
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTi2PTe2 monolayer: a promising two-dimensional anode material for sodium-ion batteries†
Jie Liua,
Man Qiaoa,
Xiaorong Zhua,
Yu Jing *bc and
Yafei Li*a
*bc and
Yafei Li*a
aJiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: liyafei@njnu.edu.cn
bCollege of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: yujing@njfu.edu.cn
cJiangsu Co-Innovation Centre of Efficient Processing and Utilization of Forest Resources, Nanjing 210037, China
First published on 17th May 2019
Abstract
Developing efficient anode materials with a good electrochemical performance has been a key scientific issue in the development of sodium ion batteries (SIBs). In this work, by means of density functional theory (DFT) computations, we demonstrate that two-dimensional (2D) Ti2PTe2 monolayer is a promising candidate for this application. The exfoliation of Ti2PTe2 monolayer from its experimentally known layered bulk phase is feasible due to the moderate cohesive energy. Different from many binary 2D transitions metal chalcogenides (TMCs) that are semiconducting, Ti2PTe2 monolayer is metallic with considerable electronic states at the Fermi level. Remarkably, Ti2PTe2 monolayer has a considerably high theoretical capacity of 280.72 mA h g−1, a rather small Na diffusion barrier of 0.22 eV, and a low average open circuit voltage of 0.31 eV. These results suggest that Ti2PTe2 monolayer can be utilized as a promising anode material for SIBs with high power density and fast charge/discharge rates.
Introduction
In the past few decades, the large-scale exploitation and utilization of fossil fuels has not only led to energy shortages, but also caused serious environmental pollution. Therefore, in recent years, green renewable energy such as solar energy, wind energy and tidal energy has become a hot research topic in scientific community. To this end, the development of related energy storage technologies with high energy density and long cycle life to efficiently store and utilize these renewable energy sources is particularly critical.1,2 Among current various energy storage technologies, lithium ion batteries (LIBs) have occupied most of the portable electronics market and become the primary choice for energy storage systems, such as electric vehicles and portable mobile device.3,4 However, the limited lithium (Li) resources on earth would increase the cost of LIBs,5,6 thereby hindering the large-scale application of LIBs. Sodium (Na) is abundant in nature and low in price, and has similar electrochemical properties to Li. Therefore, secondary batteries based on Na hold the great potential to replace LIBs to become the next generation batteries.7–11 However, the graphite anode materials that widely used in LIBs cannot be directly applied to sodium ion batteries (SIBs) due to the weak interaction between Na and graphite.12,13 To this end, developing new anode materials with high specific capacity, high energy density, and good cycle performance is an urgent task for the application of SIBs.Since the experimental realization of graphene by Geim et al. in 2004,14,15 two-dimensional (2D) materials have gained a lot of interests in the fields of electrochemical energy storage and conversion systems due to its extraordinary properties.16–18 Especially, both sides of 2D materials can offer ultra-high surface area of for Na adsorption and short distance for Na migration, which make 2D materials rather attractive for SIBs. However, rather few experimentally realized 2D structures can be served as anode materials for SIBs. For example, graphene, which has rather high electronic conductivity, was excluded as the qualified candidate as the adsorption of Na ions on the surface of graphene is energetically unfavorable.19,20 The single-layered black phosphorus, namely phosphorene,21,22 was revealed theoretically to have a high Na capacity and a low Na diffusion barrier.23,24 However, the poor stability of phosphorene in ambient conditions would significantly constraint its application in energy storage.25 Quite recently, 2D transitions metal chalcogenides (TMCs) were also proposed to be the promising anode materials for SIBs.26–28 However, many TMCs anodes (e.g., MoS2) suffer from rather inferior capacities and low electronic conductivity. At present, searching for new 2D layered anodes with high Na capacity, low Na diffusion barrier, and high electronic conductivity remains a big challenge.29–35
Experimentally, many 2D structures (e.g. graphene,14 MoS2,36 black phosphorene22) have been realized via exfoliation strategies from there bulk counterparts, which provides us a feasible way to explore new 2D electrodes for SIBs. Recently, several layered ternary TMCs (e.g. Ti2PTe2,37–40 Zr2PTe2 (ref. 41)) with more than two chalcogens of pnictogen and tellurium with zirconium or titanium cations were synthesized and revealed to be metallic. Inspired by the extensive studies of developing 2D anodes for SIBs, one may naturally wonder whether 2D ternary TMCs could be exfoliated and attractive for SIBs. However, at present the energy storage performance of 2D ternary TMCs remain yet to be explored. Theoretical advances in recent years have made it is possible to evaluate a wide range of essential battery-related properties, such as voltage, ion diffusion, and theoretical capacity.42–44 Therefore, a theoretical study on this issue will shed some lights on developing novel anodes for SIBs.
In this work, we systematically studied the structural and electronic properties of Ti2PTe2 monolayer and explored the possibility of using it as an anode material for SIBs. It is found that Ti2PTe2 monolayer is a stable 2D metal and can be easily obtained via exfoliation strategies. Remarkably, our results revealed that Ti2PTe2 monolayer can provide moderate Na binding strength, low Na diffusion barrier, and higher theoretical capacity than 2D binary TMCs, thus holding great potentials to be utilized as an anode material for SIBs.
