
A soft anti-virulence liposome realizing the explosive release of antibiotics at an infectious site to improve antimicrobial therapy†
Shudong
Zhang‡
abc,
Xiang
Lu‡
abc,
Binghua
Wang
a,
Ge
Zhang
a,
Mengyuan
Liu
a,
Shizhen
Geng
a,
Lulu
Sun
a,
Jingyi
An
a,
Zhenzhong
Zhang
 a and
Hongling
Zhang
*abcd
a and
Hongling
Zhang
*abcd
aSchool of Pharmaceutical Sciences, Zhengzhou University, No. 100, Kexue Avenue, Zhengzhou 450001, China. E-mail: zhanghongling729@zzu.edu.cn; Fax: +86 371 6778 1908; Tel: +86 371 6778 1911
bKey Laboratory of Targeting Therapy and Diagnosis for Critical Diseases, Henan Province, China
cCollaborative Innovation Center of New Drug Research and Safety Evaluation, Henan Province, Zhengzhou, China
dSchool of Materials Science and Engineering, Zhengzhou University, Zhengzhou, China
First published on 5th November 2020
Abstract
Pore-forming toxins (PFTs), the most common virulence proteins, are promising therapeutic keys in bacterial infections. CAL02, consisting of sphingomyelin (Sm) and containing a maximum ratio of cholesterol (Ch), has been applied to sequester PFTs. However, Sm, a saturated phospholipid, leads to structural rigidity of the liposome, which does not benefit PFT combination. Therefore, in order to decrease the membrane rigidity and improve the fluidity of liposomes, we have introduced an unsaturated phospholipid, phosphatidylcholine (Pc), to the saturated Sm. In this report, a soft nanoliposome (called CSPL), composed of Ch, Sm and Pc, was artificially prepared. In order to further improve its antibacterial effect, vancomycin (Van) was loaded into the hydrophilic core of CSPL, where Van can be released radically at the infectious site through transmembrane pores formed by the PFTs in CSPL. This soft Van@CSPL nanoliposome with detoxification/drug release was able to inhibit the possibility of antibiotic resistance and could play a better role in treating severe invasive infections in mice.
1. Introduction
Bacterial infections, as the most prevalent diseases in clinical medicine, seriously threaten public health.1 Since the discovery of the first antibiotic in 1940, antibacterial therapy has made rapid progress.2 However, with the widespread use and misuse of antibiotics, the growth of antibiotic resistance has significantly limited use of the full lifespan of antibiotics.3,4 Despite the fact that some antibiotics are still effective against multidrug-resistant pathogens, antibiotic resistance remains a public health crisis.5,6 Classic antimicrobials kill bacteria by affecting vital processes required for bacterial growth and survival, easily resulting in bacterial evolution and the emergence of drug resistance.7,8 Therefore, alternative antimicrobial treatment strategies are urgently required in order to prevent drug resistance.During infections, bacteria can secrete a large amount of toxins, which can be used as protective “weapons” for the bacteria.9 For example, pore-forming toxins (PFTs) play an important role in increasing morbidity and mortality.10 PFTs are able to specifically identify and combine with host cell membranes, and then oligomers are inserted directly into the lipid bilayer to form transmembrane pores, which alter the balance of the intracellular ion concentration, resulting in cell disruption and tissue damage.11,12 PFTs can also provide nutrients for bacterial reproduction, thereby increasing the amount of PFTs themselves, leading to endless attack of the host cells. Moreover, PFTs can protect the bacteria from being eliminated by interrupting the host immune response during infections.13 Therefore, PFTs are important targets for broadly applicable antimicrobial therapeutics.
In recent years, several formulations have been reported for neutralizing PFTs. Zhang et al. have designed a “nanosponge”, in which a polymeric core was coated by the membrane of red blood cells. This biodegradable and biocompatible “nanosponge” can be widely applied for detoxification of PFTs.14 However, considering the differences in blood types of people, this will become difficult for treatment purposes. In addition, as a type of biological product, it will be a challenge for the “nanosponge” to be used because of its harsh storage conditions. Consequently, there is an urgent need to exploit a novel broad-spectrum antibacterial agent for PFT neutralization.
It is well known that artificial liposomes have a similar lipid bilayer structure to animal plasmalemma (such as an erythrocyte membrane). In addition, liposomes have been widely used as carriers for drug delivery, possessing many merits such as good biocompatibility, low immunogenicity, an efficient drug-loading ability, and a long half-life.15 Therefore, it will be a unique mode of action to use liposomes as an antimicrobial therapeutic strategy by capturing PFTs. Compared to the cell membranes of bacteria, cholesterol (Ch) and sphingomyelin (Sm), which specifically exist in the membranes of animal cells, are the basic objective molecules for PFTs.12,16,17 Therefore, some strategies have attempted to sequester PFTs using a liposome composed of Sm and a high concentration (66 mol per %) of Ch (CSL2 in this manuscript).16 In contrast to the host cells, the ratio of Ch and Sm in CSL2 is greatly increased. Thus, CSL2 possesses higher selectivity towards PFTs than the host cells, thereby exerting a detoxification effect. Furthermore, the binding efficacy of liposomes with PFTs is closely related to the fluidity of phospholipid bilayers.18,19 Sm, as a long-chain saturated phospholipid, leads to structural rigidity of liposomes, and this does not benefit the combination of PFTs.20,21 In addition, increasing the ratio of Ch in the Sm-liposome can improve the membrane fluidity. In this report, in order to further increase the affinity between the liposome and the PFTs, phosphatidylcholine (Pc), a natural unsaturated phospholipid, was added into CSL2 to weaken its structural rigidity, thereby greatly improving the fluidity of the liposome.22 Additionally, increasing the variety of phospholipids can also enhance the fluidity of the liposomes. Thus, a soft nanoliposome, composed of Ch, Sm, and Pc in the molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, was synthesized (called CSPL).
:1:1, was synthesized (called CSPL).
Furthermore, the first-line antibacterial drug, vancomycin (Van), was selected as a model drug and was encapsulated into the hydrophilic core of CSPL (Van@CSPL). When Van@CSPL reached the infected site, the CSPL captured a large amount of PFTs. At the same time, Van was released explosively to the infection site through the transmembrane pores formed by the PFTs, hence decreasing the side effects of antibiotics and simultaneously reducing the likelihood of the increase in drug resistance. It has been shown that sublethal levels of antibiotic treatment easily induce mutagenesis, thereby leading to drug resistance.23,24 In this study, the toxin-binding efficacy and antibacterial effect of CSPL were tested. As a result, this soft CSPL displayed excellent toxin-binding efficacy in vitro and potent antibacterial efficacy in Staphylococcus aureus (S. aureus) infections. In addition, this smart Van@CSPL nanoliposome exerts a better antibacterial effect. This work provides a novel strategy for enhancing the potency of antibiotic therapy, along with avoiding the emergence of drug resistance (Scheme 1).
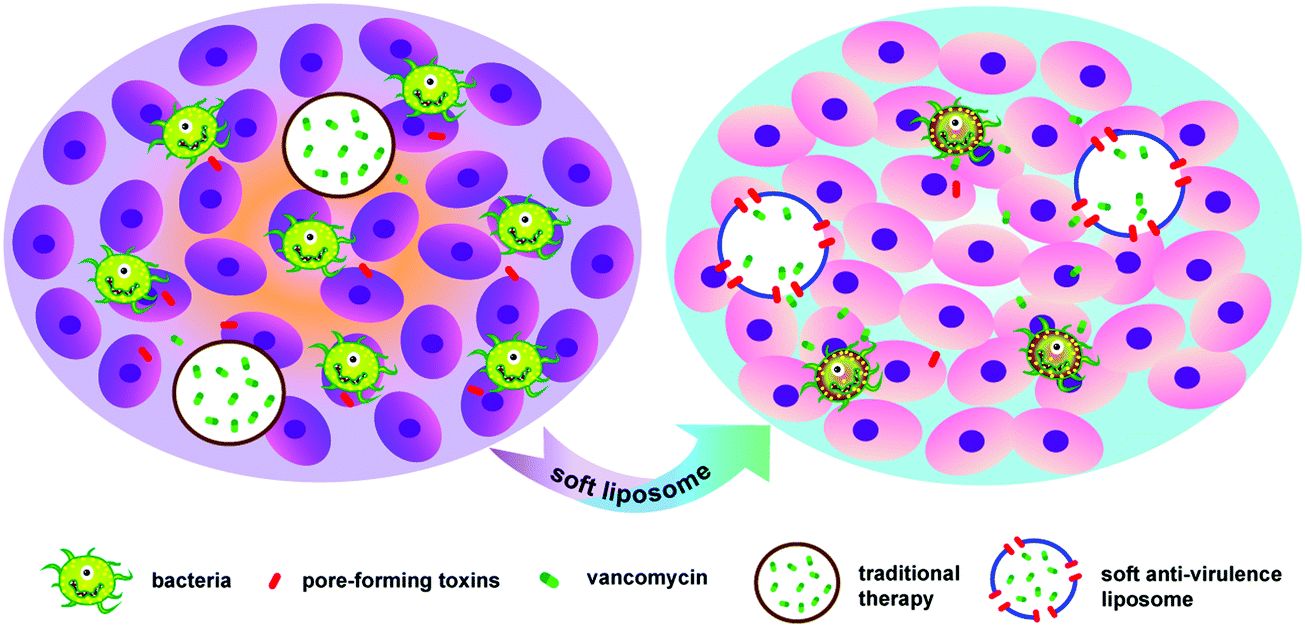 | ||
| Scheme 1 Schematic design of Van@CSPL for neutralizing PFTs, as well as for the treatment of bacterial infections. Based on the pore-forming principle between PFTs and the membranes of animal cells, CSPLs possess potent affinity for PFTs. In addition, when Van@CSPL reaches the infected site, CSPL can absorb a large amount of PFTs. At the same time, PFTs form pores on the membrane of CSPL, therefore Van can be released explosively and can develop an effective antibacterial effect. | ||
2. Materials and methods
2.1 Materials
Vancomycin (Van), dipalmitoyl phosphatidylcholine (DPPC), and L-α-phosphatidylcholine were purchased from Switzerland Solarbio Co. Ltd. Sphingomyelin and α-hemolysin (α-toxin) from Staphylococcus aureus were obtained from Sigma-Aldrich, whilst 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-poly(ethyleneglycol)2000 (mPEG2000-DSPE) was procured from Lipoid Co. Ltd (Ludwigshafen, Germany). Cholesterol was received from Chengdu Kelong Chemical Company (Chengdu, Sichuan, China). The Cell Counting Kit-8 was purchased from Dongren Chemical Technology Co. Ltd (Shanghai, China). Cyclophosphamide monohydrate (BR, 97%) was bought from Source Leaf Biotechnology Co. Ltd (Shanghai, China). Other reagents were acquired from China National Medicine Corporation Ltd.2.2 Cell culture
The human umbilical vein endothelial cells (HUVECs) used in this study were obtained from the China Center for Type Culture Collection at Wuhan University (Wuhan, Hubei, China). HUVECs were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS). All cells were cultivated at 37 °C in a humidified atmosphere with 5% CO2, and the cells used in the experiments were at the stage of cell index growth.2.3 Preparation of conventional liposome (CL)
The conventional liposome was fabricated by a solvent evaporation method. Briefly, DPPC (14 mg), Ch (2 mg) and mPEG2000-DSPE (1 mg) were dissolved in chloroform (5 mL). Evaporation to remove the chloroform was carried out, and then the thin film that was formed was dispersed in deionized water (2 mL). The obtained nanosuspension was sonicated twenty times for 5 s at 6 °C using a Ti probe (Branson 450 Sonifier, Danbury, CT) with a power of 150 W. The resulting CL was stored at 4 °C until further use.2.4 Preparation of CSL1, CSL2, and CSPL
Increasing the ratio of Ch can strengthen the fluidity of CSPLs. In addition, a high content of Ch can guarantee the structural stability of a liposome,22 and the maximum ratio of Ch in a liposome is 66 mol per %.16 As the other basic target for PFTs existing on the membranes of mammalian cells, Sm is another important component in CSPLs. In addition, as an unsaturated phospholipids, Pc can enrich the lipid composition and further increase the fluidity of CSPLs. Combined with the above, we finally designed a CSPL, which was composed of Ch, Sm, and Pc in the molar ratio of 4:1:1.
