
Defect-rich and ultrathin N doped carbon nanosheets as advanced trifunctional metal-free electrocatalysts for the ORR, OER and HER†
Hao
Jiang
ab,
Jinxing
Gu
c,
Xusheng
Zheng
d,
Min
Liu
e,
Xiaoqing
Qiu
 a,
Liangbing
Wang
f,
Wenzhang
Li
*ag,
Zhongfang
Chen
*c,
Xiaobo
Ji
a and
Jie
Li
*ag
a,
Liangbing
Wang
f,
Wenzhang
Li
*ag,
Zhongfang
Chen
*c,
Xiaobo
Ji
a and
Jie
Li
*ag
aSchool of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: liwenzhang@csu.edu.cn; lijieliu@csu.edu.cn; Fax: +86-731-88879616; Tel: +86-731-88879616
bCollege of Materials and Chemical Engineering, Hunan City University, Yiyang, 413000, China
cDepartment of Chemistry, University of Puerto Rico Rio Piedras Campus, San Juan, PR 00931, USA. E-mail: zhongfangchen@gmail.com
dHefei National Laboratory for Physical Sciences at the Microscale, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230026, China
eInstitute of Super-microstructure and Ultrafast Process in Advanced Materials, School of Physics and Electronics, Central South University, Changsha, Hunan, China
fSchool of Materials Science and Engineering, Central South University, Changsha, Hunan, China
gHunan Provincial Key Laboratory of Chemical Power Sources, Central South University, Changsha 410083, Hunan, China
First published on 21st November 2018
Abstract
Rational design and facile preparation of non-noble trifunctional electrocatalysts with high performance, low cost and strong durability for the oxygen reduction reaction (ORR), oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) are highly demanded, but remain as a big challenge. Herein, we report a spontaneous gas-foaming method to prepare nitrogen doped ultrathin carbon nanosheets (NCNs) by simply pyrolysing a mixture of citric acid and NH4Cl. Under the optimized pyrolysis temperature (carbonized at 1000 °C) and mass ratio of precursors (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the synthesized NCN-1000-5 sample possesses an ultrathin sheet structure, an ultrahigh specific surface area (1793 m2 g−1), and rich edge defects, and exhibits low overpotential and robust stability for the ORR, OER and HER. By means of density functional theory (DFT) computations, we revealed that the intrinsic active sites for the ORR, OER and HER are the carbon atoms located at the armchair edge and adjacent to the graphitic N dopants. When practically used as a catalyst in rechargeable Zn–air batteries, a high energy density (806 W h kg−1), a low charge/discharge voltage gap (0.77 V) and an ultralong cycle life (over 330 h) were obtained at 10 mA cm−2 for NCN-1000-5. This work not only presents a versatile strategy to develop advanced carbon materials with ultrahigh specific surface area and abundant edge defects, but also provides useful guidance for designing and developing multifunctional metal-free catalysts for various energy-related electrocatalytic reactions.
:1), the synthesized NCN-1000-5 sample possesses an ultrathin sheet structure, an ultrahigh specific surface area (1793 m2 g−1), and rich edge defects, and exhibits low overpotential and robust stability for the ORR, OER and HER. By means of density functional theory (DFT) computations, we revealed that the intrinsic active sites for the ORR, OER and HER are the carbon atoms located at the armchair edge and adjacent to the graphitic N dopants. When practically used as a catalyst in rechargeable Zn–air batteries, a high energy density (806 W h kg−1), a low charge/discharge voltage gap (0.77 V) and an ultralong cycle life (over 330 h) were obtained at 10 mA cm−2 for NCN-1000-5. This work not only presents a versatile strategy to develop advanced carbon materials with ultrahigh specific surface area and abundant edge defects, but also provides useful guidance for designing and developing multifunctional metal-free catalysts for various energy-related electrocatalytic reactions.
Broader contextFor the commercial application of fuel cells, metal–air batteries and water splitting, the fundamental obstacles are the high cost of noble metal-based catalysts (Pt or IrO2) for the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER). Therefore, exploring advanced non-noble metal catalysts to replace Pt or IrO2 is highly desired, but remains a challenge. In this research, we report a spontaneous gas-foaming strategy to synthesize N doped ultrathin carbon nanosheets by carbonizing a uniform mixture of citric acid and NH4Cl. The optimized product possesses prominent structural characteristics and exhibits outstanding catalytic performance for the ORR, OER and HER. Experiments and density functional theory (DFT) revealed that the intrinsic active sites for the ORR, OER and HER are the carbon atoms located at the armchair edge and adjacent to the graphitic N dopants. The present findings could provide a universal strategy and useful guidance for designing and developing multifunctional metal-free catalysts for various energy-related electrocatalytic reactions. |
Introduction
High energy consumption, serious pollution and non-renewable fossil fuels are not in conformity with the requirements of sustainable development of human society. Clean, high capacity and renewable energy storage and conversion techniques such as fuel cells, metal–air batteries and water splitting systems are promising alternatives to traditional fossil fuels.1–4 The electrochemical oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER) are at the heart of the above mentioned renewable energy technologies. However, efficient catalysts are desired to overcome the sluggish kinetics and large overpotential of these electrochemical reactions. To date, the Pt noble metal remains the most efficient electrocatalyst for the ORR and HER, whereas IrO2 and RuO2 are highly active towards the OER.5–8 Nevertheless, the scarcity, high cost and poor tolerance of these platinum group metals (PGMs) severely hinder their large-scale applications. Therefore, developing low cost, highly active and robust catalysts based on earth-abundant non-noble metals for these important electrocatalytic processes is paramount and highly rewarding.To date, tremendous efforts have been made in developing advanced non-noble metal catalysts (NNMCs) to substitute Pt and Ru-based catalysts,3,9,10 among which heteroatom (N, P, S, or B) doped carbon materials distinguish themselves as one of the most promising substitutes of NNMCs for either the ORR, OER or HER due to their low cost, high activities and robust stability.11 For example, we have recently demonstrated that dual atom (N and P) co-functionalized porous carbon networks possess superior ORR activity over a broad pH range of aqueous media.12 Qiao's group prepared a self-supported N-doped carbon hydrogel film, which is greatly favorable for the OER.13 As for the HER, N and P-doped graphite carbon coupled with carbon cloth showed a low HER overpotential of 240 mV at 10 mA cm−2.14 Moreover, Dai and coworkers developed a series of doped nanocarbon materials, including N-doped porous carbon nanofiber films, N and P co-doped mesoporous carbon foam and porous carbon networks, which have novel structures, high specific surface area, and more importantly, outstanding bifunctional activity toward the ORR/OER or ORR/HER.1,15–17 Although so many doped carbon materials show remarkable electrocatalytic activity in a separate ORR, or OER, or HER, they can scarcely function well concurrently toward the OER, HER and ORR due to their unstable or inactive nature in an unfavorable pH environment. Recently, Hu et al. prepared N,S co-doped graphitic sheets (SHG) with a unique hierarchical structure, which exhibited remarkable trifunctional electrocatalytic activity for the ORR, OER and HER.18 However, the synthesis of SHG suffers from removing the template with aqua-regia and a multi-step pyrolysis process, which has the shortcomings of being multistep and time-consuming and having a high cost. Therefore, exploring facile and universal methods to fabricate carbon materials with unique structure, high specific surface area and multifunctional electrocatalytic features is highly desired, but remains a challenge. Furthermore, the research on the catalytic mechanisms and activity sites of heteroatom doped carbon materials toward the OER, HER and ORR is also insufficient. To the best of our knowledge, experimental and theoretical research of N doped carbon materials concurrently toward the ORR, OER and HER has never been reported.
In this work, we put forward a spontaneous gas-foaming strategy to synthesize N doped ultrathin carbon nanosheets (NCNs) through simple one-step carbonization of citric acid and NH4Cl. This method introduces rich defects and a large quantity of micropores into the NCNs. Especially, the sample (NCN-1000-5) obtained at 1000 °C with a mass ratio of 1:1 possesses an ultrathin sheet structure (the edge thickness is only 4 graphitic layers), an ultrahigh specific surface area, rich edge defects and extraordinary trifunctional activity. It shows superior ORR activity (onset potential) even better than commercial Pt/C and comparable OER activity to IrO2 in alkaline electrolytes. Moreover, it is also highly active and stable towards the HER in acidic media. Our DFT computations revealed that graphitic N dopants and the abundant carbon edge defects in the porous structure are responsible for the trifunctional electrocatalytic activity of NCN-1000-5. As a showcase for its application in energy devices, we assembled a conventional Zn–air battery with NCN-1000-5 as the air cathode catalyst, which exhibits outstanding rechargeable performance and ultrastrong durability.
