
Dithienobenzoxadiazole-based wide bandgap donor polymers with strong aggregation properties for the preparation of efficient as-cast non-fullerene polymer solar cells processed using a non-halogenated solvent†
Haiying
Jiang
a,
Guoming
Qin
a,
Lianjie
Zhang
 *a,
Feilong
Pan
a,
Zhuhao
Wu
a,
Qian
Wang
a,
Guanzhao
Wen
b,
Wei
Zhang
*b,
Yong
Cao
a and
Junwu
Chen
*a
*a,
Feilong
Pan
a,
Zhuhao
Wu
a,
Qian
Wang
a,
Guanzhao
Wen
b,
Wei
Zhang
*b,
Yong
Cao
a and
Junwu
Chen
*a
aInstitute of Polymer Optoelectronic Materials & Devices, State Key Laboratory of Luminescent Materials & Devices, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: lianjiezhang@scut.edu.cn
bSchool of Physics and Materials Science, Guangzhou University, Guangzhou 510006, China
First published on 14th November 2020
Abstract
The efficient as-cast polymer solar cells processed using a non-halogenated solvent are suitable for roll-to-roll printing. In this study, three dithienobenzoxadiazole-based wide bandgap polymers PBOffDT, PBOTT and PBOTVT, which have different backbone structures, were designed and synthesized to reveal the relationship between the backbone structure and photovoltaic performance. The backbone structure had a great influence on the energy level, absorption behavior, aggregation and packing behavior of the polymers. PBOffDT exhibited the deepest HOMO level, the strongest aggregation and the most ordered crystallinity among the three polymers. IT-4Cl was used as the acceptor to fabricate PSC devices, and power conversion efficiencies (PCEs) of 10.56, 8.25 and 3.12% were obtained for PBOffDT, PBOTT and PBOTVT based as-cast devices using non-halogenated 1,3,5-trimethylbenzene (1,3,5-TMB) as the solvent, respectively. PBOffDT:IT-4Cl based devices showed the highest open circuit voltage (Voc), which can be ascribed to the deepest HOMO level of PBOffDT. Meanwhile, as revealed by the surface energy measurement, PBOffDT had suitable compatibility with IT-4Cl, and thus a favorable morphology can be achieved leading to efficient exciton dissociation, suppressed recombination and balanced charge transport for the PBOffDT:IT-4Cl devices. Our studies showed that the enhancement and retention of polymer crystallinity can be valuable to enhance the photovoltaic performance of the DTfBO-based polymer in non-halogenated solvent processed as-cast devices.
Introduction
Solution-processed polymer solar cells (PSCs) have attracted considerable attention due to their promising advantages of light weight, flexibility, and suitability for roll-to-roll printing.1–7 The active layer, composed of a P-type material and an N-type material, is the most important component of PSCs, where light absorption, exciton generation and charge production take place. Generally, the active layer of highly efficient PSCs should have a broad absorption range to maximize the photon-harvesting for achieving high short current density (Jsc). Meanwhile, the energy level of the donor and acceptor should be appropriately aligned to provide sufficient driving force for exciton dissociation as well as for obtaining high open circuit voltage (Voc) simultaneously. Moreover, the exciton dissociation and charge transport are highly dependent on the morphology of bulk heterojunctions, and thus a suitable domain size accompanied by a nanoscale bicontinuous interpenetrating network is essential for realizing high Jsc and fill factor (FF). Recently, considerable efforts have been devoted to developing non-fullerene acceptors (NFAs), and a number of highly efficient NFAs including ITIC, IT-4F, IT-4Cl, IEICO-4F, and Y6 have been developed, which hold the advantages of strong absorption in the visible and near-infrared region, tunable energy levels, good crystallinity and excellent morphology stability.8–17 As a result, power conversion efficiencies (PCEs) of over 17% have been demonstrated both in single-junction and tandem non-fullerene PSCs indicating the great potential of non-fullerene PSCs.18–21Apart from developing novel NFAs, it is also important to develop donor polymers that could match well with NFAs for achieving high PCEs.22–24 Typically, wide bandgap donor–acceptor (D–A) copolymer donors are promising candidates for constructing the active layer with NFAs to achieve efficient non-fullerene PSCs, due to their strong absorption in the short wavelength region and a relatively low-lying highest occupied molecular orbital (HOMO) energy level, which is beneficial for achieving high short-circuit current density (Jsc) and open circuit voltage (Voc).25–28 For donor polymer design, the optical and electronic properties of the D–A copolymer can be easily tuned by altering the electron donating and electron withdrawing units to adjust the intramolecular charge transfer (ICT) effect. Furthermore, side chain engineering and halogenation as molecular modification strategies have been proved to effectively tune the physiochemical properties, crystallinity, aggregation properties and charge transportability of the polymers.23,29–33 However, most of the reported highly efficient wide bandgap D–A polymers are mainly based on benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD),34–37 difluorobenzo[d][1,2,3]triazole (FTAZ),38–41 or ester modified thiophene as the A unit and benzodithiophene (BDT) as the D unit.8,42–44 Recently, Ding et al. reported a dithieno[3′,2′:3,4;2′′,3′′:5,6]benzo[1,2-c][1,2,5]thiadiazole (DTBT) based donor polymer D18 and reported a PCE of over 18% when incorporated with Y6 as the acceptor, which has been the highest PCE value for PSCs to date.20 Therefore, developing novel wide bandgap polymers based on the new building block for matching the NFAs is highly desired in the field of PSCs. On the other hand, most of the efficient PSC devices were fabricated using halogenated solvents, such as chloroform (CF), chlorobenzene (CB), and dichlorobenzene (DCB). The high toxicity of the halogenated solvent would become an issue for massive production and hinder the commercialization of PCSs.45–47 Besides, post-treatments including thermal annealing, solvent vapor annealing and vacuum drying to remove solvent additives are usually adopted to optimize the BHJ morphology of the blend films in most efficient PSCs. However, these post treatment processes also increase the cost of device fabrication and decrease the compatibility with the roll-to-roll processing. Thus, developing efficient PSCs which can be processed using non-halogenated solvents without any post-treatment would further promote the practical application of the PSCs.
Previously, our group reported a highly crystalline wide bandgap donor polymer PBODT based on dithienobenzoxadiazole (DTfBO) as the electron withdrawing unit and tetrathiophene (4T) as the electron donating unit.48 Polymer PBODT showed a relatively deep energy level, distinct edge-on packing orientation and high hole mobility.48 With strong temperature dependent aggregation properties, the as-cast devices based on the PBODT:IDIC blend showed a decent PCE of over 8% processed by using non-halogenated solvent 1,2,4-trimethylbenzene (1,2,4-TMB). The results indicated that polymer PBODT could be a promising candidate for efficient as-cast non-fullerene PSCs. In general, the coplanarity of the polymer has great impact on the polymer aggregation behavior, charge transport ability and the BHJ morphology. Therefore, judicious backbone design that regulates the polymer coplanarity can be an effective way to improve the performance of the DTfBO-based polymer.
Herein, we modulate the main chain structure of PBODT via the modification of the bithiophene moiety in order to alter the bond length alternation of the electron-donating comonomer, respectively. And three DTfBO-based polymers PBOffDT, PBOTT and PBOTVT were designed and synthesized. The effect of the non-covalent and covalent planarization strategies on the photovoltaic performance of DTfBO-based polymer was systematically studied. It was found that the different D units had great impact on the energy level, absorption, aggregation behavior and the crystalline properties of the polymers. As a result, polymer PBOffDT showed the strongest aggregation in the solution state due to the strong intermolecular interactions resulting from the F⋯S and F⋯H noncovalent force. Meanwhile PBOffDT also showed the highest crystallinity along with the dense and more ordered π–π stacking with the edge-on packing orientation. Benefitting from the suitable compatibility with IT-4Cl as the acceptor, a favorable morphology was obtained for the PBOffDT-based blend. Therefore, efficient exciton dissociation, balanced charge transport and suppressed recombination were achieved, leading to higher Jsc and FF. Consequently, a PCE of 10.56% was obtained for the PBOffDT-based devices processed by using a non-halogenated solvent without any post-treatment. Our results suggest that rationally modifying the backbone structure of the DTfBO-based donor polymers is an effective way to tune the aggregation behavior toward efficient as-cast devices which can be processed by using a non-halogenated solvent.
