
Room temperature ferroelectricity and blue photoluminescence in zero dimensional organic lead iodine perovskites†
Yiming
Wang
a,
Zheng
Tang
a,
Chunlin
Liu
b,
Junjie
Jiang
a,
Wenlong
Liu
b,
Binbin
Zhang
 c,
Kaige
Gao
*b,
Hong-Ling
Cai
a and
Xiaoshan
Wu
a
c,
Kaige
Gao
*b,
Hong-Ling
Cai
a and
Xiaoshan
Wu
a
aCollaborative Innovation Center of Advanced Microstructures, Laboratory of Solid State Microstructures, School of Physics, Nanjing University, Nanjing 210093, P. R. China
bCollege of Physical Science and Technology, Yangzhou University, Jiangsu 225009, P. R. China. E-mail: kggao@yzu.edu.cn
cState Key Laboratory of Solidification Processing & Key Laboratory of Radiation Detection Materials and Devices & School of Materials Science and Engineering, Northwestern Polytechnical University, Xi’an 710072, P. R. China
First published on 19th November 2020
Abstract
Organic–inorganic hybrid perovskite materials have attracted great attention due to their great application potential in photovoltaics and optoelectronics. Among them, some 2D and 1D lead-iodide-based perovskites were found to exhibit ferroelectricity at room temperature. Yet, no 0D lead-iodide-based perovskites were reported to be room temperature ferroelectric. Here, we report a new lead-iodide-based perovskite material, (DMA)4PbI6, which is ferroelectric at room temperature. The spontaneous polarization of (DMA)4PbI6 is about 0.3 μC cm−2. (DMA)4PbI6 undergoes a ferroelectric–ferroelectric phase transition around 252 K in the heating process. The ferroelectric–ferroelectric phase transition is a first order phase transition with a hysteresis of 18 K between the heating process and the cooling process. The absorption spectra show that (DMA)4PbI6 is a direct band material with a band gap of 2.80 eV. A broadband blue photoluminescence centered at 2.7 eV was observed, which may be attributed to the self-trapped excitons.
1. Introduction
Hybrid organic–inorganic perovskites (HOIPs) have attracted great attention due to their potential application in photovoltaics, optoelectronics, and pizoelectrics.1–6 MAPbI3 is one of the most famous HOIP materials as a good photovoltaic material.7,8 The power conversion efficiency of MAPbI3 (CH3NH3PbI3) based solar cells has boosted from 3.8% to 24.2% in just a few years. The ferroelectricity of MAPbI3 was proved by different methods by different groups.9–13 Polarization in MAPbI3 films is believed to be helpful for the separation of photo generated electron–hole pairs in solar cells. The bulk ferroelectric photovoltaic effect may contribute largely to the power conversion efficiency in MAPbI3 solar cells. MAPbI3 is a three-dimensional perovskite, which is similar to BaTiO3. The PbI6 octahedron shares one I site with nearby six PbI6 octahedra, forming a three-dimensional grid structure. The three dimensional perovskite structure makes MAPbI3 have a small band gap of 1.5–1.6 eV. Meanwhile, MAPbI3 behaves as a semiconductor with good conductivity, which makes it hard to study the ferroelectricity in MAPbI3. Therefore, the ferroelectricity of MAPbI3 is still controversial.14 In addition to MAPbI3, very recently two 3D perovskites that crystallize in polar structures comprising methylhydrazinium cations have been discovered,15,16 which may be potential ferroelectric materials with good photoluminescence properties.Although it is hard to study the ferroelectricity in 3D organic–inorganic halide lead perovskites, some 2D and 1D organic–inorganic halide lead perovskites are found to exhibit room temperature ferroelectricity.17–19 The band gaps of the 2D/1D ferroelectrics are usually smaller than those of other molecular ferroelectrics, making them good candidates for optoelectronic device materials. Of all the reported hybrid halide lead perovskite ferroelectrics, no one was reported to have blue light emission. In this study, we synthesized a new hybrid halide lead perovskite ((DMA)4PbI6(CH3)4PbI6) with a general formula of A4BX6. (DMA)4PbI6 is a 0 dimensional perovskite, where the individual lead halide octahedra (PbI6) are completely isolated from each other and surrounded by the wide-band-gap organic ligands (CH3NH2CH3+). This isolation allows the bulk crystals to exhibit the intrinsic properties of the individual lead halide units/clusters by inhibiting interactions between the lead halide octahedra.20–22 0D lead iodide hybrid perovskites have been significantly underexplored, although some early literature studies have demonstrated lead iodide hybrid perovskites with a 0D structure. For example, MA4PbI6·2H2O is reported to have a 0D structure with isolated PbI6 octahedra.23 However, this crystal phase is unstable and slowly decomposes into PbI2.24 The most studied 0D metal halide hybrid perovskites are Sn4+, Sn2+, Bi3+, and Te4+ based octahedra. The properties of 0D organic lead iodine hybrid perovskites have not been reported. Pb2+ based 0D iodine hybrid perovskites require four positive monovalent organic molecules to form a stable crystal structure. In the DMA4PbI6 structure, the PbI64− octahedra are completely decoupled by wide band DMA+ molecules in all dimensions. The optical properties of the DMA4PbI6 crystals may closely resemble those of individual PbI64− clusters. The unique photophysical properties of PbI6 based 0D perovskites are of interest for a variety of potential applications, such as optically pumped and electrically driven LEDs, lasers, scintillators, etc. in addition to the intrinsic optical properties, we found that DMA4PbX6 is also a room temperature 0D ferroelectric material. Ferroelectricity in 0D perovskites may be helpful to regulate the optical and electrical properties.
