have been revealed with no inhibitors being identified.20 In addition, effort has been mostly focused on screening large synthetic chemical libraries, omitting stereochemically and structurally diverse NPs.21–26 Several reviews have been published to date on anti-TB NPs,27,28 however the molecular targets and modes of action have not been discussed. There is an unmet need to further investigate NPs with well-defined mechanisms of action.
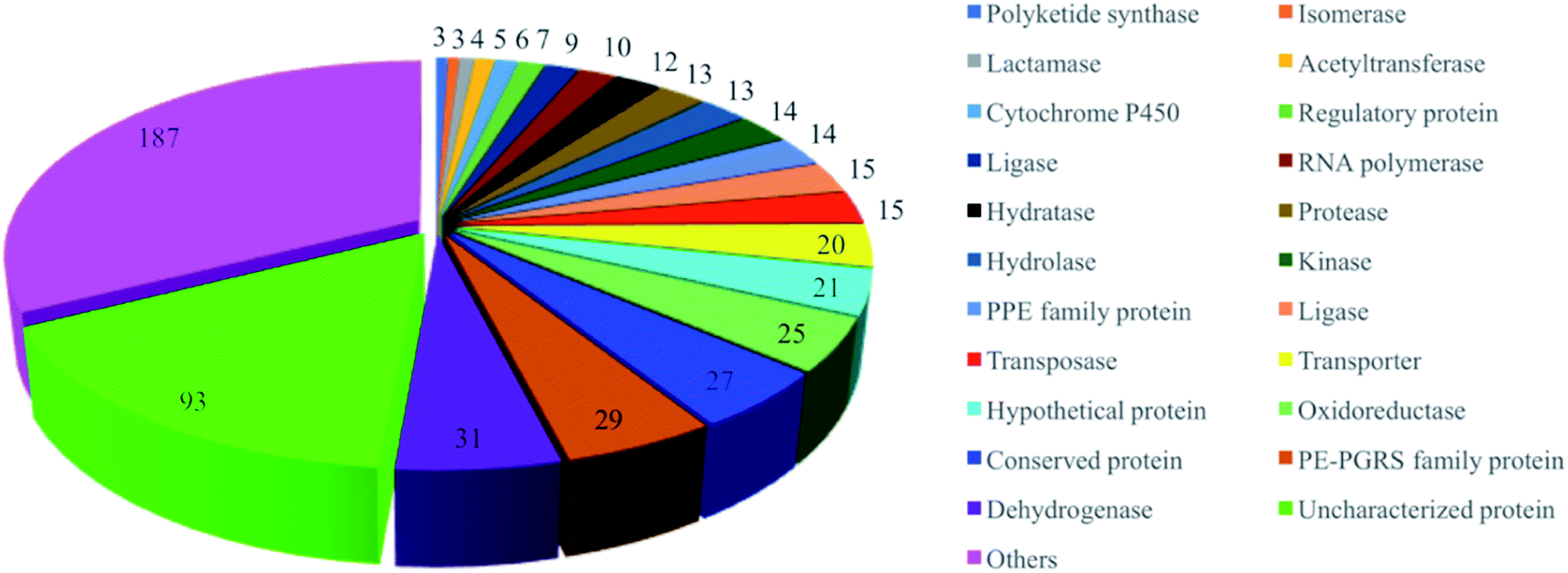 | ||
| Fig. 2 Gene-family distribution of the potential molecular targets of M. tuberculosis. | ||
A literature search in Google Scholar and PubMed with an end date of June, 2020 resulted in 147 NPs with activities against mycobacterial enzymes (Table S3†). Further data analysis revealed that 53 NPs possessed cellular antimycobacterial activity with defined molecular targets (Table 1). In this review, we summarized these 53 active NPs based on their molecular targets in cell wall biosynthesis, protein synthesis, nucleic acid synthesis, energy production, and other emerging targets.29,30
| No | Compound | Originated organism | Target | Enzyme inhibition IC50/KD/Ki (μM) | Growth inhibition MIC/IC50 (μM) |
|---|---|---|---|---|---|
| a MIC: minimum inhibitory concentration; IC50: half maximal inhibitory concentration; NA: not available; NPs showing activity against drug-resistant TB are indicated in bold. | |||||
| 1 | Cycloserine31,33,34 | Streptomyces sp. | Alr and Ddl | 14–900 | 97.9–195.9 |
| 2 | 3-Methoxynordomesticine35 | Ocotea macrophylla | MurE | 67 | 187.5 |
| 3 | N-Methoxycarbonyl-3-methoxynordomesticine35 | Ocotea macrophylla | MurE | 75 | 320.5 |
| 4 | Sansanmycin analogues36 | Streptomyces sp. | MurX | 0.016 | 0.037 |
| 5 | Teixobactin37 | Eleftheria terrae | Lipid II, lipid III | NA | 0.10 |
| 6 | 3-O-Methyl-butylgallate38 | Loranthus micranthus | DesA3 | NA | 6.25 |
| 7 | Cerulenin39 | Cephalosporium caerulens | KasA, KasB | NA | 13.4–28.0 |
| 8 | Platensimycin40 | Streptomyces platensis | KasA, KasB | 2, 4.2 | 27.2 |
| 9 | Thiolactomycin 41,42 | Nocardia sp. | KasA, KasB | 226 | 118.9 |
| 10 | Butein43 | Rhus verniciflua | Dehydratase | NA | 157 |
| 11 | Isoliquirtigenin43 | Dalbergia odorifera | Dehydratase | NA | 195 |
| 12 | 2,2′,4′-Trihydroxychalcone43 | Dalbergia odorifera | Dehydratase | NA | 214 |
| 13 | Fisetin43 | Rhus cotinus | Dehydratase | NA | 220 |
| 14 | Pseudopyronine B44 | Pseudomonas sp. F92S91 | InhA | 12.9 | 2.65–5.30 |
| 15 | Pyridomycin 45 | Dactylosporangium fulvum | InhA | 6.5 | 0.72 |
| 16 | Micrococcin P1 (ref. 46) | Staphylococcus equorum | RplK | NA | 0.032–0.063 |
| 17 | Fusidic acid47 | Fusidium coccineum | FusA1, FusA2 | NA | 1.0 |
| 18 | Trichoderin A48 | Trichoderma sp. | ATP synthase | NA | 0.10 |
| 19 | Aurachin RE49 | Rhodococcus erythropolis | MenA | NA | 63.2 |
| 20 | Novobiocin 50,51 | Streptomyces niveus | GyrB | 0.82 | 6.53 |
| 21 | Griselimycin52 | Streptomyces sp. | DnaN | 1 × 10−4 | 0.90 |
| 22 | Doxorubicin53 | Streptomyces sp. | DnaG | 7.7 | 5 |
| 23 | Daunorubicin53 | Streptomyces sp. | DnaG | 7.2 | 1.25 |
| 24 | Aloe emodin53 | Frangula alnus | DnaG | 19 | >320 |
| 25 | Lipiarmycin A3 (ref. 54) | Catellatospora sp. | RNAP | 0.3 | 0.014 |
| 26 | Gladiolin 55 | Burkholderia gladioli | RNAP | 32 | 0.51 |
| 27 | Kanglemycin A 56 | Amycolatopsis DEM3035 | RNAP | 0.044 | 0.34 |
| 28 | (S)-(−)-Acidomycin 57 | Streptomyces sp. | BioB | 1 | 0.096–6.2 |
| 29 | Ophiobolin K58 | Emericella variecolor | Biofilm | 4.1 | 33 |
| 30 | Rufomycin NBZ1 (ref. 59) | Streptomyces atratus | ClpC1 | 0.5 | 0.25 |
| 31 | Rufomycin NBZ2 (ref. 59) | Streptomyces atratus | ClpC1 | 1.3 | 0.44 |
| 32 | Rufomycin NBZ3 (ref. 59) | Streptomyces atratus | ClpC1 | 1.1 | 0.84 |
| 33 | Rufomycin NBZ4 (ref. 59) | Streptomyces atratus | ClpC1 | 5.7 | >10 |
| 34 | Rufomycin NBZ5 (ref. 59) | Streptomyces atratus | ClpC1 | 0.45 | 0.11 |
| 35 | Rufomycin NBZ6 (ref. 59) | Streptomyces atratus | ClpC1 | 1.5 | 0.57 |
| 36 | Rufomycin NBZ7 (ref. 59) | Streptomyces atratus | ClpC1 | 0.28 | 0.1 |
| 37 | Rufomycin NBZ8 (ref. 59) | Streptomyces atratus | ClpC1 | 0.05 | 0.03 |
| 38 | Ecumicin 60,61 | Nonomuraea sp. | ClpC1 | 0.6 | <0.12–0.19 |
| 39 | Lassomycin 62 | Lentzea kentuckyensis sp. | ClpC1 | 0.41 | 0.41–0.83 |
| 40 | Cyclomarin A 63 | Streptomycete CNB-982 | ClpC1 | 0.1 | 2.5 |