Result and discussion
Synthesis of the polymer donors
The chemical structures of polymers PBOffDT, PBOTT and PBOTVT are shown in Fig. 1a. The synthesis route for the three analogous polymers is shown in Scheme S1 (ESI†). The key dibromo-monomer DTfBO-DT-2Br was synthesized according to our previous reports.48 The polymers were prepared by Stille polymerization with Pd2(dba)3 as the catalyst and P(o-toy)3 as the ligand and purified by Soxhelt-extraction. The final PBOffDT product from CB fraction was precipitated in the methanol. PBOffDT shows poor solubility in chlorobenzene (CB) and o-xylene at room temperature but it can be well dissolved in CB, o-dichlorobenzene (DCB) and 1,3,5-trimethylbenzene (1,3,5-TMB) at 80 °C. While PBOTT and PBOTVT from the chloroform (CF) fraction were precipitated, both of them can be dissolved in common solvents, such as CF, CB, DCB and TMB at room temperature. The molecular weight of the three polymers was measured by high temperature gel permeation chromatography (GPC) at 150 °C using 1,2,4-trichlorobenezene as the eluent. The number-average molecular weights (Mn) of PBOffDT, PBOTT and PBOTVT are 50.8, 38.1 and 41.7 kg mol−1 with polydispersity values of 1.62, 2.19 and 1.75, respectively. The thermal stability of the polymers was estimated by thermogravimetric analysis (TGA). As shown in Fig. S1 (ESI†), polymers PBOffDT, PBOTT and PBOTVT manifest excellent thermal stability with decomposition temperatures (Td) for 5% weight loss of 386, 390 and 398 °C, respectively. Differential scanning calorimetry (DSC) was carried out to characterize the thermal transition of the polymer (Fig. S1, ESI†). As shown in Fig. S1b and c (ESI†), no obvious endothermic or exothermic transitions were observed for PBOffDT and PBOTT during the heating or cooling process. But for the PBOTVT, endothermic and exothermic transitions peak for the melting and crystallization can be observed during the heating and cooling process, respectively. The melting temperature (Tm) and the crystallization temperature (Tc) for PBOTVT are 264 and 237 °C, respectively.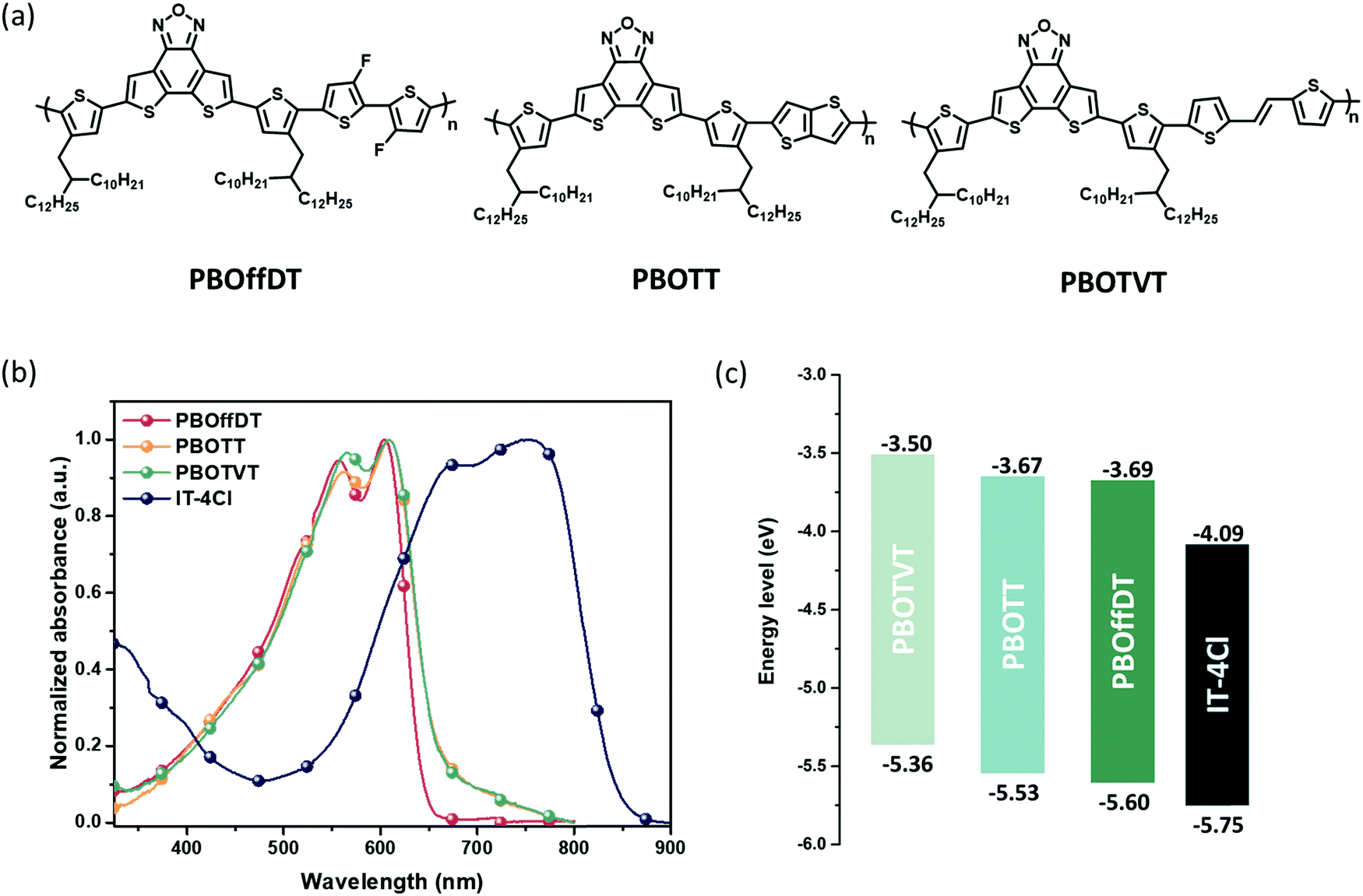 | ||
| Fig. 1 (a) Chemical structures of PBOffDT, PBOTT and PBOTVT. (b) Normalized absorption spectra of neat films based on PBOffDT, PBOTT, PBOTVT and IT-4Cl. (c) Energy level diagrams of the components. | ||
The UV-vis absorption spectra of the three polymers and IT-4Cl pristine films are shown in Fig. 1b. All three polymers show strong absorption in the range of 400–650 nm. Meanwhile, two well distinct 0–0 and 0–1 vibrational transition peaks at ∼600 and 550 nm can be observed for the three polymer pristine films, which indicate strong π–π stacking interactions between the polymer backbones in the solid state. Polymers PBOTT and PBOTVT display almost the same absorption edge of 660 nm corresponding to the optical band gap (Eg) of 1.88 eV, while polymer PBOffDT shows slightly blue-shifted and sharp absorption edge at 650 nm with a larger Eg of 1.91 eV. It can be found that the three polymers showed complementary absorption with the narrow band gap nonfullerene acceptor IT-4Cl, which is beneficial for harvesting the photons and then improving the Jsc in PSC devices.
The highest occupied molecular orbital (HOMO) levels of the polymers were determined by electrochemical cyclic voltammetry (CV) using Ag/Ag+ as the reference electrode and ferrocene as the internal standard (Fig. S3, ESI†). The HOMO levels of PBOTVT, PBOTT and PBOffDT are −5.36, −5.53 and −5.60 eV, respectively. The lowest unoccupied molecular orbital (LUMO) energy levels of the polymers was derived from the HOMO levels and the corresponding optical band gaps. Thus, the LUMO energy levels of PBOTVT, PBOTT and PBOffDT are −3.50, −3.67 and −3.69 eV, respectively. The HOMO and LUMO levels for IT-4Cl are −5.75 and −4.09 eV, respectively. Therefore, the ΔELUMO (LUMOD–LUMOA) for PBOTVT, PBOTT and PBOffDT is 0.59, 0.42 and 0.40 eV, respectively. For the three combinations, the ΔELUMO values are larger than the empirical value of 0.3 eV which should be able to provide sufficient driving force for the electron transfer.49,50 However, the ΔEHOMO (HOMOD–HOMOA) for PBOTVT, PBOTT and PBOffDT is calculated to be 0.39, 0.22 and 0.15 eV, respectively. The ΔEHOMO values of PBOTT and PBOffDT are smaller than 0.3 eV which is a common phenomenon in nonfullerene solar cells (Table 1).13
| Polymers | M n (kg mol−1) | M w (kg mol−1) | PDI | T d (°C) | E g (eV) | HOMO (eV) | LUMO (eV) |
|---|---|---|---|---|---|---|---|
| PBOffDT | 50.8 | 82.5 | 1.62 | 386 | 1.91 | −5.60 | −3.69 |
| PBOTT | 38.1 | 83.9 | 2.19 | 390 | 1.86 | −5.53 | −3.67 |
| PBOTVT | 41.7 | 73.0 | 1.75 | 398 | 1.86 | −5.36 | −3.50 |
To study the aggregation behavior of the polymers, the temperature-dependent absorption spectra of the three polymers in diluted (1 × 10−5 M) CB solution have been recorded (Fig. 2). For the PBOffDT solution, the distinct 0–0 and 0–1 absorption peaks can be retained at room temperature, implying the strong aggregation tendency of polymer PBOffDT in the CB solution. However, for polymers PBOTT and PBOTVT in CB solution, the 0–0 absorption peak almost disappeared at room temperature, suggesting that polymers PBOTT and PBOTVT have almost fully disaggregated in the CB solution at room temperature. As the temperature increases from 20 to 95 °C, an obvious blue-shift and intensity decrease could be observed in the absorption spectra of the polymers. For polymer PBOffDT, the 0–0 absorption peak can be retained even though the temperature has been increased up to 95 °C. The result indicated that polymer PBOffDT possessed strong temperature-dependent aggregation properties and hardly fully disaggregated even at 95 °C, which could be attributed to the high degree of polymer backbone planarity and strong interchain π–π interactions of the polymer resulting from the F⋯S and F⋯H non-covalent bonds. However, polymers PBOTT and PBOTVT show relatively weak aggregation properties in the solution. The orders of the aggregation ability are PBOffDT > PBOTVT > PBOTT. We also recorded the temperature-dependent absorption spectra of the three polymers in diluted 1,3,5-TMB. As shown in Fig. S3 (ESI†), the polymers showed stronger aggregation behavior in 1,3,5-TMB compared to that in CB, which implied that the pre-aggregation ability of the polymers in 1,3,5-TMB was higher than that in CB. Therefore, by tuning the D unit, the polymers can present different aggregation properties, which may be relative to the backbone planarity and the interchain interactions. For polymer PBOffDT, its strong temperature-dependent aggregation properties may tend to control the morphology of BHJ during the spin coating.