Results and discussion
The NCNs were prepared by a spontaneous gas-foaming approach using citric acid and NH4Cl as raw materials (Scheme 1). Due to its abundant carbon atoms and powerful combining capacity to form complexes, citric acid was selected as the carbon source to prepare NCNs. As for NH4Cl, it not only served as a nitrogen source for NCNs, but also acted as a foaming agent to construct the cross-linked 3D porous network structure, since NH4Cl can be decomposed into NH3 and HCl during the carbonization process. On one hand, the resulting NH3 can further combine with the carboxyl from citric acid through a dehydration–condensation reaction, which in situ introduces the N atoms uniformly into the graphite lattice after carbonization; on the other hand, the large amounts of released NH3 and instantaneous high internal pressure can blow the carbon matrix to form defect-rich and ultrathin carbon nanosheets (Fig. S1, ESI†). The optical images of the preparation process for NCNs are presented in Fig. S2 (ESI†). Besides, it needs to be emphasized that the spontaneous gas-foaming strategy is versatile and highly valuable for preparing advanced carbon materials with ultrahigh specific surface area and abundant porous structures (Fig. S3, ESI†). | ||
| Scheme 1 The schematic procedure for synthesizing the NCNs. | ||
To investigate the evolution process of NCNs, a series of conditional experiments on carbonization temperature and the mass ratio of precursors are implemented. Fig. S4 and S5 (ESI†) present the scanning electronic microscopy (SEM) and transmission electron microscopy (TEM) images of the obtained products carbonized at different temperatures. Without the presence of NH4Cl, only large carbon blocks (C-1000) can be obtained even carbonizing at 1000 °C (Fig. 1a and Fig. S4a, ESI†). After carbonizing the precursors (the mass ratio is 1:1) at a relatively low temperature of 800 °C, the rudiment of thick carbon nanosheets (NCN-800-5) with porous architecture emerged (Fig. 1b and Fig. S4b, ESI†). When the carbonization temperature rapidly rises to 1000 °C, cross-linked 3D porous carbon networks (NCN-1000-5) can be clearly observed, which are composed of interconnected ultrathin carbon nanosheets with a thickness of ∼2 nm (Fig. 1e and h). The corresponding TEM images (Fig. 1d and f) further reveal the existence of numerous mesopores and nanopores on the graphene-like ultrathin carbon nanosheets. The high-resolution TEM (HRTEM) image shows that the edges of NCN-1000-5 are only 4 graphitic layers thick (Fig. 1g). A lattice spacing of 0.42 nm and distinct diffraction rings can be observed from the high-resolution TEM (HRTEM) image (Fig. 1g) and selected-area electron diffraction (SAED) pattern (Fig. S6, ESI†) of NCN-1000-5, which correspond to the (002) plane of the typical hexagonal pattern for graphitic carbon.12,19 The elemental mapping images of NCN-1000-5 (Fig. S7, ESI†) indicate the even distribution of C, O and N elements in this sample. Atomic force microscope (AFM) images clearly demonstrate that the thickness of NCNs gradually decreases from 800 to 1000 °C (Fig. S8, ESI†). Thus, the carbonization temperature is crucial for regulating the porosity and thickness of carbon nanosheets. Moreover, the mass ratio of citric acid to NH4Cl also plays a significant role in influencing the morphology and structure of the NCNs (Fig. S9, ESI†).
 | ||
| Fig. 1 TEM images of (a) C-1000, (b) NCN-800-5, (c) NCN-900-5 and (d) NCN-1000-5; (e) SEM, (f) TEM, (g) HRTEM and (h) AFM images of NCN-1000-5. | ||
We employed the X-ray diffraction (XRD) pattern and Raman spectra to examine the crystal texture of the pyrolytic carbon products. The two broad diffraction peaks at 2θ ≈ 24.8 and 44° in XRD (Fig. S10a, ESI†) are well indexed to the (002) and (101) facets of graphite.20,21 The corresponding Raman spectra clearly show characteristic D and G bands at 1340 cm−1 and 1591 cm−1, which are assigned to disordered carbon atoms and sp2 hybridized graphitic carbon atoms, respectively (Fig. S10b, ESI†).22,23 The gradually increased values of ID/IG for NCNs, which imply the insertion of N atoms under high pyrolytic temperature, indicate the formation of more edge and topological defects in NCNs.12,24 These abundant defect sites can modulate and tune the electronic and surface properties of NCN-1000-5, and thus optimize the adsorption energies of electrochemical catalysis steps.24,25
The surface area and porous structure of the resultant samples were further characterized by N2 adsorption/desorption measurements. The type I isotherm curves and rapid nitrogen uptake (P/P0 < 0.1) (Fig. 2a) confirm the existence of numerous micropores and mesopores in NCNs.26,27 The specific surface area of NCNs increases remarkably with increasing the carbonization temperature and the mass of NH4Cl (Table S1, ESI†). Particularly, NCN-1000-5 has the highest ultrahigh specific surface area of 1793 cm2 g−1 among all the synthesized NCN samples (nearly 48 times larger than that of C-1000, 37 cm2 g−1), and this value outperforms the vast majority of doped carbon materials, e.g. N,P-doped mesoporous nanocarbon foams (1663 cm2 g−1),1 P,S-codoped carbon nitride sponges (1474 cm2 g−1),28 N,S-enriched porous carbon (830 cm2 g−1),29 N,P-doped porous carbon networks (743 cm2 g−1)12 and N,S-doped graphitic sheets (576 cm2 g−1).18 The corresponding pore size distribution curves indicate that the NCNs possess large pore volume and are dominated by micropores and mesopores (Fig. 2b and Table S1, ESI†). The existence of micropores greatly enhances the specific surface areas of NCNs and provides a high density of active sites, while the mesopores are conducive to creating more structural defects and transporting the reaction intermediates. The ultrahigh specific surface area and large pore volume of NCN-1000-5 are expected to facilitate the mass transport and expose more active surface sites for electrochemical reactions.12,30,31
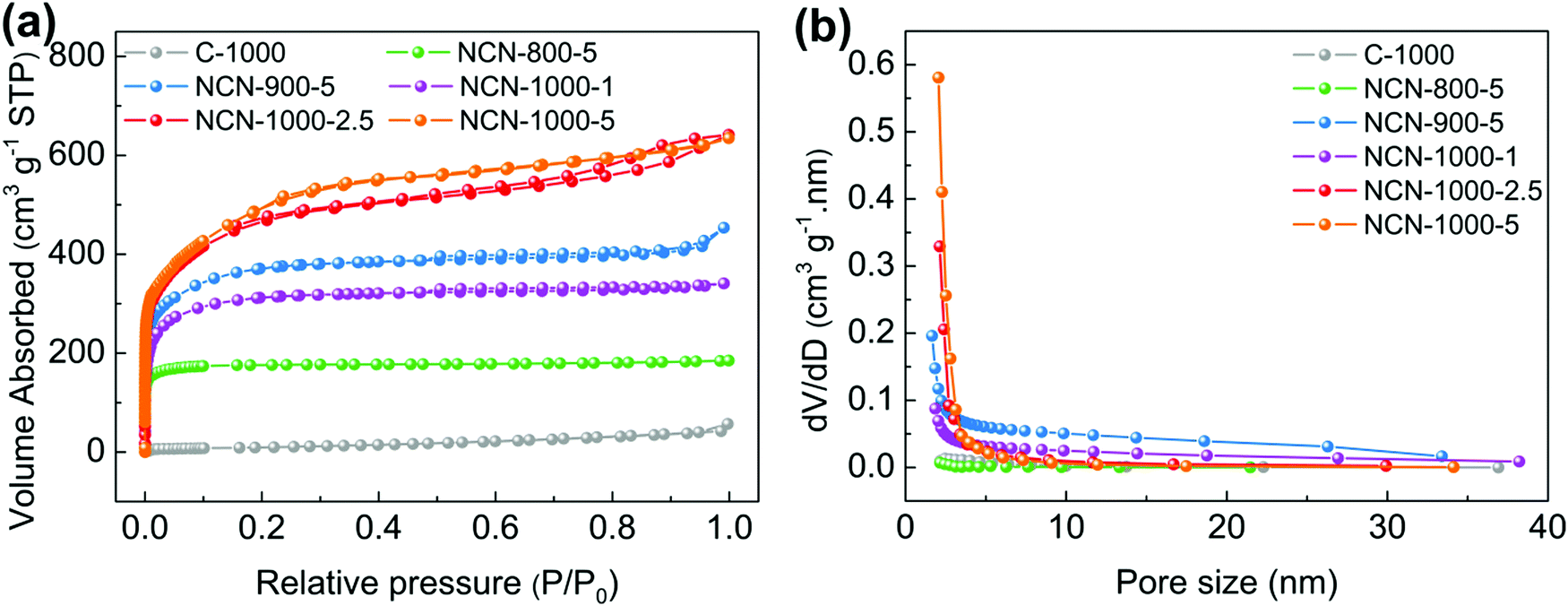 | ||
| Fig. 2 (a) N2 absorption/desorption isotherms and (b) pore size distribution of C-1000, NCN-800-5, NCN-900-5, NCN-1000-1, NCN-1000-2.5 and NCN-1000-5. | ||
X-ray photoelectron spectroscopy (XPS) was executed to investigate the chemical composition and bonding configurations of the NCNs. Fig. 3a and Fig. S11 (ESI†) confirm the presence of C, O and N elements in the NCNs, which are in accordance with the elemental mapping results (Fig. S7, ESI†). The corresponding atomic percentages of the above elements are listed in Table S2 (ESI†). The fitted high-resolution C 1s spectrum can be classified into four peaks at about 284.8, 285.7, 286.2 and 288.3 eV, corresponding to C–C, C–N, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively (Fig. 3b).18,32 The successful doping of N atoms into the carbon skeleton is verified by the corresponding high-resolution N 1s spectrum. As displayed in Fig. 3d and Fig. S11 (ESI†), N 1s of the NCNs can be classified into pyridinic N (398.3 eV), pyrrolic N (399.8 eV), graphitic N (401.1 eV) and pyridinic N oxide (402–406 eV), respectively.33,34 It has been generally accepted that pyridinic N, pyrrolic N and graphitic N play significant roles in the reversible oxygen reduction/evolution reaction and HER processes.18,35 Further investigations reveal that the percentage of pyridinic N and pyrrolic N remarkably decreased, whereas the content of graphitic N increases from 800 to 1000 °C (Fig. 3c and Table S3, ESI†). This is because pyridinic N and pyrrolic N are thermolabile and are inclined to transform into graphitic N with increasing temperature. Elemental analysis of the possible existence of transition metals in NCN catalysts was further conducted using inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The detected total transition metal (Fe, Co and Ni) content of NCN-1000-5 is below 0.01 wt% (Table S4, ESI†), indicating that the amount of metal Fe, Co and Ni is negligible. In addition, the functional groups on the surface of NCNs were further identified by Fourier transform infrared spectra (FT-IR), as displayed in Fig. S12 (ESI†).