DMA halide hybrid perovskites have been reported previously, such as DMAPbI3,25,26 DMAPbBr3,27 DMAPbCl3,27 DMA7PbBr15,28 and DMA7PbCl15.28 0D DMA4PbI6 has not been reported before. The optical energy gap energy is 2.80 eV, which is larger than 2.59 eV of DMAPbI3.25 A broadband photoluminescence centered at 2.7 eV was observed, which may be attributed to the self-trapped excitons.
2. Experimental section
2.1. Crystal growth
1 mmol PbI2 and 4 mmol MAI or FAI or NH4I were mixed with 20 ml of DMF. The solution was stirred until the solution became clear. Then 10 ml of hydroiodic acid (57%) and 2 ml of 50% phosphorous acid solution were added dropwise into the solution. Phosphorous acid can protect hydroiodic acid from oxidation. The solution was stirred for about 2 hours. 0D DMA4PbI6 single crystals were obtained by slow evaporation of the solution at 60 °C. About two weeks later, crystals appeared in the solution. About a month later, large single crystals were collected.2.2. Characterization
The elemental analysis was performed on a Heraeus CHN-0-Rapid elemental analyzer. The absorption spectrum was recorded by using a PV Measurements Model QEX10 through using an Integral Sphere Method from 300 nm to 1100 nm. Single crystal X-ray diffraction was carried out by using a Bruker D8 QUEST. Powder X-ray diffraction (PXRD) was conducted on a Rigaku D/MAX 2000 PC X-ray diffractometer. DSC measurements of single crystals were recorded by using a NETZSCH DSC 200F3 in the temperature range of 100–300 K. All electrical tests were carried out on single crystal flakes as shown in Fig. S1 (ESI†). The silver paste is coated on the largest crystal surface as an electrode. The complex permittivity was measured using a Tonghui TH2828A LCR meter. The photoluminescence spectra were detected by time resolved fluorescence spectroscopy (FLSP20, Edinburgh, lhstrcamenss). The absorption spectra were recorded by using a Quantum efficiency tester. Raman spectra were collected using a Horiba Jobin Yvon HR800 spectrometer device by means of a 488 nm laser line. P–E hysteresis loops were recorded on a Precision Premier II (Radiant Technologies, Inc.). Pyroelectric current was measured using a Keithley 6517B. Silver glue is applied on the largest surface of the crystal as an electrode. The temperature is controlled by blowing liquid nitrogen to cool down and by using heating plates to heat. During the pyroelectric current measurement, the heating and cooling rate is 10 K min−1. The IV tests are conducted using a Keithley 2400 at room temperature.3. Results and discussion
As 0D DMA4PbI6 is not stable in PbI2 solution, it is hard to eliminate 0D DMA4PbI6 single crystals in the traditional methods. Furthermore, when DMA was added into the solution, it is easier to grow 1D DMAPbI3 rather than 0D DMA4PbI6. Though 0D DMA4PbI6 crystals can appear in the solution, 0D DMA4PbI6 tends to decompose to form 1D DMAPbI3 crystals. In the typical method, DMAI and PbI2 were used as reactants. Here, DMAI comes from the reaction of DMF, HI and H2O. MAI or FAI or NH4I was added to the solution to form clusters with PbI2 in the solution. The growth of 3D MAPbI3 in DMF solution is hard. So we do not need to worry about growing MAPbI3 impurities. In this way, single crystals of DMA4PbI6 were obtained by slow evaporation at 60 °C. Due to the consumption of MAI or FAI or NH4I by PbI2, DMA+ is greatly excessive in comparison to the free PbI64−, which contributes to the single crystal growth of DMA4PbI6. Fig. S1 (ESI†) shows the photograph of single crystals we acquired. The single crystals present parallelogram-shaped flakes. The side of the parallelogram is about two millimeters long. The thickness of the crystal is 0.5–1 mm.To ascertain the structure and purity of compounds (DMA)4PbI6, we quantitatively measured the mass fractions of carbon, and nitrogen in the compound by means of CHN elemental analysis. The results are C 8.28%, H 2.84%, and N 4.84% for the compound, which are in accordance with the theoretical values (C 8.32%, H 2.78%, and N 4.85%). The measurement error of the mass fractions is about 0.3%. The Raman spectrum was recorded on the surface of single crystal of (DMA)4PbI6 (Fig. S3, ESI†). The Raman spectrum corresponds to the vibration spectrum of dimethylamine ions, proving that the organic molecules in the crystal are dimethylamine ions.
