
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient one-pot green synthesis of tetrakis(acetonitrile)copper(I) complex in aqueous media†
Ilya S. Kritchenkov ,
Julia R. Shakirova and
Sergey P. Tunik*
,
Julia R. Shakirova and
Sergey P. Tunik*
St. Petersburg State University, 7/9 Universitetskaya emb., 199034 St. Petersburg, Russia. E-mail: sergey.tunik@spbu.ru
First published on 17th May 2019
Abstract
New simple, fast, effective and environmentally friendly one-pot method for the synthesis of extensively used tetrakis(acetonitrile)copper(I) complexes with BF4−, PF6− and ClO4− counterions is invented and optimized. The approach suggested allows using water as solvent and minimizes amounts of toxic organic reagents in the synthetic protocol.
Introduction
Labile tetrakis–acetonitrile complexes of the [Cu(CH3CN)4]X type, where X = BF4−, PF6−, ClO4− (ref. 1 and 2) are widely used in research laboratories and industrial processes as catalysts in cycloaddition3,4 and Ullmann5 reactions, intramolecular aromatic annulations,6,7 oxidation of primary alcohols8 and many other popular reactions of organic synthesis.9–13 At the same time they serve as important precursors for the synthesis of overwhelming majority of mononuclear14–16 and homo-17 and hetero-polynuclear18,19 copper(I) complexes, which also have numerous practical applications,20–22 one of which is large-scale production of emitting materials for OLED fabrication.23 Total number of references related to these compounds as reagents/reactants exceeds 3100 (over 100 of them during 2018 year), including the papers in journals of highest rank. It is obvious that production of these compounds is of great importance for research and industry and simple, economical and environmentally friendly method of synthesis is of high demand.Complexes of the [Cu(CH3CN)4]X type with bulky counterions are known for many decades since the 1923 when the first representative of this family was isolated by the reduction of silver nitrate with copper powder in acetonitrile.24 Since then the synthetic procedures were modified to exclude expensive Ag(I) salts from the reaction scheme. At present time there are two major ways for the synthesis of this complexes: the first one consists in direct interaction of copper(I) oxide with the corresponding acid in acetonitrile (Scheme 1A),25 and the second in comproportionation of the corresponding CuX2 salt with copper powder or wire in refluxing acetonitrile (Scheme 1B).26
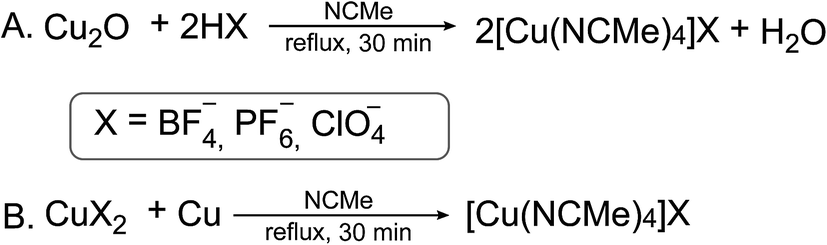 | ||
| Scheme 1 Syntheses of [Cu(CH3CN)4]X described in literature. | ||
These approaches suffer of significant drawbacks such as using of toxic solvent – acetonitrile (A and B) and application of corrosive toxic acids (A) that is not desirable from the safety point of view. Additionally, availability of the corresponding CuX2 salts or HX acids are required for the synthesis of the complexes with desired counterions. Herein we report on the improved procedure for the synthesis of [Cu(CH3CN)4]X (X = BF4−, PF6−, ClO4−) making it cheaper, safer and “greener”.