Computational details
Our spin-polarized density functional theory (DFT) computations were performed using the plane-wave technique as implemented in Vienna ab initio simulation package (VASP).45 The ion–electron interaction was described using the projector-augmented plane wave (PAW) approach.46,47 The structural optimization was conducted using the generalized gradient approximation (GGA) expressed by PBE functional48 while the electronic structure computations were performed by employing the HSE06 hybrid functional method.49 A 500 eV cutoff energy for the plane-wave basis set was adopted in all computations. The weak interactions were described by adopting the DFT-D3 (D stands for dispersion) method with the Grimme vdW correction.50 The convergence threshold was set as 10−4 eV in energy and 10−3 eV Å−1 in force. We set the x and y directions parallel and the z direction perpendicular to the layer plane, and adopted a supercell length of 20 Å in the z direction. The 2D Brillouin zone of Ti2PTe2 monolayer was sampled with a 4 × 4 × 1 Γ centered k points grid.The ab initio molecular dynamics (AIMD) simulations were carried out to assess the thermal stability of the Ti2PTe2 monolayer. The temperature was controlled by using the Nosé–Hoover method.51 At each temperature, the AIMD simulation with the mount of substance (N), volume (V), and temperature (T) being conserved (NVT ensemble) lasts for 10 ps with a time step of 1.0 fs. The phonon band structure of Ti2PTe2 monolayer was computed using the density functional perturbation theory (DFPT)52 as implemented in the Phonopy program.53 The climbing-image nudged elastic band (CI-NEB) method54 as implemented in VASP was employed to simulate the Na diffusion energy barrier on Ti2PTe2 monolayer.
Results and discussion
Fig. 1a shows the geometrics of Ti2PTe2 bulk, which crystallizes in a hexagonal structure with the space group R3m (no. 166). In Ti2PTe2 bulk, Ti2PTe2 layers are stacked together with interlayer vdW binding along the z-axis in ABC sequence. The equilibrium lattice parameters of Ti2PTe2 bulk were optimized to be a = 3.66 Å, b = 3.66 Å, and c = 29.81 Å at PBE-D3 level of theory, which are in good agreements with the experimentally measured values (a = 3.64 Å, b = 3.64 Å, c = 28.49 Å).37 The optimized lattice parameters of Ti2PTe2 monolayer (a = 3.64 Å, b = 3.64 Å) are slightly smaller than those of the bulk phase. Each Ti2PTe2 monolayer consists of five atomic layers (two Ti layers, one P layer, and two Te layers), which is different to the typical 2D binary TMCs with three atomic layers. In Ti2PTe2 monolayer (Fig. 1b), each Ti atom binds with three Te atoms and three P atoms while each P atom solely binds with six Ti atoms. Therefore, the structure of Ti2PTe2 can be described as layers of isolated telluride and phosphide anions form an ordered close sphere-packing with titanium cations filling two-thirds of the octahedral voids. The Te–Ti and Ti–P bond lengths are 2.79 and 2.48 Å, respectively. According to the Bader charge population analysis, the Ti, P, and Te atoms in Ti2PTe2 monolayer possess a charge of +1.17, −1.36, and −0.49 |e|, respectively, which is in accordance with its ionic formula (Ti4+)2(P3−)(Te2−)2(e−) deduced experimentally.37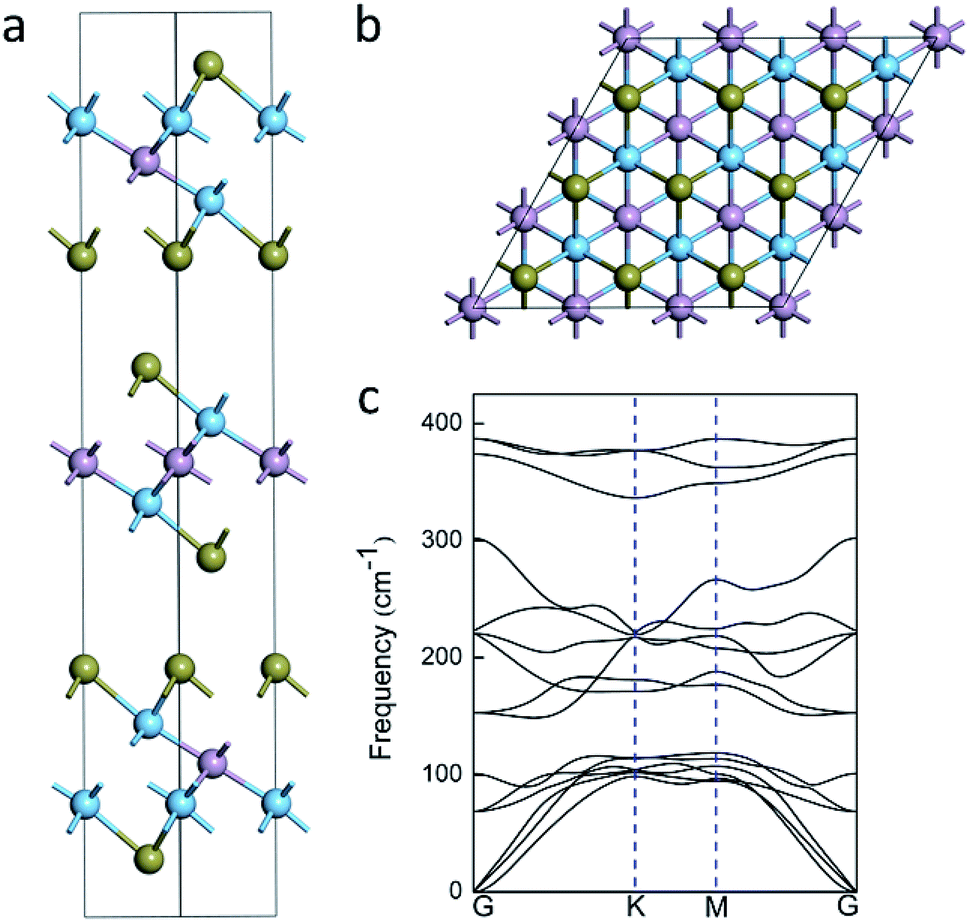 | ||
| Fig. 1 (a) Side view of the crystal structure of Ti2PTe2 bulk. The brown, blue and pink balls represent Te, Ti, and P atoms, respectively. (b) Top view of the optimized structure of Ti2PTe2 monolayer. (c) Phonon dispersion curves of Ti2PTe2 monolayer. | ||
Since Ti2PTe2 monolayer has not been realized experimentally yet, it is necessary for us to access its stability before revealing the properties and potential applications. The stability of Ti2PTe2 monolayer can be firstly endorsed by the phonon dispersion curves shown in Fig. 1c, where no imaginary phonon modes can be observed and the highest frequency can reach up to 390 cm−1. Therefore, Ti2PTe2 monolayer is a kinetically stable 2D structure. We also carried out the AIMD simulations to assess the thermodynamic stability of Ti2PTe2 monolayer. As shown in Fig. S1,† the geometry of Ti2PTe2 monolayer is not pronouncedly distorted at the temperature of 500 K, suggesting that Ti2PTe2 monolayer has good thermodynamic stability and can work at harsh environments.