Single crystal X-ray diffraction (SCXRD) was performed to obtain the crystal structure of the 0D organic metal hybrid perovskites. The structure of the obtained crystal was solved with Shelxtl 97 package. It is hard to choose the room temperature space group based on the single crystal X-ray diffraction data. Two space groups were suggested, Cmca and Aba2. The CFOM of the two space group is close. According to the room temperature ferroelectric properties, the crystal structure of (DMA)4PbI6 was solved in polar space group (Aba2). Fig. 1 shows the crystal structure of (DMA)4PbI6. In (DMA)4PbI6, the individual PbI64− octahedra are surrounded by eight DMA+ cations. The iodine ions in (DMA)4PbI6 are no longer shared between PbI64− octahedra. At room temperature, N atoms in the DMA cations and 2/3 iodine atoms are disordered. All N atoms in DMA have a 50% probability of appearing in each of the two positions. The disorder of DMA molecules may be caused by the rotation of half of the DMA molecules along the C–C axis. The disorder of 2/3 iodine atoms may be caused by the rotation of half of the PbI64− along the rest of the I–Pb–I axis. Disorder makes the crystals crystallize in a relatively high symmetry space group Aba2. Compared with Cs4PbI6, (DMA)4PbI6 could be considered as true 0D organometal halide perovskites due to the complete isolation of the photoactive PbI64− octahedra by the wide band DMA+ cations.4,22,29 The powder X-ray diffraction pattern of the ball milled crystal powder is in well accordance with the simulated patterns from SCXRD, indicating the purity of the synthesized 0D (DMA)4PbI6 crystals.
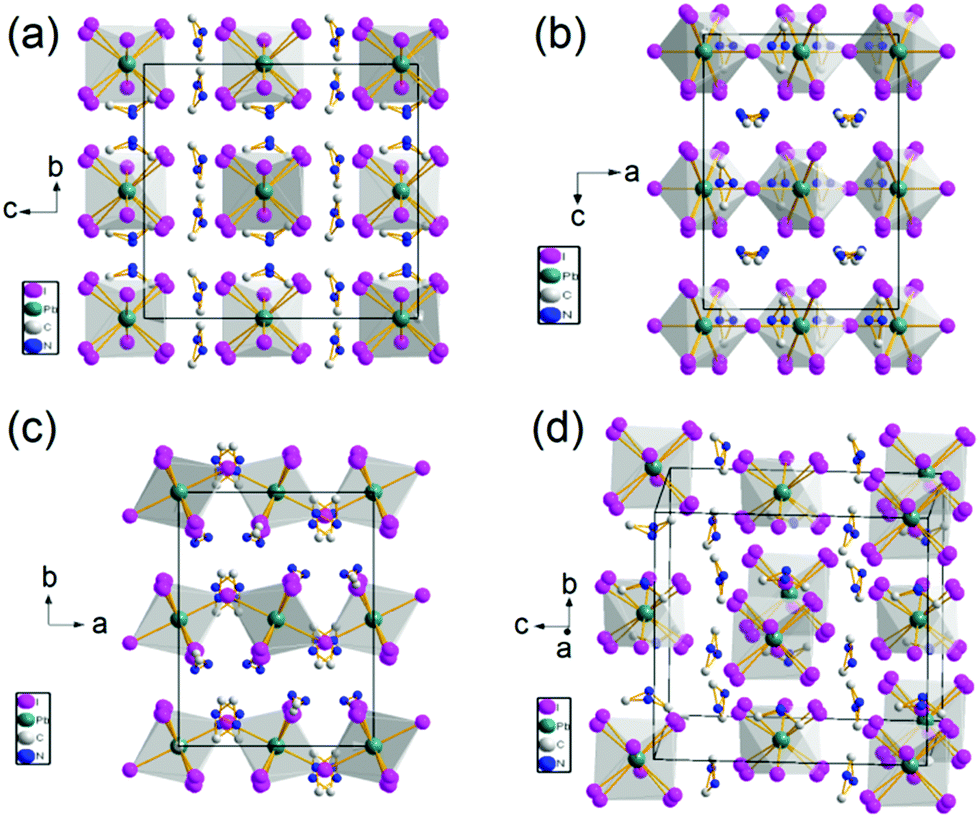 | ||
| Fig. 1 Crystal structure of 0D (DMA)4PbI6 shown with polyhedron. | ||
Most ferroelectrics exhibit phase transitions. Differential scanning calorimetry (DSC) is sensitive to phase transition. Fig. 2a shows the DSC curve of the powder of 0D (DMA)4PbI6. An endothermic peak at 252.5 K and an exothermic peak at 234.2 K can be found from the DSC when scanning at a temperature rate of 10 K min−1, indicating the reversible phase transition happening at around 252 K. This phase transition displays a large hysteresis of 18.3 