O, respectively (Fig. 3b).18,32 The successful doping of N atoms into the carbon skeleton is verified by the corresponding high-resolution N 1s spectrum. As displayed in Fig. 3d and Fig. S11 (ESI†), N 1s of the NCNs can be classified into pyridinic N (398.3 eV), pyrrolic N (399.8 eV), graphitic N (401.1 eV) and pyridinic N oxide (402–406 eV), respectively.33,34 It has been generally accepted that pyridinic N, pyrrolic N and graphitic N play significant roles in the reversible oxygen reduction/evolution reaction and HER processes.18,35 Further investigations reveal that the percentage of pyridinic N and pyrrolic N remarkably decreased, whereas the content of graphitic N increases from 800 to 1000 °C (Fig. 3c and Table S3, ESI†). This is because pyridinic N and pyrrolic N are thermolabile and are inclined to transform into graphitic N with increasing temperature. Elemental analysis of the possible existence of transition metals in NCN catalysts was further conducted using inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The detected total transition metal (Fe, Co and Ni) content of NCN-1000-5 is below 0.01 wt% (Table S4, ESI†), indicating that the amount of metal Fe, Co and Ni is negligible. In addition, the functional groups on the surface of NCNs were further identified by Fourier transform infrared spectra (FT-IR), as displayed in Fig. S12 (ESI†).
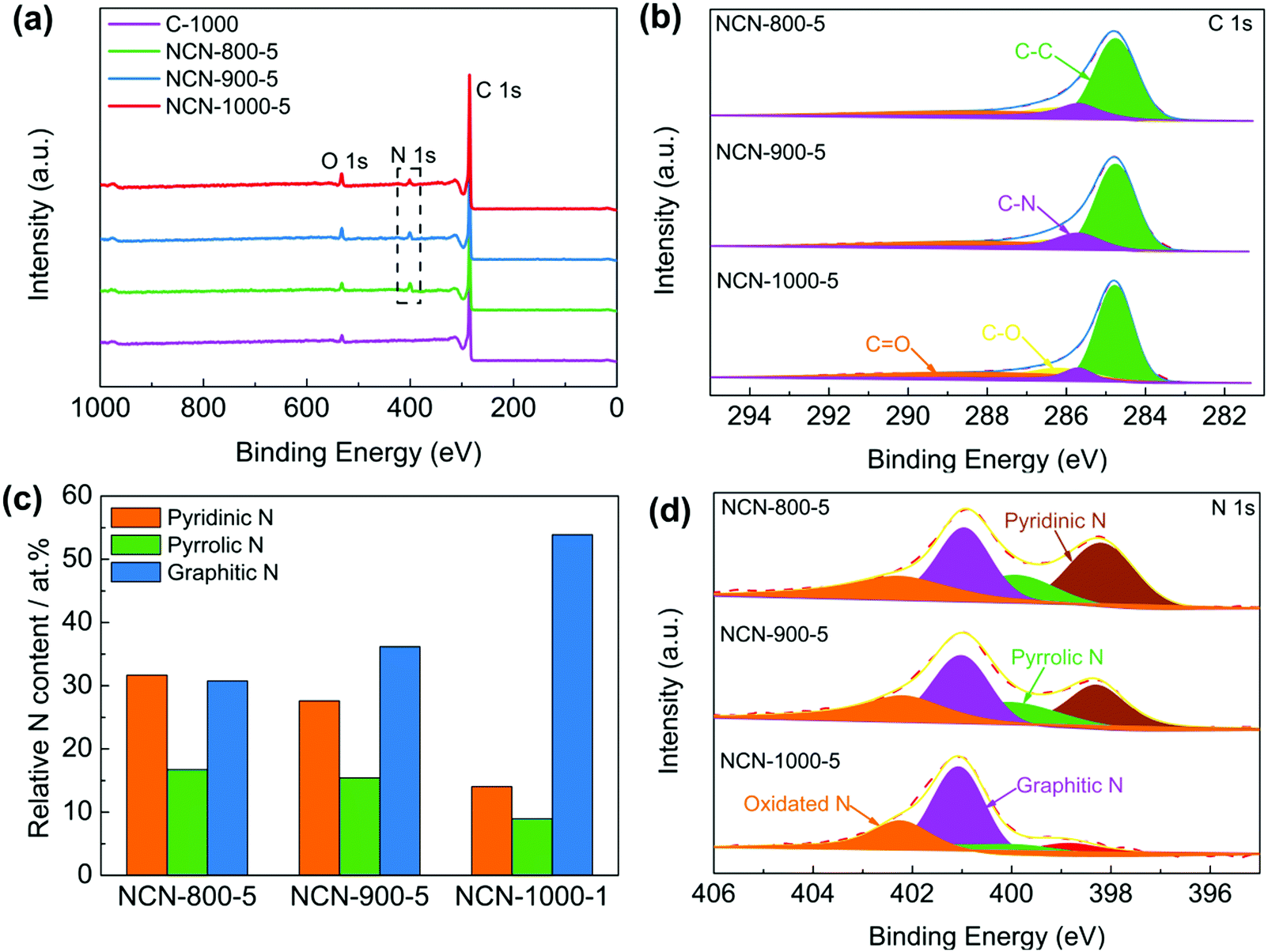 | ||
| Fig. 3 (a) The wide XPS survey spectra of C-1000, NCN-800-5, NCN-900-5 and NCN-1000-5; (b) high resolution C 1s spectra, (c) the contents of different kinds of nitrogen and (d) N 1s spectra of NCN-800-5, NCN-900-5 and NCN-1000-5. | ||
To further investigate the electronic structure and local chemical configuration of the carbon-based catalysts, near edge X-ray absorption fine structure (NEXAFS) experiments were carried out. The obtained C K-edge spectra of C-1000 and NCNs are extremely similar (Fig. 4a), indicating similar environments of carbon skeletons present. Defects at 284.0 eV are assigned to the low-coordination carbon atoms at the edges of NCNs, which can lead to the rehybridization of C atoms and break the integrity of π conjugation.36–38 In addition, a portion of active unpaired π electrons locate at each edge carbon atom, which can effectively facilitate electron transfer to O2.39 The excitations of π* at 285.4 eV (peak C1) and σ* at 291–293 eV (peak C3) are attributed to CC and C–C species in C-1000 and the NCNs, respectively. The peak C2 at 288.4 eV suggests the formation of C–N–C or C–O–C.40,41 Due to the decomposition of oxygen-containing functional groups and the loss of N atoms at high pyrolysis temperature, the intensity of peak C2 from NCN-800-5 to NCN-1000-5 displays a decrease. In contrast to the overlapping peaks of XPS analysis (Fig. 3d), three well-defined peaks below the ionisation potential of N corresponding to transitions from 1s into the π* orbitals and a broad peak in the energy range 407–408 eV corresponding to the σ* resonance can be observed in N K-edge NEXAFS spectra (Fig. 4b). Among them, the resonances of π* at 398.1 (N1), 399.2 (N2) and 401.4 eV (N3) are contributed by pyridinic N, pyrrolic N and graphitic N, respectively.42 The excitations of σ* at 407–408 eV are attributed to C–N–C or C–N.41 The existence of N species improves the charge mobility of the carbon matrix by introducing electron-donor characteristics and enhancing the carbon catalytic activity in electron-transfer reactions.43
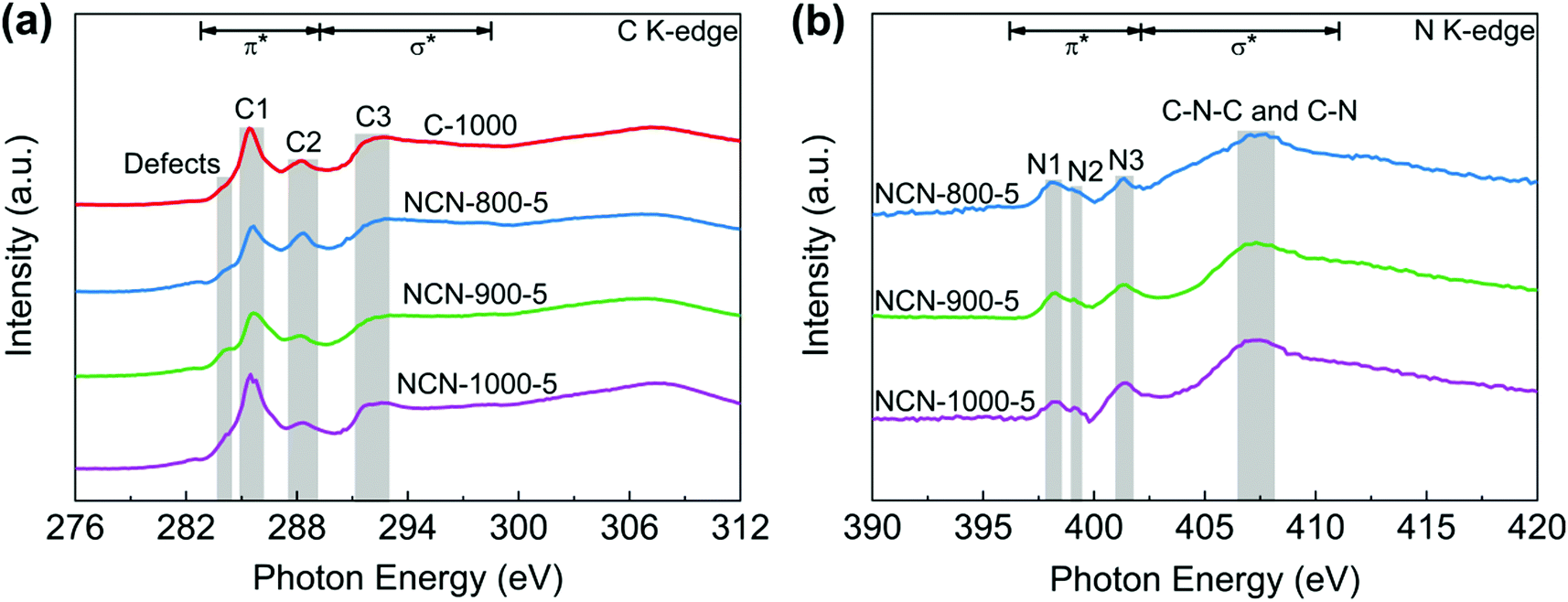 | ||
| Fig. 4 (a) C K-edge and (b) N K-edge NEXAFS spectra of C-1000, NCN-800-5, NCN-900-5 and NCN-1000-5. | ||
Catalytic activity towards the ORR