The feasibility of producing Ti2PTe2 monolayer via the exfoliation strategy was then evaluated by computing the cleavage energy (Ecl) of Ti2PTe2, which is defined as the minimum energy required to isolate a monolayer from the bulk. Specifically, the exfoliation process was simulated by imposing a fracture in a five-layer slab model of Ti2PTe2, in which the top layer is removable while the rest four layers are fixed. The distance between two fractures in the equilibrium configuration was defined as zero, and the Ecl can be determined via increasing the separation distance. As shown in Fig. 2, when the separation distance is larger than 6 Å, the Ecl of Ti2PTe2 monolayer gradually converges to a constant value of ∼0.38 J m−2. Remarkably, the Ecl of Ti2PTe2 monolayer is quite close to that of graphene (0.37 J m−2)55 and much lower than the experimentally realized Ga2N monolayer (1.09 J m−2),56 suggesting the high feasibility to extract 2D Ti2PTe2 from its bulk phase in the laboratory.
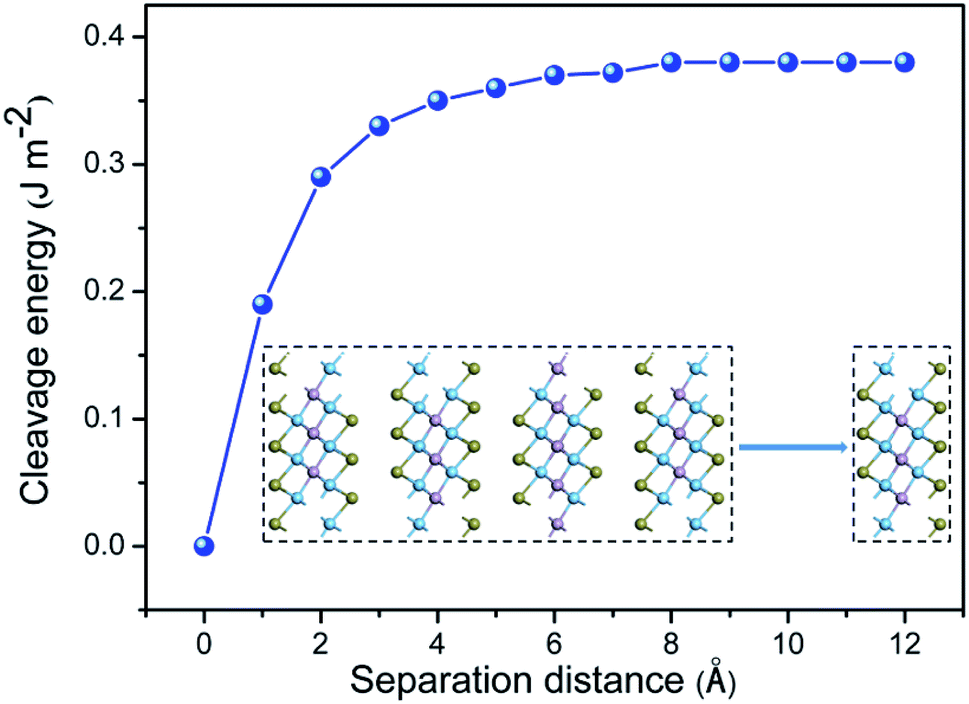 | ||
| Fig. 2 Cleavage energy as a function of the separation distance for a fracture in bulk Ti2PTe2. Inset is the schematic of separating a Ti2PTe2 monolayer from its neighboring four-layer. | ||
The good electrical conductivity is essentially required for the excellent electrochemical performance of electrode materials. However, most binary 2D TMCs (e.g. MoS2, TiS2) are semiconducting with poor conductivity, which usually requires the addition of conducting materials (e.g. graphene) to facilitate the electrochemical reactions. The electronic properties of Ti2PTe2 monolayer were then investigated by computing its electronic band structure and density of states (DOS). As shown in Fig. 3, Ti2PTe2 monolayer is metallic with several energy levels crossing the Fermi level computed at the HSE06 level of theory. According to the partial DOS analysis, the metallic states at the Fermi level are mainly contributed by Ti-3d states. Therefore, the metallic Ti2PTe2 monolayer stands out to be a highly desirable candidate of anode material for SIBs.