K, suggesting a first order phase transition. The enthalpy (ΔH) is 3.07 × 104 J mol−1 in the cooling process and 3.32 × 104 J mol−1 in the heating process. The entropy change is 131.1 J mol−1 K−1 (cooling) and 131.5 J mol−1 K−1 (heating), as determined from the area under the heat flow/temperature curve and the peak temperature Tmax. The large entropy change indicates that this phase transition belongs to order–disorder phase transition. Considering the disorder of DMA+ and iodine atoms in DMA4PbI6 at room temperature, the order–disorder phase transition may originate from the rotation of DMA+ molecules and PbI64−.
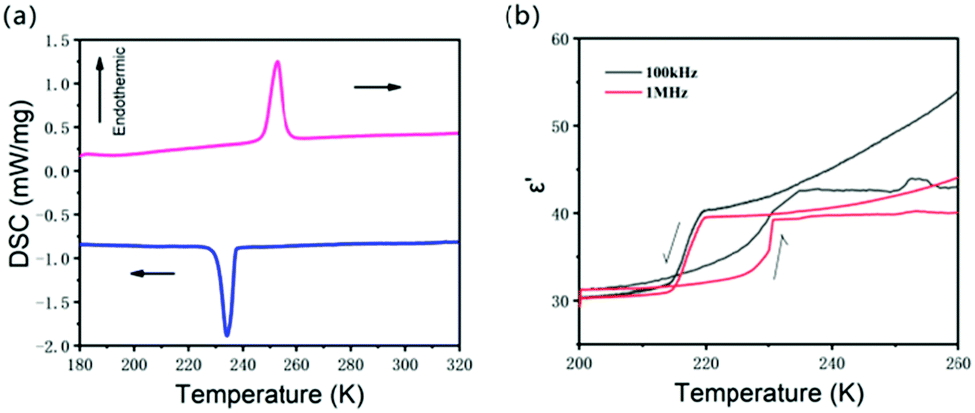 | ||
| Fig. 2 (a) DSC of (DMA)4PbI6 measured with 10 K min−1. (b) The temperature dependent real part of the dielectric constant in the heating process and in the cooling process. | ||
SCXRD of (DMA)4PbI6 was also performed below the phase transition temperature. However, the diffraction point of the low temperature phase is so poor that we cannot obtain the low temperature crystal structure of (DMA)4PbI6. Twins and defects induced by the phase transition may be responsible for the poor crystal diffraction data.
The complex dielectric constants (ε = ε′ − ε′′, where ε′ and ε′′ are the real and imaginary parts, respectively) of DMA4PbI6 were measured on the single crystal. The heating and cooling temperature rate is about 5 K min−1. The temperature dependent dielectric ε′ (Fig. 2b and Fig. S4, ESI†) shows a step-like dielectric anomaly at around phase transition temperature (T1). As the phase transition has a hysteresis of 18.3 K in the heating and cooling process, the dielectric is bistable around T1 with a temperature range of about 15 K. As a switchable dielectric, DMA4PbI6 can undergo a transition between high and low dielectric states at T1. Due to the existence of polar DMA molecules, dipolar reorientation in DMA4PbI6 may contribute significantly to the dielectric response.25,30–32 In DMA4PbI6, all the DMA molecules are disordered above T1. The phase transition of DMA4PbI6 is an order–disorder phase transition according to the DSC result. The motion of DMA may contribute a lot to the dielectric transition in DMA4PbI6.