| 41 | Erythromycin64 | Streptomyces erythreus | MABP-1 | 285 | 34.1 |
| 42 | Manzamine A65 | Acanthostrongylophora sp. | MtSK | 120 | 2.7 |
| 43 | Hydroxymanzamine A65 | Acanthostrongylophora sp. | MtSK | 11 | 1.6 |
| 44 | Manzamine E65 | Acanthostrongylophora sp. | MtSK | 63 | 6.7 |
| 45 | Manzamine F65 | Acanthostrongylophora sp. | MtSK | 40 | 4.5 |
| 46 | 6-Deoxymanzamine X65 | Acanthostrongylophora sp. | MtSK | 21 | 3.2 |
| 47 | Clorobiocin66 | Streptomyces sp. | SerB2 | 16.84 | 2.34 |
| 48 | Betulinic acid67 | Betula sp. | Serine protease | NA | 183.1 |
| 49 | Plumbagin68 | Plumbago indica | ThyX | 9 | 21.3 |
| 50 | Agelasine D69 | Agelas nakamurai | BCG3185c | 2.42 | 3.69 |
| 51 | Melophlin A70 | Melophlus sp. | BCG1083, BCG1321c | NA | 2.28–71.7 |
| 52 | Ursolic acid71 | Ocimum sanctum | Mycolic acid | NA | 21.9 |
| 53 | Altholactone72 | Polyalthia sp. | Rv1466 | 42 | 64 |
1.1. Cell wall synthesis
Mtb has a unique cell wall and envelope structure, consisting of three distinct layers of arabinogalactan (AG), mycolic acid (MA), and peptidoglycan (PG). This complex structure is indispensable in supporting cell growth, virulence and most importantly providing a barrier to antibiotics. The assembly of this structure requires numerous catalytic enzymes, some of which have served as attractive targets for TB drug discovery. Of the 53 NPs, 15 show inhibitory activities against Mtb cell wall enzymes, as summarized in Fig. 3 and structures in Fig. 4.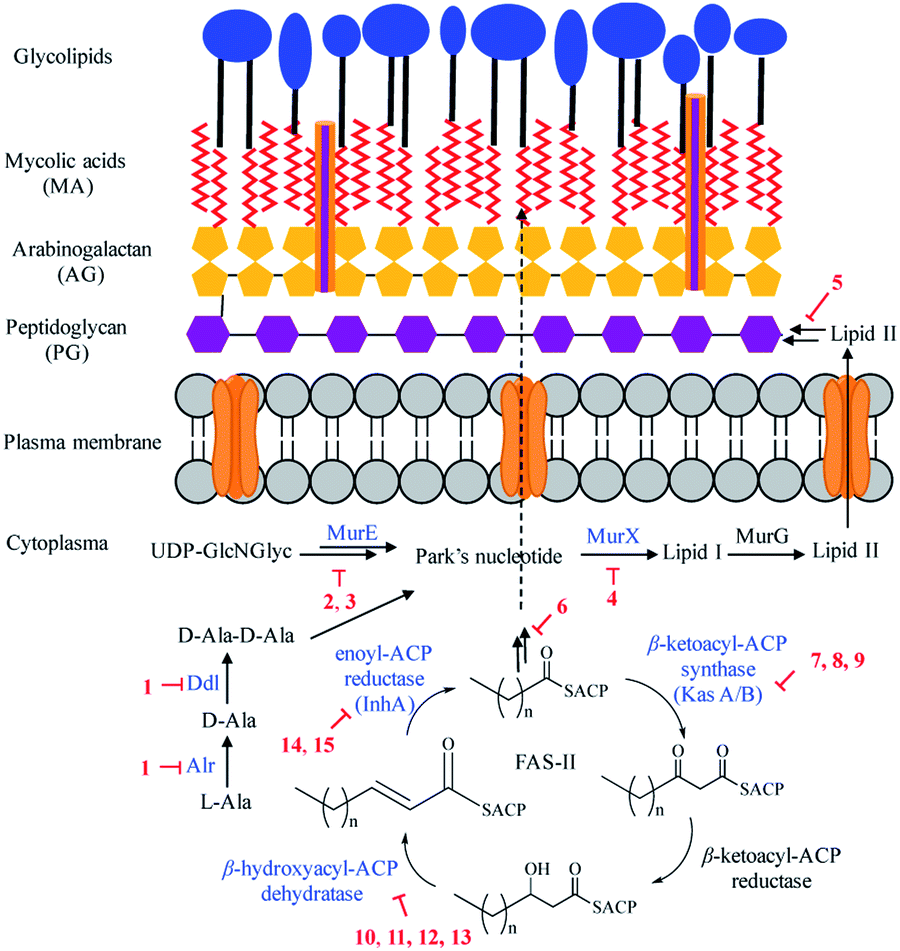 | ||
| Fig. 3 Natural product inhibitors targeting Mtb cell wall. | ||
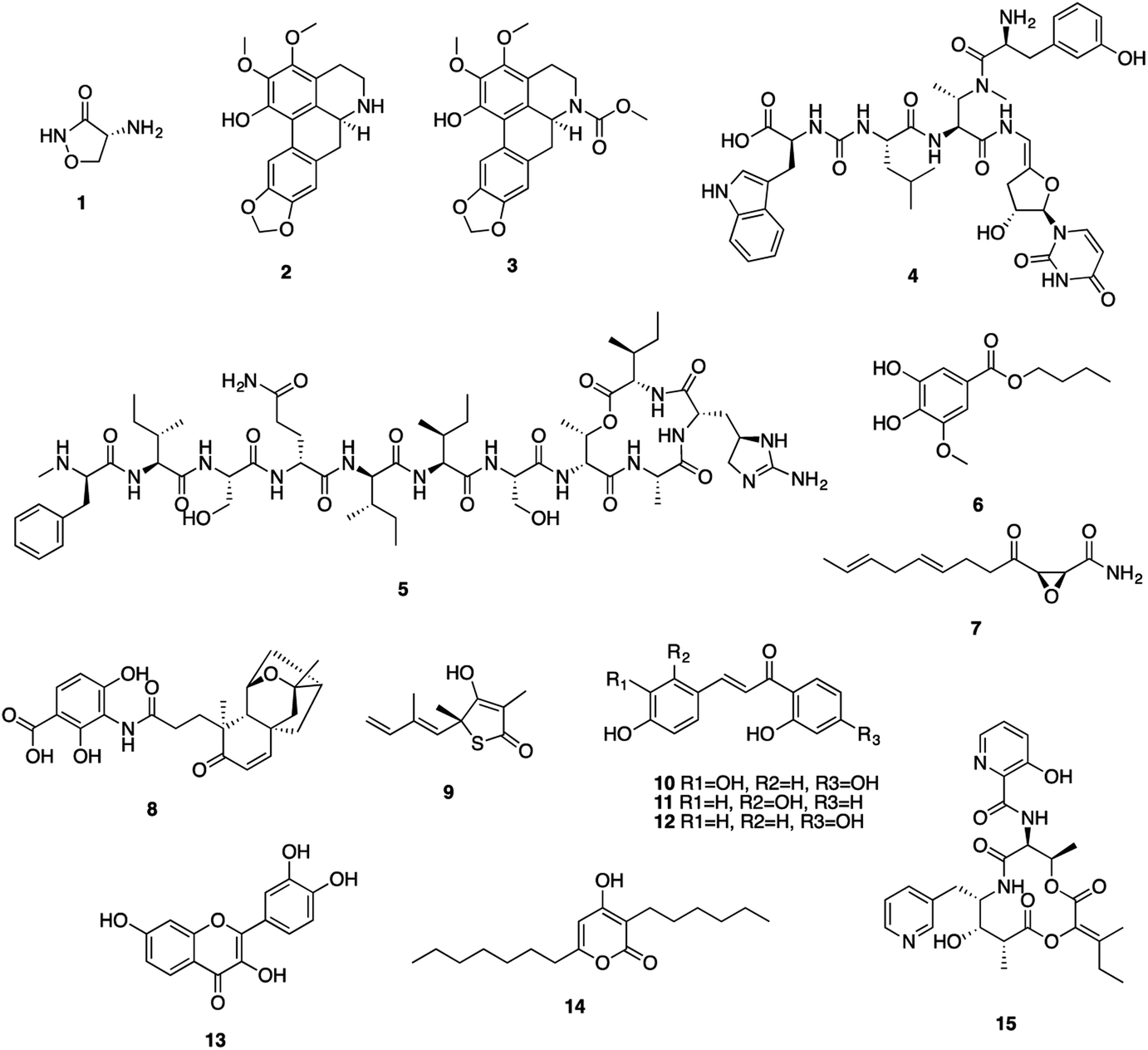 | ||
| Fig. 4 Natural product inhibitors targeting Mtb cell wall synthesis. | ||
Peptidoglycan (PG), a complex polymer composed of long glycan chains cross-linked via short peptides, is an essential component of Mtb cell wall. L-Alanine racemase (Alr) and D-alanyl-D-alanine ligase (Ddl) are involved in PG synthesis. Cycloserine (1), a cyclic analogue of D-alanine, was initially isolated from Streptomyces in the 1950s, and is currently used to treat MDR-TB.31 Cycloserine competitively inhibits Ddl and blocks the connectivity of two D-alanine residues, leading to the inhibition of the PG synthesis.32
ATP-dependent MurE ligase and MurX translocase are two crucial enzymes responsible for the biosynthesis of lipid I, a key intermediate in Mtb PG synthesis (Fig. 3). 3-Methoxynordomesticine (2) and N-methoxycarbonyl-3-methoxynordomesticine (3), isolated from Colombian plants, showed activity against Mtb H37Rv with MICs of 187.5 and 320.5 μM, respectively. The in vitro inhibition on overexpressed and purified recombinant Mtb protein MurE confirmed these aporphine alkaloids as the first natural product inhibitors of MurE ligase, providing an initial hit for exploring related chemical space.35 As a promising new TB drug lead, sansanmycin B (4) possesses antimycobacterial activity with a MIC value of 9.5 μM. The mechanism of action study revealed that the sansanmycin analogues potently inhibited the enzyme MurX.36 Teixobactin (5), discovered in a screen of uncultured bacteria, was isolated from a new species of the β-proteobacteria Eleftheria terrae in 2015.37 It showed excellent activity against M. tuberculosis H37Rv (MIC: 0.1 μM). Teixobactin was also found to inhibit the synthesis of cell wall by binding to a highly-conserved motif of lipid II and lipid III. It is the first member of a new class of lipid II binding antibiotics.37 DesA3 (Rv3229c) is a membrane-bound stearoyl CoA desaturase that reacts with the oxidoreductase Rv3230c to produce oleic acid.73 3-O-Methyl-butylgallate (6), a natural product isolated from Loranthus micranthus was found to be the first specific inhibitor of Mtb DesA3.38 Compound 6 showed specific inhibition against Mtb with a MIC90 (minimum inhibitory concentration required to reduce bacterial growth by 90% relative to controls) value of 6.25 μM. Observation of Mtb XDR strains resistant to 3-O-methyl-butylgallate indicated that this compound might only be used for limited clinical treatment of drug-sensitive Mtb strains. Nevertheless, combination of rifampicin or isoniazid with 3-O-methyl-butylgallate interestingly caused a strong synergistic killing effect, indicating an alternative TB therapy with potentially shortened treatment duration.