Results and discussion
In our tactic we combined the methodology of Scheme 1B and basic principles of ion metathesis to develop simple, fast and effective one-pot comproportionation/metathesis method for the synthesis of [Cu(CH3CN)4]X complexes (Scheme 2). As starting materials, we chose the copper(II) sulfate pentahydrate and the corresponding MX metal salts with desired counterions (MX: NaBF4, KPF6, LiClO4) since they are inexpensive, stable and low toxic. As a source of copper(0) we use copper wire instead of powder as it could be easily removed from the reaction mixture that makes product separation technology particularly convenient. All the compounds used in this reaction and the byproduct (M2SO4 salt) are water-soluble that makes possible to carry out the process in water using only a small excess of acetonitrile. At the same time the final products are low-soluble in water that allows isolation of the products as pure solids from the reaction mixture by simple centrifugation with a considerable yields (82–87%).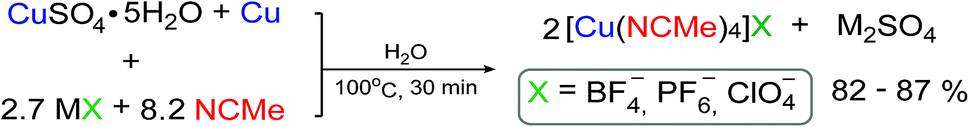 | ||
| Scheme 2 Suggested reaction scheme. | ||
By varying the molar ratio of reagents, time and temperature we found optimal conditions for obtaining highly pure [Cu(CH3CN)4]X (X = BF4−, PF6−, ClO4−) complexes in high yields (>80%). The protocol in brief can be described in the following way (see Experimental for details): copper(II) sulfate pentahydrate, alkali metal salt with bulky anion (2.7 eq.) were placed in a plastic vial followed by addition of water and acetonitrile (8.2 eq.); the salt and MeCN were added in a slight excess. The vial was then charged with copper wire and closed with cap. The reaction mixture was kept at 100 °C in water bath for ca. 30 minutes, showing gradual disappearance of blue color of the starting solution (Fig. 1). The mixture obtained was then cooled to room temperature to give precipitate of white crystalline product, the copper wire was removed from the system and suspension was additionally cooled in fridge to complete crystallization of the product. The precipitate was then centrifuged, decanted, successively washed with water containing ca. 2% of CH3CN (slight amounts of acetonitrile were added at this and the next steps in order to prevent complex degradation), with ethyl acetate–ethanol mixture 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 containing ca. 4% of CH3CN, with ethyl acetate containing ca. 2% of CH3CN and dried.
:1 containing ca. 4% of CH3CN, with ethyl acetate containing ca. 2% of CH3CN and dried.
 | ||
| Fig. 1 Photographs showing the reaction mixture color changes during reaction. | ||
The presence of copper metal, Cu(0), in the reaction mixture prevents disproportionation of the product and/or its oxidation, which are typical detrimental channels of the copper(I) ions degradation in the presence of water. This approach is not new and was previously used by other researchers.27 The reaction products are poorly soluble in water that also shifts the reaction equilibrium toward the desired Cu(I) complex. Nevertheless, solubility varies for different counterions, we suppose it is slightly higher for the perchlorate complex compared to the tertafluoroborate and hexafluorophosphate compounds that result in lower yield of the reaction in the former case.
The products obtained were fully characterized by elemental analysis, X-ray powder diffraction analysis, 1H, 19F NMR and IR spectroscopy, UV-VIS spectroscopy. All these analyses confirm formation of high purity targeted compounds. XRD powder diffraction patterns for the complexes with BF4− and ClO4− counterions are in good agreement with those simulated from the data obtained for single crystal XRD (Fig. 2, S1 and S2†). In the case of PF6− there is some inconsistency that may be ascribed to the presence of acetonitrile molecules in all the structures uploaded to the CCDC, which we used for simulation. Additionally, we synthesized the corresponding complexes with BF4− and PF6− counterions using “classic”25 methodology and compared their powder diffraction patterns with those for complexes synthesized under the framework of the current study (Fig. S1 and S2†).