The ferroelectricity of DMA4PbI6 was confirmed by the P–E (polarization–electric field) hysteresis loop. A typical ferroelectric hysteresis loop was observed at 269 K and 210 K with a Sawyer–Tower circuit (Fig. 3). The ferroelectric hysteresis loop measured at 269 K and 210 K indicates that 0D DMA4PbI6 is ferroelectric below T1 and above T1. At 269 K, the spontaneous polarization Ps is about 0.29 μC cm−2, the coercive field Ec is about 1.34 kV cm−1, and the remanent polarization Pr is about 0.19 μC cm−2. The PS of 0D DMA4PbI6 is comparable to that of Rochelle salt. At 210 K, Ps, Pr and Ec are 0.24 μC cm−2, 0.11 μC cm−2, and 0.89 kV cm−1, respectively. The spontaneous polarization of DMA4PbI6 at 210 K is smaller than that at 269 K, accompanied by a smaller coercive field and remnant polarization. So the high temperature phase is the high polarization state and the low temperature phase is the low polarization state. The switchable dielectric behavior is caused by the ferroelectric–ferroelectric phase transition.
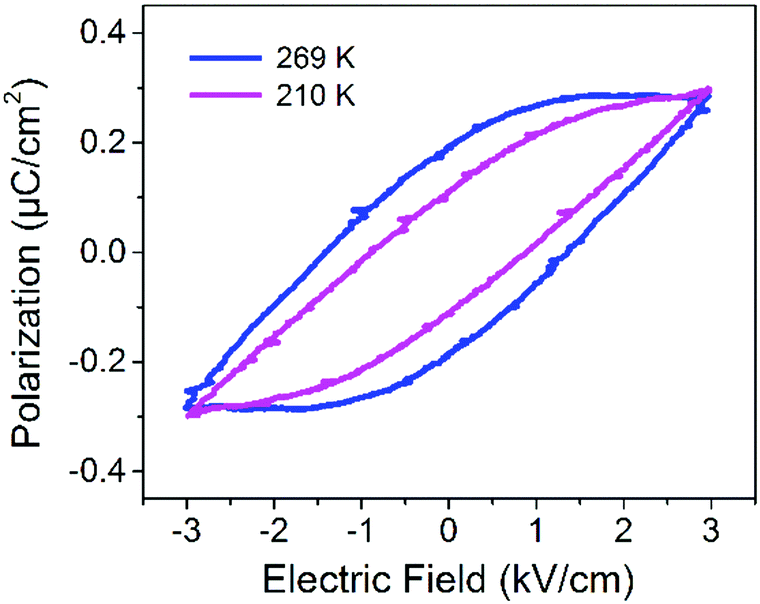 | ||
| Fig. 3 The ferroelectric hysteresis loop of (DMA)4PbI6 measured on the 001 face. | ||
To further determine the ferroelectricity, we measured the I–V characteristics of the material. As shown in Fig. S10 (ESI†), 0-dimensional materials exhibit the I–V characteristic curve of ferroelectric materials at room temperature. Obvious polarization reversal current peaks can be observed around 35 V. The P–E hysteresis loop and I–V characteristic curve of ferroelectrics are adequate to prove the ferroelectricity of 0D DMA4PbI6, which is different from the “banana effect”. The I–V characteristic curve of ferroelectric cannot be observed in the banana effect.
The pyroelectric properties of single crystals of DMA4PbI6 were determined to confirm the ferroelectricity (Fig. S8 and S9, ESI†). Across the phase transition temperature range, a broad pyrolelectric current peak was detected implying that 0D DMA4PbI6 is an improper ferroelectric compound. Polarization changes acquired from the pyroelectric current measurements are larger than that acquired from P–E hysteresis loop measurements, which may be caused by the high conductive properties induced by high defect concentrations.
The absorption spectrum of DMA4PbI6 was recorded by using an integral sphere method. Fig. 4a shows the absorption spectrum of DMA4PbI6 powder. The optical coefficient α of DMA4PbI6 around the optical absorption band can be fitted well with Kubelka–Munk function ((αhv)2 = C(hv − Eg), Eg is the band gap), indicating that DMA4PbI6 is a direct band gap compound. The optical band gap Eg of DMA4PbI6 is determined to be 2.80 eV by the Kubelka–Munk function. The band gap of DMA4PbI6 is smaller than that of Cs4PbI6 (3.38 eV).33 Due to the correlation between Cs and PbI6, Cs4PbI6 does not exhibit intrinsic luminescence of individual PbI6 octahedra. Intrinsic luminescence of individual PbI6 octahedra is characteristic of 0-dimensional materials.