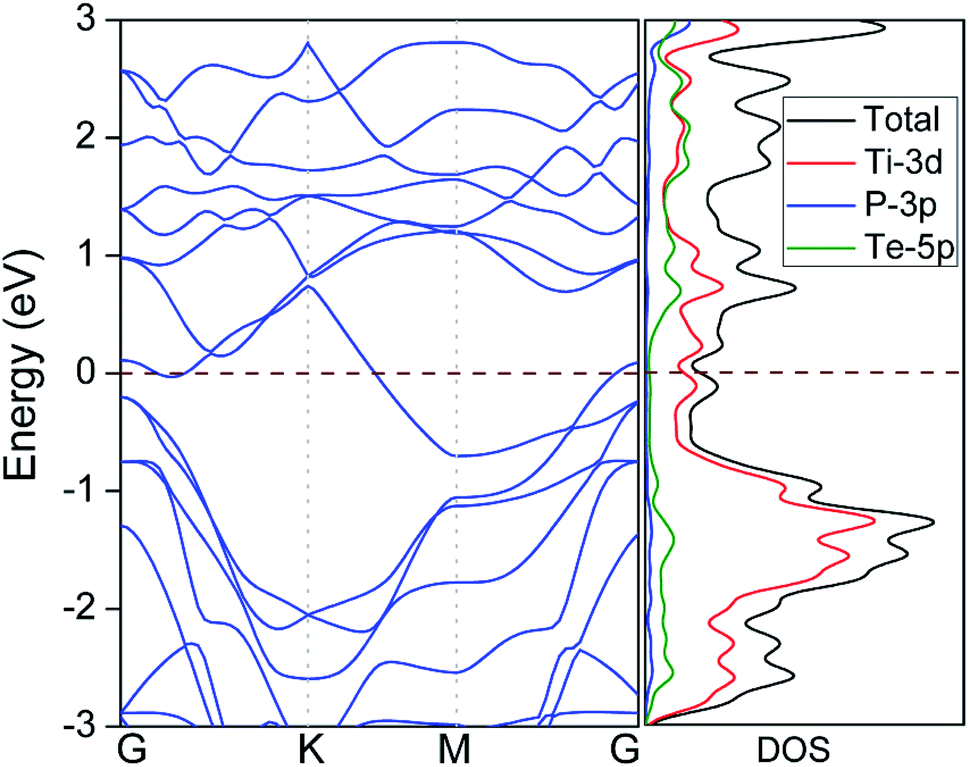 | ||
| Fig. 3 Band structure (left) and density of states (DOS) (right) of Ti2PTe2 monolayer. The pink dashed lines denote the position of Fermi level. | ||
After the thorough understanding on the basic electronic properties of Ti2PTe2 monolayer, it is next accessed whether this novel 2D structure can be served as an anode material for SIBs. To this end, we firstly studied the adsorption of one isolated Na atom on the surface of Ti2PTe2 monolayer by constructing a 3 × 3 supercell. The adsorption energy of Na (Ea) is defined as:
| Ea = ETi2PTe2Na − ETi2PTe2 − μNa |
Considering the symmetry of Ti2PTe2 monolayer, three possible Na adsorption sites can be identified (Fig. 4), including the hollow site (H) above the center of a Ti3Te3 hexagon, the top site above one Ti atom (T1), and the top site directly above one Te atom (T2). Actually, the Na adsorption on H and T1 sites are expected to have interactions with three surrounding Te atoms. The adsorption energies of all these three sites were computed to determine the most favorable adsorption configuration for Na. According to our computations, the three considered sites all have a negative adsorption energy, indicating that the Na storage on Ti2PTe2 monolayer could be exothermic and spontaneous. In particular, the T1 site has the most negative adsorption energy (−1.17 eV), followed by H site (−1.14 eV) and T2 site (−0.56 eV). The distance between the adsorbed Na atom and the neighboring Te atoms were optimized to be 2.69, 2.64, and 2.86 Å for T1, H, and T2, respectively. Remarkably, the Na adsorption on Ti2PTe2 monolayer has not lead to appreciable geometric change or lattice expansion, indicative of great stability that is favorable for SIBs applications. According to the Bader charge population analysis, the adsorbed Na atom possesses a positive charge of 0.76, 0.75, and 0.81|e| at T1, H, and T2 site, respectively, indicating that the adsorbed Na are pronouncedly ionized and the interaction between Ti2PTe2 monolayer and Na is mainly of the Coulomb interaction.