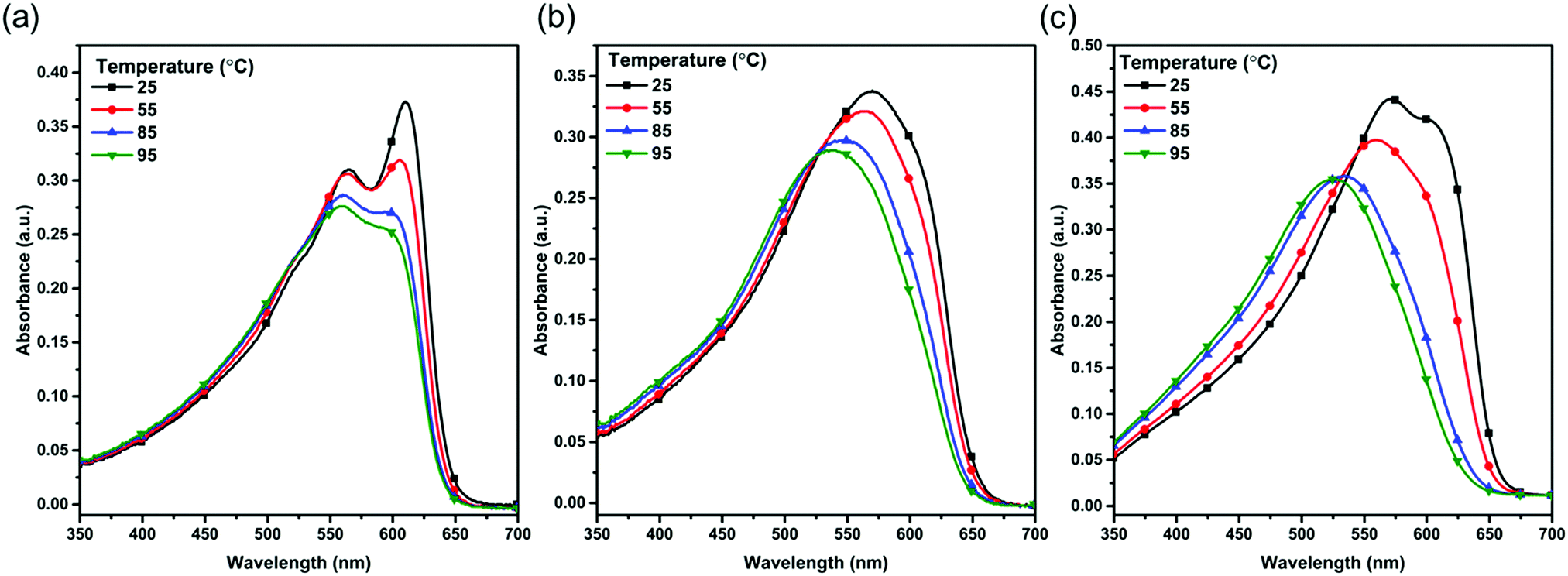 | ||
| Fig. 2 Temperature dependent UV-vis absorption spectra of (a) PBOffDT, (b) PBOTT and (c) PBOTVT in CB diluted solution. | ||
To study the molecular packing structures of the polymers in the neat film, grazing-incident wide angle X-ray scattering (GIWAXS) measurement was conducted. The 2D patterns are shown in Fig. 3a and the corresponding line-cut profile is depicted in Fig. 3b. For polymer PBOffDT and PBOTT neat films, three pronounced (n00) diffraction peaks in the out-of-plane (OOP) direction with a distinct (010) diffraction peak in the in-plane (IP) direction can be observed, indicating that both polymers PBOffDT and PBOTT adopted the edge-on predominant orientation in the neat film, which is similar to that of the polymer PBODT in our previous report.48 However, the polymer PBOTVT neat film possessed very dispersed (100) and (010) diffraction peaks in the OOP direction, which indicated that polymer PBOTVT exhibited the face-on predominant orientation and relatively weak crystallinity. Based on the (100) diffraction peak position, the lamellar stacking distances of polymers PBOffDT, PBOTT and PBOTVT were calculated to be 21.9, 22.5 and 22.8 Å, respectively. Three polymers employed the same alkyl side chains thus having a similar lamellar stacking distance. Meanwhile, the π–π stacking distances of polymers PBOffDT, PBOTT and PBOTVT were determined to be 3.52, 3.55 and 3.78 Å according to the position of the (010) diffraction peak. Polymer PBOffDT had the closest π–π stacking distances among the three polymers which can be ascribed to its strong interchain interactions resulting from the F⋯H and F⋯S noncovalent interactions. It should be noted that polymer PBOTVT adopted the face-on packing orientation but possessed relatively large π–π stacking distances and weak crystallinity, which may weaken the carrier transport.
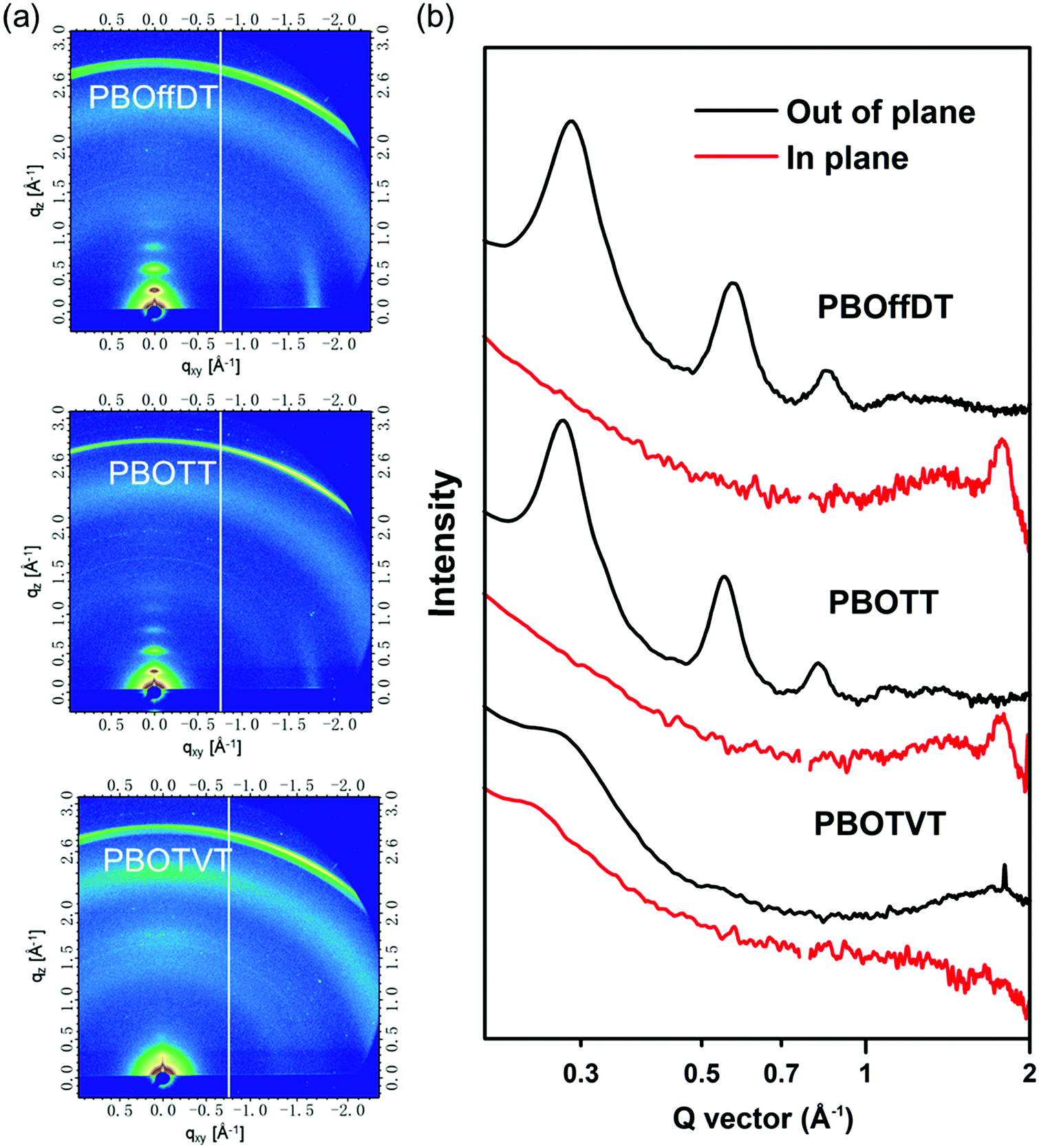 | ||
| Fig. 3 (a) 2D-GIWAXS patterns of neat polymer films; (b) line-cut profiles along IP and OOP directions for the corresponding films. | ||
To investigate the photovoltaic performance of the three polymers, the inverted PSC device with device configuration of ITO/ZnO/polymer:IT-4Cl/MoO3/Al was fabricated. The detailed device fabrication processes and the full name of the materials can be found in the supporting information. The photovoltaic performances of devices were investigated under illumination of AM1.5G simulated solar light at 100 mW cm−2. The active layers were cast from the non-halogen solvent 1,3,5-TMB without any solvent additive or post-treatments. The current density–voltage (J–V) curves of the champion devices are shown in Fig. 4a and the relevant photovoltaic parameters are listed in Table 2. PBOffDT and PBOTT based devices exhibited high Voc values of 0.86 and 0.84 V due to their deep HOMO levels, while PBOTVT based devices shows a lower Voc of 0.66 V. The results are well consistent with the CV measurement of the HOMO energy levels of the polymers. The