Mycolic acids (MA) are one of the most distinctive features of the Mtb cell wall and diverse enzymes are involved in the formation of MA. The biosynthesis of MA starts with the de novo synthesis and elongation of FAs catalysed by the Mtb FAS-I and FAS-II synthases. FAS-II catalyses short-chain acyl CoA from FAS-I by β-ketoacyl-ACP synthase (KasA/B). The newly formed β-ketoacyl-ACP is reduced by β-ketoacyl-ACP reductase to form β-hydroxyl-acyl-ACP. This product is then dehydrated by β-hydroxyacyl-ACP dehydratase, followed by further reduction by an enoyl-ACP reductase (InhA) to complete the FAS-II cycle (Fig. 3).74 Cerulenin (7), an antibiotic isolated from Cephalosporium caerulens in the 1960s, was originally found as an antifungal antibiotic.75 Cerulenin also inhibited the growth of a variety of mycobacteria species including multi-drug resistant strains of M. tuberculosis, with MICs ranging from 6.7 to 56.0 μM.39 Effects of cerulenin on lipid synthesis suggest that cerulenin targets the β-ketoacyl-ACP synthase of FAS system.39,76 Platensimycin (8), a secondary metabolite of S. platensis, showed activity against M. smegmatis and Mtb with MIC values of 31.7 and 27.2 μM, respectively. In vitro assays using purified protein demonstrated that platensimycin inhibited Mtb KasA and KasB.40 Thiolactomycin (9), a unique thiolactone isolated from Nocardia sp. was also reported to target Mtb KasA and KasB.41 The crystal structures of Mtb KasA revealed an intricate binding mode of thiolactomycin. Two methyl groups are positioned in two hydrophobic pockets and isoprenoid moiety points toward an extended lipophilic pocket.77 The flavonoids butein (10), isoliquirtigenin (11), 2,2′,4′-trihydroxychalcone (12) and fisetin (13) inhibited the growth of M. bovis BCG with MIC99 (MIC required to inhibit the growth of 99% of tested cultures) values ranging from 157 μM to 220 μM.43 Bioinformatic analysis and overexpression of Rv0636 in M. smegmatis suggest Rv0636, the unidentified β-hydroxyacyl-ACP dehydratase, as the potential target.43 Pseudopyronines were isolated from Pseudomonas sp. and showed broad biological activity.44,78 Pseudopyronines A and B (14) displayed strong activity against M. tuberculosis H37Rv with MIC values of 11.7 and 2.65 μM, respectively. Pseudopyronine B also showed in vitro activity against fatty acid biosynthesis enzyme InhA with IC50 value of 12.9 μM. Pseudopyronine A showed 35% InhA inhibitory activity at 10 μM.44 Pyridomycin (15) showed specific activity against mycobacteria.79 It was isolated from a Streptomyces species in the 1950s and Dactylosporangium in the 1980s. InhA was identified as the principal target by whole-genome sequencing of pyridomycin-resistant mutants and genetic validation.45 Intriguingly, no cross resistance was observed between pyridomycin and isoniazid, providing promising avenues to develop pyridomycin as treatment of isoniazid-resistant tuberculosis.45 Pyridomycin competitively inhibited NADH and blocked both the NADH cofactor- and lipid substrate-binding pockets of InhA. The crystal structures showed that pyridomycin occupied the InhA active site and hijacked the interactions between InhA and NADH.80
1.2. Protein synthesis
Inhibitors targeting protein synthesis are another mainstay in the treatment of Mtb infections. Thiopeptide antibiotics, with their complex molecular architectures, are potent inhibitors of protein synthesis, as exemplified by micrococcin P1 (16) (Fig. 5).81 Biochemical experiments revealed that this antibiotic inhibited ribosomal protein synthesis by interacting with the L11 binding domain (RplK) of the 23S ribosomal RNA, validating RplK as a drug target. Micrococcin P1 exhibited very strong antimycobacterial activity with MIC of 0.032–0.063 μM,46 while its precise structure remained undefined until mid-2009.82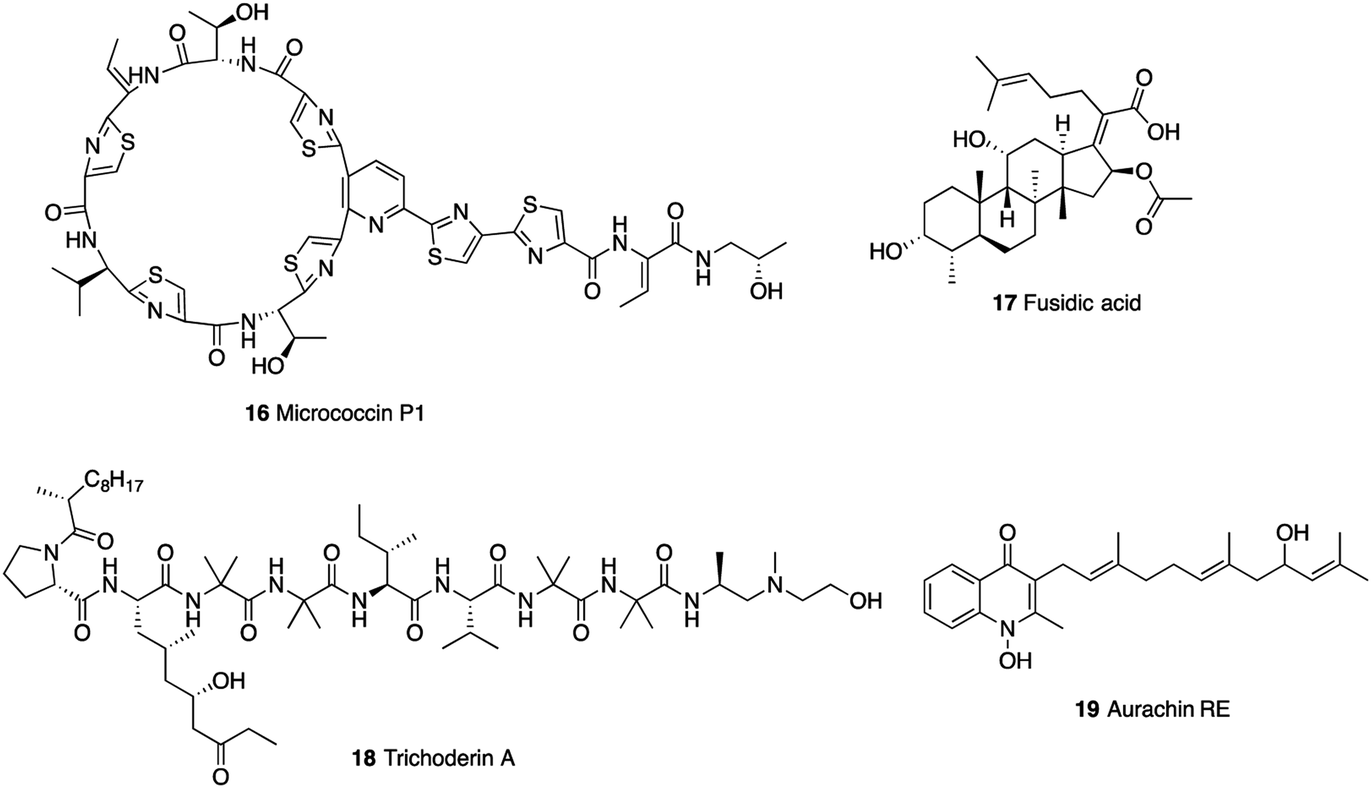 | ||
| Fig. 5 Natural product inhibitors targeting Mtb protein synthesis and energy production. | ||
The elongation factor G (EF-G) FusA1, and FusA2 play important roles in the efficient synthesis of proteins.47 Fusidic acid (17) was shown to target FusA1 and FusA2 and be effective in vitro against Mtb (MIC = 15.5–40.6 μM).83
1.3. Energy production
Energy metabolism in Mtb has emerged as a novel target pathway in anti-TB drug discovery since the approval of the ATP synthase inhibitor bedaquiline. Trichoderin A (18), a novel aminolipopeptide isolated from a marine sponge-derived Trichoderma sp., was found to target mitochondrial ATP synthase (Fig. 6).48 This was verified by the inhibitory effect of trichoderin A on the ATP contents of M. bovis BCG.48 Trichoderin A displayed activity against M. smegmatis (MIC = 0.086 μM), M. bovis BCG (MIC = 0.017 μM), and Mtb H37Rv (MIC = 0.10 μM) under standard aerobic growth conditions as well as dormancy-inducing hypoxic conditions.84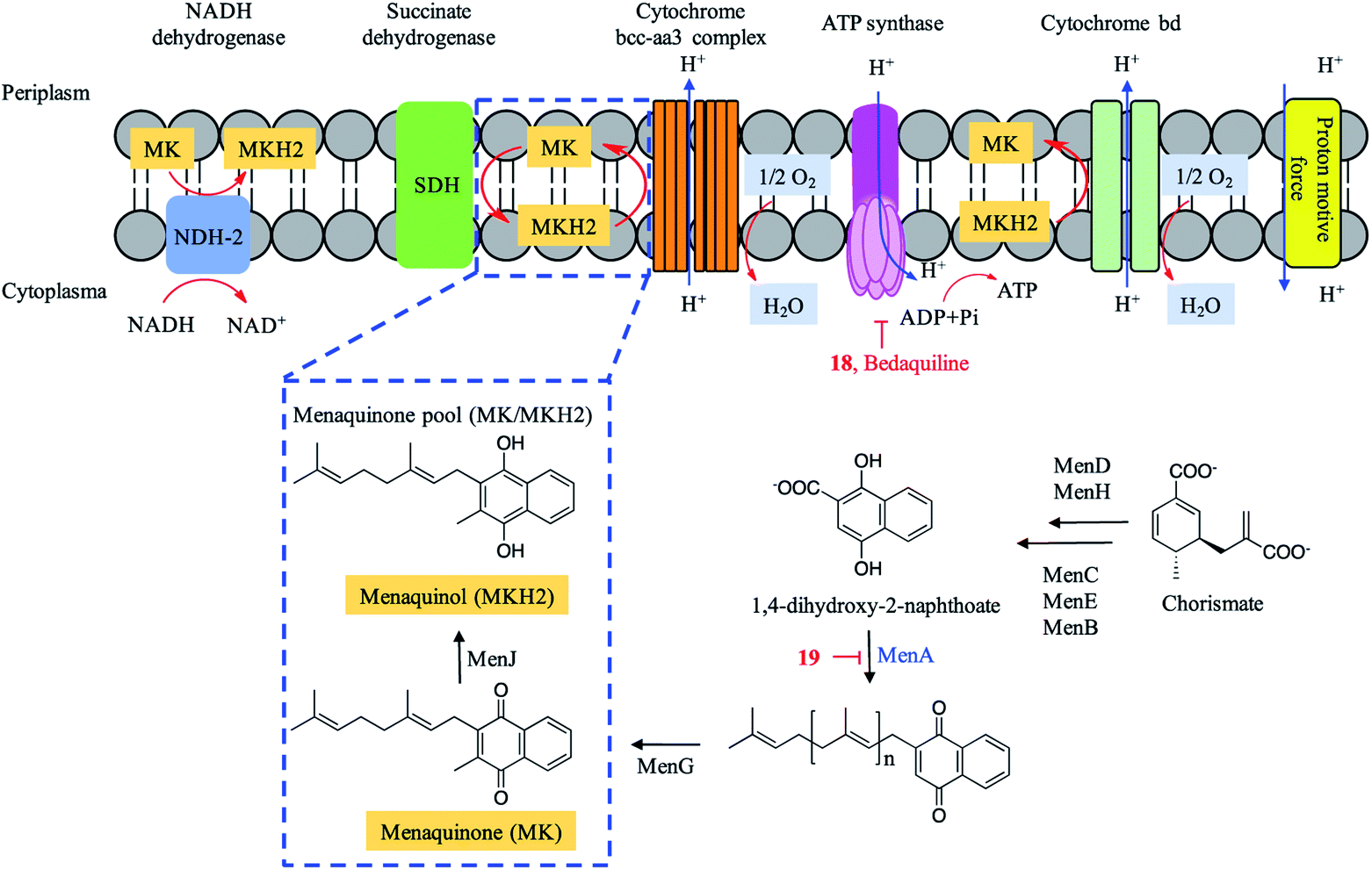 | ||
| Fig. 6 M. tuberculosis electron transport chain (ETC) and biosynthesis of menaquinone (MK). | ||
Menaquinone transfers two electrons in a process of either aerobic or anaerobic respiration and plays an important role in the electron transport chain (ETC) of Mtb. The menaquinone pool (MK/MKH2) is involved in the inflow of electrons and maintenance of the proton motive force, resulting in the synthesis of ATP (Fig. 6). Aurachin RE (19), a new quinolone natural product isolated from Rhodococcus erythropolis JCM 6824, was found to specifically target 1,4-dihydroxy-2-naphthoate prenyltransferase (MenA) in the biosynthesis of menaquinone (MK) and possess mycobacterial electron transport inhibitory activities. Aurachin RE exhibited a wide antimicrobial spectrum including Mtb (MIC = 63.2 μM).49,85 A series of chiral molecules based on MenA inhibitor 19 were synthesized, and revealed as promising menaquinone biosynthesis inhibitors.49
1.4. Nucleic acid synthesis
Enzymes involved in nucleic acid synthesis are potential drug targets as these enzymes are vital for Mtb growth. DNA gyrase is a clinically validated therapeutic target, which is an essential enzyme that catalyses negative supercoiling of DNA and regulates the efficient DNA replication and transcription.50 Novobiocin (20), an aminocoumarin antibiotic isolated from Streptomyces sp., was an effective agent against methicillin-resistant Staphylococcus aureus (MRSA) infections (Fig. 7). However, the clinical use was withdrawn due to the toxicity and side effects.86 Novobiocin showed in vitro inhibitory activity on gyrase of M. smegmatis with maximal non-effective concentration (MNEC, the highest concentration of the inhibitor that failed to show any detectable inhibition of the supercoiling activity) value of 0.10 μM and IC100 (the minimum concentration that produced complete inhibition) value of 0.82 μM.51 Novobiocin competed with ATP for binding to the GyrB subunit, thus inhibiting the ATP-dependent step in the enzyme catalytic cycle.87 It exhibited activity against resistant Mtb strains with MICs ranging from 1.01 to 13.06 μM.50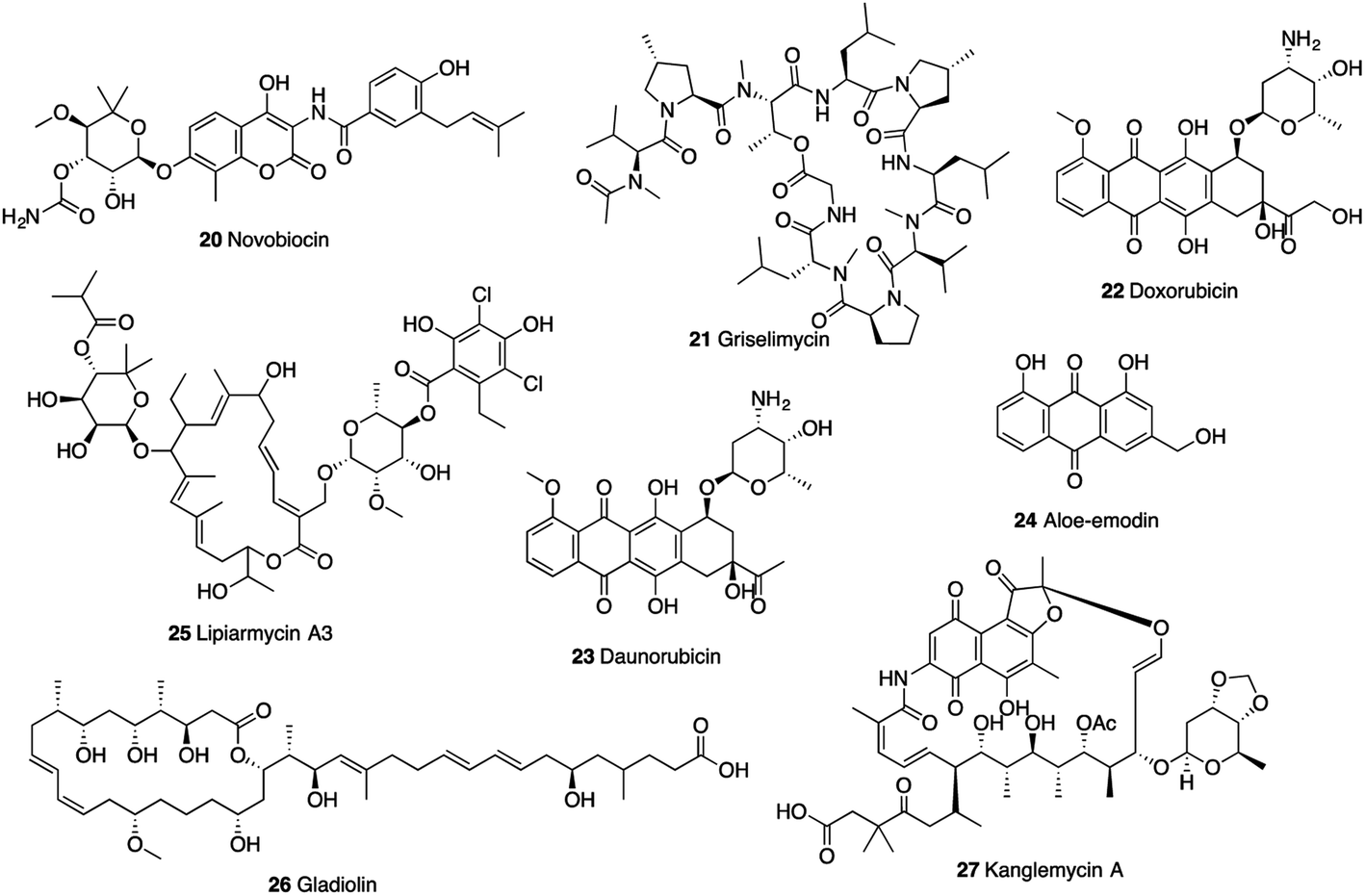 | ||