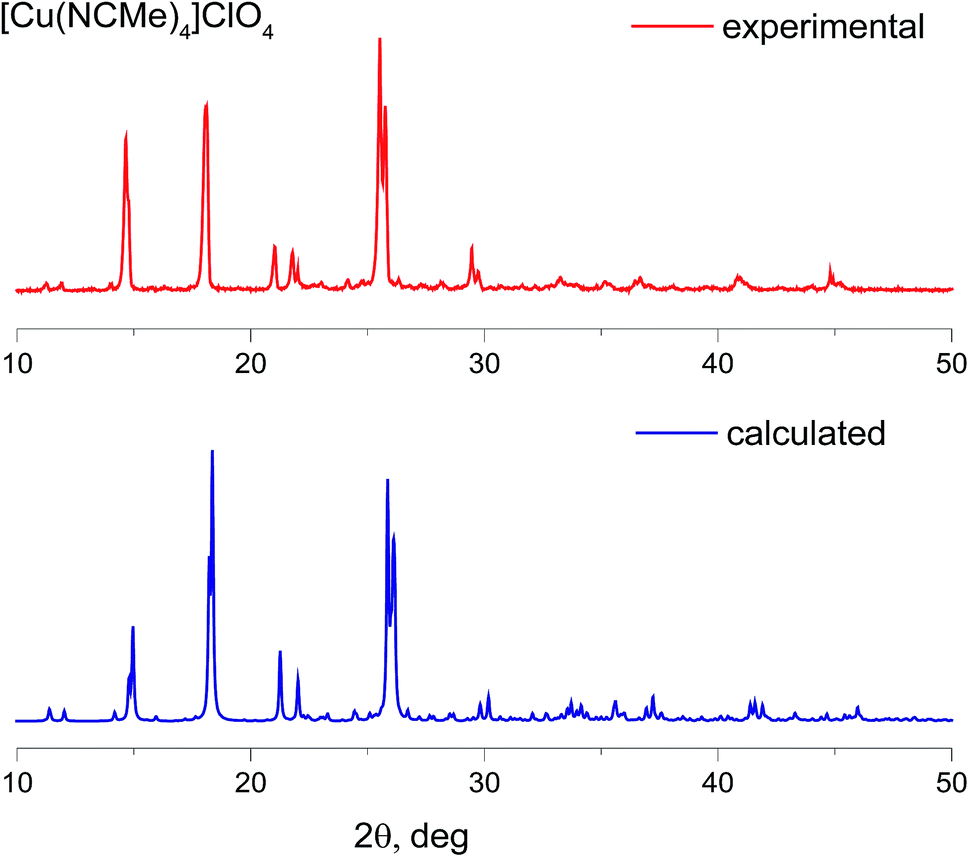 | ||
| Fig. 2 Powder X-ray diffraction patterns of [Cu(CH3CN)4][ClO4]: experimental (red) and calculated from the single-crystal data CCDC 772755 (blue). | ||
The IR spectra recorded in Nujol display the bands characteristic of the coordinated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond1 together with the bands assigned to vibration frequencies of the corresponding counterion (Fig. 3).1,28
N bond1 together with the bands assigned to vibration frequencies of the corresponding counterion (Fig. 3).1,28
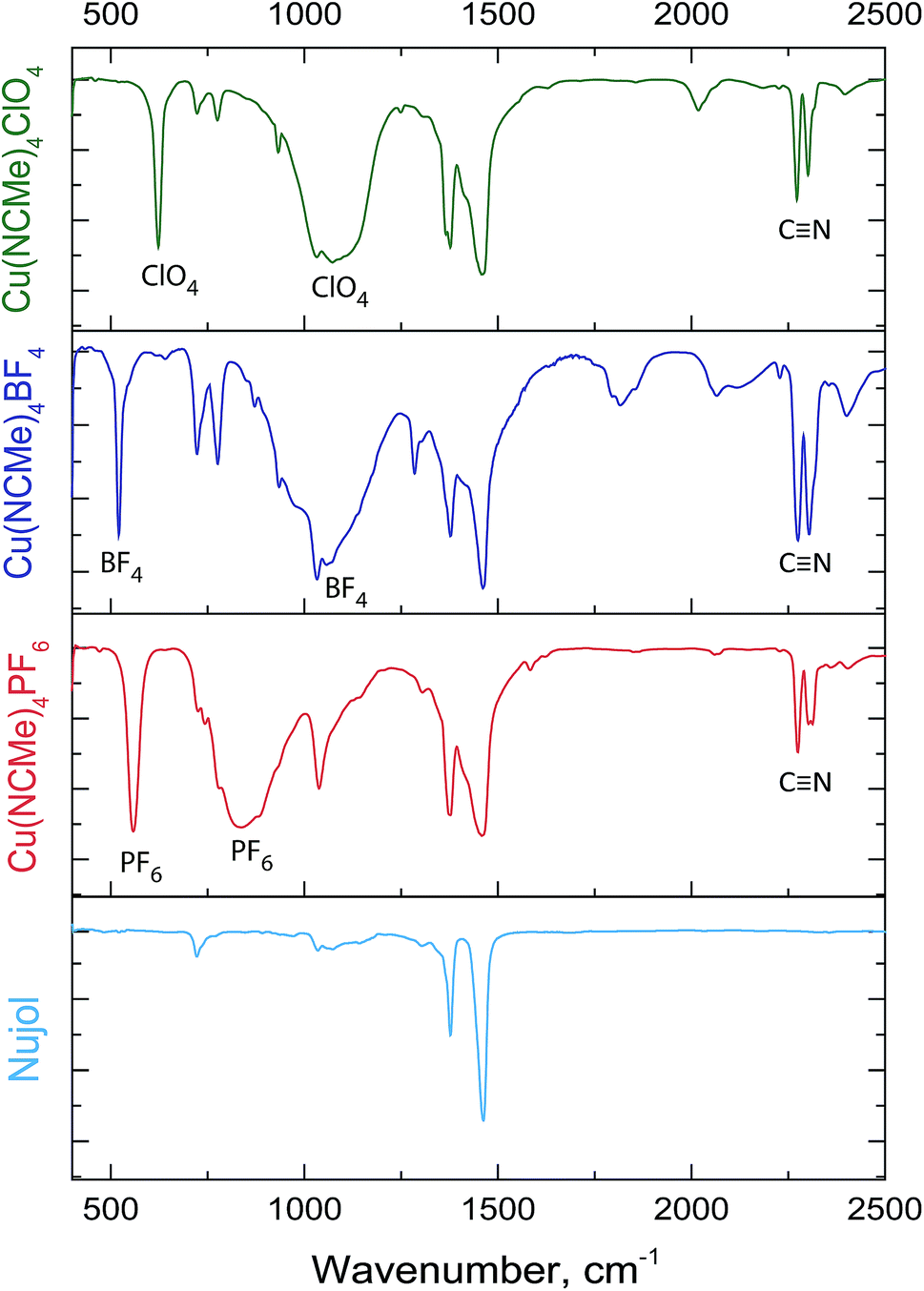 | ||
| Fig. 3 IR spectra of [Cu(NCMe)4]X complexes in Nujol. | ||
To make sure the completion of metathesis of the counterions we examine the 1H and 19F NMR (CDCl3) spectra of the complexes obtained with addition of 2 equivalents of 4-iodobenzotrifluoride (as an internal standard), in all the cases integral intensities of the signals of counterions and p-I–C6H4–CF3 are in good agreement with relative amount of fluorine in these compounds (Fig. 4, S3–S5†). The “BaSO4 test” for the SO42− anion detection also gave negative result that confirms completion of the metathesis. In the 1H NMR spectra one sharp singlet signal at ca. 2.22 ppm occur with integral intensity corresponding to four acetonitrile molecules coordinated to copper(I) ion (Fig. 4, S3 and S4†).