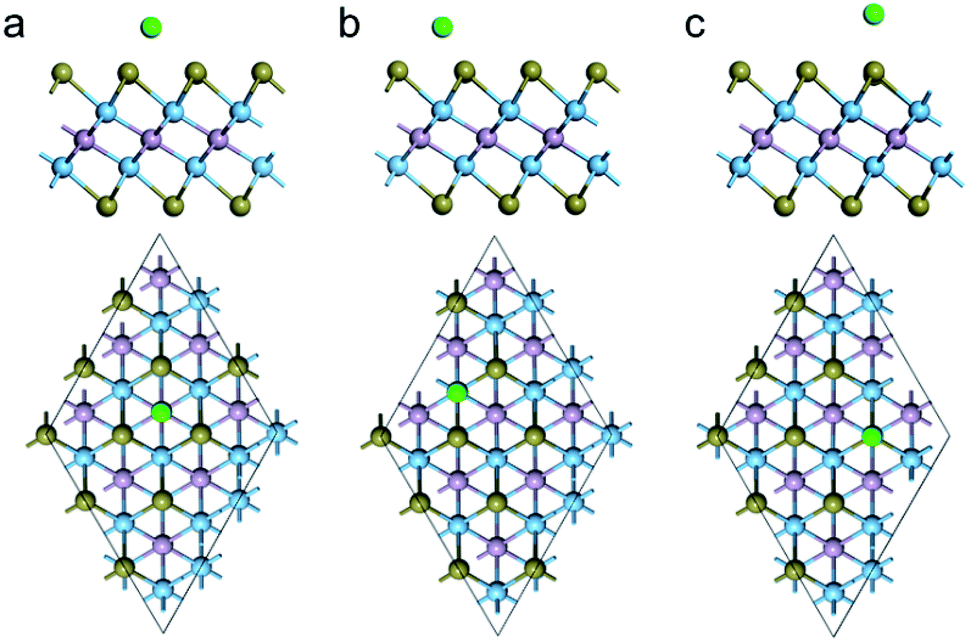 | ||
| Fig. 4 Side (upper) and top (bottom) views of the optimized structures for Na adsorption on the (a) H, (b) T1, and (c) T2 sites of Ti2PTe2 monolayer. The green ball represents Na atom. | ||
The density of states (DOS) of Ti2PTe2 monolayer with the adsorbed Na at three different sites were then examined. As clearly shown in Fig. S2,† the adsorption of Na does not significantly change the overall electronic properties of Ti2PTe2 monolayer except for the upshift of Fermi level. Therefore, Ti2PTe2 monolayer can maintain its metallic nature after the adsorption of Na, which is an advantage for its applications as battery electrodes.
We then turned our attention to the motion of Na on the surface of Ti2PTe2 monolayer as the rate performance of a battery electrode is mainly determined by the mobility of intercalated metal ions. Here the motion of Na on Ti2PTe2 monolayer was evaluated by using the CI-NEB method to determine the minimum-energy pathways and estimate the diffusion barriers. The adsorption configuration of Na atom on Ti2PTe2 monolayer (T1 site) was used as the initial state while the configuration with Na adsorption at a neighboring T1 site was chosen to be the final state. As shown in Fig. 5, two possible migration pathways, which correspond to the diffusion of Na between two neighboring T1 sites passing through a P atom (viewed from top) (path 1) or passing through a Te atom (path 2) were identified. According to our CI-NEB computations, the diffusion barrier of path 1 (0.22 eV) is much lower than that of path 2 (0.67 eV). These results are in good agreements with our computations of the adsorption energy. Encouragingly, the obtained Na diffusion barriers on the surface of Ti2PTe2 monolayer are actually rather low and comparable to those of MoS2,28 phosphorene,24 and graphene materials.57 Therefore, Na can diffuse rather fast on the surface of Ti2PTe2 monolayer, and good rate capability can be expected for Ti2PTe2 monolayer when it is utilized as the anode material in SIBs.
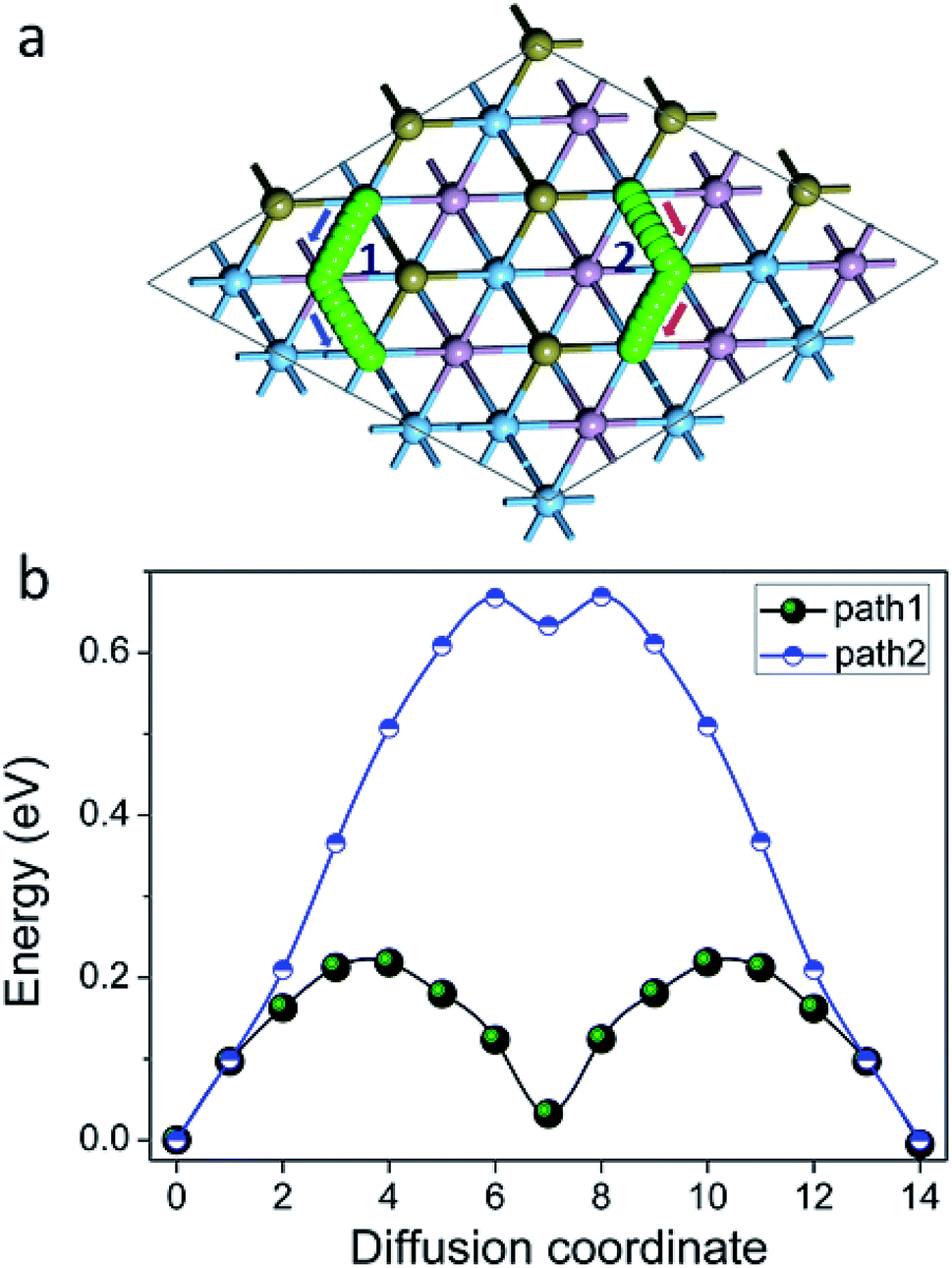 | ||
| Fig. 5 (a) Migration paths and (b) diffusion barrier profile for Na on the surface of Ti2PTe2 monolayer. | ||