| Fig. 7 Natural product inhibitors targeting Mtb nucleic acid synthesis. | ||
DNA polymerase sliding clamp (DnaN) is a protein essential for Mtb DNA replication and DNA repair. Griselimycin (21, GM), a cyclic peptide isolated from Streptomyces in the 1960s, was found to target DnaN, which was verified by genome sequence comparisons between the wild-type (WT) parent Mycobacterium strain and mutants.52 Surface plasmon resonance (SPR) revealed high binding affinity of griselimycin to DnaN of M. smegmatis and Mtb (KD: 8.3 × 10−11 M and 1.0 × 10−10 M, respectively).52 It showed specific antimycobacterial activity against Mycobacterium species, including the drug-resistant strains. The MIC values of griselimycin against Mtb H37Rv in broth culture and within macrophage-like (RAW264.7) cells were 0.90 and 5.57 μM, respectively.52
DnaG, a DNA-dependent RNA polymerase, is an attractive potential target for novel antibacterial agent discovery.53 Anthracyclines, doxorubicin (22), daunorubicin (23), and anthranoid aloe-emodin (24), were found to be potent inhibitors of Mtb DnaG (IC50: 7.7, 7.2, and 19 μM, respectively). Doxorubicin and daunorubicin also displayed potent growth inhibition on Mtb, with MIC values of 5.0 and 1.25 μM, respectively.53
The transcription process of RNA polymerase (RNAP) is a crucial point to control gene expression and a preferred target for antimycobacterial chemotherapy. Lipiarmycin A3 (25), a macro-cyclic antibiotic isolated from Catellatospora sp., gladiolin (26), a novel macrolide antibiotic isolated from Burkholderia gladioli, and kanglemycin A (27), an ansamycin natural product isolated from Nocardia sp. were identified to target RNAP.54–56 Lipiarmycin A3 (25) exhibited excellent bactericidal activity against MDR-Mtb strains with MIC values less than 0.014 μM and potently inhibited RNAP from genus Mycobacterium with IC50 value of 0.3 μM.54,88 It was found that lipiarmycin A3 had no cross-resistance to rifampicin (also targeting RNAP), which indicated that lipiarmycin A3 could serve as a good starting point for novel transcription inhibitors.54 The cryo-EM structure of Mtb RNAP-lipiarmycin A3 complex showed that lipiarmycin A3 bound at the base of the RNAP clamp in a fully extended conformation, leading to the induction or stabilization of an open-clamp conformation.88 Gladiolin (26) and kanglemycin A (27) showed mycobacterial inhibitory activity against Mtb H37Rv strain with MIC values of 0.51 μM and 0.34 μM, respectively.55,56 Concentration-dependent inhibition of M. smegmatis RNAP by gladiolin (26) and kanglemycin A (27) occurred with IC50 values of 32 μM and 0.044 μM, respectively. The crystal structure showed that kanglemycin A binds to RNAP at the rifampicin-binding pocket with an altered binding conformation due to two large ansa-bridge substituents. The sugar side chain in the molecule increased the interaction surface and helped maintain a high affinity in a broad context of rifampicin resistance.56
1.5. Emerging targets
Biotin serves as an essential cofactor of enzymes (pyruvate carboxylase and acyl-CoA carboxylase) involved in cellular metabolism, influencing cell growth.89 Mechanism of action studies revealed that (S)-(−)-acidomycin (28) (Fig. 8), originally isolated from a Streptomyces sp., targeted the biotin biosynthetic pathway and competitively inhibited Mtb biotin synthase (BioB).57 Acidomycin possesses excellent antitubercular activity against a series of drug susceptible and drug-resistant Mtb strains (MICs: 0.096–6.2 μM).57 (S)-(−)-Acidomycin can stimulate the cleavage of S-adenosyl-L-methionine (SAM) in BioB to generate toxic 5′-deoxyadenosine. Cellular accumulation studies demonstrated that acidomycin selectively accumulates in the intracellular environment in Mtb.57 Acidomycin lacked in vivo activity in mice because of poor PK properties. However, BioB is a promising target.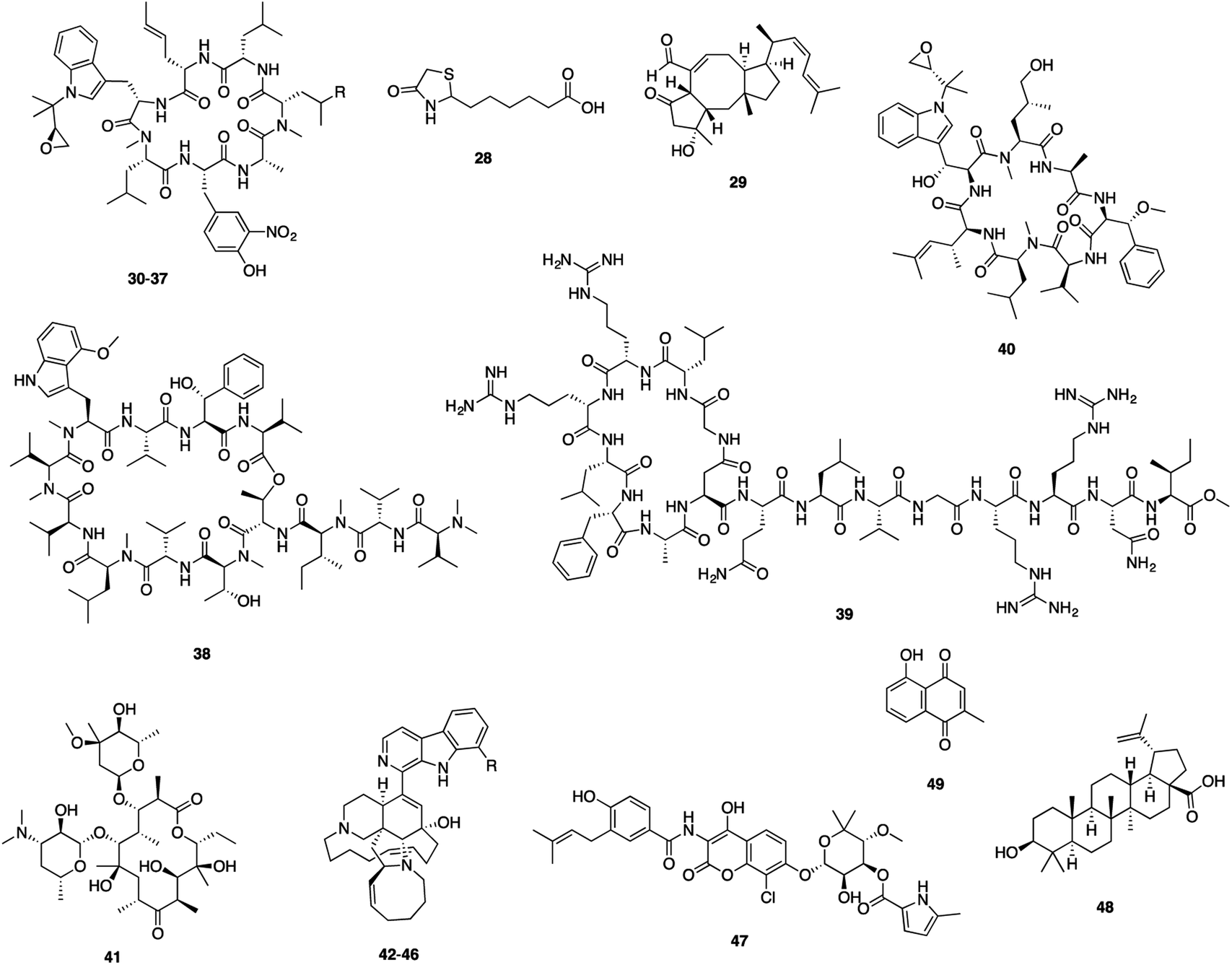 | ||
| Fig. 8 Natural product inhibitors targeting Mtb emerging targets. | ||
Formation of biofilms in bacteria defends them from antibiotics, and are associated with antibiotic tolerance.90,91 Effective control of biofilm formation requires development of therapeutic agents that target the biofilm phenotype or prevent the formation of biofilms.92 Ophiobolin K (29), one sesterterpenoid isolated from marine-derived Emericella variecolor, showed anti-biofilm formation activity against M. smegmatis and M. bovis BCG with MIC values of 4.1 and 8.2 μM, respectively and antimycobacterial activities with MIC values of 33 and 64 μM, respectively.58 However, the molecular target of these compounds is still unclear. The identification of the target would advance the discovery of novel inhibitors with potential pharmaceutical use.58
Caseinolytic protein C1 (ClpC1) showing an inherent ATPase activity is important for protein homeostasis, and the clpC1 gene was identified as essential for Mtb cell growth.93,94 Cyclic peptides rufomycins (RUFs, 30–37), ecumicin (ECUs, 38), lassomycin (39) and cyclomarin A (40) have been found to target ClpC. Rufomycins (syn. ilamycins), a family of cyclic heptapeptides, exhibited excellent antimycobacterial activity, as exemplified by ilamycins E1 and E2 identified from S. atratus (MIC: 0.0098 μM) and rufomycins NBZ1–NBZ8 from S. atratus (MICs from 0.03 μM to >10 μM).59,95 The binding affinity of rufomycins with the N-terminal domain (NTD) of mycobacterial ClpC1 were assessed by using surface plasmon resonance (SPR) with KD values ranging from 0.06 μM to 1.6 μM.59 The X-ray structure of the ClpC1-NTD-RUFI complex revealed the epoxide moiety of RUFI opened and covalently bound to ClpC1-NTD via the sulfur atom of Met1.94 Ecumicin (38), a cyclic tridecapeptide, was isolated from Nonomuraea sp.60 Ecumicin retains selective activity against MDR and XDR Mtb strains (MIC: 0.12–0.62 μM) and was nontoxic for a mammalian cell line (selectivity index >640). Ecumicin also inhibited growth of Mtb in the mouse model of acute TB infection.61 The structure of ClpC1-ecumicin complex revealed an unexpected binding mode with a unique 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (target:ligand) stoichiometry, leading to a significant conformational change of the N-terminus. Structurally similar analogues had decreased binding, indicating that N-terminal interactions were important for the binding of ECU to ClpC1.96