 | ||
| Fig. 4 1H and 19F (inset) NMR spectrum of [Cu(CH3CN)4][PF6] in presence of 2 equivalents of 4-iodobenzotrifluoride, acetone-d6, 298 K. | ||
The completion of copper(II) reduction in the course of reaction was monitored by visible color change of reaction solution from blue to colorless. To make sure that final products have no admixture of corresponding copper(II) compounds we measured UV-VIS absorbance spectra of all the complexes obtained in acetonitrile. The Cu(ClO4)2·6H2O salt was used as a reference since it forms [Cu(MeCN)4][ClO4]2 complex upon dissolving in acetonitrile.29 For all the compounds 0.01 M solutions in acetonitrile were prepared and their UV-VIS absorbance spectra were measured in the range 250–1100 nm (Fig. 5).
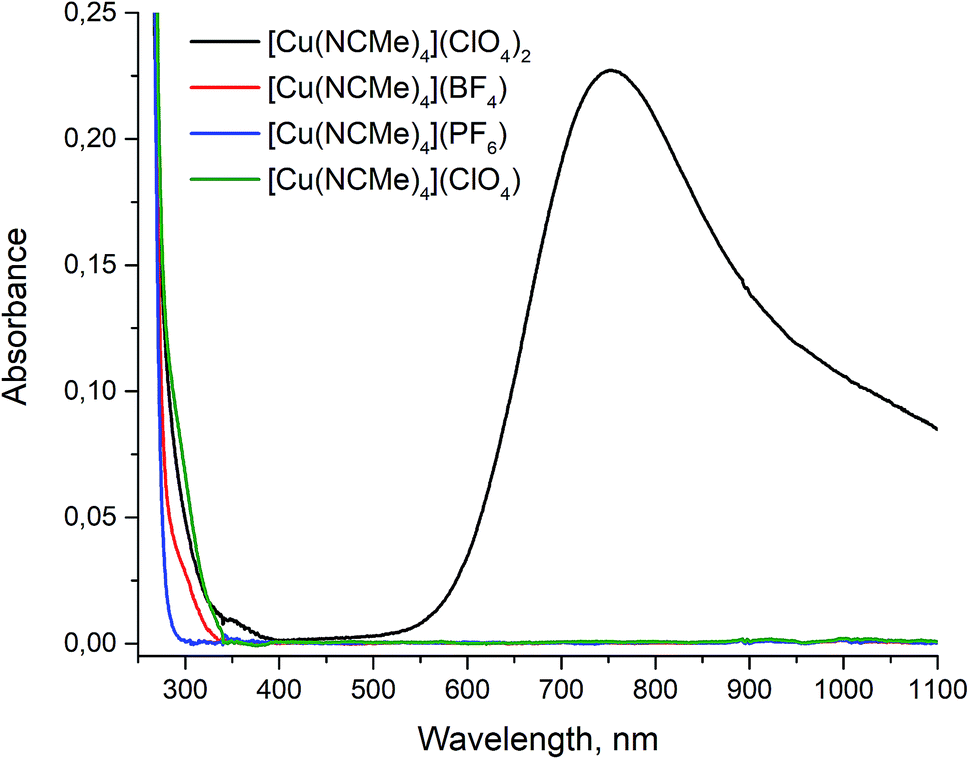 | ||
| Fig. 5 UV-VIS spectra of 0.01 M solutions of compounds in acetonitrile. | ||
The Cu(II) has clearly detectable strong absorption band in the 600 ≥ 1100 nm interval. On the contrary, we did not find any absorption for the samples obtained in the same spectral range, that clearly point to the absence of detectable amount of Cu(II) ions in the final products.