The catalytic activity of NCNs toward the ORR was initially investigated in 0.1 M KOH electrolyte. The most positive reduction peak potential (Ep) at 0.86 V was observed in the cyclic voltammetry (CV) curves (Fig. 5a and Fig. S13a, ESI†), implying that NCN-1000-5 possesses superior ORR activity. The linear scan voltammogram (LSV) curves (Fig. 5b and Fig. S13b, ESI†) further confirm the excellent ORR performance of NCN-1000-5, with a positive onset potential (Eonset) of 0.95 V, a half-wave potential (E1/2) of 0.82 V, and a high limiting current density of 6.43 mA cm−2 (Table S5, ESI†). Remarkably, these performance indexes of NCN-1000-5 are comparable to those of Pt/C (Table S5, ESI†) and some advanced metal free ORR catalysts reported in the literature (Table S6, ESI†). The comparable Tafel slopes for NCN-1000-5 (86 mV dec−1) and Pt/C (84 mV dec−1) signify that the transfer of the first electron may be the rate-determining step in the ORR catalyzed by them (Fig. 5c).44–46 The linear and parallel Koutecky–Levich (K–L) plots (Fig. S15, ESI†) derived from LSV curves (Fig. S14, ESI†) reflect a first-order reaction with respect to the dissolved O2.47 The calculated electron transfer number (n) for NCN-1000-5, 3.92 at 0.5 V based on K–L plots, confirms an apparent 4e− pathway to reduce O2. The corresponding kinetic current density (Jk) of NCN-1000-5 is 50 mA cm−2 at this potential (Fig. 5d), which is even slightly larger than that of Pt/C (46.3 mA cm−2).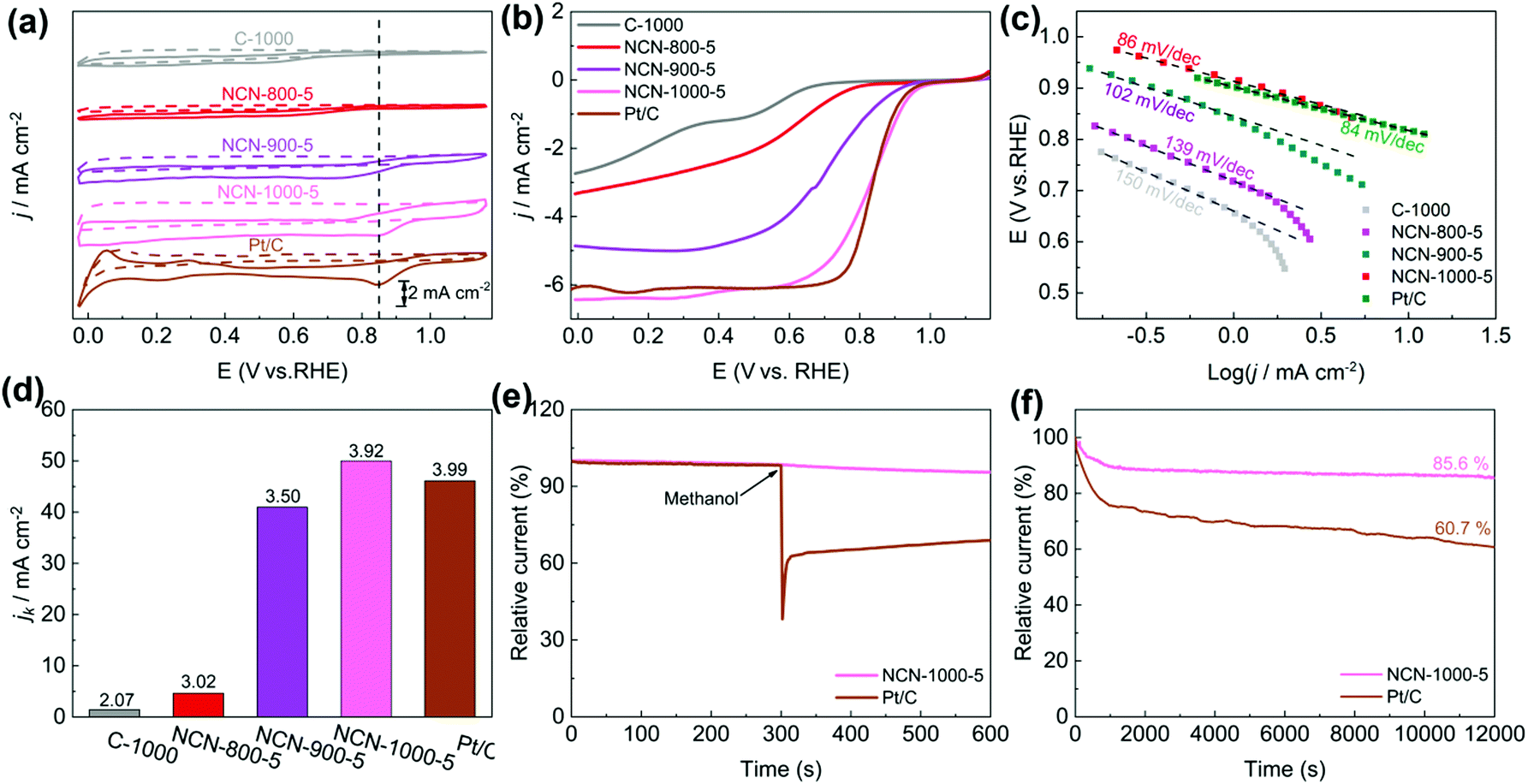 | ||
| Fig. 5 (a) CV curves of C-1000, NCN-800-5, NCN-900-5, NCN-1000-5 and Pt/C in N2 (dotted line) and O2 (solid line)-saturated 0.1 M KOH (50 mV s−1); (b) LSV curves and (c) Tafel plots of various catalysts (1600 rpm, 5 mV s−1); (d) electrochemical activity given as the kinetic current density (Jk) at 0.5 V (vs. RHE) for various catalysts, the numeral on the bar represents the corresponding electron transfer number; and (e) methanol crossover tolerance and (f) durability tests of NCN-1000-5 and Pt/C at 0.67 V (1600 rpm). | ||
Besides the high ORR activity, NCN-1000-5 also exhibits super immunity to methanol crossover (Fig. 5e). The ORR relative current of Pt/C shows a cliff-like drop after 0.5 mL methanol (3 M) is added into the electrolyte at 300 s, whereas the current of NCN-1000-5 remains almost unchanged. Furthermore, a high current retention (85.6%) after 12000 s continuous testing demonstrated the long-term stability of NCN-1000-5 (Fig. 5f). These results clearly demonstrate that NCN-1000-5 possesses excellent activity, strong stability and high selectivity toward the ORR in alkaline media, all of which are extremely significant for practical applications. XPS results reveal that the oxygen content of NCN-1000-5 increases after long-term i-t testing (Fig. S16 and Table S7, ESI†). It is possible that the nitrogen-doped sample has been partly oxidized by oxygen atoms and some oxygen containing functional groups were formed on the N doped carbon nanosheets.
More surprisingly, the NCN-1000-5 sample also displayed preeminent ORR activity and stability in acidic media, as depicted in Fig. S17 (ESI†). Similar to the situation described in alkaline media, NCN-1000-5 undergoes first-order reaction kinetics and exhibits an apparent 4e− pathway to reduce O2 in acidic media, as indicated by the LSV curves (Fig. S18, ESI†) and linear K–L plots (Fig. S19, ESI†). The Eonset of NCN-1000-5 is 0.78 V, only 20 mV more negative than that of Pt/C in 0.5 M H2SO4 electrolyte (Table S8, ESI†). The slightly lower activity in acidic media may be due to the protonation of N atoms and the subsequent adsorption of SO42− anions on the surface of NCN-1000-5, which covers up some active sites.48 Although the ORR activity of NCN-1000-5 is slightly worse than that of Pt/C in acidic media, it still outperforms some metal-free catalysts reported in the literature (Table S9, ESI†).