:2 (target:ligand) stoichiometry, leading to a significant conformational change of the N-terminus. Structurally similar analogues had decreased binding, indicating that N-terminal interactions were important for the binding of ECU to ClpC1.96
Lassomycin (39), a ribosomally encoded cyclic peptide, was isolated from Lentzea kentuckyensis. This molecule showed potent activity against a variety of Mtb strains, including drug-resistant isolates (MIC: 0.41–1.65 μM).62 Cyclomarin A (40), a cyclic heptapeptide isolated from a marine Streptomyces sp. under saline culture conditions proved to be bactericidal against Mtb by targeting ClpC1.63,97,98 Cyclomarin-induced restriction of ClpC1 dynamics can modulate the chaperone enzymatic activity leading eventually to cell death.99 The findings demonstrated the significance of Mtb ClpC1 as a potential drug target and the potential for designing ClpC1 inhibitors using macrocyclic peptides as a scaffold.
Rv3197, a non-canonical ABC protein, is a factor responsible for erythromycin (41) resistance and was identified as the first macrolide antibiotic binding protein (MABP-1) in M. tuberculosis.64 The crystal structure revealed that MABP-1 has a preference for 14-membered ring macrolides including erythromycin and confers inducible resistance to macrolides.64 Erythromycin showed weak antimycobacterial activity against Mtb (MIC: 128 μM).100 Structural modifications of erythromycin and SAR studies increased the in vitro anti-TB activity and gave significant dose-dependent inhibition of Mtb growth in mice.100,101
Mtb shikimate kinase (MtSK), the fifth enzyme in the shikimate pathway, catalyzes the conversion of shikimate into shikimate-3-phosphate (S3P) using ATP as the co-substrate. MtSK represents a favorable target for the development of inhibitors specific to the Mtb, since its function is essential for the survival of Mtb and there is no mammalian counterpart.102,103 During screening a library of 26 marine-derived alkaloids against MtSK, manzamine A (42), 8-hydroxymanzamine A (43), manzamine E (44), manzamine F (45), and 6-deoxymanzamine X (46) isolated from an Indo-Pacific sponge Acanthostrongylophora sp. were found to be non-competitive inhibitors of MtSK. All compounds displayed a strong activity against M. tuberculosis H37Rv (MIC50: 1.6–6.7 μM). A study on the kinetic profiling indicated manzamines as promising candidates for further studies.65 This unique class of manzamine alkaloids also exhibit a diverse range of bioactivities including cytotoxic, insecticidal, antibacterial, antiviral, and antimalarial activity.104–107
The serine production pathway is considered essential for bacterial growth, and Mtb phosphoserine phosphatase (SerB2) is involved in conversion of O-phospho-L-serine to L-serine.108L-Serine biosynthesis is an attractive and unexplored target, indicating that targeting the Mtb serine pathway is a promising approach for new antitubercular compound discovery.66,108 Clorobiocin (47) was reported to inhibit SerB2 (IC50: 16.84 μM) as well as mycobacterial growth (MIC99: 2.34 μM) with no toxicity against THP-1 cells.66 Further validation of SerB2 as a drug target should be addressed and could lead to identification of scaffolds with a novel mechanism.66
The first biochemical and pathological analysis of Mtb serine protease (Rv3194c) determined that Rv3194c promotes the persistence of M. smegmatis in the lung and induces lung lesions. Screening of Rv3194c inhibitors showed that betulinic acid (48) had significant protease inhibitory activity on Rv3194c.67 Betulinic acid (BA) is a pentacyclic triterpenoid and has been shown to exhibit a broad range of bioactivities.109 BA also displayed antimycobacterial activity against Mtb H37Ra with IC50 value of 183.1 μM.110 Rv3194c may serve as a novel target for anti-TB drug discovery. BA derivatives have been shown to exhibit a variety of biological activities including anti-HIV-1, antibacterial, antimalarial, and anthelmintic properties.111–114
Mtb thymidylate synthase (ThyX), responsible for the synthesis of dTMP from dUMP is considered as an attractive target as it is absent in the humans.115 Enzyme inhibition assay revealed the non-competitive inhibition of plumbagin (49) on ThyX (IC50: 3.3 μM).68 Plumbagin derived from Plumbaginaceae family can inhibit Mtb growth, including drug-resistant Mtb strains (MIC: 1.33–42.55 μM).116
2. Discussion, conclusion and outlook
In the post-genomic era, multi-omics techniques have enabled the identification of many potential drug targets, and it is vital for discovery and further optimization of lead compounds against TB. In this review, we summarized NP inhibitors of mycobacterial enzymes, and particularly focused on the compounds with in vitro antimycobacterial activity.Mechanisms of action study of 53 NPs revealed a wide range of molecular targets for TB drug discovery. They include the enzymes involved in Mtb cell wall synthesis: L-alanine racemase (Alr), D-alanyl-D-alanine ligase (Ddl), stearoyl CoA desaturase (DesA3), and ATP-dependent Mur ligases MurE and MurX; enzymes for protein synthesis represented by L11 binding domain of the 23S ribosomal RNA (RplK) and elongation factor G (EF-G) FusA1 and FusA2; enzymes in energy production such as ATP synthase and 1,4-dihydroxy-2-naphthoate prenyltransferase (MenA); and a series of enzymes on nucleic acid synthesis such as DNA gyrase (GyrB), DNA polymerase sliding clamp (DnaN), DNA-dependent RNA polymerase (DnaG), and RNA polymerase (RNAP). Some emerging targets are also revealed, including biotin synthase (BioB), caseinolytic protein C1 (ClpC1), macrolide antibiotic binding protein (MABP-1), shikimate kinase (MtSK), phosphoserine phosphatase (SerB2), serine protease (Rv3194c), and thymidylate synthase (ThyX). The majority of the 53 NPs showed activity against the enzymes, they also exhibited inhibitory activity in cells, indicating these enzymes are promising molecular targets for TB drug discovery.