Experimental
General comments
CuSO4·5H2O, Cu(ClO4)2·6H2O, NaBF4, KPF6, LiClO4 and all the solvents were used as received. The solution 1D 1H, 19F NMR spectra were recorded on a Bruker Avance 400 spectrometer. Microanalyses were carried out in the analytical laboratory of the University of Eastern Finland. The IR spectra were recorded in Nujol using a Fourier transform infrared spectrophotometer IRAffinity-1. X-ray powder diffraction analysis was carried out using a Rigaku Miniflex II (Cu Kα) diffractometer, equipped with a high-speed solid state Dtex type detector within the 2θ angle range of 5–100° in the Bragg–Brentano geometry. The measurements were done with a scan step 0.02° of 2θ and speed of 2° min−1. UV/VIS spectra were recorded using a Shimadzu UV-1800 spectrophotometer.General synthetic procedure exemplified by tetrakis(acetonitrile)copper(I) tetrafluoroborate
:1 mixture (containing 0.2 g of acetonitrile) and twice with 5 ml of ethyl acetate (containing 0.1 g of acetonitrile). The obtained precipitate was dried at 50 °C in air for 2 hours and then in vacuum at RT to give 4.34 g of the product as white crystalline solid (yield 86%). 19F NMR (acetone-d6, 298 K; δ): −151.35 (s, BF4). 1H NMR (acetone-d6, 298 K; δ): 2.22 (s, Cu–NC–Me), Anal. calcd for C8H12BCuF4N4: C, 30.16; H, 3.85; N, 17.81 found: C, 30.62; H, 3.83; N, 17.78. IR (in Nujol, ν) 2275 (m), 2304 (m) (CN); 1050 (vs), 521 (s) (BF4) cm−1.C–Me), Anal. calcd for C8H12CuF6N4P: C, 25.78; H, 3.25; N, 15.03 Found: C, 25.61; H, 3.24; N, 15.14. IR (in Nujol, ν) 2275 (m), 2311 (m) (CN); 850 (vs), 557 (s) (PF6) cm−1.C–Me), Anal. calcd for C8H12ClCuN4O4: C, 29.37; H, 3.70; N, 17.12 found: C, 29.44; H, 3.75; N, 17.07. IR (in Nujol, ν) 2272 (m), 2301 (m) (CN); 1083 (vs), 623 (s) (ClO4) cm−1.Conclusions
In conclusion, we elaborated an efficient protocol for the synthesis of [Cu(CH3CN)4]X complexes (X = BF4−, PF6−, ClO4−) using stable, inexpensive and low toxic materials. The protocol makes possible to carry out the reaction in aqueous media and minimizes the amount of toxic acetonitrile in the synthesis. According to this protocol the targeted complexes could be obtained in high yield (82–87%) as pure crystalline material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by grant of the Russian Foundation for Basic Research 16-33-60109. The work was carried out using equipment of the Research park of St. Petersburg State University (Centres for Magnetic Resonance, for Chemical Analysis and Materials Research, and for X-ray Diffraction Studies).Notes and references
- E. J. Parish, H. Qin, B. H. Lipshutz and C. Lee, Encycl. Reagents Org. Synth., 2007 DOI:10.1002/9780470842898.rt039.pub2.
- F. Wang, P. Chen and G. Liu, Encycl. Reagents Org. Synth., 2017 DOI:10.1002/047084289X.rn02002.
- B. Feng, L.-Q. Lu, J.-R. Chen, G. Feng, B.-Q. He, B. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 5888–5892 CrossRef CAS PubMed.
- J. García-Álvarez, J. Díez and J. Gimeno, Green Chem., 2010, 12, 2127–2130 RSC.
- S. M. Soria-Castro, D. M. Andrada, D. A. Caminos, J. E. Arguello, M. Robert and A. B. Penenory, J. Org. Chem., 2017, 82, 11464–11473 CrossRef CAS.
- X.-H. Ouyang, C. Hu, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 4659–4662 CrossRef CAS PubMed.
- S. Seo and M. C. Willis, Org. Lett., 2017, 19, 4556–4559 CrossRef CAS PubMed.
- J. M. Hoover, J. E. Steves and S. S. Stahl, Nat. Protoc., 2012, 7, 1161 CrossRef CAS PubMed.