Catalytic activity towards the OER
To demonstrate the trifunctional activity, we then evaluated the OER performance of the NCNs. Similar to the case for the ORR, NCN-1000-5 exhibits much better OER activity than that of the other NCN samples and pristine carbon blocks (C-1000). The Eonset and measured potential at a current density of 10 mA cm−2 (Ej=10) of NCN-1000-5 (Fig. 6a and b) are 1.55 and 1.64 V, respectively, which are slightly inferior to those of IrO2 (1.48 V and 1.59 V) but comparable to many advanced NNMCs for the OER (Table S10, ESI†). A small current was detected in the LSV curves of NCNs before the OER occurred (Fig. 6a), which may be caused by the side reactions of some oxygenic functional groups.13 The enhanced OER kinetics of NCN-1000-5 was further demonstrated by its small Tafel slope of 142 mV dec−1 at low overpotential (Fig. 6c). NCN-1000-5 also keeps high stability with a consistent OER current after 12000 s continuous operation (Fig. 6d). The strong durability of NCN-1000-5 is reconfirmed by its LSV curves with little variation after 500 cycles (the insert in Fig. 6d). Moreover, NCN-1000-5 shows the smallest Nyquist circle in electrochemical impedance spectroscopy (EIS) compared with the other NCN samples (Fig. S20, ESI†), demonstrating its quickest charge transfer ability.49 The potential difference (ΔE, defined in eqn (S4), ESI†) of the OER and ORR is generally used to estimate the performance of bifunctional oxygen electrode catalysts.50,51 A better bifunctional catalyst tends to have a smaller value of ΔE. Remarkably, NCN-1000-5 shows a low ΔE value of 0.81 V (Fig. 6e), which is smaller than that of RuO2 (1.51 V) and some reported bifunctional electrocatalysts (Fig. 6f and Table S11, ESI†).
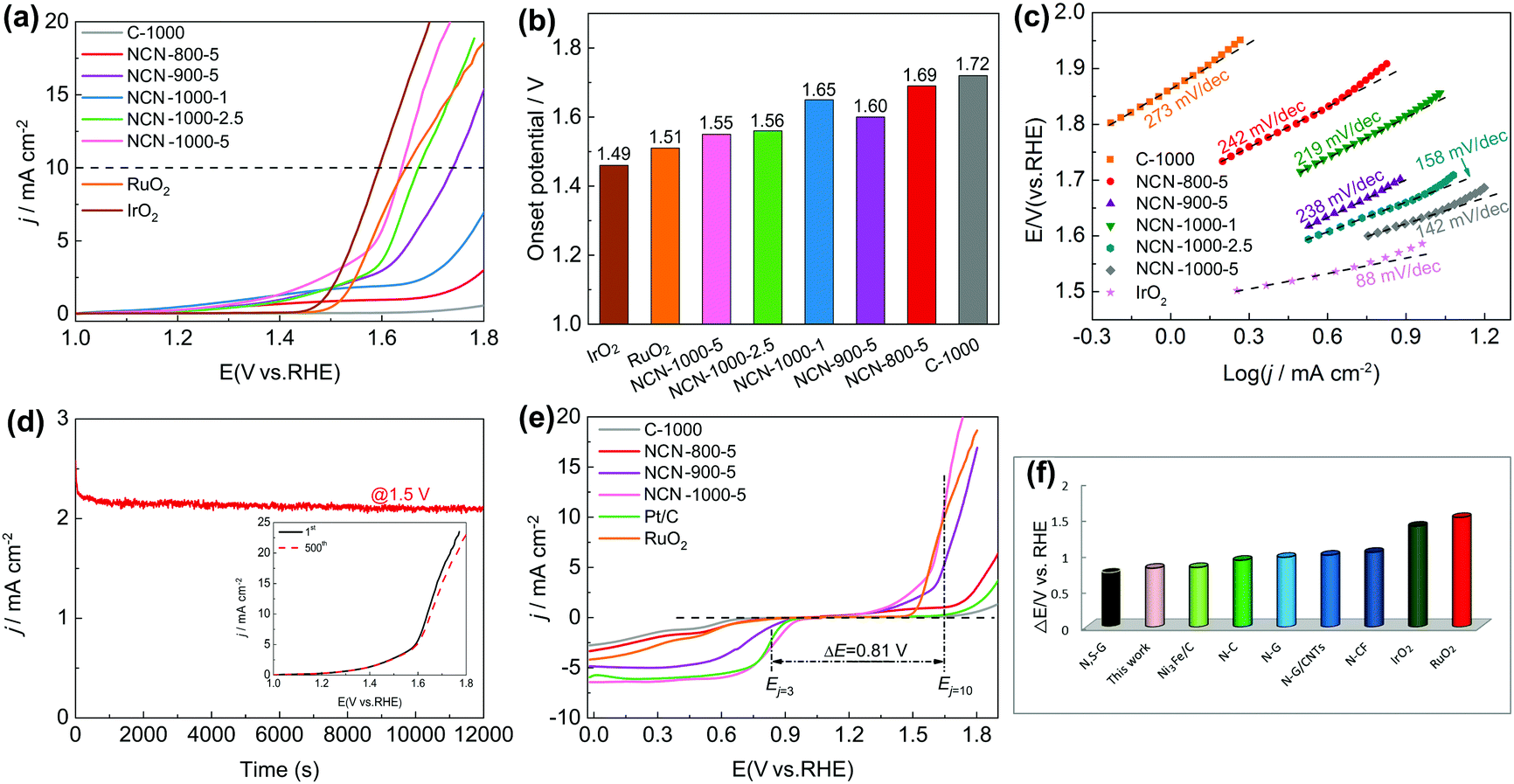 | ||
| Fig. 6 (a) LSV curves of C-1000, NCN-800-5, NCN-900-5, NCN-1000-1, NCN-1000-2.5, NCN-1000-5, RuO2 and IrO2 in O2-saturated 0.1 M KOH electrolyte (1600 rpm, 5 mV s−1); (b) onset potentials and (c) Tafel plots of various catalysts; (d) the i–t plot at the applied potential of 1.5 V (vs. RHE), the inset shows the LSV curves of NCN-1000-5 before and after 500 CV cycles; (e) the overall LSV curves for the ORR and OER of various catalysts at 1600 rpm; and (f) comparison of ΔE for NCN-1000-5 and various carbon-based materials. | ||
Catalytic activity towards the HER
Furthermore, we observed exciting HER activity of the NCNs in N2-saturated 0.5 M H2SO4 electrolyte. As displayed in Fig. 7a and Fig. S21a (ESI†), the NCN samples exhibit greatly improved HER activity as compared with the pristine carbon blocks (C-1000) and glass carbon (GC). Remarkably, NCN-1000-5 shows superior HER performance with almost the same positive Eonset as Pt/C (−0.03 V), which is much positive than that of the other as-prepared NCN samples in this work. At a current density of 10 mA cm−2 (Ej=10), the obtained potential for NCN-1000-5 (−0.09 V) is only 51 mV more negative than that of Pt/C (Fig. 7b). The excellent kinetic property of NCN-1000-5 is confirmed by its small Tafel slope of 43 mV dec−1 (Fig. 7d and Fig. S21b, ESI†).52 In fact, the HER performance of NCN-1000-5 is also better than that of many reported carbon materials, as presented in Fig. 7c and Table S12 (ESI†). Fig. 7e displays similar LSV curves at sweep speeds from 5 to 100 mV s−1 for NCN-1000-5, suggesting its robust stability for highly active electrochemical processes. The excellent stability of NCN-1000-5 was further verified by its almost unchanged LSV curves after 500 CV cycles (Fig. 7f) and a consistent HER current after 12000 s continuous operation at −0.15 V (the insert in Fig. 7f).