CSL1 (Ch:Sm, 1:1), CSL2 (Ch:Sm, 2:1), and CSPL (Ch:Sm:Pc, 4:1:1) were fabricated by a solvent evaporation method. Briefly, Ch and Pc were individually dissolved in chloroform. Sm was dissolved in a mixture of chloroform and a small amount of methanol (200 μl), and the final concentration of these three solutions was 1 mg ml−1. Next, the solutions were mixed according to their specific composition and proportions. After removing the solution, thin films of CSL1, CSL2, and CSPL were dispersed in deionized water, followed by sonication for 15 min in a bath sonicator (Fisher Scientific FS30D, Pittsburgh, PA). Then, the obtained nanosuspension was sonicated twenty times for 5 s at 6 °C using a Ti probe (Branson 450 Sonifier, Danbury, CT) with a power of 150 W. Finally, the resulting liposomes were stored at 4 °C until further use.
2.5 Preparation of vancomycin-loaded liposomes (Van@Lipo) and characterization of Van@CSPL
Van@Lipo, which includes Van@CL, Van@CSL1, Van@CSL2 and Van@CSPL, were fabricated in a similar way to the above method. However, there were some differences in the method detail. After removing the solvent, thin films of CL, CSL1, CSL2 and CSPL were dispersed in a solution of vancomycin. Then, the nanosuspensions were dialyzed against PBS buffered solution for 1 day to remove free vancomycin (molecular weight cut off: 12000–14000 Da). The resulting nanosuspensions were stored at 4 °C until further use.
In addition, DLS (Zetasizer Nano ZS-90, Malvern, UK) was carried out to examine the average size of Van@CSPL. Furthermore, in order to obtain an optimal mass ratio of Van@CSPL, we set a series of mass ratios of Van and CSPL to 3:5, 2:5 and 1:5. The Van-loading efficiency was detected by ultra-violet-visible (UV-VIS) spectrometry (Lambda 35, PerkinElmer, USA). The absorbance of Van at 280 nm was measured, in order to quantify the concentration of Van in PBS using a pre-developed calibration curve. Then, the loading content and efficiency of Van in CSPL were calculated.
2.6 Characterization of CL, CSL1, CSL2 and CSPL
To identify the morphologies of CSL1 (5 mg mL−1), CSL2 (5 mg mL−1), and CSPL (5 mg mL−1), transmission electron microscopy (TEM) was performed at an acceleration voltage of 200 kV. DLS (Zetasizer Nano ZS-90, Malvern, UK) was applied to examine the average size and zeta potential of CL, CSL1, CSL2 and CSPL.In addition, the stability of CSPL was investigated. CSPLs were stored at 4 °C, and the average size and zeta potential were measured over the next 15 days. In addition, NTA (Nanosight NS300, Malvern, UK) was carried out to measure the size distribution and concentration of the particles of CSPL (1 mg mL−1). Each measurement was carried out in triplicate and a mean value was obtained.
2.7 In vitro hemolysis assay
The ability of CSPL to neutralize PFTs was tested using a hemolysis assay, which included five groups: CL, CSL1, CSL2 and CSPL, and the group with or without α-toxin was used as a control. First, fresh blood from mice was collected, this was then centrifuged (3000 rpm, 10 minutes) and washed 3 times with PBS buffer. Next, the red blood cells (RBCs) were resuspended in PBS to obtain a 5% RBC suspension. After that, 1 μg of α-toxin was mixed with CL, CSL1, CSL2 or CSPL (1 mg mL−1, 200 μL). PBS with or without α-toxin was also included. The mixtures of each group were incubated at 37 °C for 30 min, followed by the addition of 5% RBCs (1 mL) to each group, and incubation under the same conditions.14 Subsequently, the mixtures were centrifuged (3000 rpm, 10 min). The degree of release of haemoglobin in the supernatant was detected at 540 nm. To further test the anti-hemolytic efficiency of CSPL, a series of different amounts of α-toxin (4, 8, 16, 32, 64, 128, and 256 μg) were produced and then experiments carried out in the same way as above.2.8 In vitro cytotoxicity testing
The cellular cytotoxicity of α-toxin was studied in HUVECs using a Cell Counting Kit (CCK-8).14,16 For the experiment, 100 μL of cell suspension was seeded at a density of 5 × 103 per well in a 96-well plate and incubated for 24 h. CL, CSL1, CSL2 and CSPL (20 μg mL−1) were mixed with a series of concentrations of α-toxin (0.2, 2.0, and 20 μg mL−1), respectively, and incubated at 37 °C for 30 min. A sample with α-toxin alone was also included. Then, the mixtures were added to each well, and the plate was incubated for another 24 h. After that, 10 μL of CCK-8 was added into each well and the plate was incubated at 37 °C for 2 h. The absorbance was determined with a microplate reader at 450 nm. Cell viability was calculated using the following formula:| Cell viability (%) = OD (sample)/OD (control) × 100% |
2.9 Cellular internalization
The interactions of CSPL and HUVECs were examined to test the ability of CSPL to absorb PFTs.14 First, DiI-labeled CL (DiI@CL) and DiI-labeled CSPL (DiI@CSPL) were prepared. The HUVECs were seeded into 6-well plates (15 × 104 cells per well) for 24 h cultivation. Next, DiI@CL and DiI@CSPL (100 μL, 1 mg mL−1) were mixed with α-toxin, and incubated at 37 °C for 30 min, respectively. Then, the mixtures were added to each well with final α-toxin concentrations of 4 μg mL−1, 8 μg mL−1, and 16 μg mL−1, and the plates were incubated at 37 °C for 4 h. Thereafter, the cell nuclei were stained with Hoechst 33342 (10 μg mL−1) solution. After fifteen minutes, excess Hoechst 33342 was removed by washing with PBS buffer solution. Finally, the samples were imaged by Laser Scanning Confocal Microscopy (Zeiss LSM 800).2.10 Skin injury assay of CSPL
Female Kunming mice (18–22 g) were divided into three groups. Briefly, for the CSPL group, 100 μL of CSPL (1 mg mL−1) was incubated with 50 μL of α-toxin (60 μg mL−1) at 37 °C for 30 min. Then, the mixture was injected subcutaneously beneath the right flank of skin of the mice. For the groups of α-toxin and the control, 150 μL of α-toxin (20 μg mL−1 in PBS) or PBS were injected in the same way as above, respectively. At 72 h after injection, the skins were collected, sectioned and then stained with haematoxylin and eosin (H&E) for histological analyses.142.11 In vivo distribution of CSPL
To clarify the fate of CSPL in mice, the biodistribution was studied. CSPL were labeled with Dir (a fluorescent dye) before being used. Firstly, 150 μL of PBS or a bolus lethal dose of α-toxin (75 μg kg−1) was intravenously injected into the mice. Two minutes later, 200 μL of Dir-labeled CSPL (8 mg mL−1) was injected through the tail vein. Finally, whole-body fluorescence images (2D FRI) were recorded at desired times (1, 2, 4, 8, 12, and 24 h).2.12 The integrity influence of S. aureus cells induced by Van@CSPL
Vancomycin (Van) can effectively inhibit the growth of Gram-positive pathogens by obstructing the formation of cell wells. We explored whether Van@CSPL could affect the morphology and ultrastructure of S. aureus, included in the four groups: control, Van, Van@CL and Van@CSPL. Firstly, a single colony on a Mueller–Hinton Agar (MHA) plate was transferred into 2 mL of Mueller–Hinton Broth (MHB) culture medium, and this was grown at 37 °C for 10 h. The suspension was centrifuged (3000 rpm) at room temperature for 10 min, and the S. aureus cells were resuspended with 2 mL MHB culture medium. Next, 1.5 mL suspension was added into 6-well plates, and then free Van, Van@CL and Van@CSPL (256 μg mL−1) were added to each well, respectively. As a control, 1.7 mL liquid MHB and 300 μL suspension were added into 6-well plates. The compound cavity was hence set up for each group. The bacteria were incubated at 37 °C for 8 h,25 and then centrifuged as well as washed three times with deionized water. Thereafter, the bacteria were fixed in 2.5% glutaraldehyde for 4 h and processed for scanning electron microscopy (SEM).2.13 In vitro pore-forming studies
To verify the pore-forming activity of α-toxin (the main PFTs secreted by S. aureus), rhodamine B-loaded CL (RB@CL) and RB@CSPL were prepared, and the concentration of RB was 0.512 mg mL−1. The release behavior of RB from RB@CL or RB@CSPL was determined in phosphate buffer (10 mM, pH 7.4). Briefly, RB@CL or RB@CSPL were transferred into dialysis bags (molecular weight cut off: 12000–14000 Da) and immersed into 10 mL release media at 37 °C. At predetermined time points (30 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h, 8 h, 9 h, 10 h, 11 h, 12 h, 24 h, and 36 h), 500 μL dialysis solution was collected, and then 500 μL of the release media was added back into the sample for continued incubation. In addition, at the time point of 2 h, 10 μL of α-toxin (500 μg mL−1) or PBS were added into dialysis bags. The absorbance of rhodamine B in the released media was determined at 556 nm by UV-visible spectrophotometry. The release experiments were conducted in triplicate.