For practical application, the Na storage capacity is the key indicator for the performance of electrode materials for SIBs. Therefore, we further estimated the theoretical Na storage capacity for Ti2PTe2 monolayer, which is defined as the maximum capacity that still shows a negative average adsorption energy. Here we employed a 3 × 3 × 1 supercell as the model for extra Na storage. Considering that previous studies have demonstrated that the multilayer Na adsorption can significantly enhance the capacities of 2D anodes,30,31 we also allowed that the concentration of adsorbed Na on Ti2PTe2 monolayer increasing layer by layer on both sides simultaneously. Specifically, the first layer of Na atoms are adsorbed on the T1 sties, which are the most stable sites for Na adsorption. After the T1 sites are occupied completely, the Na atoms will be adsorbed at H sites to form the second layer. Similarly, Na atoms in the third layer would be adsorbed on the top of Se atoms. To evaluate the interaction between Na atoms and the host material, we computed the average adsorption energies of Na atoms in each layer (Eave), which is defined as:
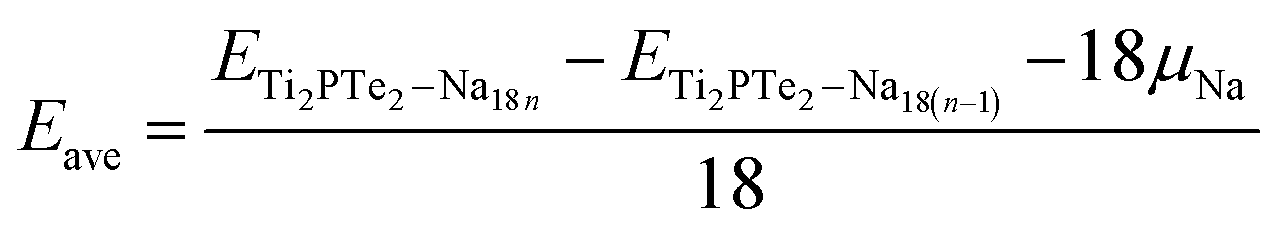
According to our computations, the average adsorption energy of first Na layer is −0.49 eV, implying the strong interaction between these Na ions and Ti2PTe2 monolayer. In contrast, the average adsorption energy of the second Na layer rapidly decreases to −0.13 eV. Nevertheless, the negative average adsorption energy can still ensure good intercalation stability of these Na ions on Ti2PTe2 monolayer. However, the average adsorption energy of the third Na layer becomes positive, indicating that the adsorption of these Na ions is energetically unfavorable and they would tend to form undesirable Na dendrites. Therefore, our computations reveals that the maximum number of Na storage for a 3 × 3 × 1 supercell of Ti2PTe2 monolayer is 36, corresponding to the chemical stoichiometry of Na4Ti2PTe2 (Fig. 6). As a result, the theoretical specific capacity of Ti2PTe2 monolayer as anode of SIBs is ∼280.72 mA h g−1, which is much higher than those of MoS2 monolayer and Mxene. Especially, during the intercalation of Na ions on Ti2PTe2 monolayer, the lattice constants in the x–y plane of intercalated compounds only increase slightly (∼1%). This very small strain is another great advantage of Ti2PTe2 monolayer to be used as an electrode for rechargeable batteries. Moreover, it is found that the metallic feature of Ti2PTe2 monolayer can be well kept after adding two Na layers in both sides (Fig. S3†).