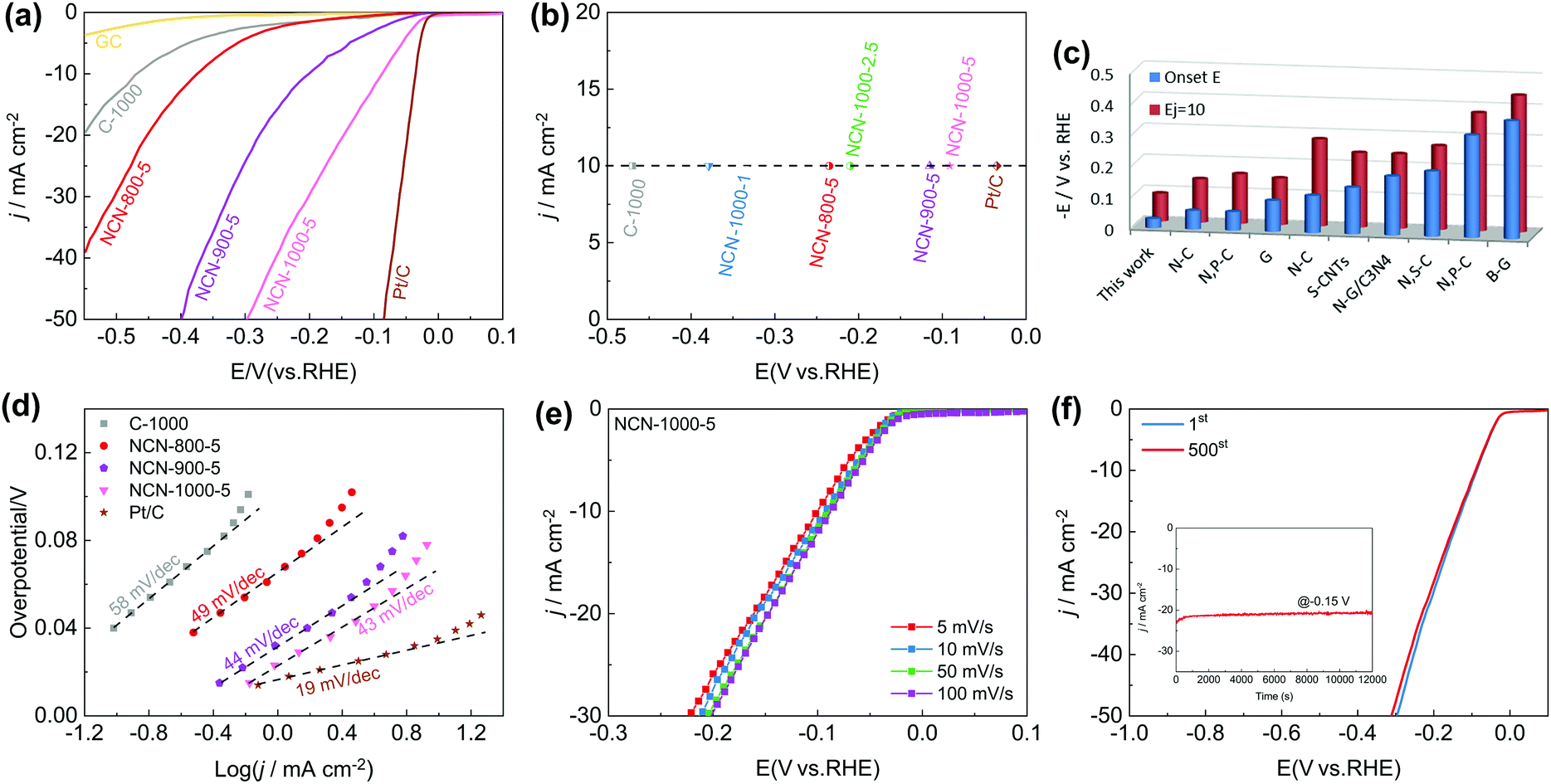 | ||
| Fig. 7 (a) LSV curves of GC, C-1000, NCN-800-5, NCN-900-5, NCN-1000-5 and Pt/C in N2-saturated 0.5 M H2SO4 (5 mV s−1); (b) the measured potential at a current density of 10 mA cm−2 (Ej=10) for various catalysts; (c) comparison of the activities (Eonset and Ej=10) for NCN-1000-5 and various carbon-based materials; (d) Tafel curves of various catalysts; (e) LSV curves of NCN-1000-5 with scan rates from 5 to 100 mV s−1; and (f) the i–t plot at an applied potential of −0.15 V (vs. RHE), the inset shows the LSV curves of NCN-1000-5 before and after 500 CV cycles. | ||
Catalytic mechanisms revealed by DFT computations
Our experimental studies strongly demonstrate that the hierarchical porous and defect-rich NCN-1000-5 is a promising cost-effective and trifunctional electrocatalyst for the ORR, OER and HER simultaneously. Then, we performed systematic DFT computations to reveal the underlying catalytic mechanisms and the active sites of NCNs for the ORR, OER and HER.To examine the effect of doping type (graphitic, pyridinic or pyrrolic N), doping concentration, and the distance of active sites away from the hole edges, we constructed two types of structural models (in total 14 models) in our theoretical studies. The first is a graphene monolayer doped by N atoms, which serves as a model to study the active sites far away from the hole edges; the second is graphene nanoribbons doped by N atoms, which are used to simulate the active sites on or close to the hole edges. The models are sketched in Fig. 8, and the 14 different models are detailed in the ESI.†
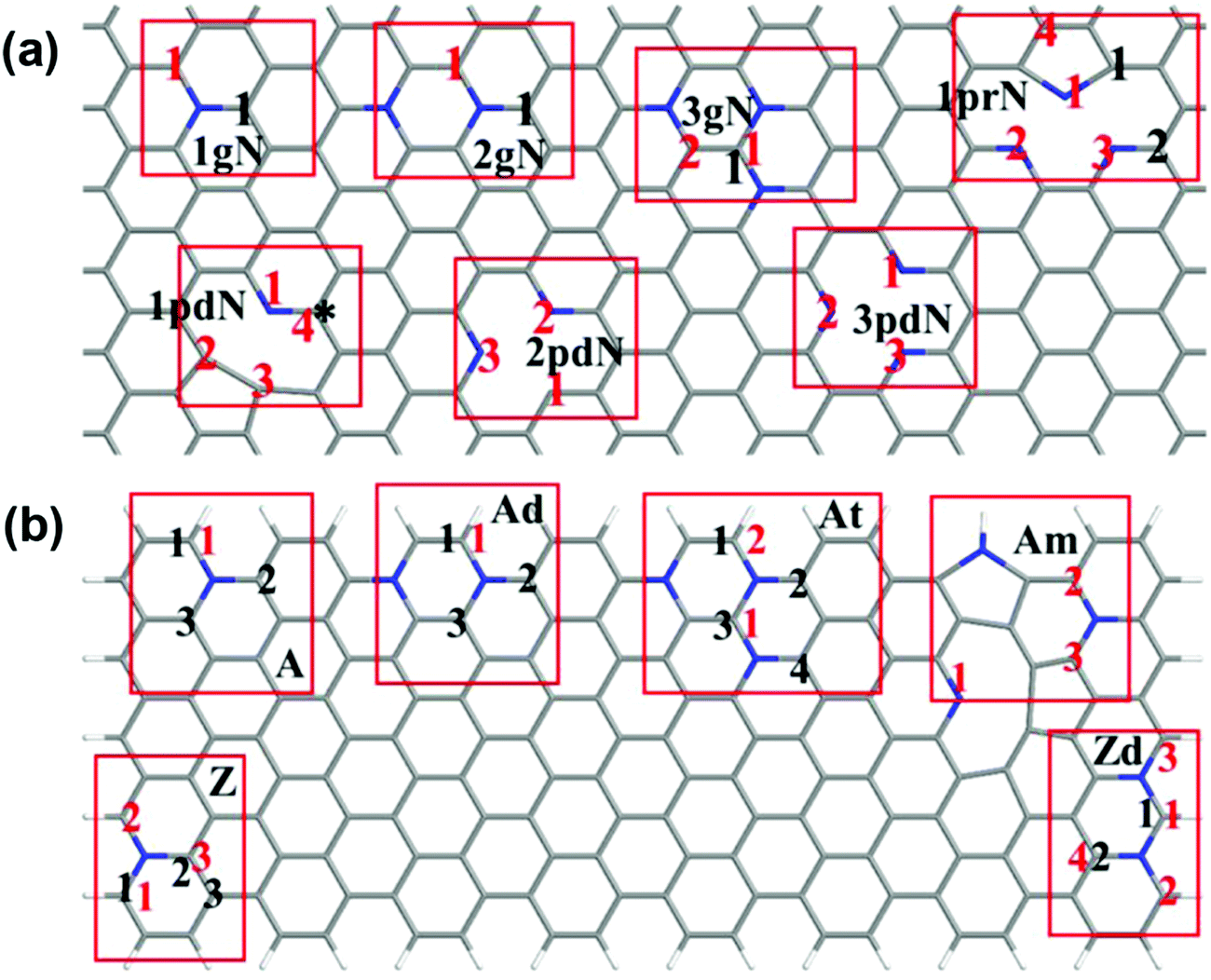 | ||
| Fig. 8 Schematic presentation of the constructed models. (a) Doped graphene monolayer; (b) doped graphene nanoribbons. The pristine undoped carbon nanosheet is labeled as PC, while the graphitic N, pyridinic N, and pyrrolic N doping models are marked as ngN, npdN, and nprN respectively, where n (1–3) is the number of a specific N dopant in the selected supercell. Labels A, Ad, At, Z, Zd and Am are used for graphene nanoribbons: A and Z denote the armchair and zigzag nanoribbons, d and t mean double and ternary N doping, and m refers to the mixed doping where three N dopants are close to each other. Herein, the star (*) and black numbers relate to studied active sites for the ORR/OER, whereas the red numbers relate to potential HER active sites. More structural details are in Fig. S22 and S23 (ESI†). | ||
The possible active sites for the ORR and OER on N-doped graphene are basically the carbon atoms adjacent to the N dopant.53–55 These carbon atoms have either high positive atomic charge or high spin density, and are labelled as M-x, where M is the model, and x refers to the numbering of the active site where the reaction occurs. For instance, site 2 of the one-graphitic-N doped armchair nanoribbon (A) is termed as A-2.
Firstly, we investigated the catalytic performance of various active sites for the ORR and OER in acidic media (pH = 0) by examining their overpotentials (η) according to the 4e− mechanism and computational hydrogen electrode model.56 More computational details are included in the ESI.†Fig. 9a presents the volcano plot of ORR/OER overpotential versus ΔG(*O) for various active sites on the N-doped graphene monolayer and armchair and zigzag graphene nanoribbons. Note that the left/right side of the x axis indicates strong/weak interaction between the O atom and the substrates. Obviously, PC has the weakest interaction with O, leading to the poor performance for the ORR or OER. Once doped by N atoms, this kind of interaction becomes stronger, thus decreasing the overpotential for the ORR/OER in some cases. Because the optimal catalytic active site should possess the lowest overpotential, we confirmed that among all the examined possible active sites, the most efficient active site for the ORR is the carbon atom that is at the armchair nanoribbon edge and adjacent to the graphitic N dopant (A-1, Fig. 9b). Meanwhile, the most efficient active site for the OER is the carbon atom that is near the graphitic N dopant, but 3.34 Å away from the armchair ribbon edge (A-3, Fig. 9c). We also compared the monolayer models with three different N dopant types (1gN-1, 1pdN-1 and 1prN-1 for the graphitic, pyridinic, and pyrrolic N, respectively). Among these models, 1gN-1 has the smallest overpotential, indicating that the graphitic N plays the key role in enhancing the ORR/OER activities for carbon nanosheets. These results are consistent with previous reports.57,58 Then, the HER activity of NCNs in acidic media (pH = 0) was evaluated by the descriptor ΔG(*H), the Gibbs free energy of hydrogen adsorption.59 An ideal HER catalyst has a nearly zero ΔG(*H) value. Interestingly, the best active site for the ORR, namely the carbon atoms at the armchair nanoribbon edge adjacent to the graphitic N dopants (A-1), also has the highest HER activity, and its ΔG(*H) value is 0.07 eV at a hydrogen coverage of 2.27% (Fig. S25, ESI† and Fig. 9d). Typically, N-doping leads to different local structures, and thus affects the electronic properties of carbon nanosheets, and finally alters the adsorption behavior for H and O atoms. The band structure computation (Fig. S26, ESI†) reveals that graphitic N introduces n-type doping, while pyridinic and pyrrolic N introduce p-type doping in the graphene nanosheets/nanoribbons. According to the plots of Fig. 9a and Fig. S25 (ESI†), it is concluded that p-type doping favors the adsorption of H atoms, and n-type doping favors the adsorption of O atoms, because O is highly electronegative.