- X.-Y. Yu, Q.-Q. Zhao, J. Chen, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505–15509 CrossRef CAS.
- H.-J. Zhang and L. Yin, J. Am. Chem. Soc., 2018, 140, 12270–12279 CrossRef CAS.
- T. C. Malig, D. Yu and J. E. Hein, J. Am. Chem. Soc., 2018, 140, 9167–9173 CrossRef CAS PubMed.
- P. K. Chikkade, Y. Kuninobu and M. Kanai, Chem. Sci., 2015, 6, 3195–3200 RSC.
- L. Maestre, E. Ozkal, C. Ayats, Á. Beltrán, M. M. Díaz-Requejo, P. J. Pérez and M. A. Pericàs, Chem. Sci., 2015, 6, 1510–1515 RSC.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69–115 CrossRef CAS.
- M. Mohankumar, M. Holler, E. Meichsner, J.-F. Nierengarten, F. Niess, J.-P. Sauvage, B. Delavaux-Nicot, E. Leoni, F. Monti, J. M. Malicka, M. Cocchi, E. Bandini and N. Armaroli, J. Am. Chem. Soc., 2018, 140, 2336–2347 CrossRef CAS PubMed.
- A. F. Chaudhry, S. Mandal, K. I. Hardcastle and C. J. Fahrni, Chem. Sci., 2011, 2, 1016–1024 RSC.
- M. Trose, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2018, 355, 380–403 CrossRef CAS.
- I. O. Koshevoy, A. J. Karttunen, J. R. Shakirova, A. S. Melnikov, M. Haukka, S. P. Tunik and T. A. Pakkanen, Angew. Chem., Int. Ed. Engl., 2010, 49, 8864–8866 CrossRef CAS PubMed.
- J. R. Shakirova, E. V Grachova, V. V Gurzhiy, S. K. Thangaraj, J. Jänis, A. S. Melnikov, A. J. Karttunen, S. P. Tunik and I. O. Koshevoy, Angew. Chem., Int. Ed., 2018, 57, 14154–14158 CrossRef CAS PubMed.
- M. J. Leitl, D. M. Zink, A. Schinabeck, T. Baumann, D. Volz and H. Yersin, Top. Curr. Chem., 2016, 374, 25 CrossRef.
- E. Fresta and R. D. Costa, J. Mater. Chem. C, 2017, 5, 5643–5675 RSC.
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 RSC.
- X.-L. Chen, C.-S. Lin, X.-Y. Wu, R. Yu, T. Teng, Q.-K. Zhang, Q. Zhang, W.-B. Yang and C.-Z. Lu, J. Mater. Chem. C, 2015, 3, 1187–1195 RSC.
- H. H. Morgan, J. Chem. Soc., Trans., 1923, 123, 2901–2907 RSC.
- G. J. Kubas, Inorg. Synth., 1979, 19, 90–92 CAS.
- H.-C. Liang, K. D. Karlin, R. Dyson, S. Kaderli, B. Jung and A. D. Zuberbühler, Inorg. Chem., 2000, 39, 5884–5894 CrossRef CAS PubMed.
- J. S. Ovens, P. R. Christensen and D. B. Leznoff, Chem.–Eur. J., 2016, 22, 8234–8239 CrossRef CAS PubMed.
- N. E. Heimer, R. E. Del Sesto, Z. Meng, J. S. Wilkes and W. R. Carper, J. Mol. Liq., 2006, 124, 84–95 CrossRef CAS.
- A. S. Mereshchenko, P. K. Olshin, A. M. Karimov, M. Y. Skripkin, K. A. Burkov, Y. S. Tveryanovich and A. N. Tarnovsky, Chem. Phys. Lett., 2014, 615, 105–110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns, NMR and IR spectra. See DOI: 10.1039/c8ra10564b |
| This journal is © The Royal Society of Chemistry 2019 |