Of these 53 NPs more than half (30 compounds, 57%) were isolated from bacteria, especially Streptomyces (Fig. 9A). Other sources include plants (12 compounds, 23%), marine sponges (7 compounds, 13%), and fungi (4 compounds, 7%). In terms of activity, bacteria-derived compounds showed stronger antimycobacterial activity (<0.1 μM, 7, 23.3%; 0.1–1 μM, 11, 36.7%) than plant- and sponge-derived compounds (<0.1 μM, 0; 0.1–1 μM, 0) (Fig. 9B). To further evaluate the drug-likeness of these compounds, their physicochemical property was calculated using Instant JChem (version 18.8.0) against Lipinski's rule of five (RO5) parameters.117 log P values of all compounds revealed 76.9% had a log P value less than 5. Overall, 17 compounds have no violation against Lipinski's RO5 (32.7%), and 12 compounds have one violation (23.1%) (Fig. 10). A large proportion of peptides possess large molecular weight, which is consistent with the recent analysis of oral drugs and clinical candidates.118,119
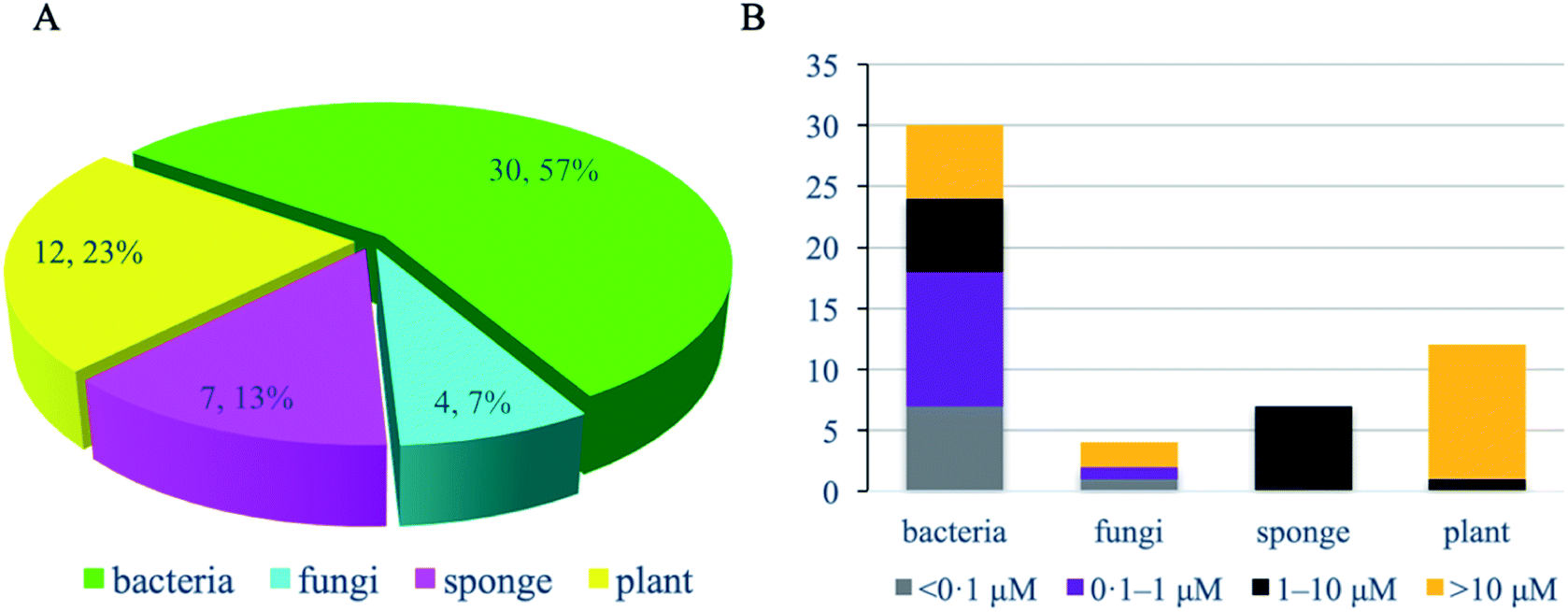 | ||
| Fig. 9 Antitubercular natural product inhibitors of mycobacterial enzymes. Source category of antitubercular natural product inhibitors of mycobacterial enzymes. (A) MIC category (whole cell-based assay) of antitubercular natural product inhibitors of mycobacterial enzymes by source (B). | ||
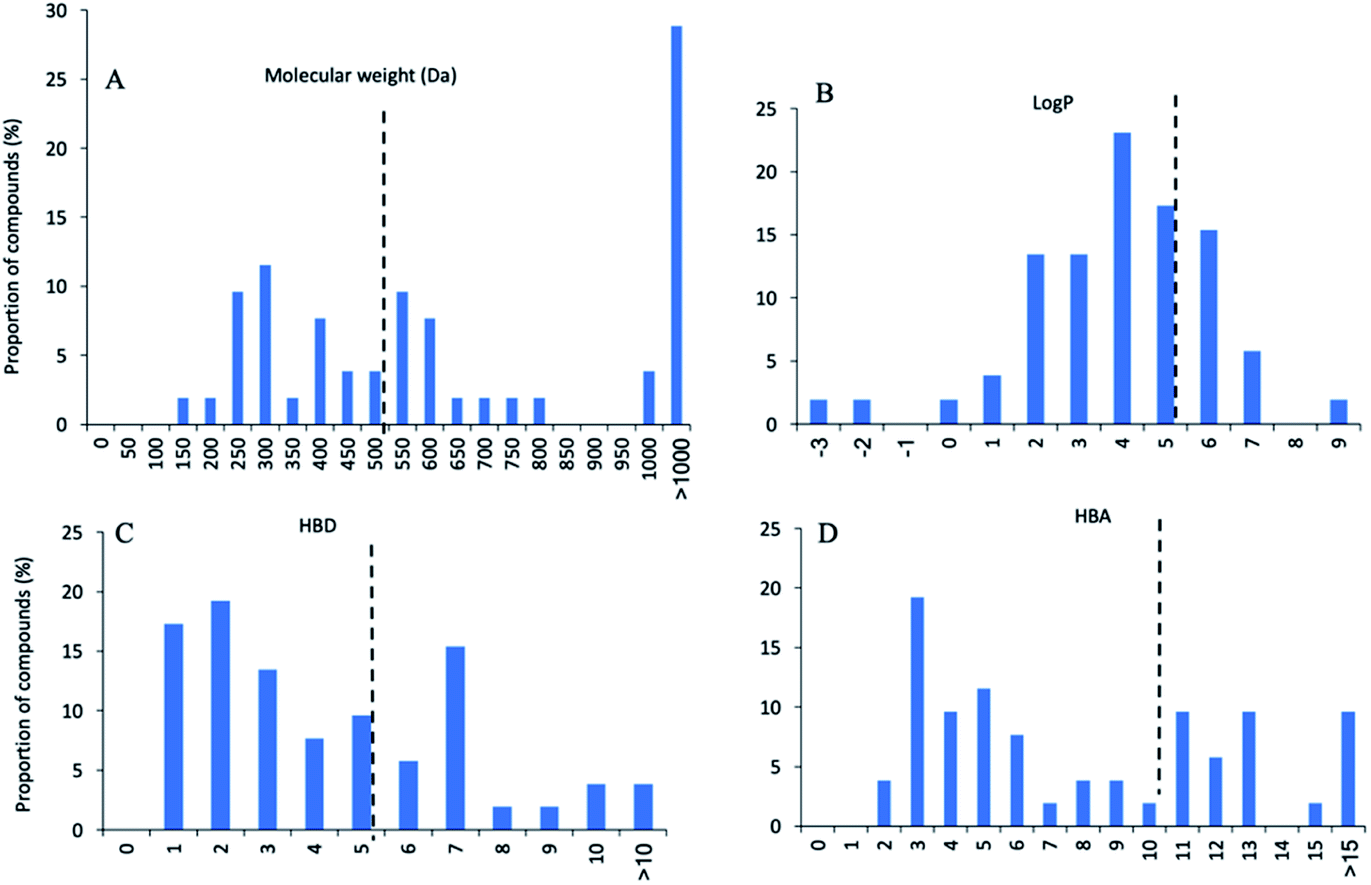 | ||
| Fig. 10 Physicochemical property histograms of 53 natural product inhibitors of mycobacterial enzymes. (A) Molecular weight (MW), (B) calculated log P, (C) hydrogen bond donors (HBD), (D) hydrogen bond acceptors (HBA). | ||
The compounds in the review can be divided into four structural classes: peptide, polyketide, terpenoid, and alkaloid. Peptides ares the largest group with more than 20 compounds. The antimycobacterial activity of peptides are more potent compared to polyketides, terpenoids, and alkaloids (0.03–6.53 μM, 0.014–320.5 μM, 1–183.1 μM, and 1.6–6.7 μM, respectively). Encouragingly, one-fifth of compounds showed activity against drug-resistant Mtb strains, including thiolactomycin (9), pyridomycin (15), novobiocin (20), lipiarmycin A3 (25), gladiolin (26), and kanglemycin A (27), (S)-(−)-acidomycin (28), ecumicin (38), lassomycin (39), and cyclomarin A (40), most of which are cyclic peptides. Among these compounds, lassomycin (39) and cyclomarin A (40) also showed activity against dormant mycobacteria. These compounds could be expected to become important weapons in the therapeutical arsenal against drug-resistant and latent Mtb infection.
The above highlighted compounds with activity against drug-resistant strains and dormant mycobacteria were all isolated from bacteria, and the biosynthesis of most compounds has been elucidated (Table 2), offering the ability to supply larger amounts for further study through large scale fermentation experiments. The biosynthetic genes also provide a template to mine more related compounds with better druggability and activity against drug-resistant Mtb strains.
Finding new anti-TB drugs and shortening the duration of treatment have been and continue to be a challenge with growing resistance to Mtb. Expanding druggable targets and validating the targets of active compounds are crucial steps for TB drug discovery. The anti-TB NPs described in this review have demonstrated excellent in vitro activity and mycobacterial enzyme activity. Several NPs showed strong activity on novel targets, such as (S)-(−)-acidomycin (28) on MtBioB, trichoderin A (18) on ATP synthase, aurachin RE (19) on 1,4-dihydroxy-2-naphthoate prenyltransferase. Development of these compounds into new drug leads requires follow-up structural optimization and pharmacokinetic studies.
Future anti-TB drug discovery, to develop new regimens, should have a focus on molecules inhibiting novel targets and resistant Mtb. These molecules will potentially be more effective to address TB treatment challenges. The PhenoTarget approach has shown great potential in identifying active natural products and their targets. The approach starts with phenotypic screening of fractions followed by molecular target screening, as demonstrated by the discovery of altholactone (53).72,126 With developments in structural biology (102 crystal structures for M. tuberculosis H37Rv in PDB) and in bioinformatics, computer-assisted virtual docking becomes accessible to screen the vast chemical space for specific targets, especially the novel and emerging targets, such as MtBioB and ATP synthase. The hits identified by virtual screening can then be experimentally validated by PhenoTarget screening. Many techniques, such as imaging, have been developed for further validation the in vivo activities and drug target interaction. This will ensure an improve clinical success.127
3. Conflicts of interest
There are no conflicts to declare.4. Acknowledgements
Jianying Han would like to acknowledge Griffith University for the provision of the PhD scholarships (GUPRS and GUIPRS).5. References
- WHO, https://www.who.int/en/, accessed Apr. 16, 2020.
- WHO, Global tuberculosis report 2019, World Health Organization, 2019 Search PubMed.
- W. Moreira, G. J. Ngan, J. L. Low, A. Poulsen, B. C. Chia, M. J. Ang, A. Yap, J. Fulwood, U. Lakshmanan and J. Lim, mBio, 2015, 6, e00253-15 CrossRef.
- C. Lienhardt, M. Raviglione, M. Spigelman, R. Hafner, E. Jaramillo, M. Hoelscher, A. Zumla and J. Gheuens, J. Infect. Dis., 2012, 205, S241–S249 CrossRef PubMed.
- R. Shi, N. Itagaki and I. Sugawara, Mini-Rev. Med. Chem., 2007, 7, 1177–1185 CrossRef CAS.
- S. K. Mishra, G. Tripathi, N. Kishore, R. K. Singh, A. Singh and V. K. Tiwari, Eur. J. Med. Chem., 2017, 137, 504–544 CrossRef CAS.
- H. N. Jnawali and S. Ryoo, Tuberculosis-Current Issues in Diagnosis and Management, 2013, pp, 163–180 Search PubMed.