 | ||
| Fig. 9 (a) The volcano plot for the ORR and OER by plotting the overpotential as a function of ΔG(*O) at various possible active sites. The top and side views of the active site (b) A-1 for the ORR, (c) A-3 for the OER with OOH adsorbed, and (d) A-1 for the HER, the green ball representing the adsorbed H (θ = 2.27%). | ||
The above computations show that the carbon atoms close to the graphitic N dopants are the most active sites for the ORR, OER and HER reactions, and graphitic N dopants are more vital than the pyridinic and pyrrolic N dopants. These most active sites are at the armchair edges of our computed models, which correspond to the armchair edge of the large holes found in experiments. These findings echo the experimental results that the sample prepared at high temperature (1000 °C), which has a greater percentage of graphitic N and abundant micropores as well as edge defects, has more enhanced catalytic performance than those prepared at low temperature (800 °C).
Application in a Zn–air battery
As superior ORR/OER bifunctional electrocatalysts are demanded by metal–air batteries,60 we fabricated a conventional Zn–air battery employing NCN-1000-5 as the catalyst in the air-cathode (Fig. S27, ESI†). The assembled battery affords a high open circuit voltage of 1.44 V (Fig. 10a), close to that of Pt/C + RuO2 (1.47 V). Fig. 10b presents the discharge polarization (V–j) curves and the corresponding power density plots. The peak power density of the Zn–air battery equipped with the NCN-1000-5 catalyst is calculated to be 207 mW cm−2, which is slightly larger than that of Pt/C + RuO2 (196 mW cm−2). As revealed in Fig. 10c, the small charge/discharge voltage gap of NCN-1000-5 is approximately equal to that of Pt/C + RuO2, indicating its excellent recharge ability when used as an oxygen electrode bifunctional catalyst in Zn–air batteries. The battery with the NCN-1000-5 air cathode exhibits a voltage platform of 1.21 V (Fig. 10d and e) at 10 mA cm−2, approximating that of Pt/C + RuO2 (1.25 V). After normalizing to the weight of the dissipative Zn plate, a high specific capacity and energy density, 672 mA h g−1 and 805 W h kg−1, are achieved for NCN-1000-5, which are slightly smaller than those of Pt/C + RuO2 (724 mA h g−1 and 890 W h kg−1). Gratifyingly, the battery can be successfully regenerated by replacing a new Zn plate after full discharge. Moreover, we do not observe any distinct voltage decline after five cycles at 10 mA cm−2, and it maintains continuous discharge over 127 h (Fig. 10f), revealing remarkable long-term durability of NCN-1000-5 for the ORR. Furthermore, the cycling performance of the batteries was assessed employing galvanostatic 10 min discharging and 10 min charging at 10 mA cm-2 (Fig. 10g). Initially, the battery equipped with the NCN-1000-5 catalyst affords a discharge voltage of 1.21 V and a charge voltage of 1.98 V, with a diminutive voltage gap of 0.77 V and a high initial round-trip efficiency of 61.1%. After 1000 continuous charge/discharge cycles (about 330 h), the voltage gap of the NCN-1000-5 air cathode is almost unchanged (Fig. S28, ESI†). Whereas, a distinct deterioration is observed for the Pt/C + RuO2 air cathode in a shorter cycling period (436 cycles, about 145 h), with an enlarged voltage gap of 0.47 V. More importantly, the preeminent discharging capability and cycling performance of the Zn–air battery equipped with NCN-1000-5 are also comparable to or even better than those of reported advanced bifunctional catalysts (Table S14, ESI†). To demonstrate the feasibility for energy supply in some practical power devices, several Zn–air batteries connected in series were constructed and integrated into circuits. As exemplified in Fig. 10h, two concatenated Zn–air batteries equipped with the NCN-1000-5 catalyst can provide sufficient voltage to power a red LED (2.0 V). Hence, these results sufficiently illustrate that NCN-1000-5 can be used as a promising bifunctional oxygen electrode catalyst for metal–air batteries.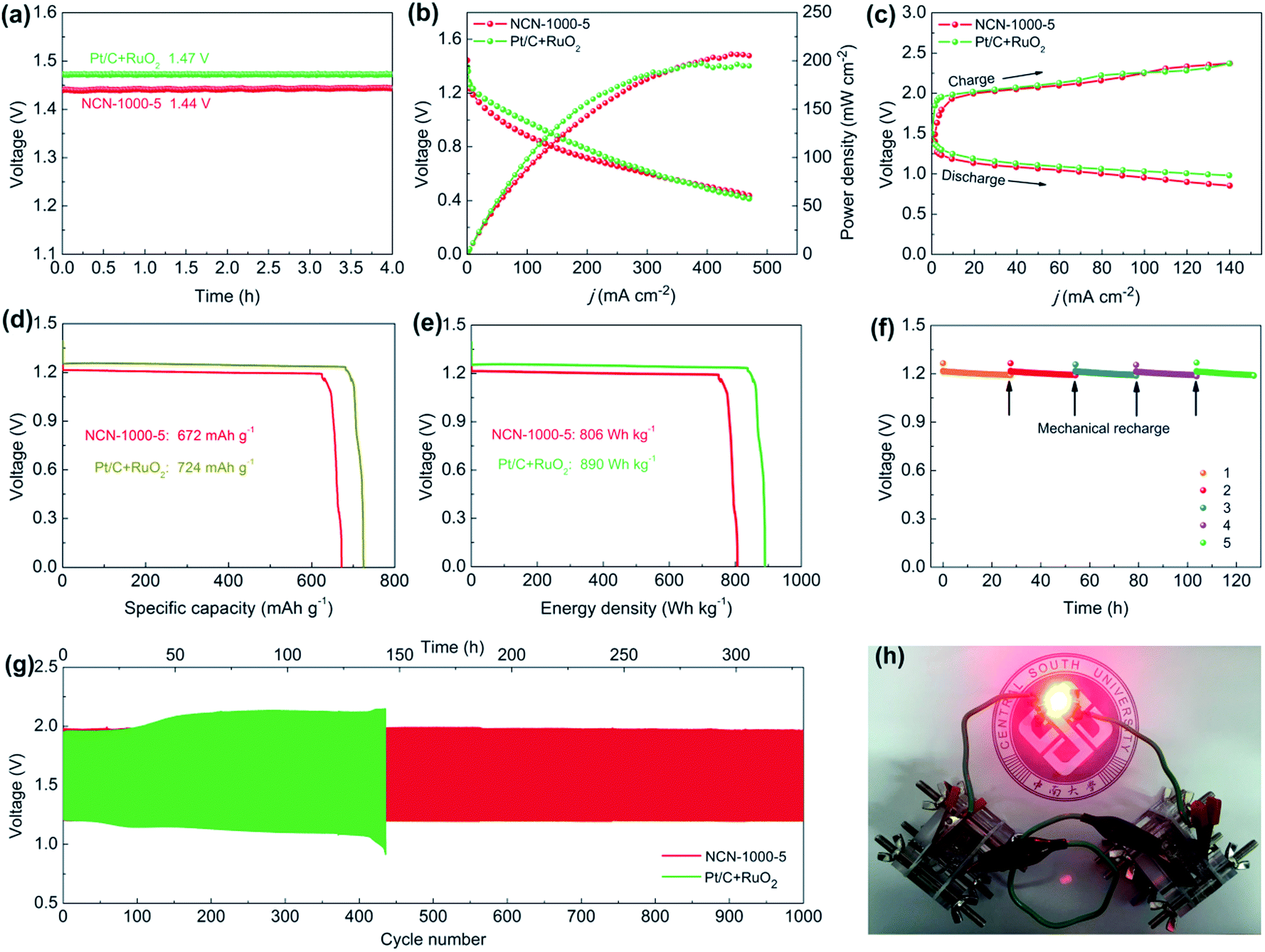 | ||
| Fig. 10 Zn–air battery performance with NCN-1000-5 acting as the air cathode in comparison with the coupled noble metal Pt/C + RuO2 catalysts. (a) Open circuit plots, (b) discharge polarization (V–j) curves and the corresponding power density curves, (c) charge and discharge polarization (V–j) curves, (d) specific capacities and (e) the corresponding energy density plots at 10 mA cm−2; (f) long-term stability of the primary Zn–air battery with the NCN-1000-5 cathode at 10 mA cm−2; (g) galvanostatic discharge and charge cycling curves of the Zn–air battery at 10 mA cm−2 with 10 min discharge and 10 min charge; and (h) photograph of a red LED (2.0 V) powered by two Zn–air batteries based on NCN-1000-5. | ||
Conclusions
In summary, we have developed a spontaneous gas-foaming strategy to fabricate neoteric nitrogen doped carbon nanosheets (NCN-1000-5) with abundant micropores and edge defects, a unique ultrathin nanosheet architecture and an ultrahigh specific surface area. These exceptional advantages endow NCN-1000-5 with superior trifunctional electrocatalytic activities and strong durability for simultaneous ORR, OER and HER. DFT calculations revealed that the carbon atoms located at the armchair edge and adjacent to the graphitic N dopants act as the most active sites for the ORR, OER and HER. The theoretical results well explain why NCN-1000-5 performs the best among our synthesized samples. Due to the outstanding activity of NCN-1000-5, the rechargeable Zn–air battery using NCN-1000-5 as the cathode catalyst exhibited a high energy density, a low charge/discharge voltage gap, ultrastrong reversibility and long cycling stability. These findings are encouraging for researchers to synthesize catalysts with high specific surface area and rich edge defects as much as possible for the ORR, OER and HER. This work not only presents an innovative and universal strategy to prepare advanced carbon materials with ultrahigh specific surface area and abundant edge defects, but also provides useful guidance for designing and developing multifunctional metal-free catalysts for various energy-related electrocatalytic reactions.Experimental section
The synthesis of NCNs, instruments and measurements, and fabrication and measurements of Zn–air batteries are described in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (No. 21878340), the Hunan Provincial Science and Technology Plan Project, China (No. 2017TP1001), Project of Innovation-Driven Plan in Central South University (2017CX003), State Key Laboratory of Powder Metallurgy in Central South University, Thousand Youth Talents Plan of China, Hundred Youth Talents Program of Hunan, Shenzhen science and technology innovation project (630) and in USA by NSF-CREST Center for Innovation, Research and Education in Environmental Nanotechnology (CIRE2N, No. HRD-1736093). We thank the Photoemission Endstations (BL10B) in the National Synchrotron Radiation Laboratory (NSRL) for help with characterizations.References
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326–1330 CrossRef CAS PubMed.