2.14 Antibacterial efficacy of CSPL and Van@CSPL in vivo
S. aureus, being one of the pathogens most related to human health, can cause various diseases ranging from mild skin and soft tissue infections to life-threatening infections, such as bone and joint tissue infections, endocarditis, sepsis, pneumonia, and toxic shock syndrome.26–28 These invasive diseases are closely related to virulence factors (including multitudinous PFTs), which are secreted by S. aureus.14 As a PFT, α-toxin plays a significant role in multiple common infections, including skin and soft tissue infections, sepsis and pneumonia.29,30 Hence, in the following step, the antibacterial efficacies of CSPL and Van@CSPL were studied with the model of S. aureus ATCC29213.After 4 h of infection, the mice were treated with CSPL, CSL2, CSL2, or CL with a 50 mg kg−1 dose by intravenous injection through the tail vein, and the PBS-treated group was also included. Twenty-four hours later, their blood was obtained, and the levels of TNF-α were measured by an enzyme-linked immunosorbent assay (ELISA), according to the manufacturer's instructions. In addition, the blood was coated onto MHA agar plates to calculate the colony forming units (CFUs). In order to investigate the timeliness of the treatment of CSPL, the mice were given an intravenous administration of CSPL (50 mg kg−1) at time points of 6, 10, or 16 h after infection. After twenty-four hours, the CFUs were calculated.16
In addition, the mice were administered with CSPL, CSL2, CSL2, or CL (50 mg kg−1) at time points of 4 and 10 h after infection, and a PBS group was also created. Then, the survival rate of the mice was monitored every day over a 6 days period and the rate was plotted as survival curves. For histological analysis, the main organs (heart, liver, spleen, lung, and kidney) were obtained and fixed with 4% formaldehyde, then they were embedded in paraffin, sliced and stained with H&E. Finally, we investigated the treatment effect of Van@CSPL in S. aureus septicemia. The mice were given intravenous administrations of Van@CSPL, Van@CSL1, Van@CSL2, Van@CL and free Van with doses of 10 mg kg−1 at the time points of 4, 12, and 24 h after infection. In addition, the CSPL group was also included, and the survival rates of the mice were monitored.
In order to assess the therapeutic efficacy of CSPL, the mice were infected with a lethal dose of S. aureus. To summarize in brief, S. aureus cells were washed and resuspended in PBS solution (100 μL), and the mice were intranasally injected with the suspension (40 μL). Four hours later, the mice were treated with CSPL, CSL1, CSL2, or CL intranasally with a dose of 50 mg kg−1. The mortality of the mice was monitored for 3 days.
Finally, we studied the treatment effect of Van@CSPL. The mice were given intravenous administration of Van@CSPL, Van@CSL1, Van@CSL2, Van@CL or free Van (10 mg kg−1)28,31 at time points of 4 h and 10 h after injection, and a CSPL group was also included. Then, the survival rate was monitored and the lung tissues were collected for histological analysis.
2.15 Statistical analysis
Data were presented as the mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was performed for statistical evaluation. A p-value below 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001) was considered to be statistically significant.3. Results and discussion
3.1 Characterization of CSL1, CSL2, and CSPL
First, we synthesized CSL1, CSL2, and CSPL. The resulting nanosuspension was sonicated to obtain particles with good dispersibility. Fig. 1 shows the TEM images of CSL1 (Fig. 1A), CSL2 (Fig. 1B), and CSPL (Fig. 1C), and all three liposomes exhibited an obvious spherical shape. DLS analysis showed that the average size of CSPL was 148.5 nm with a PDI of 0.217 (Fig. 1D). The size distributions of CL, CSL1 and CSL2 were also measured by DLS. As Fig. S1A–C (ESI†) reveal, the average sizes of CL, CSL1 and CSL2 were around 150 nm. In addition, DLS analysis showed that CL, CSL1, CSL2 and CSPL possessed negative ζ potential (Fig. 1E and Fig. S1D–F, ESI†). Furthermore, as shown in Fig. 1F, the soft CSPL possess good stability.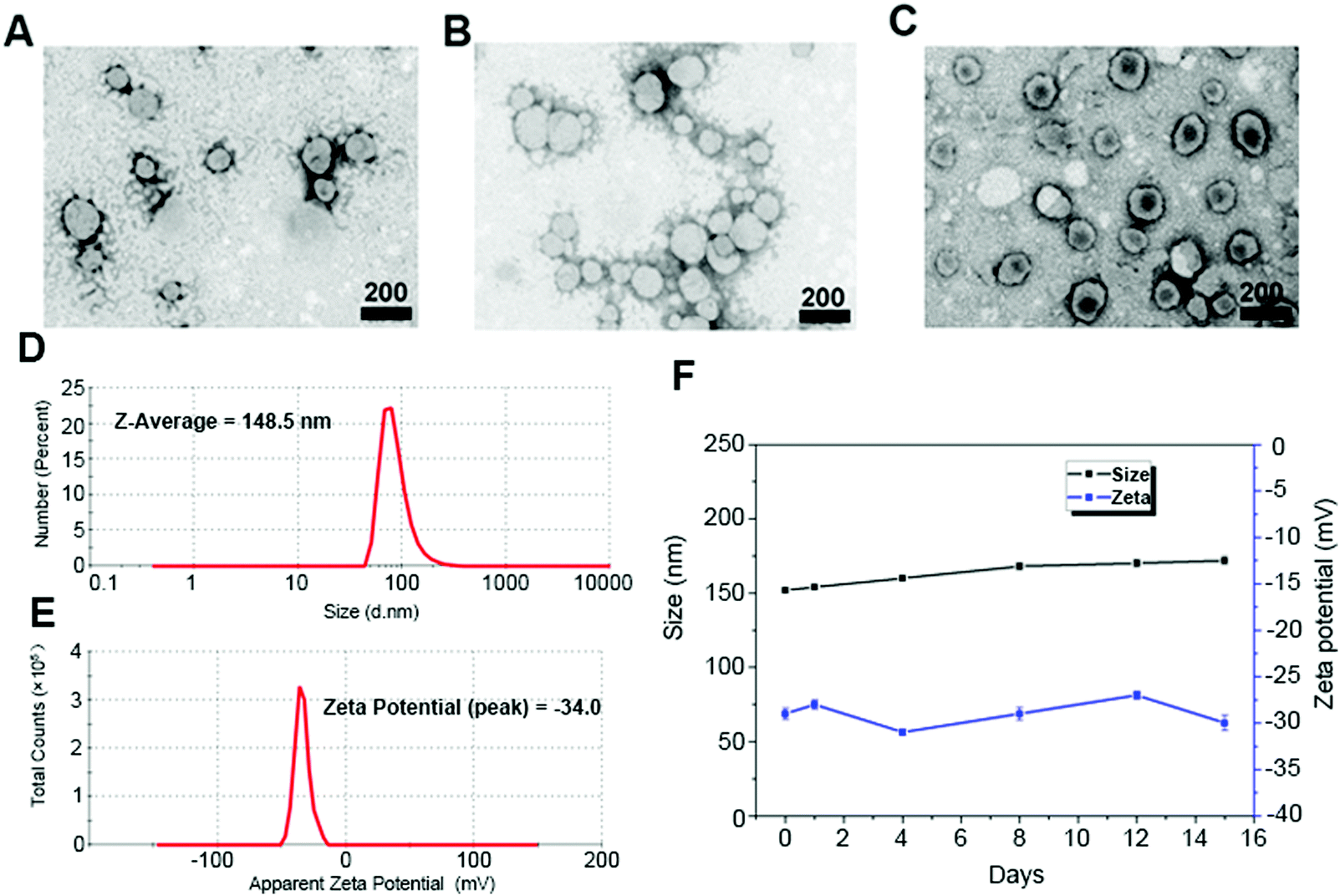 | ||
| Fig. 1 Characterization of CSL1, CSL2, and CSPL. (A) Transmission electron microscopy (TEM) image of CSL1. (B) TEM image of CSL2. (C) TEM image of CSPL. (D) Size distribution of CSPL. (E) Zeta potential of CSPL. (F) The stability of CSPL at 4 °C. Scale bar = 200 nm. | ||
3.2 Characterization of Van@CSPL
As shown in Fig. S2 (ESI†), the average size of Van@CSPL was around 150 nm. In addition, we chose a ratio of Van to CSPL of 2:5 as the optimal mass ratio. The drug-loading content (DLC) and drug encapsulation efficiency (DEE) of Van@CSPL were around 7.92% and 29.8%, respectively.