- K. Quissell and G. Walt, Health Policy and Planning, 2016, 31, i17–i32 CrossRef PubMed.
- C. Nathan, Nat. Rev. Microbiol., 2015, 13, 651–657 CrossRef CAS PubMed.
- L. A. Mitscher and W. Baker, Med. Res. Rev., 1998, 18, 363–374 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- D. Quan, G. Nagalingam, R. Payne and J. A. Triccas, Int. J. Infect. Dis., 2017, 56, 212–220 CrossRef CAS PubMed.
- M. Dong, B. Pfeiffer and K.-H. Altmann, Drug Discovery Today, 2017, 22, 585–591 CrossRef CAS PubMed.
- L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS PubMed.
- J. W.-H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 CrossRef CAS.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29–40 CrossRef CAS PubMed.
- I. Smith, Clin. Microbiol. Rev., 2003, 16, 463–496 CrossRef CAS PubMed.
- H. N. Jnawali and S. Ryoo, Tuberc.: Curr. Issues Diagn. Manage., 2013, 20, 163–180 Search PubMed.
- https://www.ssgcid.org/, accessed Aug. 24, 2020.
- J. B. Billones, M. C. O. Carrillo, V. G. Organo, S. J. Y. Macalino, J. B. A. Sy, I. A. Emnacen, N. A. B. Clavio and G. P. Concepcion, Drug Des., Dev. Ther., 2016, 10, 1147 CrossRef CAS.
- J. Herrmann, J. Rybniker and R. Müller, Curr. Opin. Biotechnol., 2017, 48, 94–101 CrossRef CAS PubMed.
- A. Kumar, A. Casey, J. Odingo, E. A. Kesicki, G. Abrahams, M. Vieth, T. Masquelin, V. Mizrahi, P. A. Hipskind and D. R. Sherman, PLoS One, 2013, 8, e72786 CrossRef CAS PubMed.
- S. Galandrin, V. Guillet, R. S. Rane, M. Léger, N. Radha, N. Eynard, K. Das, T. S. Balganesh, L. Mourey and M. Daffé, J. Biomol. Screening, 2013, 18, 576–587 CrossRef.
- Y. Lin, N. Zhu, Y. Han, J. Jiang and S. Si, J. Antibiot., 2014, 67, 671–676 CrossRef CAS PubMed.
- M. Yoshida, Biosci., Biotechnol., Biochem., 2019, 83, 1–9 CrossRef PubMed.
- A. García, V. Bocanegra-García, J. P. Palma-Nicolás and G. Rivera, Eur. J. Med. Chem., 2012, 49, 1–23 CrossRef.
- B. Lechartier, J. Rybniker, A. Zumla and S. T. Cole, EMBO Mol. Med., 2014, 6, 158–168 CrossRef CAS PubMed.
- S. Wellington and D. T. Hung, ACS Infect. Dis., 2018, 4, 696–714 CrossRef CAS.
- Y. B. Lorraine, C. V. Sundeep, L. B. Tom and M. Grace, Drug Discovery Dev., 2019, 49–68 Search PubMed.
- Z. Feng and R. G. Barletta, Antimicrob. Agents Chemother., 2003, 47, 283–291 CrossRef CAS.
- J. B. Bruning, A. C. Murillo, O. Chacon, R. G. Barletta and J. C. Sacchettini, Antimicrob. Agents Chemother., 2011, 55, 291–301 CrossRef CAS PubMed.
- J. Chen, S. Zhang, P. Cui, W. Shi, W. Zhang and Y. Zhang, J. Antimicrob. Chemother., 2017, 72, 3272–3276 CrossRef CAS.
- S. Halouska, R. J. Fenton, D. K. Zinniel, D. D. Marshall, R. l. G. Barletta and R. Powers, J. Proteome Res., 2014, 13, 1065–1076 CrossRef CAS PubMed.
- J. D. Guzman, A. Gupta, D. Evangelopoulos, C. Basavannacharya, L. C. Pabon, E. A. Plazas, D. R. Munoz, W. A. Delgado, L. E. Cuca and W. Ribon, J. Antimicrob. Chemother., 2010, 65, 2101–2107 CrossRef CAS PubMed.
- A. T. Tran, E. E. Watson, V. Pujari, T. Conroy, L. J. Dowman, A. M. Giltrap, A. Pang, W. R. Wong, R. G. Linington and S. Mahapatra, Nat. Commun., 2017, 8, 1–9 CrossRef.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes and S. Epstein, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- N. Rehberg, E. Omeje, S. S. Ebada, L. van Geelen, Z. Liu, P. Sureechatchayan, M. U. Kassack, T. R. Ioerger, P. Proksch and R. Kalscheuer, Antimicrob. Agents Chemother., 2019, 63, e00136-19 CrossRef PubMed.
- N. M. Parrish, F. P. Kuhajda, H. S. Heine, W. R. Bishai and J. D. Dick, J. Antimicrob. Chemother., 1999, 43, 219–226 CrossRef CAS.
- A. K. Brown, R. C. Taylor, A. Bhatt, K. Fütterer and G. S. Besra, PLoS One, 2009, 4, e6306 CrossRef PubMed.
- L. Kremer, J. D. Douglas, A. R. Baulard, C. Morehouse, M. R. Guy, D. Alland, L. G. Dover, J. H. Lakey, W. R. Jacobs and P. J. Brennan, J. Biol. Chem., 2000, 275, 16857–16864 CrossRef CAS.
- K. Kapilashrami, G. R. Bommineni, C. A. Machutta, P. Kim, C.-T. Lai, C. Simmerling, F. Picart and P. J. Tonge, J. Biol. Chem., 2013, 288, 6045–6052 CrossRef CAS.
- A. K. Brown, A. Papaemmanouil, V. Bhowruth, A. Bhatt, L. G. Dover and G. S. Besra, Microbiology, 2007, 153, 3314–3322 CrossRef CAS.
- A. C. Giddens, L. Nielsen, H. I. Boshoff, D. Tasdemir, R. Perozzo, M. Kaiser, F. Wang, J. C. Sacchettini and B. R. Copp, Tetrahedron, 2008, 64, 1242–1249 CrossRef CAS.
- R. C. Hartkoorn, C. Sala, J. Neres, F. Pojer, S. Magnet, R. Mukherjee, S. Uplekar, S. Boy-Röttger, K. H. Altmann and S. T. Cole, EMBO Mol. Med., 2012, 4, 1032–1042 CrossRef CAS PubMed.
- G. Degiacomi, Y. Personne, G. Mondésert, X. Ge, C. S. Mandava, R. C. Hartkoorn, F. Boldrin, P. Goel, K. Peisker and A. Benjak, Tuberculosis, 2016, 100, 95–101 CrossRef CAS.
- G. Ramakrishnan, N. R. Chandra and N. Srinivasan, Mol. BioSyst., 2015, 11, 3316–3331 RSC.
- P. Pruksakorn, M. Arai, L. Liu, P. Moodley, W. R. Jacobs Jr and M. Kobayashi, Biol. Pharm. Bull., 2011, 34, 1287–1290 CrossRef CAS.
- J. Debnath, S. Siricilla, B. Wan, D. C. Crick, A. J. Lenaerts, S. G. Franzblau and M. Kurosu, J. Med. Chem., 2012, 55, 3739–3755 CrossRef CAS.
- S. Chopra, K. Matsuyama, T. Tran, J. P. Malerich, B. Wan, S. G. Franzblau, S. Lun, H. Guo, M. C. Maiga and W. R. Bishai, J. Antimicrob. Chemother., 2012, 67, 415–421 CrossRef CAS.
- M. Chatterji, S. Unniraman, S. Mahadevan and V. Nagaraja, J. Antimicrob. Chemother., 2001, 48, 479–485 CrossRef CAS.
- A. Kling, P. Lukat, D. V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. C. Wenzel and C. König, Science, 2015, 348, 1106–1112 CrossRef CAS PubMed.
- C. Gajadeera, M. J. Willby, K. D. Green, P. Shaul, M. Fridman, S. Garneau-Tsodikova, J. E. Posey and O. V. Tsodikov, J. Antibiot., 2015, 68, 153–157 CrossRef CAS.
- M. Kurabachew, S. H. Lu, P. Krastel, E. K. Schmitt, B. L. Suresh, A. Goh, J. E. Knox, N. L. Ma, J. Jiricek and D. Beer, J. Antimicrob. Chemother., 2008, 62, 713–719 CrossRef CAS.
- L. Song, M. Jenner, J. Masschelein, C. Jones, M. J. Bull, S. R. Harris, R. C. Hartkoorn, A. Vocat, I. Romero-Canelon and P. Coupland, J. Am. Chem. Soc., 2017, 139, 7974–7981 CrossRef CAS PubMed.
- H. Mosaei, V. Molodtsov, B. Kepplinger, J. Harbottle, C. W. Moon, R. E. Jeeves, L. Ceccaroni, Y. Shin, S. Morton-Laing and E. C. L. Marrs, Mol. Cell, 2018, 72, 263–274 CrossRef CAS.
- M. R. Bockman, C. A. Engelhart, J. D. Cramer, M. D. Howe, N. K. Mishra, M. Zimmerman, P. Larson, N. Alvarez-Cabrera, S. W. Park and H. I. Boshoff, ACS Infect. Dis., 2019, 5, 598–617 CrossRef CAS.
- M. Arai, H. Niikawa and M. Kobayashi, J. Nat. Med., 2013, 67, 271–275 CrossRef CAS.
- B. Zhou, G. Shetye, Y. Yu, B. D. Santarsiero, L. L. Klein, C. Abad-Zapatero, N. M. Wolf, J. Cheng, Y. Jin and H. Lee, J. Nat. Prod., 2020, 83, 657–667 CrossRef CAS.
- W. Gao, J.-Y. Kim, S.-N. Chen, S.-H. Cho, J. Choi, B. U. Jaki, Y.-Y. Jin, D. C. Lankin, J.-E. Lee and S.-Y. Lee, Org. Lett., 2014, 16, 6044–6047 CrossRef CAS.
- W. Gao, J.-Y. Kim, J. R. Anderson, T. Akopian, S. Hong, Y.-Y. Jin, O. Kandror, J.-W. Kim, I.-A. Lee and S.-Y. Lee, Antimicrob. Agents Chemother., 2015, 59, 880–889 CrossRef PubMed.
- E. Gavrish, C. S. Sit, S. Cao, O. Kandror, A. Spoering, A. Peoples, L. Ling, A. Fetterman, D. Hughes and A. Bissell, Chem. Biol., 2014, 21, 509–518 CrossRef CAS PubMed.
- E. K. Schmitt, M. Riwanto, V. Sambandamurthy, S. Roggo, C. Miault, C. Zwingelstein, P. Krastel, C. Noble, D. Beer and S. P. Rao, Angew. Chem., 2011, 123, 6011–6013 CrossRef.
- Q. Zhang, H. Liu, X. Liu, D. Jiang, B. Zhang, H. Tian, C. Yang, L. W. Guddat, H. Yang and K. Mi, Protein & Cell., 2018, 9, 971–975 Search PubMed.