short-circuit current density (Jsc) of PBOffDT, PBOTT and PBOTVT based PSC devices is 18.27, 14.95 and 8.87 mA cm−2, respectively. Intriguingly, from PBOTVT to PBOTT to PBOffDT as the HOMO and LUMO energy offset values decrease, the Jsc showed an opposite tendency. Typically, a higher energy offset would provide a larger driving force for the exciton dissociation which is beneficial for achieving higher Jsc in the devices. Therefore, for DTfBO-based polymers, the energy offset may not be a crucial factor leading to the huge Jsc difference. The fill factor (FF) of PBOffDT, PBOTT and PBOTVT based devices is 67.1%, 65.5% and 53.1%, respectively. PBOffDT- and PBOTT-based devices showed much higher FF than PBOTVT-based devices which may be related to the better crystallinity of polymers PBOffDT and PBOTT leading to a more favorable morphology in the BHJ blends. Finally, the power conversion efficiencies (PCEs) of 10.56%, 8.25% and 3.12% were achieved for PBOffDT-, PBOTT- and PBOTVT-based devices, respectively. Moreover, the CB-processed devices were also fabricated and the detailed photovoltaic parameters are shown in Table S1 (ESI†). PBOffDT-, PBOTT- and PBOTVT-based devices showed a lower PCE of 8.24, 6.89 and 3.09%, respectively. The result suggested that the non-halogenated solvent 1,3,5-TMB might be more suitable for the polymers with a higher retention degree of pre-aggregation ability.
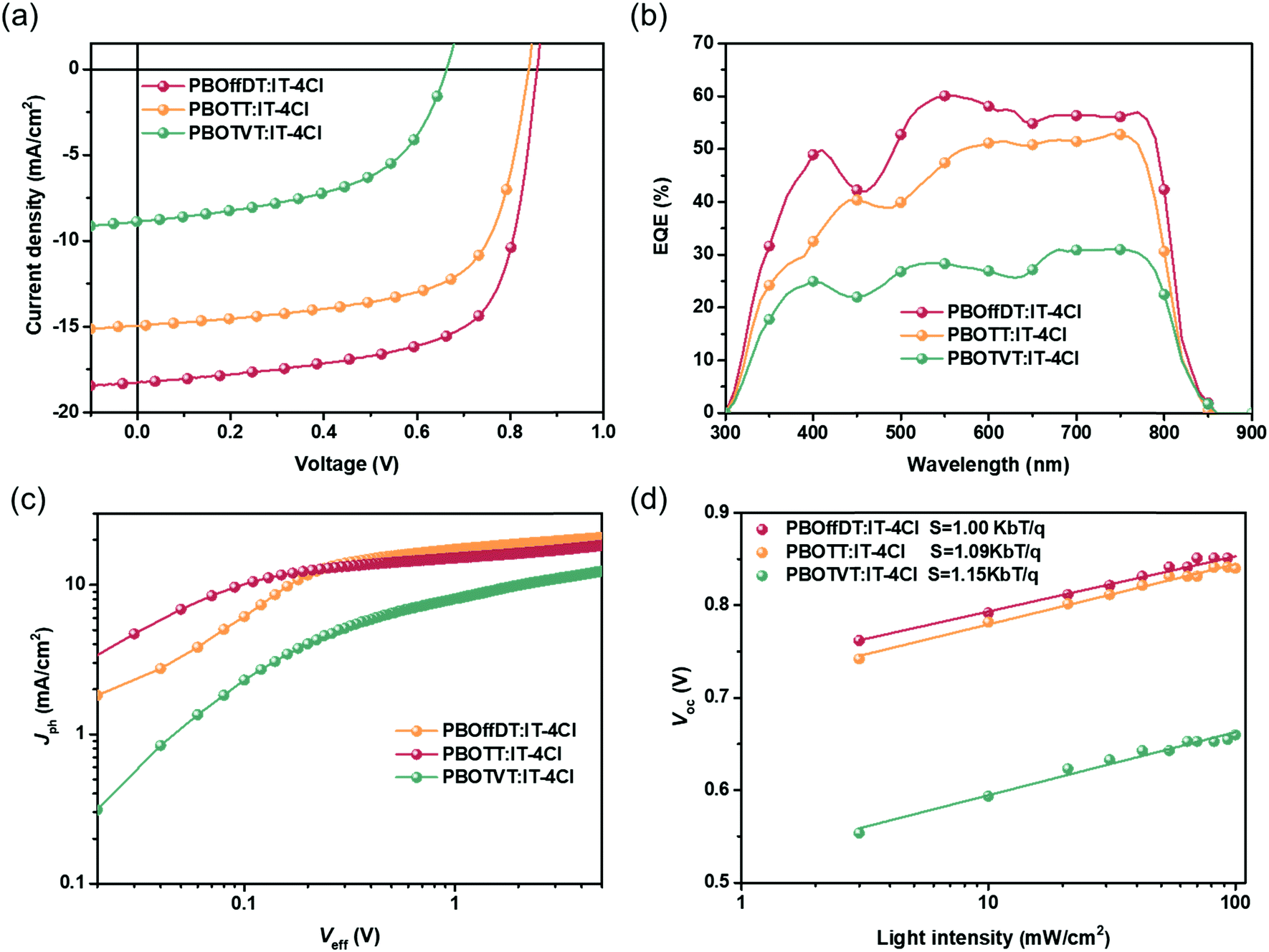 | ||
| Fig. 4 (a) The J–V curves, (b) external quantum efficiency (EQE) curves, (c) Jphversus Veff plots and (d) Vocversus light intensity plots for the optimized devices based on PBOffDT:IT-4Cl, PBOTT:IT-4Cl and PBOTVT:IT-4Cl blends. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IT-4Cl (1:1) based devices using 1,3,5-TMB as the process solvent under AM1.5G conditions, 100 mW cm−2
:IT-4Cl (1:1) based devices using 1,3,5-TMB as the process solvent under AM1.5G conditions, 100 mW cm−2
| Polymer | V oc (V) | J sc (mA cm−2) | FF (%) | PCEa (%) | J cal (mA cm−2) |
|---|---|---|---|---|---|
| a The average values and the standard deviation in the parentheses are obtained from over 10 devices. | |||||
| PBOffDT | 0.86 | 18.27 | 67.1 | 10.56 (10.42 ± 0.17) | 17.73 |
| PBOTT | 0.84 | 14.95 | 65.5 | 8.25 (8.13 ± 0.26) | 14.47 |
| PBOTVT | 0.66 | 8.87 | 53.1 | 3.12 (2.98 ± 0.17) | 8.52 |
The external quantum efficiency (EQE) spectra of the optimized devices are presented in Fig. 4b. Owing to the complementary absorption between the polymers and IT-4Cl, three blends showed wide spectral responses from 300 to 800 nm which is in well agreement with the corresponding blend film absorption spectra. The maximum EQE values of PBOffDT-, PBOTT- and PBOTVT-based devices are 70%, 59% and 34%, respectively. The PBOffDT-based devices showed the highest EQE response, contributing to the highest Jsc. The integrated current densities obtained from the EQE spectra are of 17.73, 14.47 and 8.52 mA cm−2, which agreed well with the Jsc value achieved from the J–V curve within 5% mismatch.