3.3 In vitro anti-hemolysis assay of CSPL
In order to test the neutralizing abilities of CSPL towards PFTs, a preliminary anti-hemolysis assay was conducted with α-toxin (1 μg). As shown in Fig. 2A, the samples of CSPL, CSL2, and CSL1 displayed a noticeably clear supernatant, indicating that α-toxins were sequestrated after mixing with the three types of liposomes. However, for the groups of control (+) and CL, obvious hemolysis could be seen in the supernatant. These results illustrated that CSPL, CSL2, and CSL1 are able to protect RBCs from being damaged by α-toxin. Using the results of the relative haemolysis degrees (Fig. 2B), we could easily see the severity of haemolysis in the group of CL, and this was the same as in the control (+). Furthermore, the PFT-neutralization abilities of CSPL, CSL2, and CSL1 were measured by mixing them with a range of different amounts of α-toxin from 4 μg to 256 μg. As shown in Fig. 2C, the RBCs were completely ruptured with 100% haemoglobin release under the condition where 32 μg of α-toxin was added. However, compared to other groups with equivalent amounts of α-toxin, the relative hemolysis of the CSPL group maintained the lowest level. It is also worth noting that the relative hemolysis of the CSPL group was still less than 20% when the amount of α-toxin reached 256 μg. The color tendency can be seen from Fig. 2D, indicating that this soft CSPL has the optimal binding efficacy with α-toxin.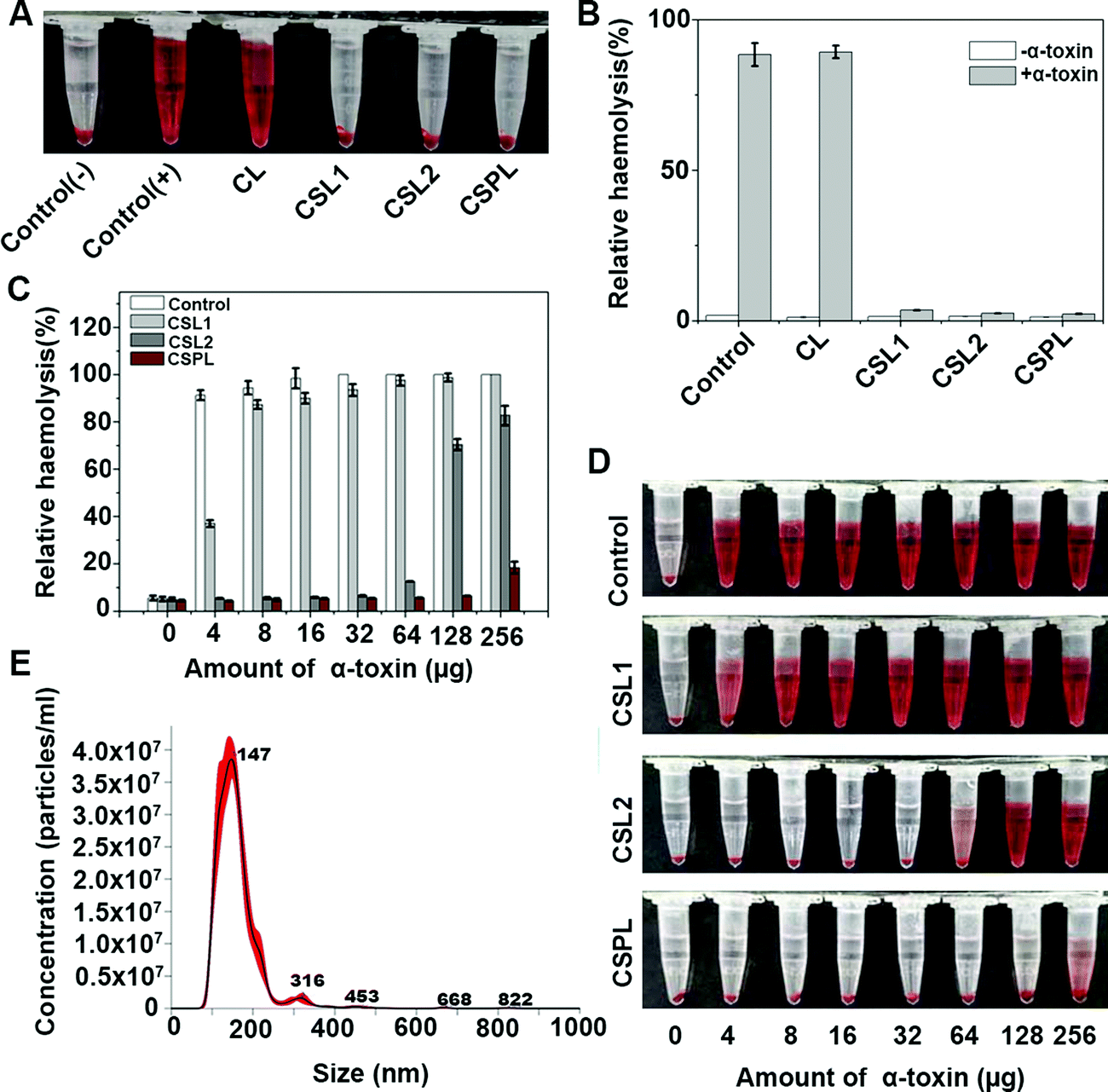 | ||
| Fig. 2 In vitro anti-hemolysis assay of CSPL. (A) The results of centrifuged RBCs of CL, CSL1, CSL2, and CSPL groups after incubating with α-toxin (1 μg); the groups with or without α-toxin were used as the control (+) and control (−), respectively. (B) The relative hemolysis of the samples in A; these types of liposomes alone were used as negative controls in each group. (C) The relative hemolysis of CSL1, CSL2 or CSPL groups under the action of different amounts of α-toxin. The amount of CSL1 (1 mg mL−1), CSL2 (1 mg mL−1) or CSPL (1 mg mL−1) was 200 μg, and the group treated with α-toxin alone was set as the control. (D) The results of centrifuged RBCs of CSL1, CSL2, and CSPL groups after incubating with a series of different amounts of α-toxin (4, 8, 16, 32, 64, 128, and 256 μg). The group of the control was also included. (E) The concentration distribution of CSPL particles with corresponding size, measured by NTA. | ||
To further study the toxin-binding efficacy of this soft CSPL, α-toxin and CSPL were quantified. As shown in Fig. 2C, the relative hemolysis of the CSPL group showed a notable rise when the dose of α-toxin reached 256 μg. Therefore, we concluded that the toxin binding capacity of CSPL becomes saturated when the dose of α-toxin reaches 128 μg. NTA (Fig. 2E) showed that 1 mg mL−1 of CSPL contained 3.42 × 1011 particles, and 128 μg of α-toxin (60%, 34 kDa) contained 1.356 × 1015 protein molecules. Therefore, we eventually obtained the result that approximately one CSPL can absorb 1.98 × 104 protein molecules of α-toxin, and this is much better than the absorption of “nanosponges”.11,14
Finally, in order to test whether CSPLs possess this characteristic against a broad-spectrum of toxins, melittin was selected, and the anti-hemolysis assay was conducted in the same way as for α-toxin. As Fig. S3 (ESI†) reveals, CSPL showed the best efficacy for neutralizing melittin, compared to CSL1 and CSL2.