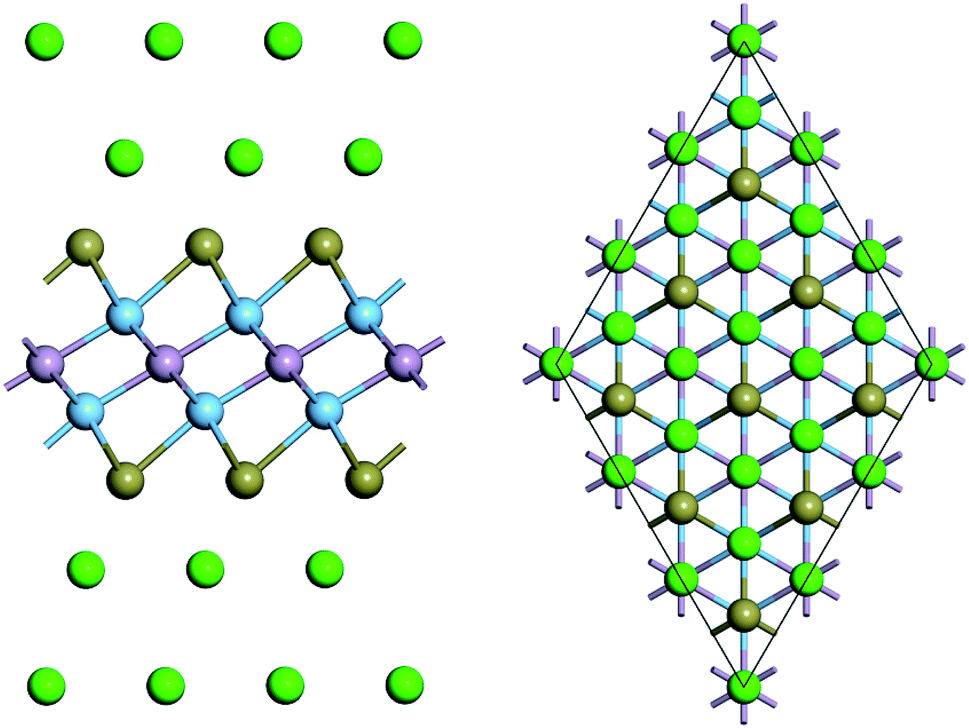 | ||
| Fig. 6 Side (left) and top (right) views of optimized geometric structure of Na4Ti2PTe2. | ||
Experimentally, the voltage has been well employed as a discriminator in screening the suitable anode materials for SIBs, and it is accepted that a voltage in the range of 0.0–1.0 V is suitable to maximize the energy density as well as avoid the formation of Na dendrites. To this end, we also computed the average open circuit voltage (OCV) for Na intercalation on Ti2PTe2 monolayer. Typically, the anode charge/discharge processes assume the following half–cell reaction vs. Na/Na+
| Ti2PTe2 + xNa+ + xe− ↔ NaxTi2PTe2 |
With the effects of volume and entropy being both neglected, the OCV for Na intercalation on Ti2PTe2 monolayer can be computed from the total energy difference on the basis of following equation by neglecting the:58
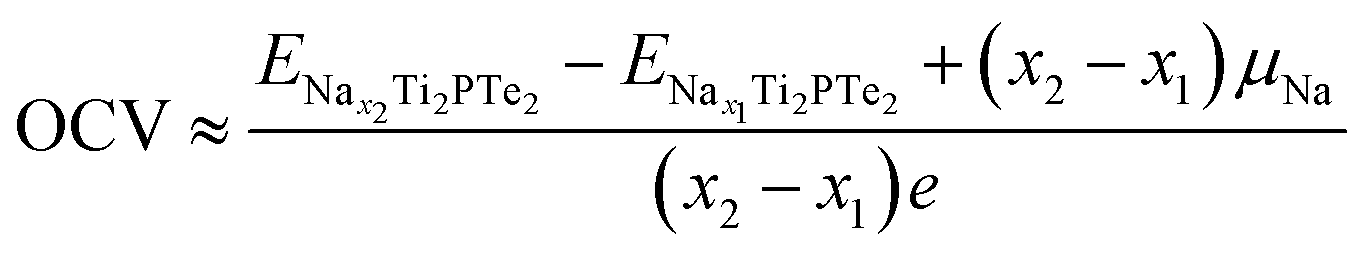
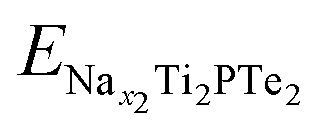 and
and 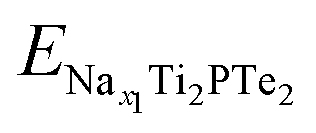 are the total energies of Ti2PTe2 monolayer with Na concentrations limits of x2 and x1, respectively. According to our computations, when Ti2PTe2 monolayer reaches the highest Na capacity, corresponding to the case of x1 = 0 and x2 = 4, the average OCV is 0.31 V, which is lower than those of most 2D binary TMCs.28 Therefore, Ti2PTe2 monolayer should be a rather good candidate of electrode material for SIBs.
are the total energies of Ti2PTe2 monolayer with Na concentrations limits of x2 and x1, respectively. According to our computations, when Ti2PTe2 monolayer reaches the highest Na capacity, corresponding to the case of x1 = 0 and x2 = 4, the average OCV is 0.31 V, which is lower than those of most 2D binary TMCs.28 Therefore, Ti2PTe2 monolayer should be a rather good candidate of electrode material for SIBs.
Conclusion
To summarize, by means of comprehensive DFT computations, we systematically studied the structural, electronic, and energy storage properties of Ti2PTe2 monolayer. According to our computational results, Ti2PTe2 monolayer is a stable 2D structure and could be realized experimentally via exfoliation strategies. Remarkably, Ti2PTe2 monolayer has a rather small Na diffusion barrier and it can stably accommodate Na up to Na4Ti2PTe2 with an open circuit voltage of 0.31 V. Especially, Ti2PTe2 monolayer is metallic with excellent electronic conductivity. With all of these extraordinary properties, Ti2PTe2 monolayer would have a great potential to be utilized as an anode material for SIBs. Due to the high experimental feasibility, it is expected that Ti2PTe2 monolayer could be realized in the lab in the quite near future. We also hope our studies could stimulate more theoretical and experimental efforts on developing 2D ternary TMCs for energy storage applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support in China by the Natural Science Foundation of China (No. 21873050), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX18_1193), and the Priority Academic Program Development of Jiangsu Higher Education Institutions. The computational resources utilized in this research were provided by Shanghai Supercomputer Center.References
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Chem. Soc. Rev., 2015, 44, 1777 RSC.