To gain deep insight into the exciton dissociation, the relationship between photocurrent density (Jph) and effective voltage (Veff) was obtained. The Jph is defined as Jph = JL − JD, where JL and JD are the current densities of the devices under illumination and in the dark, respectively. Veff is defined as Veff = V0 − Vbias where V0 is the voltage when Jph is zero and Vbias is the applied bias voltage. The charge dissociation probability (P(E,T)) can be obtained from the value of Jph/Jsat. When Veff reaches 5 V, the Jph of the PBOffDT and PBOTT based devices reached saturation, while that of PBOTVT based devices cannot reach saturation, indicating sever recombination in PBOTVT blends. Under the short circuit conditions, the P(E,T) values of PBOffDT, PBOTT and PBOTVT based devices are 93%, 87% and 80%, respectively. The highest exciton dissociation probability in the PBOffDT:IT-4Cl based device contributes to the highest Jsc of the three blends.
| Film | A 1 | τ 1 (ps) | A 2 | τ 2 (ps) |
![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) (ps) (ps) |
H |
|---|---|---|---|---|---|---|
| IT-4Cl (830 nm) | 1.13 | 207 ± 4.0 | — | — | — | — |
| PBOffDT (640 nm) | 1.15 | 114 ± 2.0 | — | — | — | — |
| PBOTT (650 nm) | 1.03 | 184 ± 4.0 | — | — | — | — |
| PBOffDT:IT-4Cl (830 nm) | 1.09 | 25 ± 1.0 | — | — | — | — |
| PBOffDT:IT-4Cl (640 nm) | 2.29 | 2.3 ± 0.1 | — | — | — | — |
| PBOTT:IT-4Cl (820 nm) | 1.11 | 27 ± 1.0 | — | — | — | — |
| PBOTT:IT-4Cl (650 nm) | 1.10 | 3.0 ± 0.5 | 0.63 | 27 ± 2.0 | 11.54 | 93.7% |
We note that the driving forces for the exciton dissociation of the donor (LUMO level offsets) are 0.4, 0.42, and 0.59 eV in PBOffDT:IT-4Cl, PBOTT:IT-4Cl and PBOTVT:IT-4Cl blend films, respectively, while the driving forces for the exciton dissociation of the acceptor (HOMO level offsets) are 0.15, 0.22, and 0.39 eV. The effect of the driving force on the exciton dissociation in blend films is an important factor for the mechanism of charge photogeneration in polymer solar cells and has been widely studied in the past few decades.51–53 It was once widely believed that the minimum driving force for the exciton dissociation in polymer solar cells is ∼0.3 eV.54 However, many types of polymer solar cells with high PCE showing a driving force of <0.3 eV have been reported in recent studies.55–57 For the solar cells developed in this work, the driving force of the acceptors in PBOffDT:IT-4Cl and PBOTT:IT-4Cl blend films was significantly lower than 0.3 eV. To examine the dissociation of excitons in these two types of blend films, time resolved photoluminescence (TRPL) measurements were performed, as shown in Fig. 5. By comparing the PL lifetime of donor (acceptor) excitons in neat and blend films, we find that the lifetime of excitons in blend films is much shorter than that in neat donor (acceptor) films, as shown in table, suggesting that the excitons of both donors and acceptors in blend films can dissociate efficiently. The exciton dissociation efficiency of donors and acceptors in blend films can be calculated by 1 − τblend/τpure, as shown in Table 3, where τblend and τpure are the PL lifetime of donor (acceptor) excitons in the blend films and neat donor (acceptor) films. Generally, the exciton dissociation efficiency of the donor is higher than 90%, while the exciton dissociation efficiency of the acceptor is ∼87% in PBOffDT:IT-4Cl and PBOTT:IT-4Cl. We note that the exciton dissociation efficiency of the donor in PBOffDT:IT-4Cl (98.0%) is higher than that in PBOTT/IT-4Cl (93.7%), and could be a reason for the highest PCE of PBOffDT:IT-4Cl solar cells. The exciton dissociation efficiency of acceptors (IT-4Cl) in both blend films is 87%, suggesting that the hole transfer process is highly efficient even though the driving force is just ∼0.15 eV.
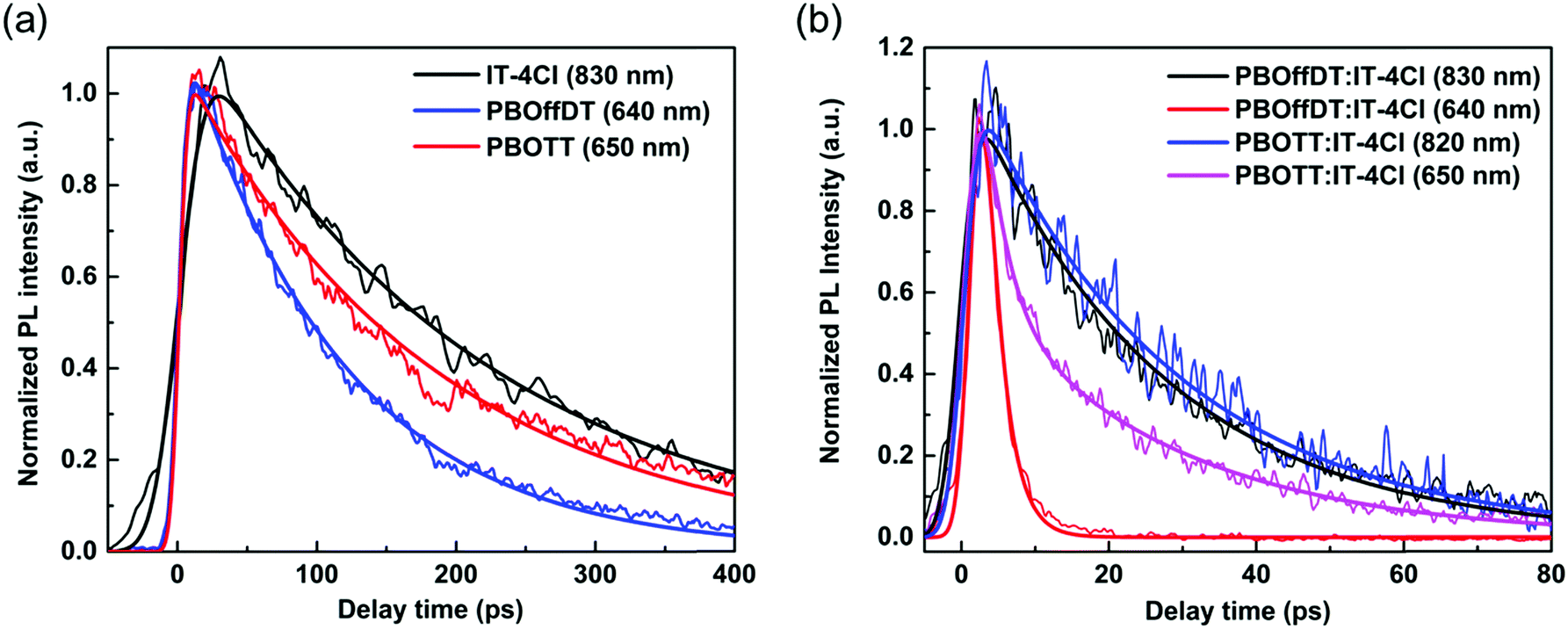 | ||
| Fig. 5 TRPL kinetics of (a) neat donor and acceptor films and (b) blend films at indicated probing wavelength after photoexcitation at 390 nm. In blend films, the kinetics at ∼650 and ∼830 nm are dominated by the emission of donors and acceptors in the blend films, respectively. The kinetics are fitted by the exponential decay function, and the fitting parameters are listed in Table 3. | ||
The dependence of the Voc on different light intensities (Plight) was measured to determine the recombination mechanism of the devices. The relationship between Voc and Plight can be described as Voc ∝ nkBT/qln(Plight) (where n, kB, T and q represent the ideal factor, Boltzmann constant, temperature and elementary charge, respectively). The slope of Vocversus ln(Plight) can be used to estimate the degree of trap-assisted recombination of the devices. By fitting the line, the slope values of 1.00kBT/q, 1.09kBT/q and 1.15kBT/q were obtained for the PBOffDT, PBOTT and PBOTVT based devices. The results indicated that the trap-assisted recombination can be neglected in the PBOffDT:IT-4Cl blend, which agrees well with its high FF in the PSC devices.