- J. Simithy, N. R. Fuanta, M. Alturki, J. V. Hobrath, A. E. Wahba, I. Pina, J. Rath, M. T. Hamann, J. DeRuiter and D. C. Goodwin, Biochemistry, 2018, 57, 4923–4933 CrossRef CAS PubMed.
- G. Arora, P. Tiwari, R. S. Mandal, A. Gupta, D. Sharma, S. Saha and R. Singh, J. Biol. Chem., 2014, 289, 25149–25165 CrossRef CAS PubMed.
- H. Li, G. Dang, H. Liu, Z. Wang, Z. Cui, N. Song, L. Chen and S. Liu, Tuberculosis, 2019, 119, 101880 CrossRef CAS.
- A. Sarkar, S. Ghosh, R. Shaw, M. M. Patra, F. Calcuttawala, N. Mukherjee and S. K. Das Gupta, PLoS One, 2020, 15, e0228657 CrossRef CAS PubMed.
- M. Arai, Y. Yamano, A. Setiawan and M. Kobayashi, ChemBioChem, 2014, 15, 117–123 CrossRef CAS PubMed.
- M. Arai, Y. Yamano, K. Kamiya, A. Setiawan and M. Kobayashi, J. Nat. Med., 2016, 70, 467–475 CrossRef CAS PubMed.
- M. A. Jyoti, T. Zerin, T.-H. Kim, T.-S. Hwang, W. S. Jang, K.-W. Nam and H.-Y. Song, Pulm. Pharmacol. Ther., 2015, 33, 17–24 CrossRef CAS PubMed.
- A. R. Elnaas, D. Grice, J. Han, Y. Feng, A. D. Capua, T. Mak, J. A. Laureanti, G. W. Buchko, P. J. Myler and G. Cook, Molecules, 2020, 25, 2384 CrossRef CAS PubMed.
- Y. Chang, G. E. Wesenberg, C. A. Bingman and B. G. Fox, J. Bacteriol., 2008, 190, 6686–6696 CrossRef CAS PubMed.
- H. Marrakchi, M.-A. Lanéelle and M. Daffé, Chem. Biol., 2014, 21, 67–85 CrossRef CAS PubMed.
- S. Omura, Bacteriol. Rev., 1976, 40, 681–697 CrossRef CAS.
- A. C. Price, K.-H. Choi, R. J. Heath, Z. Li, S. W. White and C. O. Rock, J. Biol. Chem., 2001, 276, 6551–6559 CrossRef CAS.
- S. R. Luckner, C. A. Machutta, P. J. Tonge and C. Kisker, Structure, 2009, 17, 1004–1013 CrossRef CAS PubMed.
- F. Kong, M. P. Singh and G. T. Carter, J. Nat. Prod., 2005, 68, 920–923 CrossRef CAS.
- K. Maeda, H. Kosaka, Y. Okami and H. Umezawa, J. Antibiot., 1953, 6, 140 CAS.
- R. C. Hartkoorn, F. Pojer, J. A. Read, H. Gingell, J. Neres, O. P. Horlacher, K.-H. Altmann and S. T. Cole, Nat. Chem. Biol., 2014, 10, 96–98 CrossRef CAS.
- R. A. Hughes and C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 7930–7954 CrossRef CAS PubMed.
- M. A. Ciufolini and D. Lefranc, Nat. Prod. Rep., 2010, 27, 330–342 RSC.
- P. De, K. De, D. Veau, F. Bedos-Belval, S. Chassaing and M. Baltas, Expert Opin. Ther. Pat., 2012, 22, 155–168 CrossRef CAS.
- P. Pruksakorn, M. Arai, N. Kotoku, C. Vilchèze, A. D. Baughn, P. Moodley, W. R. Jacobs Jr and M. Kobayashi, Bioorg. Med. Chem. Lett., 2010, 20, 3658–3663 CrossRef CAS PubMed.
- W. Kitagawa and T. Tamura, J. Antibiot., 2008, 61, 680–682 CrossRef CAS.
- M. Brvar, A. Perdih, M. Oblak, L. P. Mašič and T. Solmajer, Bioorg. Med. Chem. Lett., 2010, 20, 958–962 CrossRef CAS PubMed.
- B. J. Bradbury and M. J. Pucci, Curr. Opin. Pharmacol., 2008, 8, 574–581 CrossRef CAS PubMed.
- W. Lin, K. Das, D. Degen, A. Mazumder, D. Duchi, D. Wang, Y. W. Ebright, R. Y. Ebright, E. Sineva and M. Gigliotti, Mol. Cell, 2018, 70, 60–71 CrossRef CAS PubMed.
- W. Salaemae, A. Azhar, G. W. Booker and S. W. Polyak, Protein & Cell., 2011, 2, 691–695 Search PubMed.
- D. López, H. Vlamakis and R. Kolter, Cold Spring Harbor Perspect. Biol., 2010, 2, a000398 Search PubMed.
- A. K. Ojha, A. D. Baughn, D. Sambandan, T. Hsu, X. Trivelli, Y. Guerardel, A. Alahari, L. Kremer, W. R. Jacobs Jr and G. F. Hatfull, Mol. Microbiol., 2008, 69, 164–174 CrossRef CAS.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- N. P. Kar, D. Sikriwal, P. Rath, R. K. Choudhary and J. K. Batra, FEBS J., 2008, 275, 6149–6158 CrossRef CAS.
- N. M. Wolf, H. Lee, M. P. Choules, G. F. Pauli, R. Phansalkar, J. R. Anderson, W. Gao, J. Ren, B. D. Santarsiero and H. Lee, ACS Infect. Dis., 2019, 5, 829–840 CrossRef CAS PubMed.
- J. Ma, H. Huang, Y. Xie, Z. Liu, J. Zhao, C. Zhang, Y. Jia, Y. Zhang, H. Zhang and T. Zhang, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- N. M. Wolf, H. Lee, D. Zagal, J.-W. Nam, D.-C. Oh, J.-W. Suh, G. Pauli, S. Cho and C. Abad-Zapatero, Acta Crystallogr., Sect. D: Struct. Biol., 2020, 76, 458–471 CrossRef CAS.
- M. K. Renner, Y.-C. Shen, X.-C. Cheng, P. R. Jensen, W. Frankmoelle, C. A. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Am. Chem. Soc., 1999, 121, 11273–11276 CrossRef CAS.
- D. Vasudevan, S. P. Rao and C. G. Noble, J. Biol. Chem., 2013, 288, 30883–30891 CrossRef CAS PubMed.
- K. Weinhäupl, M. Brennich, U. Kazmaier, J. Lelievre, L. Ballell, A. Goldberg, P. Schanda and H. Fraga, J. Biol. Chem., 2018, 293, 8379–8393 CrossRef.
- Z. J. Zhu, O. Krasnykh, D. Pan, V. Petukhova, G. Yu, Y. Liu, H. Liu, S. Hong, Y. Wang and B. Wan, Tuberculosis, 2008, 88, S49–S63 CrossRef CAS PubMed.
- K. Falzari, Z. Zhu, D. Pan, H. Liu, P. Hongmanee and S. G. Franzblau, Antimicrob. Agents Chemother., 2005, 49, 1447–1454 CrossRef CAS PubMed.
- J. E. Nunes, M. A. Duque, T. F. de Freitas, L. Galina, L. F. Timmers, C. V. Bizarro, P. Machado, L. A. Basso and R. G. Ducati, Molecules, 2020, 25, 1259 CrossRef CAS.
- R. Bentley and E. Haslam, Crit. Rev. Biochem. Mol. Biol., 1990, 25, 307–384 CrossRef CAS PubMed.
- R. Sakai, T. Higa, C. W. Jefford and G. Bernardinelli, J. Am. Chem. Soc., 1986, 108, 6404–6405 CrossRef CAS.
- R. A. Edrada, P. Proksch, V. Wray, L. Witte, W. Müller and R. W. Van Soest, J. Nat. Prod., 1996, 59, 1056–1060 CrossRef CAS.
- K. K. Ang, M. J. Holmes, T. Higa, M. T. Hamann and U. A. Kara, Antimicrob. Agents Chemother., 2000, 44, 1645–1649 CrossRef CAS PubMed.
- T. Kubota, S.-i. Kurimoto and J. i. Kobayashi, Alkaloids: Chem. Biol., 2020, 84, 1–124 CAS.
- M. Haufroid and J. Wouters, Pharmaceuticals, 2019, 12, 66 CrossRef CAS.
- P. Yogeeswari and D. Sriram, Curr. Med. Chem., 2005, 12, 657–666 CrossRef CAS.
- H. Li, D. Webster, J. A. Johnson and C. A. Gray, J. Ethnopharmacol., 2015, 165, 148–151 CrossRef CAS PubMed.
- C. Aiken and C. H. Chen, Trends Mol. Med., 2005, 11, 31–36 CrossRef CAS.
- W. Schühly, J. Heilmann, I. Calis and O. Sticher, Planta Med., 1999, 65, 740–743 CrossRef PubMed.
- G. Bringmann, W. Saeb, L. A. Assi, G. Francois, A. S. Narayanan, K. Peters and E.-M. Peters, Planta Med., 1997, 63, 255–257 CrossRef CAS.
- N. Enwerem, J. Okogun, C. Wambebe, D. Okorie and P. Akah, Phytomedicine, 2001, 8, 112–114 CrossRef CAS.
- H. Myllykallio, G. Lipowski, D. Leduc, J. Filee, P. Forterre and U. Liebl, Science, 2002, 297, 105–107 CrossRef CAS PubMed.
- D. Dey, R. Ray and B. Hazra, Phytother. Res., 2014, 28, 1014–1021 CrossRef CAS PubMed.
- http://www.chemaxon.com/ .
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- M. D. Shultz, J. Med. Chem., 2018, 62, 1701–1714 CrossRef.
- X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS PubMed.
- T. Huang, Y. Wang, J. Yin, Y. Du, M. Tao, J. Xu, W. Chen, S. Lin and Z. Deng, J. Biol. Chem., 2011, 286, 20648–20657 CrossRef CAS PubMed.
- M. Steffensky, A. Mühlenweg, Z.-X. Wang, S.-M. Li and L. Heide, Antimicrob. Agents Chemother., 2000, 44, 1214–1222 CrossRef CAS.
- J. Peek, M. Lilic, D. Montiel, A. Milshteyn, I. Woodworth, J. B. Biggins, M. A. Ternei, P. Y. Calle, M. Danziger and T. Warrier, Nat. Commun., 2018, 9, 1–15 CrossRef CAS.
- https://mibig.secondarymetabolites.org/, accessed Aug. 12, 2020.
- A. W. Schultz, D.-C. Oh, J. R. Carney, R. T. Williamson, D. W. Udwary, P. R. Jensen, S. J. Gould, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2008, 130, 4507–4516 CrossRef CAS PubMed.
- Y. Xie, Y. Feng, A. Di Capua, T. Mak, G. W. Buchko, P. J. Myler, M. Liu and R. J. Quinn, Mar. Drugs, 2020, 18, 149 CrossRef CAS.
- T. B. Durham and M.-J. Blanco, Bioorg. Med. Chem. Lett., 2015, 25, 998–1008 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1np00011j |
| This journal is © The Royal Society of Chemistry 2022 |