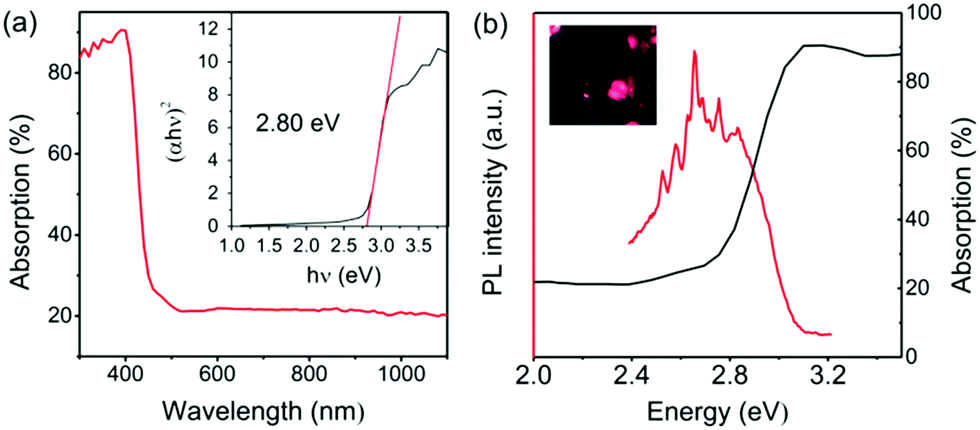 | ||
| Fig. 4 (a) The absorption spectra of (DMA)4PbI6. The inset shows the fitting curve of ((αhv)2 = C(hv − Eg)). The optical band gap is determined to be 2.80 eV. (b) The PL spectra excited with 200 nm UV light. The inset shows the digital photograph of the single crystals illuminated with 10 W 365 nm UV light. | ||
Photoluminescence spectra of 0D DMA4PbI6 were recorded to explore the intrinsic photoluminescence of PbI6 octahedra. Fig. 4b and Fig. S5 (ESI†) show the photoluminescence spectra of DMA4PbI6 excited with 200 nm UV light. Four photoluminescence peaks, peaking at 340 nm, 362 nm, 455 nm and 553 nm, were found in DMA4PbI6. Among the four photoluminescence peaks, 455 nm photoluminescence belongs to the blue emission and is the strongest photoluminescence peak of DMA4PbI6. The blue emission is a broadband emission with full width at half maximum (FWHM) of about 80 nm. This FWHM is wider than that of 1D DMAPbI3. The photoluminescence of 0D DMA4PbI6 and 1D DMAPbI3 is similar (Fig. S5, ESI†). The photoluminescence region of the 0D and 1D compounds is almost the same, except that of the broadband blue emission of the 0D compounds. This means that the main photoluminescence of the 1D and 0D compounds comes from the PbI6 octahedra. Fluorescence at 553 nm may be derived from iodine atoms.34–36 The UV and blue fluorescence may be derived from Pb ions.37 The broadband blue emission in 0D DMA4PbI6 may be ascribed to the self-trapped excitons of PbI6.38 Without intermolecular interactions or electronic band formation, 0D perovskites are expected to be the most favorable environment for the formation of self-trapped excited states.39,40 Interactions between the PbI6 octahedra in 1D DMAPbI3 may weaken the self-trapped excitation, resulting in a narrower emission peak in 1D DMAPbI3 than that in 0D DMA4PbI6.
The inset of Fig. 4b shows the microscopy photograph of DMA4PbI6 crystals under 365 nm UV light. Under 365 nm ultraviolet light, we can observe that the crystal emits blue-violet light (Fig. 4b inset). It can be observed from the photoluminescence excitation spectra of the blue emission (Fig. S5, ESI†) that deep ultraviolet (UVC) light is more effective in exciting the blue emission compared to all region UV light. 365 nm UV light can only excite weak blue emission.
4. Conclusions
The first stable 0D organic lead iodine perovskite single crystal without a crystallizing solvent was grown using MAI or FAI or NH4I in addition to PbI2 in the solution. (DMA)4PbI6 exhibits a first order phase transition at Tc (about 260 K), accompanied by a switchable dielectric behavior. The single crystal of (DMA)4PbI6 is still stable up to 450 K. PE hysteresis loops show that (DMA)4PbI6 is ferroelectric both above Tc and below Tc. (DMA)4PbI6 is a room temperature ferroelectric material with a spontaneous polarization of 0.3 μC cm−2. A broad band blue photoluminescence was found in 0D (DMA)4PbI6 which may be ascribed to the self-trapped excitation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province, China (BK20170482), the National Natural Science Foundation of China (11874200), and the Major Research Plan (2017YFA0303202).Notes and references
- Z. Xiao, Y. Yuan, Y. Shao, Q. Wang, Q. Dong, C. Bi, P. Sharma, A. Gruverman and J. Huang, Nat. Mater., 2015, 14, 193 CrossRef.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef.