- https://en.wikipedia.org/wiki/File:Elemental_abundances.svg.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS.
- H. Pan, Y. –S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 RSC.
- W. Luo, F. Shen, C. Bommier, H. Zhu, X. Ji and L. Hu, Acc. Chem. Res., 2016, 49, 231 CrossRef CAS PubMed.
- J. –Y. Hwang, S. –T. Myung and Y. –K. Sun, Chem. Soc. Rev., 2017, 46, 3529 RSC.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and C. Ceder, Energy Environ. Sci., 2011, 4, 3680 RSC.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N. –S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Gregorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and P. Kim, Nature, 2012, 490, 192 CrossRef CAS PubMed.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271 CrossRef CAS PubMed.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 2067 RSC.
- Y. Liu, B. Merinov and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3735 CrossRef CAS PubMed.
- M. S. Balogun, Y. Luo, W. Qiu, P. Liu and Y. Tong, Carbon, 2016, 98, 162 CrossRef CAS.
- H. Liu, A. T. Neal, Z. Zhu, D. Tomanek and P. D. Ye, ACS Nano, 2014, 8, 4033 CrossRef CAS.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372 CrossRef CAS PubMed.
- J. Sun, H. –W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2017, 10, 980 CrossRef PubMed.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 13921 RSC.
- J. O. Island, G. A. Steele, H. S. J. Van Der Zant and A. Castellanos-gomez, 2D Mater., 2015, 2, 011002 CrossRef.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- J. Zhou, L. Wang, M. Yang, J. Wu, F. Chen, W. Huang, N. Han, H. Ye, F. Zhao, Y. Li and Y. Li, Adv. Mater., 2017, 29, 1702061 CrossRef PubMed.
- E. Yang, H. Ji and Y. Jung, J. Phys. Chem. C, 2015, 119, 26374 CrossRef CAS.
- T. Yu, Z. Zhao, L. Liu, S. Zhang, H. Xu and G. Yang, J. Am. Chem. Soc., 2018, 140, 5962 CrossRef CAS.
- J. Hu, B. Xu, S. A. Yang, S. Guan, C. Ouyang and Y. Yao, ACS Appl. Mater. Interfaces, 2015, 7, 24016 CrossRef CAS PubMed.
- X. Zhang, Z. Yu, S. –S. Wang, S. Guan, H. Y. Yang, Y. Yao and S. A. Yang, J. Mater. Chem. A, 2016, 4, 15224 RSC.
- G. Chen, Y. Bai, H. Li, Y. Li, Z. Wang, Q. Ni, L. Liu, F. Wu, Y. Yao and C. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 6666 CrossRef CAS PubMed.
- X. Zhang, J. Hu, Y. Cheng, H. Y. Yang, Y. Yao and S. A. Yang, Nanoscale, 2016, 8, 15340 RSC.
- X. Zhang, Z. Zhang, S. Yao, A. Chen, X. Zhao and Z. Zhou, npj Comput. Mater., 2018, 4, 13 CrossRef.
- C. –S. Liu, X. –L. Yang, J. Liu and X. –J. Ye, ACS Appl. Energy Mater., 2018, 1, 3850 CrossRef CAS.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- F. Philipp, P. Schmidt, M. Ruck, W. Schnelle and A. Isaeva, J. Solid State Chem., 2008, 181, 2859 CrossRef CAS.
- F. Philipp, P. Schmidt, E. Milke, M. Binnewies and S. Hoffmann, J. Solid State Chem., 2008, 181, 758 CrossRef CAS.
- T. Yajima, M. Koshiko, Y. Zhang, T. Oguchi, W. Yu, D. Kato, Y. Kobayashi, Y. Orikasa, T. Yamamoto, Y. Uchimoto, M. A. Green and H. Kageyama, Nat. Commun., 2016, 7, 13809 CrossRef CAS PubMed.
- J. S. Oh, H. –S. Yu, C. –J. Kang, S. Sinn, M. Han, Y. J. Chang, B. –G. Park, K. Lee, B. I. Min, S. W. Kim, H. –D. Kim and T. W. Noh, Chem. Mater., 2016, 28, 7570 CrossRef CAS.
- K. Tschulik, M. Ruch, M. Binnewies, E. Milke, S. Hoffmann, W. Schnelle, B. P. T. Fokwa, M. Gillesen and P. Schmidt, Eur. J. Inorg. Chem., 2009, 21, 3102 CrossRef.
- Z. Deng, Y. Mo and S. P. Ong, NPG Asia Mater., 2016, 8, e254 CrossRef CAS.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026 CrossRef CAS PubMed.
- Q. Bai, L. Yang, H. Chen and Y. Mo, Adv. Energy Mater., 2018, 8, 1702998 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, L. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- S. Grimme, J. Comput. Chem., 2007, 27, 1787 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. E. Tuckerman, J. Chem. Phys., 1992, 97, 2635 CrossRef.
- S. Baroni, S. De Gironcoli, A. Dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS.
- R. Zacharia, H. Ulbricht and T. Hertel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 155406 CrossRef.
- S. Zhao, Z. Li and J. Yang, J. Am. Chem. Soc., 2014, 136, 13313 CrossRef CAS PubMed.
- C. Ling and F. Mizuno, Phys. Chem. Chem. Phys., 2014, 16, 10419 RSC.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Snapshots and total energy evolution of AIMD simulation of Ti2PTe2 monolayer at 500 K, DOS of Ti2PTe2 monolayer after Na adsorption. See DOI: 10.1039/c9ra01686d |
| This journal is © The Royal Society of Chemistry 2019 |