3.4 In vitro cytotoxicity assay
As a water soluble monomer, α-toxin has a powerful cytolytic capacity, including in human endothelial cells, epithelial cells, leukocytes, and platelets.32 Therefore, next we investigated the toxin-binding efficacy of this soft CSPL. Fig. 3A shows the viability of HUVECs after incubation with a mixture of CL, CSL1, CSL2, or CSPL and a series of different concentrations of α-toxin (0.2 μg mL−1, 2 μg mL−1, and 20 μg mL−1). According to the results, CL did not possess toxin-binding efficacy, as the cell viability was at the same level for every concentration of α-toxin, compared to in the control. In addition, we could intuitively see that the HUVECs of the CSPL group had the highest cell viability, compared to CSL2, CSL1, and CL groups. The survival rate of HUVECs in the CSPL group remained at 82.5% after incubation with α-toxin (0.2 μg mL−1). However, there were only 47.6% of HUVECs alive in the control group at the same concentration. Furthermore, the viability of HUVECs still displayed a distinct difference between the CSPL group and the control group, even when the concentration of α-toxin reached 20 μg mL−1, and this was better than the results for the CSL1 or CSL2 groups. Therefore, we could fundamentally infer that this soft CSPL has excellent selectivity towards toxins when competing with HUVECs, thereby protecting the HUVECs from being destroyed.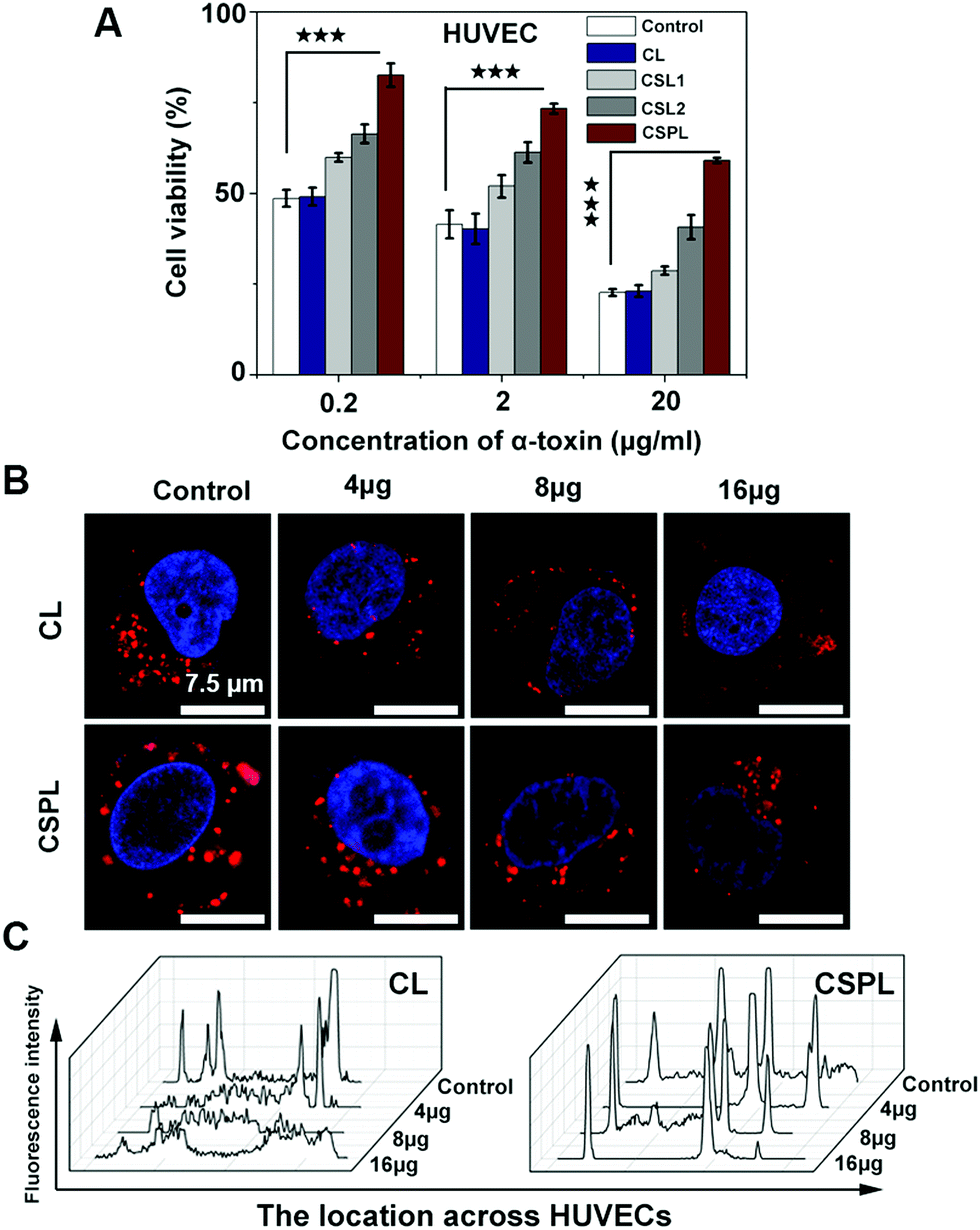 | ||
| Fig. 3 In vitro toxin neutralization assay at the cell-level. (A) Viability of HUVECs after incubating with the mixtures of CL, CSL1, CSL2, or CSPL and a range of concentrations of α-toxin (0.2 μg mL−1, 2 μg mL−1, and 20 μg mL−1). The group treated with α-toxin alone was used as a control (n = 3). (B) The cell uptake of HUVECs after adding the mixtures of CL or CSPL with varying amounts of α-toxin (4, 8, and 16 μg) for 4 h; HUVECs mixed with CL or CSPL alone as controls (scale bars, 7.5 μm). (C) The corresponding fluorescence localization analysis of Dil-labeled CSPL and Dil-labeled CL in HUVECs. The statistical significance was calculated by Student's t-test, ***P < 0.001, **P < 0.01, *P < 0.05. | ||
3.5 In vitro cellular internalization study
In order to better understand the role of CSPL during the incubation process with HUVECs, a cellular uptake assay was carried out, using CL as a control. As demonstrated by the confocal microscopy images, as shown in Fig. 3B, Hoechst 33342 was used to locate the cell nucleus (blue fluorescence), and CSPL and CL were stained with Dil (red fluorescence). The apparent red dots appeared in the intracellular region of the HUVECs (two control groups in Fig. 3B), and this showed that CSPL and CL can be “eaten” by the HUVECs in a way similar to endocytosis.33 This result indicated that the cell membranes were intact in the absence of α-toxin. However, for the CL group, the red fluorescence was partly distributed in a dispersed state when the concentration of α-toxin was 4 μg mL−1. As the concentration of α-toxin reached 16 μg mL−1, extensively distributed red fluorescence covered the entire cellular area. As shown in Fig. 3C, the fluorescence intensity was maintained at a lower level, indicating that the structure of the cell membrane was almost completely destroyed by α-toxin. In addition, it can clearly be observed that the morphology of HUVECs was gradually destroyed with increased amounts of α-toxin (Fig. S4, ESI†). In contrast, for the CSPL group, the red fluorescence that is emitted from the cell still displayed as a dot, even when the concentration of α-toxin reached 16 μg mL−1. These results show that this soft liposome has the powerful ability to absorb α-toxin, and then to protect HUVECs from being destroyed.3.6 Skin injury assay
As one of the most common pathogens, almost all types of S. aureus can induce diseases on the surface of the skin with the participation of numerous exotoxins. α-Toxin, one of the PFTs secreted by S. aureus, plays an important role in skin damage. Therefore, in the following study, the toxin-binding ability of CSPL was tested in a murine skin model. As shown in Fig. 4A, severe skin trauma appeared at the time point of 72 h after the injection of α-toxin, indicating the typical scalded skin syndrome. The H&E examination of the skin tissue revealed the disappearance of airbags, and the apoptosis and necrosis of cells in the dermis (Fig. 4B). In addition, muscle tissues presented the noticeable deformation and atrophy of muscle fibers, as well as accumulated neutrophils (Fig. 4C). In contrast, for the CSPL group, there were almost no visible skin lesions. Furthermore, normal epithelial structures and integral muscle fibers were observed in the skin and muscle histology, i.e. the same as in the control.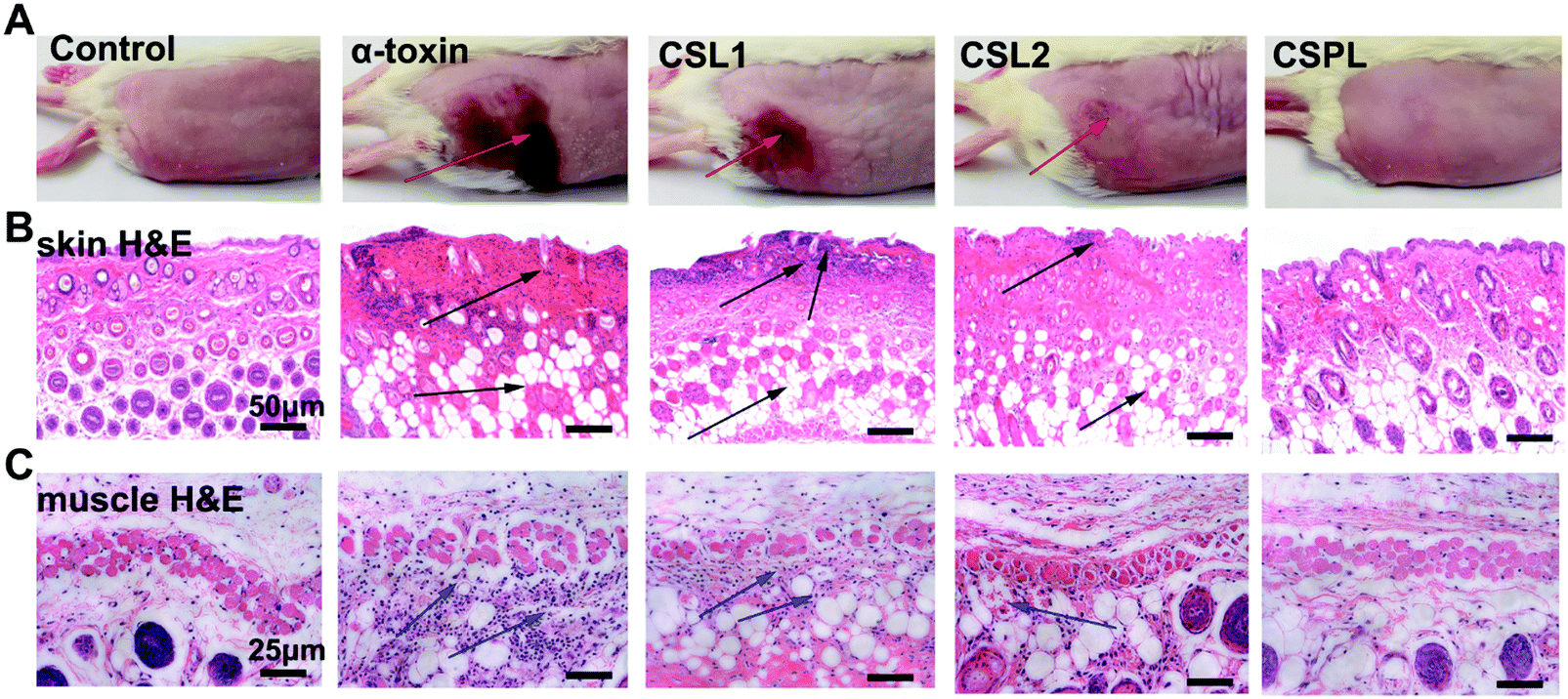 | ||
| Fig. 4 In vivo skin injury assay. Mice were subcutaneously injected with a mixture of α-toxin and CSL1, CSL2 or CSPL; mice injected with α-toxin or with PBS alone were also included. (A) The representative photographs of skin lesions 3 days post-injection. The red arrows indicate the site of skin damage. (B) Haematoxylin and eosin stained (H&E) histological sections of skin samples were collected from the mice 3 days after the injections. The black arrows indicate the lesions. Scale bars = 50 μm. (C) Corresponding H&E images of the muscle tissues of the mice in the three groups. The blue arrows indicate the aggregated inflammatory cells. Scale bars = 25 μm. | ||