- Y.-M. You, W.-Q. Liao, D. Zhao, H.-Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P.-F. Li and D.-W. Fu, Science, 2017, 357, 306–309 CrossRef.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef.
- K. Gao, C. Liu and B. Zhang, J. Mater. Chem. C, 2020, 8, 10104–10108 RSC.
- K. Gao, C. Liu, W. Zhang, K. Wang and W. Liu, R. Soc. Open Sci., 2020, 7, 200271 CrossRef.
- Z. Chen, B. Turedi, A. Y. Alsalloum, C. Yang, X. Zheng, I. Gereige, A. AlSaggaf, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2019, 4, 1258–1259 CrossRef.
- Y. Wu, F. Xie, H. Chen, X. Yang, H. Su, M. Cai, Z. Zhou, T. Noda and L. Han, Adv. Mater., 2017, 29, 1701073 CrossRef.
- L. Qiao, W.-H. Fang and R. Long, J. Phys. Chem. Lett., 2019, 10, 7237–7244 CrossRef CAS.
- W. Li, J. Zeng, L. Zheng, H. Zeng, C. Li, A. Kassiba, C. Park and G. Li, Ferroelectrics, 2019, 553, 95–102 CrossRef CAS.
- L. M. Garten, D. T. Moore, S. U. Nanayakkara, S. Dwaraknath, P. Schulz, J. Wands, A. Rockett, B. Newell, K. A. Persson and S. Trolier-McKinstry, Sci. Adv., 2019, 5, eaas9311 CrossRef.
- Y. Rakita, O. Bar-Elli, E. Meirzadeh, H. Kaslasi, Y. Peleg, G. Hodes, I. Lubomirsky, D. Oron, D. Ehre and D. Cahen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5504–E5512 CrossRef CAS.
- H.-S. Kim, S. K. Kim, B. J. Kim, K.-S. Shin, M. K. Gupta, H. S. Jung, S.-W. Kim and N.-G. Park, J. Phys. Chem. Lett., 2015, 6, 1729–1735 CrossRef CAS.
- A. Gomez, Q. Wang, A. R. Goni, M. Campoy-Quiles and A. Abate, Energy Environ. Sci., 2019, 12(8), 2537–2547 RSC.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czka, A. Gagor, J. K. Zarȩba, D. Stefanska, M. Drozd, S. Balciunas, M. Šimėnas, J. Banys and A. Sieradzki, Chem. Mater., 2020, 32, 4072–4082 CrossRef.
czka, A. Gagor, J. K. Zarȩba, D. Stefanska, M. Drozd, S. Balciunas, M. Šimėnas, J. Banys and A. Sieradzki, Chem. Mater., 2020, 32, 4072–4082 CrossRef. - M. Mczka, M. Ptak, A. Ggor, D. Stefańska, J. K. Zarȩba and A. Sieradzki, Chem. Mater., 2020, 32, 1667–1673 CrossRef.
- C.-K. Yang, W.-N. Chen, Y.-T. Ding, J. Wang, Y. Rao, W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Z.-X. Wang and R.-G. Xiong, Adv. Mater., 2019, 31, 1808088 CrossRef.
- X.-N. Hua, W.-Q. Liao, Y.-Y. Tang, P.-F. Li, P.-P. Shi, D. Zhao and R.-G. Xiong, J. Am. Chem. Soc., 2018, 140, 12296–12302 CrossRef CAS.
- T.-T. Sha, Y.-A. Xiong, Q. Pan, X.-G. Chen, X.-J. Song, J. Yao, S.-R. Miao, Z.-Y. Jing, Z.-J. Feng, Y.-M. You and R.-G. Xiong, Adv. Mater., 2019, 31(30), 1901843 CrossRef.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2017, 3, 54–62 CrossRef.
- C. Zhou, Y. Tian, M. Wang, A. Rose, T. Besara, N. K. Doyle, Z. Yuan, J. C. Wang, R. Clark and Y. Hu, Angew. Chem., Int. Ed., 2017, 56, 9018–9022 CrossRef CAS.