- J. G. Lee, J. Hwang, H. J. Hwang, O. S. Jeon, J. Jang, O. Kwon, Y. Lee, B. Han and Y.-G. Shul, J. Am. Chem. Soc., 2016, 138, 3541–3547 CrossRef CAS PubMed.
- C. Duan, D. Hook, Y. Chen, J. Tong and R. O'Hayre, Energy Environ. Sci., 2017, 10, 176–182 RSC.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma and D. Su, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- H. Wang and H. D. Abruña, J. Am. Chem. Soc., 2017, 139, 6807–6810 CrossRef CAS PubMed.
- K. A. Stoerzinger, O. Diaz-Morales, M. Kolb, R. R. Rao, R. Frydendal, L. Qiao, X. R. Wang, N. B. Halck, J. Rossmeisl, H. A. Hansen, T. Vegge, I. E. L. Stephens, M. T. M. Koper and Y. Shao-Horn, ACS Energy Lett., 2017, 2, 876–881 CrossRef CAS.
- J. Yin, Y. Li, F. Lv, Q. Fan, Y.-Q. Zhao, Q. Zhang, W. Wang, F. Cheng, P. Xi and S. Guo, ACS Nano, 2017, 11, 2275–2283 CrossRef CAS PubMed.
- Z. Pei, J. Gu, Y. Wang, Z. Tang, Z. Liu, Y. Huang, Y. Huang, J. Zhao, Z. Chen and C. Zhi, ACS Nano, 2017, 11, 6004–6014 CrossRef CAS PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- H. Jiang, Y. Wang, J. Hao, Y. Liu, W. Li and J. Li, Carbon, 2017, 122, 64–73 CrossRef CAS.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2014, 26, 2925–2930 CrossRef CAS PubMed.
- J. Lai, S. Li, F. Wu, M. Saqib, R. Luque and G. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 RSC.
- Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., 2016, 128, 2270–2274 CrossRef.
- Y. Zhang, X. Fan, J. Jian, D. Yu, Z. Zhang and L. Dai, Energy Environ. Sci., 2017, 10, 2312–2317 RSC.
- C. Hu and L. Dai, Adv. Mater., 2017, 29, 1604942 CrossRef PubMed.
- H. Hou, L. Shao, Y. Zhang, G. Zou, J. Chen and X. Ji, Adv. Sci., 2017, 4, 1600243 CrossRef PubMed.
- W.-J. Jiang, L. Gu, L. Li, Y. Zhang, X. Zhang, L.-J. Zhang, J.-Q. Wang, J.-S. Hu, Z. Wei and L.-J. Wan, J. Am. Chem. Soc., 2016, 138, 3570–3578 CrossRef CAS PubMed.
- H. Jiang, Y. Liu, J. Hao, Y. Wang, W. Li and J. Li, ACS Sustainable Chem. Eng., 2017, 5, 5341–5350 CrossRef CAS.
- Z. Liu, Z. Zhao, Y. Wang, S. Dou, D. Yan, D. Liu, Z. Xia and S. Wang, Adv. Mater., 2017, 29, 1606207 CrossRef PubMed.
- Z. Zhang, M. Dou, H. Liu, L. Dai and F. Wang, Small, 2016, 12, 4193–4199 CrossRef CAS PubMed.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- C. Tang and Q. Zhang, Adv. Mater., 2017, 29, 1604103 CrossRef PubMed.
- T. Ji, L. Chen, L. Mu, R. Yuan, M. Knoblauch, F. S. Bao and J. Zhu, Appl. Catal., B, 2016, 182, 306–315 CrossRef CAS.
- Y. Pan, Y. Zhao, S. Mu, Y. Wang, C. Jiang, Q. Liu, Q. Fang, M. Xue and S. Qiu, J. Mater. Chem. A, 2017, 5, 9544–9552 RSC.
- S. S. Shinde, C.-H. Lee, A. Sami, D.-H. Kim, S.-U. Lee and J.-H. Lee, ACS Nano, 2016, 11, 347–357 CrossRef PubMed.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 RSC.
- H. Sun, L. Mei, J. Liang, Z. Zhao, C. Lee, H. Fei, M. Ding, J. Lau, M. Li, C. Wang, X. Xu, G. Hao, B. Papandrea, I. Shakir, B. Dunn, Y. Huang and X. Duan, Science, 2017, 356, 599–604 CrossRef CAS PubMed.
- W. He, Y. Wang, C. Jiang and L. Lu, Chem. Soc. Rev., 2016, 45, 2396–2409 RSC.
- H. Jiang, C. Li, H. Shen, Y. Liu, W. Z. Li and J. Li, Electrochim. Acta, 2017, 231, 344–353 CrossRef CAS.
- S. H. Ahn and A. Manthiram, Small, 2017, 13, 1603437 CrossRef PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- D. K. Singh, R. N. Jenjeti, S. Sampath and M. Eswaramoorthy, J. Mater. Chem. A, 2017, 5, 6025–6031 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- Y. Dafeng, L. Yunxiao, H. Jia, C. Ru, D. Liming and W. Shuangyin, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- L. Zhang, X. Liang, W. Song and Z. Wu, PCCP, 2010, 12, 12055–12059 RSC.
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng, W. Yan, X. Zheng, Z. Li, X. Wang, J. Yang, D. He, Y. Wang, Z. Deng, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed.
- Y. Tong, P. Chen, T. Zhou, K. Xu, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 129, 7227–7231 CrossRef.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- K. Sakaushi, A. Lyalin, S. Tominaka, T. Taketsugu and K. Uosaki, ACS Nano, 2017, 11, 1770–1779 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- J. Park, M. Risch, G. Nam, M. Park, T. J. Shin, S. Park, M. G. Kim, Y. Shao-Horn and J. Cho, Energy Environ. Sci., 2017, 10, 129–136 RSC.
- F. Hu, H. Yang, C. Wang, Y. Zhang, H. Lu and Q. Wang, Small, 2017, 13, 1602507 CrossRef PubMed.
- L. Ye, G. Chai and Z. Wen, Adv. Funct. Mater., 2017, 27, 1606190 CrossRef.
- Y. P. Zhu, Y. Jing, A. Vasileff, T. Heine and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1602928 CrossRef.
- G.-L. Chai, K. Qiu, M. Qiao, M.-M. Titirici, C. Shang and Z. Guo, Energy Environ. Sci., 2017, 10, 1186–1195 RSC.
- D.-G. Lee, S. H. Kim, S. H. Joo, H.-I. Ji, H. Tavassol, Y. Jeon, S. Choi, M.-H. Lee, C. Kim, S. K. Kwak, G. Kim and H.-K. Song, Energy Environ. Sci., 2017, 10, 523–527 RSC.
- H. Liu, Q. He, H. Jiang, Y. Lin, Y. Zhang, M. Habib, S. Chen and L. Song, ACS Nano, 2017, 11, 11574–11583 CrossRef CAS PubMed.
- Z. Zhao, M. Li, L. Zhang, L. Dai and Z. Xia, Adv. Mater., 2015, 27, 6834–6840 CrossRef CAS PubMed.
- Z. Zhao and Z. Xia, ACS Catal., 2016, 6, 1553–1558 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66–72 CrossRef CAS.
- Z. Zhao, M. Li, L. Zhang, L. Dai and Z. Xia, Adv. Mater., 2015, 27, 6834–6840 CrossRef CAS PubMed.
- R. Parsons, Trans. Faraday Soc., 1958, 54, 1053–1063 RSC.
- I. S. Amiinu, Z. Pu, X. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. Zhang and S. Mu, Adv. Funct. Mater., 2017, 27, 1702300 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee03276a |
| This journal is © The Royal Society of Chemistry 2019 |