3.7 In vivo distribution of CSPL
In this study, we intended to explore the distribution of CSPL in vivo, and whole-body fluorescence images were employed for this. As Fig. S5A (ESI†) reveals, the fluorescence was mainly grouped together in the part around the chest. In addition, for both of the two groups, the fluorescence intensity was shown to be notably weaker at the time point of 24 h, meaning that CSPLs were gradually metabolized by the organism as time went on. In addition, the fluorescence was majorly distributed in the livers (Fig. S5B, ESI†), suggesting that CSPL can normally be eliminated, thereby protecting other organs from being impaired in the process of metabolism.3.8 Bacterial integrity influence and pore-forming assay
The obtained SEM micrographs of S. aureus are shown in Fig. 5. As shown in Fig. 5A, S. aureus displayed smooth surfaces with intact cell walls in the control. For the Van@CL group, almost the same smooth surfaces can be seen. However, after incubating with Van@CSPL for 8 h, the cell walls of S. aureus became wrinkled and impaired with distinct unusual bulges, and the same damage can be seen in the group of Van. These results indicate that Van was able to be released from Van@CSPL, thereby exerting equivalent antibacterial efficacy as free Van.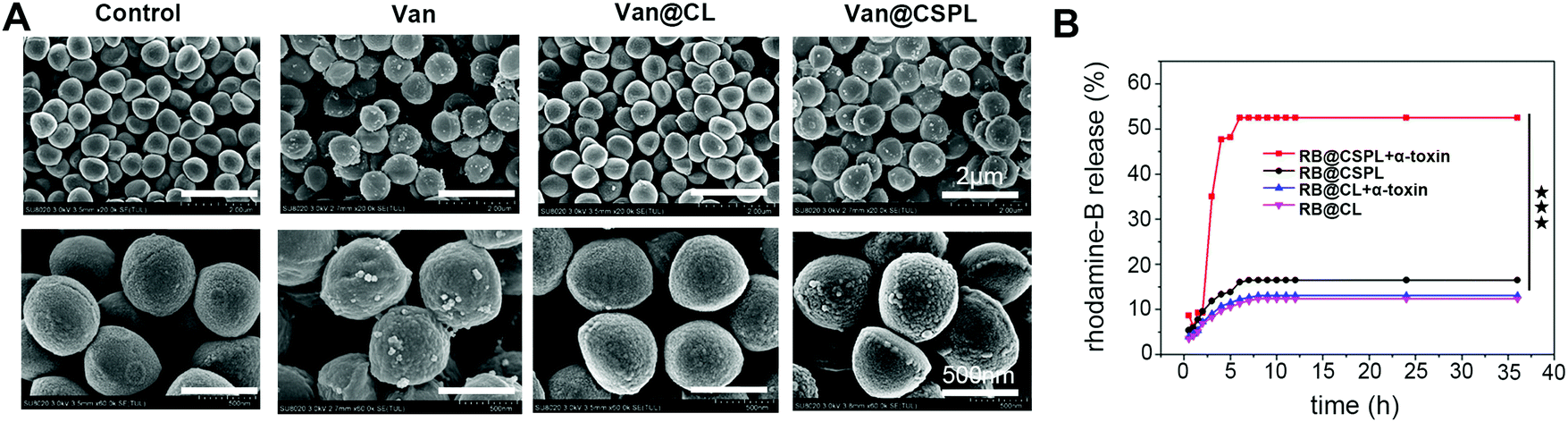 | ||
| Fig. 5 In vitro antibacterial activity of Van@CSPL. (A) Typical SEM images of S. aureus after incubation with Van, Van@CL or Van@CSPL (the concentration of vancomycin is 256 μg mL−1) for 8 h, using PBS as a control. (B) Release profiles of rhodamine B in the groups of RB@CL and RB@CSPL with α-toxin (10 μg), and RB@CL or RB@CSPL groups without α-toxin were also included. The data are presented as the mean ± SD from three independent experiments. The statistical significance was calculated by Student's t-test, ***P < 0.001, **P < 0.01, *P < 0.05. | ||
It is known that the size of a transmembrane pore caused by PFTs is around 2 nm, through which small molecules below 3 KDa are able to diffuse,34 and that the molecular weight of Van is below 1.5 kDa. Therefore, we speculated that PFTs secreted by S. aureus were in a good position to form pores in the membrane of CSPL, thereby inducing the release of Van from Van@CSPL. Therefore, in the following step, a pore-forming assay was conducted. Considering the extremely sensitive UV (ultraviolet and visible spectrum) absorption peak of rhodamine (RB), we substituted Van with RB. As shown in Fig. 5B, for the groups of Van@CL with or without α-toxin, and the group of Van@CSPL without α-toxin, the leakage of RB was maintained at a very low level over 36 h. However, for the group of Van@CSPL with α-toxin, the cumulative release of RB displayed a rapid increase after the addition of α-toxin (10 μg) at the time point of 2 hours. These results indicate that small molecules such as Van are able to be released in relative quantities from the hydrophilic phase core of CSPL. Since Van is a high resistance potential antibiotic, a sublethal level of the antibiotic dose can lead to drug resistance.7,24 Therefore, this soft Van@CSPL liposome with explosive drug release lays the foundation for reducing the possibility of antibiotic resistance in vivo.
3.9 The treatment efficacy of CSPL and Van@CSPL in septicemic mice
Septicemia is an acute systemic infection in which pathogens invade the blood circulation and produce a large number of toxins. During the infection of Staphylococcus aureus, the severity of septicemia is closely related to α-toxin, which can induce platelet aggregation and liver injury, thereby resulting in multi-organ exhaustion.29 Therefore, in the following step, the treatment efficacy of this soft CSPL was assessed in septicemic mice. As shown in Fig. 6A, all of the mice had died within 3 days in the group of PBS and CL, indicating that CL has no therapeutic effect. In contrast, for the CSPL group, the survival rate was maintained at 87.5%, which is higher than that of the CSL2 group and CSL1 group, which have survival rates of 62.5% and 50%, respectively. Furthermore, the survival rate was 50% at the time point of 6 days, and there was an improvement of 25% compared to the group of CSL2. In order to better understand the treatment efficacy of this soft CSPL, major organs including the heart, liver, spleen, lung, and kidney were stained by H&E. As Fig. S6 (ESI†) reveals, there were almost no megascopic pathological changes in the CSPL group.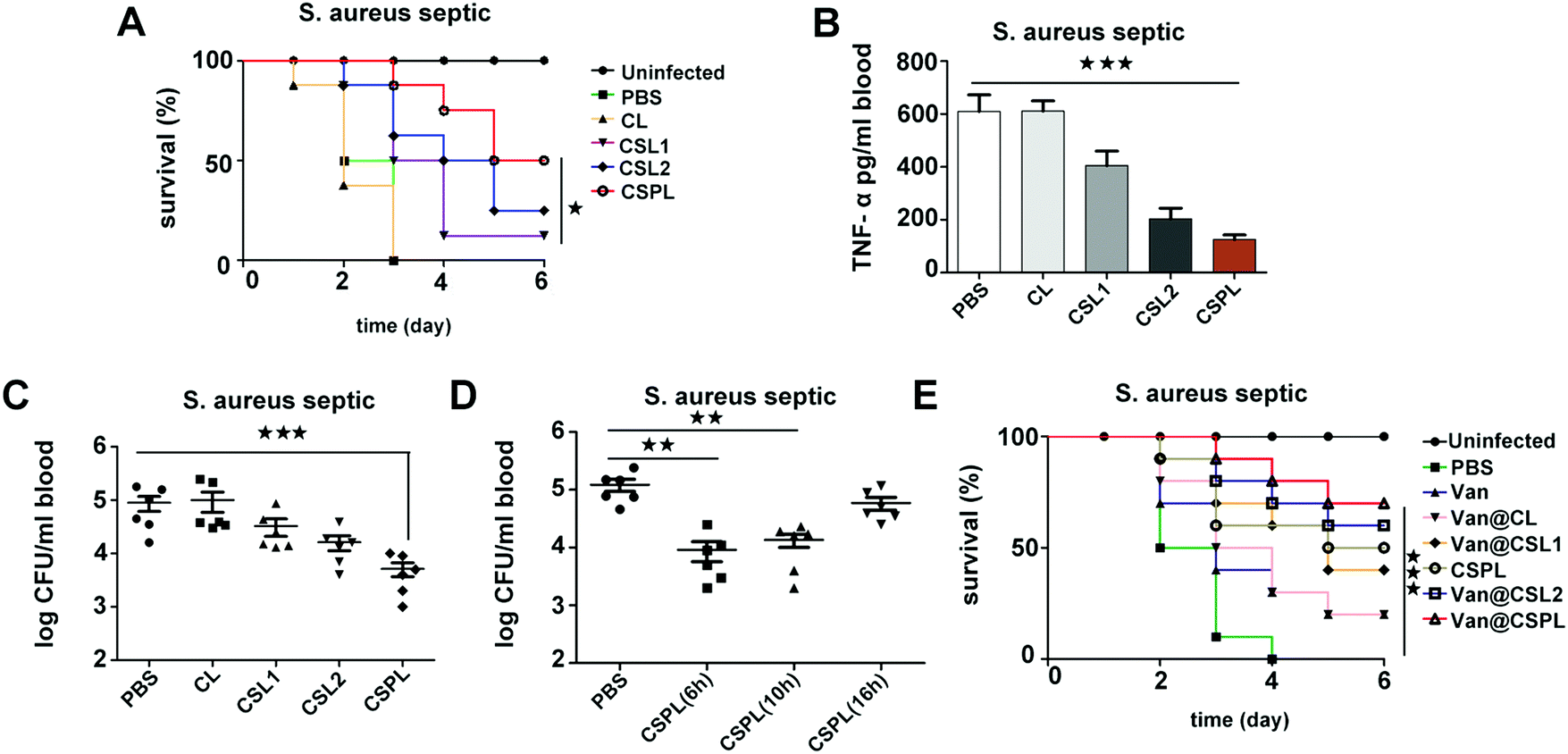 | ||
| Fig. 6 In vivo antibacterial efficacy of CSPL and Van@CSPL in mice with Staphylococcus aureus septicemia. (A) Septic mice, infected intravenously with S. aureus ATCC29213, were given an intravenous administration of 50 mg kg−1 CL, CSL1, CSL2 or CSPL both 4 h and 10 h after infection and were then evaluated for survival, using PBS as a control. The uninfected mice were also included (n = 8). (B and C) The mice were treated intravenously with CL (50 mg kg−1), CSL1 (50 mg kg−1), CSL2 (50 mg kg−1), CSPL (50 mg kg−1), or PBS 4 h after infection, and were analyzed for release of TNF-α (B; n = 4) and their bacterial count (C; n = 6) in blood. (D) Septic mice, infected intravenously with S. aureus ATCC29213, were given an intravenous administration of 50 mg kg−1 CSPL at 6, 10 or 16 h time points after infection and were then evaluated for their bacterial count in the blood (n = 6). (E) Mice were infected intravenously (i.v.) with a S. aureus strain and administered with Van@CL, Van@CSL1, Van@CSL2, Van@CSPL or free Van (10 mg kg−1) at 4 h, 12 h, and 24 h time points after infection, using PBS as a control, and the uninfected mice, as well as the mice treated with CSPL alone, were also included. The survival rates were measured (n = 10). ***P < 0.001, **P < 0.01, *P < 0.05. | ||