- C. Zhou, H. Lin, Y. Tian, Z. Yuan, R. Clark, B. Chen, L. J. van de Burgt, J. C. Wang, Y. Zhou and K. Hanson, Chem. Sci., 2018, 9, 586–593 RSC.
- B. R. Vincent, K. N. Robertson, T. S. Cameron and O. Knop, Can. J. Chem., 1987, 65, 1042–1046 CrossRef.
- G. H. Imler, X. Li, B. Xu, G. E. Dobereiner, H.-L. Dai, Y. Rao and B. B. Wayland, Chem. Commun., 2015, 51, 11290–11292 RSC.
- A. García-Fernández, J. M. Bermúdez-García, S. Castro-García, A. L. Llamas-Saiz, R. Artiaga, J. López-Beceiro, S. Hu, W. Ren, A. Stroppa, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Inorg. Chem., 2017, 56, 4918–4927 CrossRef.
- Y. Wang, Y. Liu, Y. Wu, J. Jiang, C. Liu, W.-L. Liu, K. Gao, H. Cai and X. Wu, CrystEngComm, 2020, 22, 7090–7094 RSC.
- A. García-Fernández, E. J. Juarez-Perez, J. M. Bermúdez-García, A. L. Llamas-Saiz, R. Artiaga, J. J. López-Beceiro, M. A. Señarís-Rodríguez, M. Sánchez-Andújar and S. Castro-García, J. Mater. Chem. C, 2019, 7, 10008–10018 RSC.
- A. García-Fernández, J. M. Bermúdez-García, S. Castro-García, A. L. Llamas-Saiz, R. Artiaga, J. J. López-Beceiro, M. Sánchez-Andújar and M. A. Señarís-Rodríguez, Inorg. Chem., 2018, 57, 3215–3222 CrossRef.
- D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1–12 RSC.
- W. Zhang, H.-Y. Ye, R. Graf, H. W. Spiess, Y.-F. Yao, R.-Q. Zhu and R.-G. Xiong, J. Am. Chem. Soc., 2013, 135, 5230–5233 CrossRef CAS.
- M. Šimėnas, S. Balčiūnas, A. Ciupa, L. Vilčiauskas, D. Jablonskas, M. Kinka, A. Sieradzki, V. Samulionis, M.
![[M with combining cedilla]](https://www.rsc.org/images/entities/char_004d_0327.gif) czka and J. Banys, J. Mater. Chem. C, 2019, 7, 6779–6785 RSC.
czka and J. Banys, J. Mater. Chem. C, 2019, 7, 6779–6785 RSC. - M. Simenas, S. Balciunas, J. N. Wilson, S. Svirskas, M. Kinka, A. Garbaras, V. Kalendra, A. Gagor, D. Szewczyk, A. Sieradzki, M. Maczka, V. Samulionis, A. Walsh, R. Grigalaitis and J. Banys, Nat. Commun., 2020, 11, 5103 CrossRef CAS.
- M. Nikl, E. Mihokova, K. Nitsch, F. Somma, C. Giampaolo, G. Pazzi, P. Fabeni and S. Zazubovich, Chem. Phys. Lett., 1999, 306, 280–284 CrossRef CAS.
- Q. Han, S. H. Bae, P. Sun, Y. T. Hsieh, Y. Yang, Y. S. Rim, H. Zhao, Q. Chen, W. Shi and G. Li, Adv. Mater., 2016, 28, 2253–2258 CrossRef CAS.
- H.-Y. Ye, Y.-Y. Tang, P.-F. Li, W.-Q. Liao, J.-X. Gao, X.-N. Hua, H. Cai, P.-P. Shi, Y.-M. You and R.-G. Xiong, Science, 2018, 361, 151–155 CrossRef CAS.
- A. Cingolani, M. Ferrara and M. Lugarà, Opt. Commun., 1979, 28, 97–100 CrossRef CAS.
- A. Bohun, J. Dolejší and Č. Barta, Czech. J. Phys., 1970, 20, 803–807 CrossRef CAS.
- B.-B. Zhang, B. Xiao, S. Dong and Y. Xu, J. Cryst. Growth, 2018, 498, 1–4 CrossRef CAS.
- J. Yin, J.-L. Brédas, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2020, 32, 5036–5043 CrossRef CAS.
- B.-B. Cui, Y. Han, B. Huang, Y. Zhao, X. Wu, L. Liu, G. Cao, Q. Du, N. Liu, W. Zou, M. Sun, L. Wang, X. Liu, J. Wang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 5190 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2016638. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc04813e |
| This journal is © The Royal Society of Chemistry 2021 |