The hole mobility (μh) and electron mobility (μe) of the three blend films were measured by using the space charge limited current (SCLC) method with the hole-only device structure (ITO/PEDOT:PSS/active layer/MoO3/Al) and electron-only device structure (ITO/ZnO/active layer/Al). The J1/2–V characteristics of hole-only and electron-only devices are shown in Fig. S4 (ESI†) and the results are summarized in Table 4. The PBOffDT:IT-4Cl blend film shows the highest hole mobility (μh = 7.44 × 10−4 cm2 V−1 s−1) and electron mobility (μe = 5.76 × 10−4 cm2 V−1 s−1) of the three blends leading to balanced carrier transport properties (μh/μe = 1.29). Relatively, the PBOTT:IT-4Cl blend film shows lower μh and μe values of 1.90 × 10−4 and 1.25 × 10−4 cm2 V−1 s−1 with a slight higher μh/μe value of 1.59. However, the lowest values of μh and μe are 0.23 × 10−4 and 0.64 × 10−4 cm2 V−1 s−1 for PBOTVT:IT-4Cl blend films, which indicate that the quite unbalanced hole and electron transport ability (μh/μe = 0.36) is harmful to Jsc and FF. Collectively, the good exciton dissociation probability, high and balanced carrier mobility and the suppressed recombination may lead to the superior Jsc and FF values of the PBOffDT:IT-4Cl blend, attributed to the high retention degree of the strong pre-aggregation ability of the polymer.
| Blends | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) | μ h/μe |
|---|---|---|---|
| PBOffDT:IT-4Cl | 7.44 × 10−4 | 5.76 × 10−4 | 1.29 |
| PBOTT:IT-4Cl | 1.90 × 10−4 | 1.25 × 10−4 | 1.52 |
| PBOTVT:IT-4Cl | 0.23 × 10−4 | 0.64 × 10−4 | 0.36 |
The GIWAXS measurements were further carried out to reveal the molecular packing properties of the blend films. The GIWAXS patterns of the IT-4Cl neat film as well as the blend films are shown in Fig. 6a and the corresponding line-cut of profiles are displayed in Fig. 6b. The IT-4Cl neat film exhibited the (100) diffraction peak at qxy = 0.306 Å−1 in the IP direction and the (010) diffraction peak at qz = 1.77 Å−1 in the OOP direction indicating that IT-4Cl adopted the face-on orientation. The lamellar and π–π stacking distances were calculated to be 20.53 and 3.53 Å, respectively. For PBOffDT:IT-4Cl and PBOTT:IT-4Cl blend films, the distinct (100), (200) and (300) diffraction peaks from the donor polymers as well as the (010) diffraction peaks from the IT-4Cl can be observed in the OOP direction, indicating that the blend films adopted the bimodal orientation. Meanwhile, both donors and acceptors can retain their own crystalline properties in the blend films, which is of great benefit to obtain suitable phase separation and domain purity in the bulk heterojunctions. Moreover, compared to IT-4Cl neat films, the π–π stacking distances of the IT-4Cl were decreased to 3.39 and 3.45 Å for PBOffDT:IT-4Cl and PBOTT:IT-4Cl blend films, respectively. The tighter π–π stacking distances in the PBOffDT:IT-4Cl blend film contributed to its higher electron mobility in the SCLC measurement. For PBOTVT:IT-4Cl blend films, only a dispersed (010) diffraction peak from IT-4Cl can be observed in the OOP direction, indicating polymer PBOTVT with disorder packing in the blend film. The π–π stacking distance for PBOTVT:IT-4Cl is 3.54 Å, larger than that of PBOffDT- and PBOTT-based blend films. Consequently, the disorder packing of polymer PBOTVT and relatively weak π–π interaction implied the poor charge transport and severe recombination in the blend films.
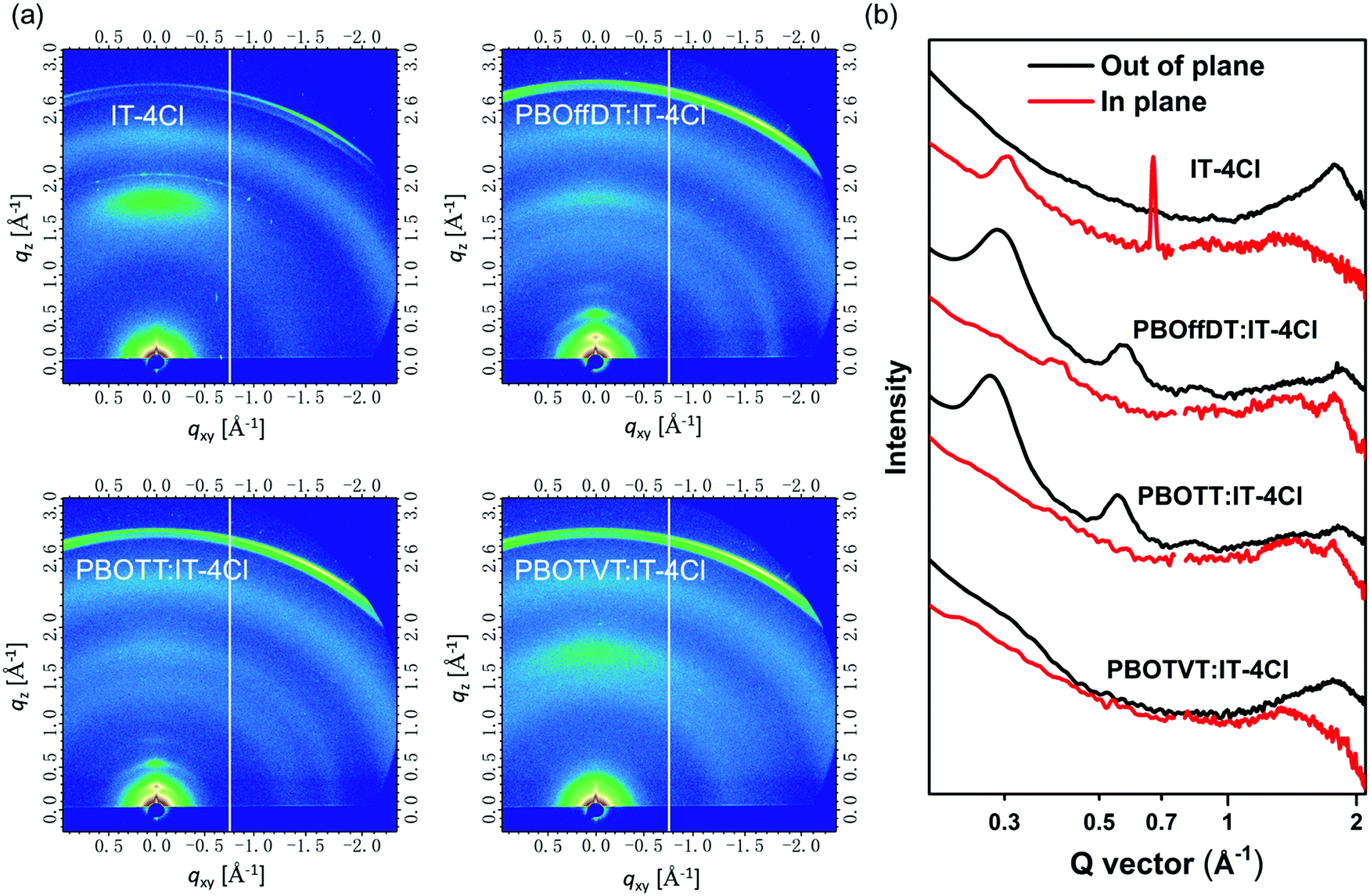 | ||
| Fig. 6 (a) GIWAXS patterns for the IT4Cl neat film and PBOffDT:IT-4Cl, PBOTT:IT-4Cl, PBOTVT:IT-4Cl blend films; (b) line-cut profiles along the IP and OOP directions of the corresponding films. | ||
Atomic force microscopy (AFM) was performed to investigate the surface morphology of the three blend films. As shown in Fig. 7a–c, the root-mean-square (RMS) roughness values for PBOffDT, PBOTT and PBOTVT based blend films are 1.27, 2.01 and 1.15 nm, respectively. PBOffDT- and PBOTT-based blend films showed slightly higher roughness which can be ascribed to their stronger crystallinity. Furthermore, transmission electron microscopy (TEM) was carried out to reveal the BHJ morphology of the blend films. As displayed in Fig. 7d and e, fibril-like structures and nanoscale bicontinuous interpenetrating network structures can be observed in the PBOffDT:IT-4Cl and PBOTT:IT-4Cl blend films, which is in favor of exciton dissociation and charge transport. Moreover, the PBOffDT:IT-4Cl blend film showed better crystallinity as well as more obvious phase separation, contributing to its high FF and Jsc in the devices. On the other hand, the PBOTVT:IT-4Cl blend film showed homogeneous texture without obvious phase separation, due to the excess miscibility between PBOTVT and IT-4Cl. Thus, the poor morphology of the PBOTVT:IT-4Cl blend film should be responsible for the low Jsc and FF of the PSC devices.
 | ||
| Fig. 7 AFM height images of (a) PBOffDT:IT-4Cl, (b) PBOTT:IT-4Cl and (c) PBOTVT:IT-4Cl; TEM images of (d) PBOffDT:IT-4Cl, (e) PBOTT:IT-4Cl and (f) PBOTVT:IT-4Cl. | ||
To explain the phase separation morphology difference of the three blend films, we calculated the surface energy of each component according to the Wu model by measuring the contact angle of water and diiodomethane on the pristine donor and acceptor films (Fig. 8).58–60 The relevant contact angles and the surface energies are listed in Table 5. Each component showed different water and oil contact angles for the different hydrophilic and oleophilic properties. The surface energy (γ) was calculated to be 42.40, 31.13, 30.59 and 31.87 mJ m−2 for IT-4Cl, PBOffDT, PBOTT and PBOTVT, respectively. The miscibility between two components in a blend can be estimated using the Flory–Huggins interaction parameter (χD,A) which can be described as 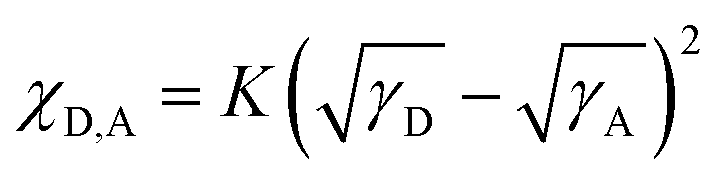 , where K is a constant while γD and γA represent the surface energy of the donor polymer and the acceptor, respectively.61–63 The larger value of χD,A indicates the poorer miscibility of the donor and acceptor and then the donor and acceptor are prone to phase separation. As listed in Table 3, the values of
, where K is a constant while γD and γA represent the surface energy of the donor polymer and the acceptor, respectively.61–63 The larger value of χD,A indicates the poorer miscibility of the donor and acceptor and then the donor and acceptor are prone to phase separation. As listed in Table 3, the values of 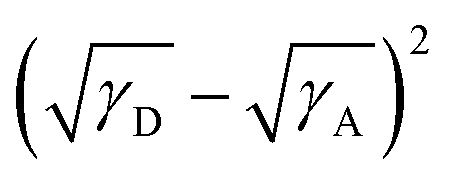 for polymers PBOffDT, PBOTT and PBOTVT are 0.87, 0.96 and 0.75, respectively. As a result, PBOTVT:IT-4Cl shows the smallest value of χD,A which resulted in a too intermixed BHJ morphology. However, PBOTT and IT-4Cl showed the highest tendency of phase separation in the blends, which may lead to an oversized domain and then cause severe exciton recombination and inferior Jsc. In comparison, the PBOffDT:IT-4Cl based blend has a middle value of χD,A, which means that PBOffDT and IT-4Cl show a more suitable phase separation tendency leading to the appropriate domain size for the exciton dissociation.
for polymers PBOffDT, PBOTT and PBOTVT are 0.87, 0.96 and 0.75, respectively. As a result, PBOTVT:IT-4Cl shows the smallest value of χD,A which resulted in a too intermixed BHJ morphology. However, PBOTT and IT-4Cl showed the highest tendency of phase separation in the blends, which may lead to an oversized domain and then cause severe exciton recombination and inferior Jsc. In comparison, the PBOffDT:IT-4Cl based blend has a middle value of χD,A, which means that PBOffDT and IT-4Cl show a more suitable phase separation tendency leading to the appropriate domain size for the exciton dissociation.
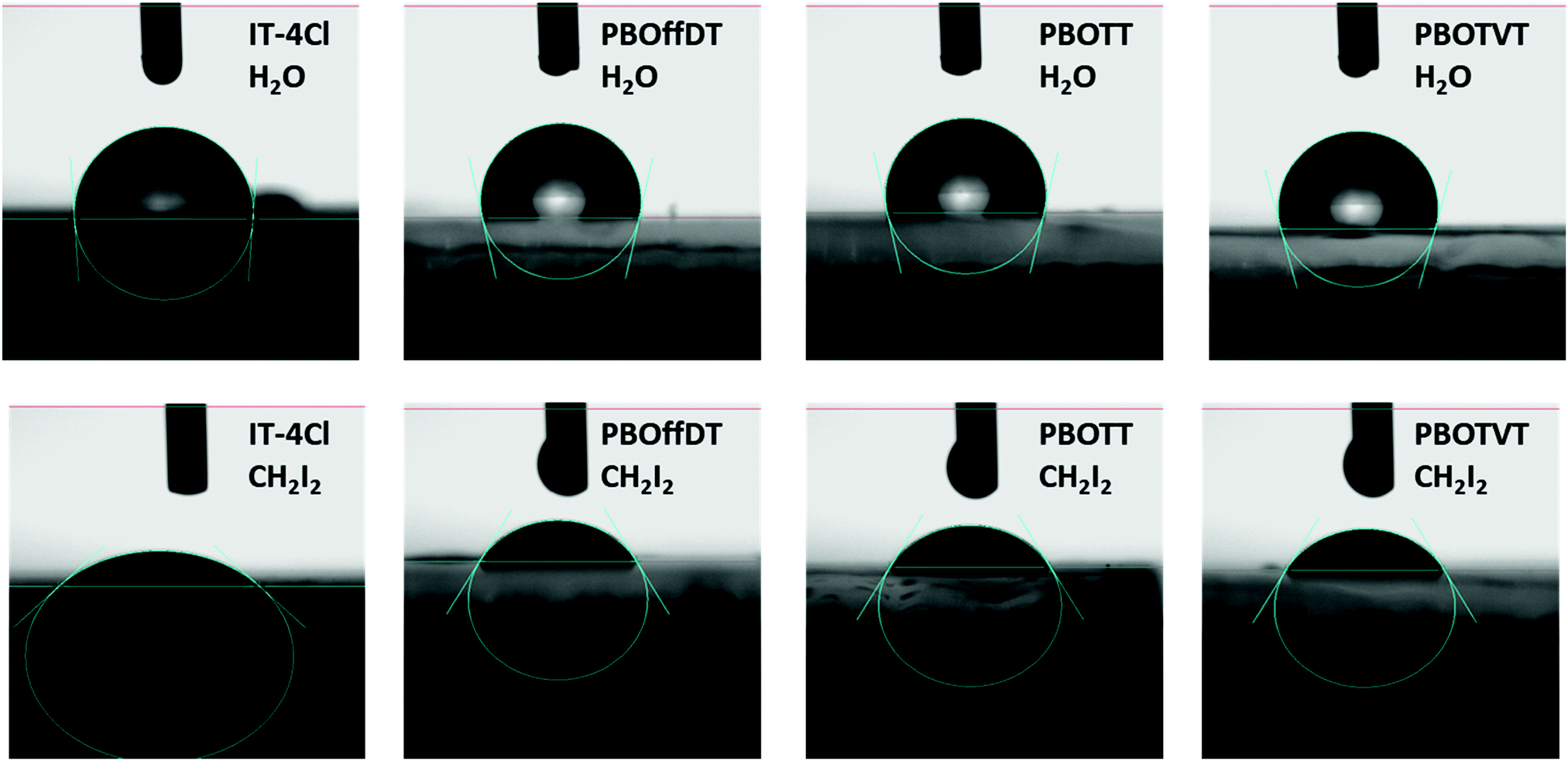 | ||
| Fig. 8 The contact angles of water and CH2I2 on the surface of IT-4Cl, PBOffDT, PBOTT and PBOTVT neat films. | ||
| Film | θ water (°) | θ oil (°) |
γ
db (mJ m−2) |
γ
pb (mJ m−2) |
γ (mJ m−2) |
|
|---|---|---|---|---|---|---|
| a θ oil represents the contact angle of diiodomethane. b γ d and γp represent the surface energy generated from the dispersion force and the polar force, respectively. | ||||||
| IT-4Cl | 94.3 | 35.4 | 39.48 | 2.92 | 42.40 | — |
| PBOffDT | 102.3 | 58.4 | 29.24 | 1.89 | 31.13 | 0.87 |
| PBOTT | 104.0 | 58.8 | 29.27 | 1.32 | 30.59 | 0.96 |
| PBOTVT | 105.7 | 59.7 | 31.48 | 0.39 | 31.87 | 0.75 |
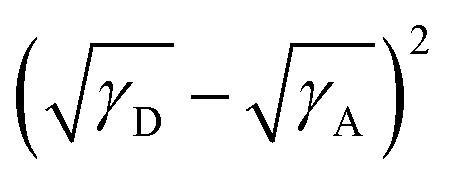
Conclusion
In conclusion, we designed and synthesized three DTfBO-4T based wide bandgap donor polymers, namely PBOffDT, PBOTT and PBOTVT, adopting three different strategies of bond length alternation. The relationship between the main chain structure and the photovoltaic performance was studied. The main chain structure had great influence on the polymer absorption, energy levels, aggregation behavior and the molecular packing properties. Polymers PBOffDT and PBOTT showed the distinct edge-on packing orientation with a more pronounced diffraction peak, while polymer PBOTVT adopts the face-on packing orientation with weak diffraction intensity from the GIWAXS measurement. Using IT-4Cl as the acceptor to construct the PSC devices and 1,3,5-TMB as the solvent, the PCEs of 10.56, 8.25 and 3.12% were obtained for PBOffDT, PBOTT and PBOTT based as-cast devices. Polymer PBOffDT has suitable compatibility with IT-4Cl, and thus a favorable morphology can be realized and further lead to efficient exciton dissociation, balanced charge transport and suppressed recombination which are responsible for the high Jsc and FF of the PBOffDT:IT-4Cl based devices. Our work suggests that the retention of crystallinity of polymer donors plays an important role in the bimodal morphology in non-halogenated solvent processed as-cast devices.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (51673070, 21903017 and U1401244), the Basic and Applied Basic Research Major Program of Guangdong Province (2019B030302007), and the National Key Research and Development Program of China (2019YFA0705900). We thank Xianshao Zou and Xiaojun Su at Lund University for helping in TRPL measurements.References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef.
- G. Li, R. Zhu and Y. Yang, Nat. Photon., 2012, 6, 153–161 CrossRef.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS.
- J. Zhang, H. S. Tan, X. Guo, A. Facchetti and H. Yan, Nat. Energy, 2018, 3, 720–731 CrossRef CAS.
- C. Cui and Y. Li, Energy Environ. Sci., 2019, 12, 3225–3246 RSC.
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, J. Am. Chem. Soc., 2018, 140, 7159–7167 CrossRef CAS.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng and P. A. Johnson, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Angew. Chem., Int. Ed., 2017, 56, 3045–3049 CrossRef CAS.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS.
- Z. Luo, R. Ma, T. Liu, J. Yu, Y. Xiao, R. Sun, G. Xie, J. Yuan, Y. Chen, K. Chen, G. Chai, H. Sun, J. Min, J. Zhang, Y. Zou, C. Yang, X. Lu, F. Gao and H. Yan, Joule, 2020, 4, 1236–1247 CrossRef CAS.
- S. Dai, J. Zhou, S. Chandrabose, Y. Shi, G. Han, K. Chen, J. Xin, K. Liu, Z. Chen, Z. Xie, W. Ma, Y. Yi, L. Jiang, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2020, 32, 2000645 CrossRef CAS.
- Q. Guo, R. Ma, J. Hu, Z. Wang, H. Sun, X. Dong, Z. Luo, T. Liu, X. Guo, X. Guo, H. Yan, F. Liu and M. Zhang, Adv. Funct. Mater., 2020, 30, 2000383 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma and C. An, Adv. Mater., 2020, 32, 1908205 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding and R. Xia, Science, 2018, 361, 1094–1098 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao and K. Sun, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- Z. Luo, R. Ma, T. Liu, J. Yu, Y. Xiao, R. Sun, G. Xie, J. Yuan, Y. Chen and K. Chen, Joule, 2020, 4, 1–12 CrossRef.
- Z. Zheng, H. Yao, L. Ye, Y. Xu, S. Zhang and J. Hou, Mater. Today, 2019, 35, 115–130 CrossRef.
- H. Yao, J. Wang, Y. Xu, S. Zhang and J. Hou, Acc. Chem. Res., 2020, 53, 822–832 CrossRef CAS.
- Y. Xu, H. Yao and J. Hou, Chin. J. Chem., 2019, 37, 207–215 CrossRef CAS.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z. G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef.
- B. Fan, X. Du, F. Liu, W. Zhong, L. Ying, R. Xie, X. Tang, K. An, J. Xin and N. Li, Nat. Energy, 2018, 3, 1051–1058 CrossRef.
- B. Fan, K. Zhang, X. F. Jiang, L. Ying, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1606396 CrossRef.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef.
- X. Xu, G. Zhang, Y. Li and Q. Peng, Chin. Chem. Lett., 2019, 30, 809–825 CrossRef.
- Q. Wang, Z. Hu, Z. Wu, Y. Lin, L. Zhang, L. Liu, Y. Ma, Y. Cao and J. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 4659–4672 CrossRef CAS.
- H. Jiang, F. Pan, L. Zhang, X. Zhou, Z. Wang, Y. Nian, C. Liu, W. Tang, Q. Ma and Z. Ni, ACS Appl. Mater. Interfaces, 2019, 11, 29094–29104 CrossRef CAS.
- X. Liu, L. Nian, K. Gao, L. Zhang, L. Qing, Z. Wang, L. Ying, Z. Xie, Y. Ma and Y. Cao, J. Mater. Chem. A, 2017, 5, 17619–17631 RSC.
- W. Li, L. Ye, S. Li, H. Yao, H. Ade and J. Hou, Adv. Mater., 2018, 30, 1707170 CrossRef.
- Q. Fan, W. Su, Y. Wang, B. Guo, Y. Jiang, X. Guo, F. Liu, T. P. Russell, M. Zhang and Y. Li, Sci. China: Chem., 2018, 61, 531–537 CrossRef CAS.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. Chesman, C. D. Easton and C. R. McNeill, Adv. Mater., 2018, 30, 1705209 CrossRef.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS.
- L. Xue, Y. Yang, J. Xu, C. Zhang, H. Bin, Z. G. Zhang, B. Qiu, X. Li, C. Sun and L. Gao, Adv. Mater., 2017, 29, 1703344 CrossRef.
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang and M. Xiao, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- Y. Lin, F. Zhao, S. K. K. Prasad, J.-D. Chen, W. Cai, Q. Zhang, K. Chen, Y. Wu, W. Ma, F. Gao, J.-X. Tang, C. Wang, W. You, J. M. Hodgkiss and X. Zhan, Adv. Mater., 2018, 30, 1706363 CrossRef.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS.
- Y. Qin, M. A. Uddin, Y. Chen, B. Jang, K. Zhao, Z. Zheng, R. Yu, T. J. Shin, H. Y. Woo and J. Hou, Adv. Mater., 2016, 28, 9416–9422 CrossRef CAS.
- D. Liu, B. Yang, B. Jang, B. Xu, S. Zhang, C. He, H. Y. Woo and J. Hou, Energy Environ. Sci., 2017, 10, 546–551 RSC.
- S. Li, W. Zhao, J. Zhang, X. Liu, Z. Zheng, C. He, B. Xu, Z. Wei and J. Hou, Chem. Mater., 2020, 32, 1993–2003 CrossRef CAS.
- L. Ye, Y. Xiong, Z. Chen, Q. Zhang, Z. Fei, R. Henry, M. Heeney, B. T. O'Connor, W. You and H. Ade, Adv. Mater., 2019, 31, 1808153 CrossRef.
- S. Zhang, L. Ye, H. Zhang and J. Hou, Mater. Today, 2016, 19, 533–543 CrossRef.
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao and K. Xian, Adv. Mater., 2019, 31, 1903441 CrossRef.
- H. Jiang, Z. Wang, L. Zhang, A. Zhong, X. Liu, F. Pan, W. Cai, O. Inganäs, Y. Liu and J. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 36061–36069 CrossRef.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef.
- Y. Xie, W. Wang, W. Huang, F. Lin, T. Li, S. Liu, X. Zhan, Y. Liang, C. Gao, H. Wu and Y. Cao, Energy Environ. Sci., 2019, 12, 3556–3566 RSC.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef.
- R. A. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef.
- S. Li, L. Zhan, C. Sun, H. Zhu, G. Zhou, W. Yang, M. Shi, C. Z. Li, J. Hou, Y. Li and H. Chen, J. Am. Chem. Soc., 2019, 141, 3073–3082 CrossRef.
- Z. Zhang, W. Liu, T. Rehman, H.-X. Ju, J. Mai, X. Lu, M. Shi, J. Zhu, C.-Z. Li and H. Chen, J. Mater. Chem. A, 2017, 5, 9649–9654 RSC.
- S. Chen, Y. Wang, L. Zhang, J. Zhao, Y. Chen, D. Zhu, H. Yao, G. Zhang, W. Ma and R. H. Friend, Adv. Mater., 2018, 30, 1804215 CrossRef.
- X. Liu, C. Zhang, C. Duan, M. Li, Z. Hu, J. Wang, F. Liu, N. Li, C. J. Brabec and R. A. Janssen, J. Am. Chem. Soc., 2018, 140, 8934–8943 CrossRef.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS.
- X. Xue, K. Weng, F. Qi, Y. Zhang, Z. Wang, J. Ali, D. Wei, Y. Sun, F. Liu and M. Wan, Adv. Energy Mater., 2019, 9, 1802686 CrossRef.
- S. Pang, R. Zhang, C. Duan, S. Zhang, X. Gu, X. Liu, F. Huang and Y. Cao, Adv. Energy Mater., 2019, 9, 1901740 CrossRef.
- S. Nilsson, A. Bernasik, A. Budkowski and E. Moons, Macromolecules, 2007, 40, 8291–8301 CrossRef CAS.
- H. Huang, Q. Guo, S. Feng, Z. Bi, W. Xue, J. Yang, J. Song, C. Li, X. Xu and Z. Tang, Nat. Commun., 2019, 10, 1–10 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04909c |
| This journal is © The Royal Society of Chemistry 2021 |