TNF-α, as an important pro-inflammatory cytokine, is mainly secreted by mononuclear-macrophages, the level of which increases in bacterial infections. As shown in Fig. 6B, the level of TNF-α is significantly decreased in the CSPL group after 24 h of infection. For CSL1 and CSL2 groups, the levels of TNF-α also decreased, but they were still higher than in the CSPL group. Therefore, we reached the conclusion that CSPL could clearly relieve the inflammatory reaction in mice with septicemia. In addition, we measured the amount of S. aureus in their blood. As shown in Fig. 6C, for the CSPL group, the bacterial cell count in the blood was significantly reduced. In addition, the bacterial load was remarkably decreased in the blood of septic mice after treatment with CSPL for 6 h or 10 h (Fig. 6D), illustrating that the administration of CSPL was effective before 10 h, while there was no effect after 16 h.
Finally, the treatment efficacy of Van@CSPL was assessed in a fatal Staphylococcus aureus septicemia model. As shown in Fig. 6E, the survival rate reached 70% after 6 days of infection, indicating the powerful antibacterial effect of the nanoliposome.
3.10 The treatment efficacy of CSPL and Van@CSPL in mice with pneumonia
During the infection of Staphylococcus aureus pneumonia, α-toxin plays an important role, inhibiting the efferocytosis of alveolar macrophages to infect neutrophils, thus preventing the clearance of S. aureus.35 Therefore, we then studied the antibacterial efficacy of this soft CSPL in mice with Staphylococcus aureus pneumonia. As shown in Fig. 7A, compared to the PBS group (with a survival rate of 37.5%), the mice administered with CSPL, CSL2, and CSL1 revealed higher survival rates (75%, 62.5% and 50%, respectively) at the time point of 48 h, and it is worth noting that half of the mice were still alive 72 h after infection in the CSPL group. In addition, the level of TNF-α in the lungs significantly decreased (Fig. 7B), indicating that CSPL was able to alleviate the drastic inflammatory response after infection in the mice. Furthermore, the bacterial loads in the blood and lungs were measured by agar plates, counting at one day post-infection. As shown in Fig. 7C, the group of CSPL displayed notably lower bacterial colonization. Comparatively, bacterial colonization counting of the mice in the PBS group showed the existence of about 6.32 × 105 cells per lung. However, for the CSPL group, there was a clear reduction to about 4.75 × 104 CFU per lung. In addition, compared to the PBS group, the bacterial cell counts in the blood were remarkably reduced by around 6.27 − log10 fold (Fig. 7D). These results indicate that this soft CSPL has excellent toxin-binding efficacy, thereby protecting the mice from attack.
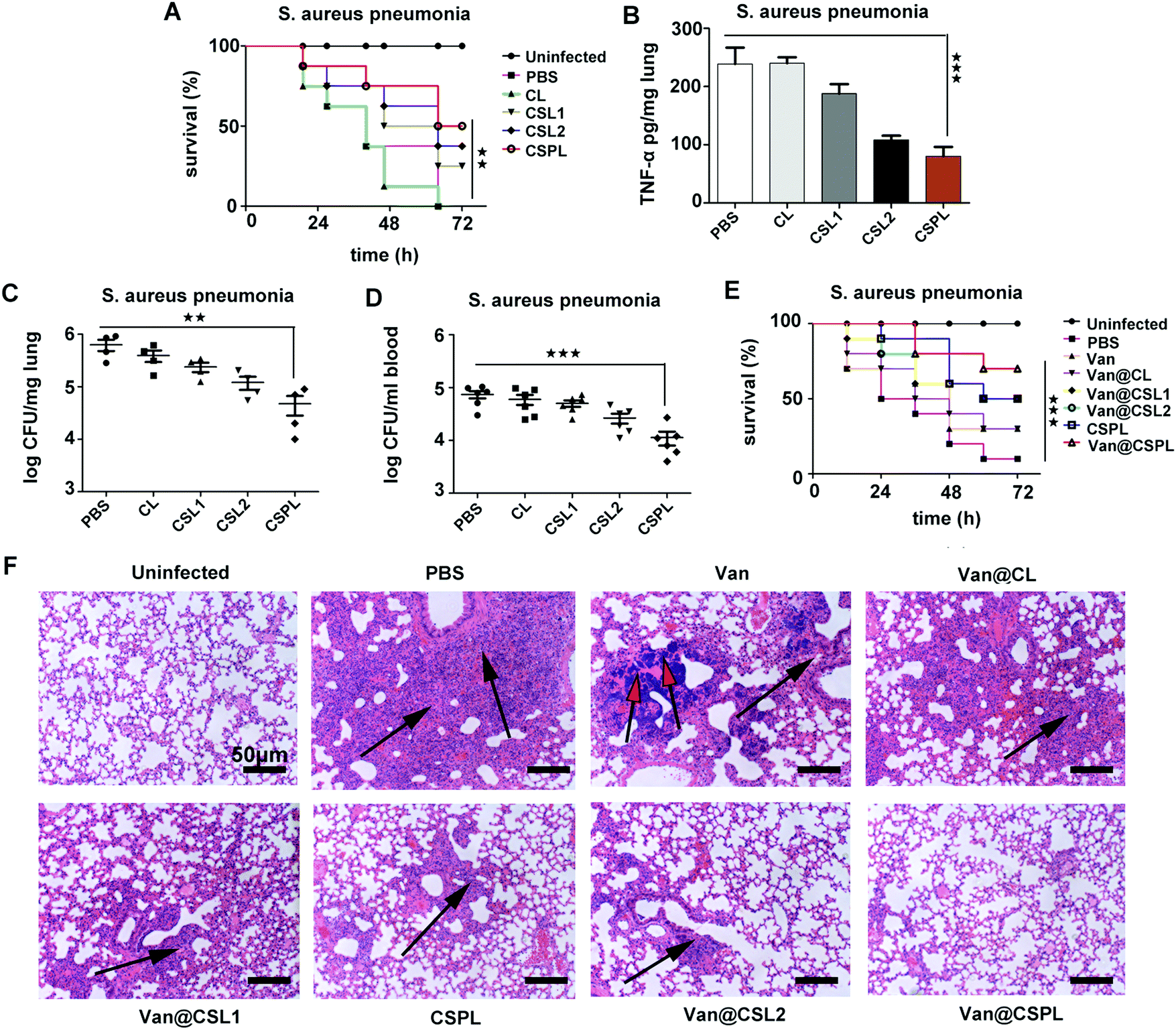 | ||
| Fig. 7 The antibacterial efficacy of CSPL and Van@CSPL in mice with Staphylococcus aureus pneumonia. (A) Survival curves of the infected mice after intranasal administration with 50 mg kg−1 of CL, CSL1, CSL2 or CSPL, using PBS as a control, and the uninfected mice were also included (n = 8). (B–D) Mice with pneumonia, infected intranasally with S. aureus ATCC29213, were given a 50 mg kg−1 dose of CL, CSL1, CSL2 or CSPL 4 h after infection, and were evaluated for their levels of TNF-α in the lungs (B; n = 3), the bacterial load in the lungs (C; n = 4), or in the blood (D; n = 6), at the time point of 24 h after infection. (E) Mice were infected intranasally with a S. aureus strain and were administered intravenously (i.v.) with 10 mg kg−1 of Van@CL, Van@CSL1, Van@CSL2, Van@CSPL or free Van 4 after 10 h of being infected, using PBS as a control. The uninfected mice and the mice treated with CSPL alone were also included. The survival rate was measured (n = 10). (F) The corresponding H&E staining images of the lungs. The black arrows indicate inflammatory infiltration in the lungs, whilst the red arrows indicate calcifications. ***P < 0.001, **P < 0.01, *P < 0.05. | ||
The treatment efficacy of this soft Van@CSPL liposome was also investigated. As shown in Fig. 7E, the survival rate of the Van@CSPL group was as high as 70% at the time point of 72 h after infection, and there was a 20% gap compared to the Van@CSL2 group. To further observe the antibacterial efficacy, histopathological examination of the lung was carried out. As shown in Fig. 7F, inflammatory infiltration and pulmonary interstitial edema covered nearly the whole lung in the PBS group. Additionally, apparent calcifications were formed in the Van group and, compared to other treatment groups, the degrees of lesion of the alveolar cavities in the mice were notably mitigated after being administered with Van@CSPL. In summary, this soft CSPL liposome significantly reduced the bacterial burden and the level of TNF-α in mice. Following the administration of Van@CSPL, the survival rate of the mice further improved.
4. Conclusions
In this report, the soft nanoliposome CSPL, composed of Ch, Sm, and Pc in the molar ratio of 4:1:1, was successfully developed. Compared with CSL2, this soft liposome (CSPL) displayed excellent toxin-binding efficacy, thus reducing the bacterial load and the levels of TNF-α. Our research results demonstrated that CSPL could enhance the potency of the therapeutic effect. Additionally, the resulting formulation (Van@CSPL) could realize the explosive release of Van at the infected site by transmembrane pores that are formed by PFTs, decreasing the side effects of the antibiotics and reducing the emergence of antibiotic resistance. In conclusion, this work provides a novel strategy to enhance the potency of antibiotic therapy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by grants from the Science and Technology Project of Henan Province (192102310420) and the National Natural Science Foundation of China (No. 81874304).References
- C. Zhang, W. Zhao, C. Bian, X. Hou, B. Deng, D. W. McComb, X. Chen and Y. Dong, ACS Appl. Bio Mater., 2019, 2, 1270–1277 CrossRef CAS.
- A. Coates, Y. M. Hu, R. Bax and C. Page, Nat. Rev. Drug Discovery, 2002, 1, 895–910 CrossRef CAS.
- J. Cheng, W. Chin, H. Dong, L. Xu, G. Zhong, Y. Huang, L. Li, K. Xu, M. Wu, J. L. Hedrick, Y. Y. Yang and W. Fan, Adv. Healthcare Mater., 2015, 4, 2128–2136 CrossRef CAS.
- C. P. Teng, T. Zhou, E. Ye, S. Liu, L. D. Koh, M. Low, X. J. Loh, K. Y. Win, L. Zhang and M. Y. Han, Adv. Healthcare Mater., 2016, 5, 2122–2130 CrossRef CAS.
- S. J. van Hal, T. P. Lodise and D. L. Paterson, Clin. Infect. Dis., 2012, 54, 755–771 CrossRef CAS.
- Y. H. Dong, L. Y. Wang and L. H. Zhang, Philos. Trans. R. Soc., B, 2007, 362, 1201–1211 CrossRef CAS.
- C. B. Cunha and S. M. Opal, Med. Clin. North Am., 2018, 102, 831–843 CrossRef.
- T. T. Tran, J. M. Munita and C. A. Arias, Ann. N. Y. Acad. Sci., 2015, 1354, 32–53 CrossRef.
- A. Kadioglu, J. N. Weiser, J. C. Paton and P. W. Andrew, Nat. Rev. Microbiol., 2008, 6, 288–301 CrossRef.
- C. F. Reboul, J. C. Whisstock and M. A. Dunstone, Biochim. Biophys. Acta, 2016, 1858, 475–486 CrossRef CAS.
- Y. He, R. Li, H. Li, S. Zhang, W. Dai, Q. Wu, L. Jiang, Z. Zheng, S. Shen, X. Chen, Y. Zhu, J. Wang and Z. Pang, ACS Nano, 2019, 13, 4148–4159 CrossRef CAS.
- M. W. Parker and S. C. Feil, Prog. Biophys. Mol. Biol., 2005, 88, 91–142 CrossRef CAS.
- F. C. Los, T. M. Randis, R. V. Aroian and A. J. Ratner, Microbiol. Mol. Biol. Rev., 2013, 77, 173–207 CrossRef CAS.
- C. M. Hu, R. H. Fang, J. Copp, B. T. Luk and L. Zhang, Nat. Nanotechnol., 2013, 8, 336–340 CrossRef CAS.
- B. Wang, H. Zhang, J. An, Y. Zhang, L. Sun, Y. Jin, J. Shi, M. Li, H. Zhang and Z. Zhang, Nano Lett., 2019, 19, 3505–3518 CrossRef CAS.
- B. D. Henry, D. R. Neill, K. A. Becker, S. Gore, L. Bricio-Moreno, R. Ziobro, M. J. Edwards, K. Muhlemann, J. Steinmann, B. Kleuser, L. Japtok, M. Luginbuhl, H. Wolfmeier, A. Scherag, E. Gulbins, A. Kadioglu, A. Draeger and E. B. Babiychuk, Nat. Biotechnol., 2015, 33, 81–88 CrossRef CAS.
- R. H. Fang, B. T. Luk, C. M. Hu and L. Zhang, Adv. Drug Delivery Rev., 2015, 90, 69–80 CrossRef CAS.
- C. Klose, C. S. Ejsing, A. J. Garcia-Saez, H. J. Kaiser, J. L. Sampaio, M. A. Surma, A. Shevchenko, P. Schwille and K. Simons, J. Biol. Chem., 2010, 285, 30224–30232 CrossRef CAS.
- D. Marsh, Biochim. Biophys. Acta, 2009, 1788, 2114–2123 CrossRef CAS.
- Y. Takechi-Haraya, K. Sakai-Kato, Y. Abe, T. Kawanishi, H. Okuda and Y. Goda, Langmuir, 2016, 32, 6074–6082 CrossRef CAS.
- E. Zant-Przeworska, M. Stasiuk, J. Gubernator and A. Kozubek, Chem. Phys. Lipids, 2010, 163, 648–654 CrossRef CAS.
- X. Liang, G. Mao and K. Y. Ng, J. Colloid Interface Sci., 2004, 278, 53–62 CrossRef CAS.
- S. A. Kelly, A. M. Rodgers, S. C. O’Brien, R. F. Donnelly and B. F. Gilmore, Trends Biotechnol., 2020, 38, 447–462 CrossRef CAS.
- M. A. Kohanski, M. A. DePristo and J. J. Collins, Mol. Cell, 2010, 37, 311–320 CrossRef CAS.
- M. Chen, S. Xie, J. Wei, X. Song, Z. Ding and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36814–36823 CrossRef CAS.
- X. Zhang, X. Hu and X. Rao, Microbiol. Res., 2017, 205, 19–24 CrossRef CAS.
- Y. Yang, Z. Hu, W. Shang, Q. Hu, J. Zhu, J. Yang, H. Peng, X. Zhang, H. Liu, Y. Cong, S. Li, X. Hu, R. Zhou and X. Rao, Microb. Drug Resist., 2017, 23, 241–246 CrossRef CAS.
- Y. Zhou, C. Niu, B. Ma, X. Xue, Z. Li, Z. Chen, F. Li, S. Zhou, X. Luo and Z. Hou, Cell Death Dis., 2018, 9, 362 CrossRef.
- B. G. J. Surewaard, A. Thanabalasuriar, Z. Zeng, C. Tkaczyk, T. S. Cohen, B. W. Bardoel, S. K. Jorch, C. Deppermann, J. Bubeck Wardenburg, R. P. Davis, C. N. Jenne, K. C. Stover, B. R. Sellman and P. Kubes, Cell Host Microbe, 2018, 24(271–284), e273 Search PubMed.
- E. S. Seilie and J. Bubeck Wardenburg, Semin. Cell Dev. Biol., 2017, 72, 101–116 CrossRef CAS.
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z.-G. She, D. Kim, A. P. Mann, T. Mölder, T. Teesalu, S. Carnazza, S. Guglielmino, M. J. Sailor and E. Ruoslahti, Nat. Biomed. Eng., 2018, 2, 95–103 CrossRef CAS.
- B. J. Berube and J. Bubeck Wardenburg, Toxins, 2013, 5, 1140–1166 CrossRef.
- W. Gao, R. Langer and O. C. Farokhzad, Angew. Chem., Int. Ed., 2010, 49, 6567–6571 CrossRef CAS.
- C. Meesters, A. Brack, N. Hellmann and H. Decker, Proteins, 2009, 75, 118–126 CrossRef CAS.
- T. S. Cohen, O. Jones-Nelson, M. Hotz, L. Cheng, L. S. Miller, J. Suzich, C. K. Stover and B. R. Sellman, Sci. Rep., 2016, 6, 35466 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb02255a |
| ‡ Shudong Zhang and Xiang Lu contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |