
In situ oxygenating and 808 nm light-sensitized nanocomposite for multimodal imaging and mitochondria-assisted cancer therapy†
Arif
Gulzar
 a,
Fei
He
ab,
Aanisa
Gulzar
c,
Ye
Kuang
a,
Fangmei
Zhang
a,
Shili
Gai
a,
Paioping
Yang
*a and
Chen
Wang
*b
a,
Fei
He
ab,
Aanisa
Gulzar
c,
Ye
Kuang
a,
Fangmei
Zhang
a,
Shili
Gai
a,
Paioping
Yang
*a and
Chen
Wang
*b
aKey Laboratory of Superlight Materials and Surface Technology, Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin, 150001, P. R. China. E-mail: Yangpaioping@hrbeu.edu.cn
bDepartment of Research Guangxi Medical University Cancer Hospital, Naning 530021, China
cDepartment of Physics, National Institute of Technology, Srinagar, J&K 190006, India. E-mail: wang2010116@hrbeu.edu.cn
First published on 3rd November 2020
Abstract
The efficiency of photodynamic therapy (PDT) is severely constrained due to the innate hypoxic environment, besides the elevated level of glutathione (GSH). To get rid of the hypoxic environment and higher concentrations of GSH in the solid tumors, we propose an approach of oxygen self-sufficient multimodal imaging-guided nanocomposite CaO2–MnO2–UCNPs–Ce6/DOX (abbreviated as CaMn–NUC), in which CaO2 nanoparticles in the hydrophobic layer were seated on the hydrophilic MnO2 sheet and conjugated with chlorin e6 (Ce6) loaded upconversion nanoparticles (UCNPs–Ce6) via the click chemistry approach. CaMn–NUC was presented to overcome hypoxia and GSH-associated photodynamic resistance due to in situ oxygen generation and GSH reduction of MnO2 upon endocytosis, and a bulk amount of Mn2+ ions generated in the process under acidic tumor environment acts as the MRI contrast agent. Moreover, the MnO2 sheet protects Ce6 from self-degradation under irradiation; thus, it can be used to switch control of cellular imaging. Afterwards, in a regulated and targeted manner, the chemotherapeutic drug (doxorubicin hydrochloride, DOX) can be released with the degradation of CaMn–NUC in the acidic tumor microenvironment (TME). Thus, we testify a competent nanoplatform employing 808 nm-excited UCNPs–Ce6 for concurrent imaging and PDT in consideration of the large anti-Stokes shifts, deep penetration into biological tissues, narrow emission bands, and high spatial-temporal resolution of the UCNPs. Thus, our proposed nanoplatform postulates a strategy to efficiently kill cancer cells in a concentration- and time-dependent manner via the in situ oxygenation of solid tumor hypoxia to enhance the efficiency of multimodal imaging-guided chemo-photodynamic therapy.
Introduction
Anomalous tumor microenvironment (TME) is backed by changes in the cancer cell metabolism. Local hypoxia is an unambiguous representation of solid tumors owing to the enhanced tumor cell growth and morphological or functional anomalies in the tumor blood vessels.1 Tumor cells employ an oxygen homeostasis watchdog, the hypoxia-inducible factor 1 (HIF-1), so as to bring the vascular endothelial growth factor (VEGF) as well as anaerobic glycolysis in order to advance the oxygen delivery and lessen the oxygen consumption to counter hypoxia.2 Accordingly, amplified glycolysis raises lactic acid, thus creating an acidic tumor environment.3 Meanwhile, the disrupted cellular metabolism because of hypoxia and abnormal mitochondria function induces reactive oxygen species (ROS) production.4 As defined, cancer cells evolved an improved antioxidant protection system to hunt the raised up ROS by enriching the transcription of glutathione (GSH) and antioxidant enzymes.5 Thus, because of intimate affiliation between their manifestation mechanisms, hypoxia, acidosis as well as elevated levels of GSH are frequently knotted, even though additional factors may also perhaps control them.6 Tumor advance and therapeutic resistance of TME have been broadly found to be encouraged by these factors.7 Antioxidative GSH may perhaps affect the effectiveness of ROS-dependent cancer therapies, for example chemotherapy and photodynamic therapy (PDT), besides radiotherapy.8,9 Since O2 is a vital component for ROS generation, PDT and radiotherapy are also restricted due to the lessened O2 accessibility in hypoxic solid tumors.10,11 Possibly, due to synergetic drug resistance established underneath concurrent hypoxic, acidic and high antioxidant circumstances might result in a much lesser antitumor efficacy.12Negligible invasiveness, site specific activation, and authorization attained for clinical bids makes PDT a propitious therapy.13–17 Nevertheless, PDT encounters several weaknesses: (1) maximum number of traditional photosensitizers are lipophilic, hence resulting in volatility inside the tumor environment and easy seepage, besides little biocompatibility.18–20 (2) Therapeutic effects of PDT are also restricted by means of the hypoxic environment of the tumor and the higher concentrations of GSH, given the fact that the hypoxic environment can destroy the photosensitizers.21 (3) Tumor selectivity is an additional influence, triggering the damage to normal tissues.22 Consequently, in PDT therapeutic applications, the oxygen source has a vital role. In a solid tumor, the oxygen concentration is lesser compared to that in a normal cell because the ingestion is greater than the extent of the source.21 Quite a few research groups have revealed that the issue of tumor hypoxia might perhaps be resolved via a technique that involves reacting manganese dioxide (MnO2) with hydrogen peroxide (H2O2) in a slightly acid environment for creating O2 [eqn (1)].23–25 Furthermore, it is known that by capping upconversion nanoparticles on the nanosheet, its fluorescence was quenched owing to the broad light absorption by MnO2; afterwards, the fluorescence may perhaps be recovered upon MnO2 degradation.26 Furthermore, the MnO2 nanosheets may well adsorb the chlorin e6 (Ce6) photosensitizer, while protecting it from self-destruction upon light irradiation. Accordingly, these properties of MnO2 can be employed for monitoring the responsive27 fluorescence imaging, in addition to limiting the side effects.
| MnO2 + H2O2 + H+ → Mn2+ + 2H2O + O2 | (1) |
| CaO2 + 2H2O → H2O2 + Ca(OH)2 | (2) |
The malignant tumor cells create disproportionate quantities of H2O2, which, in addition, has an acidic microenvironment compared to that of the normal cells.28 As we know, CaO2 is a biocompatible agent; besides, it might be a castoff for bleaching as well as disinfection because of H2O2 production.29,30 Commonly, CaO2 is termed as a “solid form” of H2O231 because it dissolves in water to form H2O2 [eqn (2)]. Thus, we developed CaO2–MnO2 nanosheets to overcome the hypoxia and GSH effects on singlet oxygen generation for PDT. Even better, the mitochondria integrate Ca2+ and oxidative stress to regulate the cellular stock of adenosine triphosphate in addition to cell death factors.32,33 A cysteine residue inside the mitochondrial uniporter is S-glutathionylated, encouraging the assemblage of mitochondrial uniporter channels into higher-order complexes that display a tenacious action, inducing enhanced mitochondrial Ca2+ uptake upon oxidation. Continuous Ca2+ uptake sequentially increases the ROS production by the mitochondria, provoking a cycle that leads to mitochondrial Ca2+ surplus, and hence, cell death.33–37
As is well known that for most of the clinically used photosensitizers, the tissue penetration depth is limited because of the fact that the activation wavelength is in the spectrum window of 630–700 nm,38 thus limiting the therapeutic effect of PDT against internal or large tumors.39–42 By changing the excitation wavelength to near the infrared (NIR) area, we can evade the above-mentioned issues. In order to confirm small photo-damage, low-autofluorescence background, in addition to the deep penetration into the biological tissue, NIR light instead of visible light is used as an excitation source, which, when blended with the extended lifetime of the lanthanide-doped UCNPs, ensures high spatial-temporal resolution.43–50 For concurrent fluorescence imaging and PDT against cancer, herein, we synthesized a nanoplatform built on the NaGdF4:Yb/Er@NaGdF4:Nd/Yb core–shell nanoparticles that convert NIR light. This exceedingly competent structure exploits an energy transfer from 808 nm NIR light to two upconversion luminescence bands at about 550 and 660 nm, which are aimed at concurrent imaging in addition to therapy. Also, the surface-functionalized core–shell nanoparticles Ce6 were covalently conjugated at a high efficiency of about 4000 molecules per nanoparticle. Afterwards, DOX was loaded on to the surface of CaO2–MnO2–UCNPs–Ce6. Upconversion luminescence spectra and luminescence decay lifetimes were employed for verifying the energy transfer from the nanoparticles to the photosensitizers, besides verifying their capability to generate singlet oxygen. Cancer theranostic applications of our nanoplatform were additionally demonstrated by means of in vitro fluorescence imaging with various cancer cells and through its killing of the cancer cells in a very efficient way in just 10 min on 808 nm irradiation. The biomedical applications of a nanoplatform exploiting CaO2–MnO2 conjugated with 808 nm sensitized UCNPs for imaging and improved chemo-photodynamic therapy by reducing the hypoxia and GSH in solid tumor environment and utilizing mitochondrial cell death is the first such study to the best of our knowledge.
Results and discussion
The synthetic procedure and subsequent antitumor mechanism of our nanocomposite CaMn–NUC have been schematically represented in Scheme 1. At first, in the presence of MES, the CaO2–MnO2 nanocomposite was prepared by the reduction of KMnO4. This procedure can be employed for the simplified preparation of the MnO2 nanosheet. The SEM and TEM images confirmed the excellent dispersity of CaO2 and the CaO2–MnO2 nanosheet. The size of CaO2 nanoparticles was found to be 50 nm and the size of the CaO2–MnO2 nanosheet was found to be 100 nm. The size of CaO2 nanoparticles was further evaluated by TEM imaging as 50 nm (Fig. 1a–c). As a control, the size of pristine MnO2 was found to be 50 nm (Fig. 1d). To further reduce the size of CaO2–MnO2 for better biomedical applications, the nanocomposite was subjected to ultra-sonication and using TEM imaging, the nanocomposite was found to be 50 nm in size. The height of the CaO2–MnO2 nanosheet was found to be 7.3 nm by the AFM image (Fig. S1a, ESI†). To confirm the successful formation of the CaO2–MnO2 nanocomposite, TEM elemental mapping was carried out, which proves the existence of the Ca, Mn, and O elements (Fig. 1f). To further support the successful formation of our nanocomposite, XPS analysis was carried out on CaO2 and the CaO2–MnO2 nanocomposite, as shown in Fig. 1g and h, where the peak at 347.2 eV corresponds to the Ca 2p region and the peak at about 439 eV corresponds to the Ca 2s region of calcium in CaO2. Also, the peak at about 641.8 eV corresponds to the Mn 2p region in CaO2–MnO2, thereby further supporting the idea of successful synthesis of our nanocomposite. To further confirm the crystalline phase and the successful synthesis of the nanocomposite, the XRD analysis of the composite was carried out (Fig. 1i). The diffraction peaks 18.061° and 28.642° symbolize the (200) and (111) crystallographic faces of MnO2, while the peaks at 29.355° and 35.814°, in addition to the peak at 47.509°, correspond to the (011), (110), and (112) indices of the CaO2 crystallographic faces. Zeta potential, besides the dynamic light scattering (DLS) analysis of CaO2 and CaO2–MnO2, was carried out to determine the solubility and size of the nanocomposite. The zeta potential of CaO2 attained was −2.7 mV while, as in case of CaO2–MnO2, the zeta potential at pH 7.4 was recorded to be 4.84 mV and at pH 5.8, the zeta potential was recorded to be 6.68 mV, which signifies the successful synthesis of our nanosheet and its high solubility (Fig. 1j). The DLS measurement confirmed the size of CaO2 and CaO2–MnO2 to be about 100 nm along the Z axis (Fig. 1k and l). To further support the successful loading of the CaO2 nanoparticles on the MnO2 nanosheet, EDS was carried out and the results authenticate the above-mentioned characterization techniques for the successful loading of CaO2 on MnO2 (Fig. S1b, ESI†).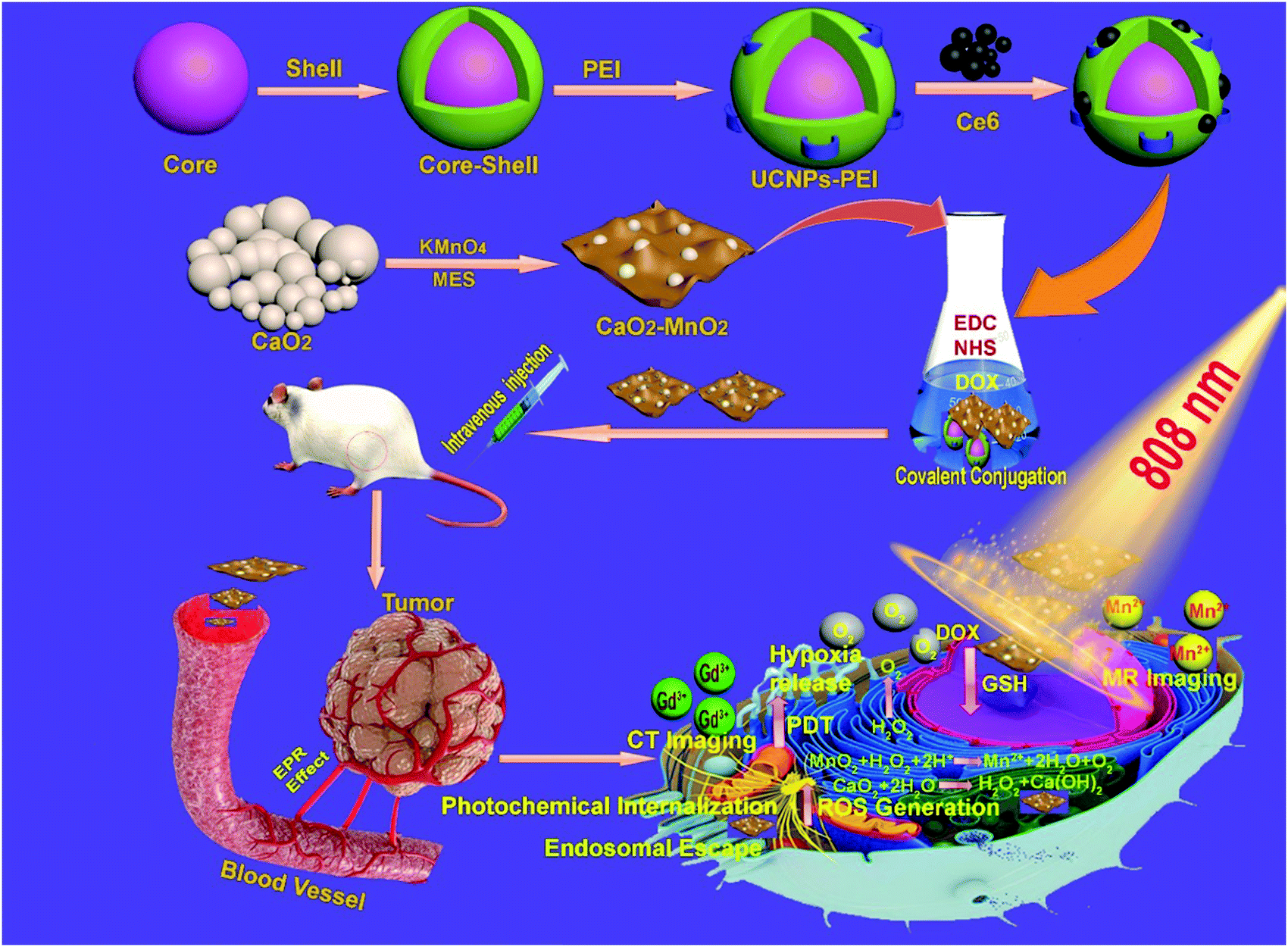 | ||
| Scheme 1 Schematic illustration of the synthesis and antitumor performance of CaMn–NUC nanoplatform upon 808 nm laser irradiation. | ||
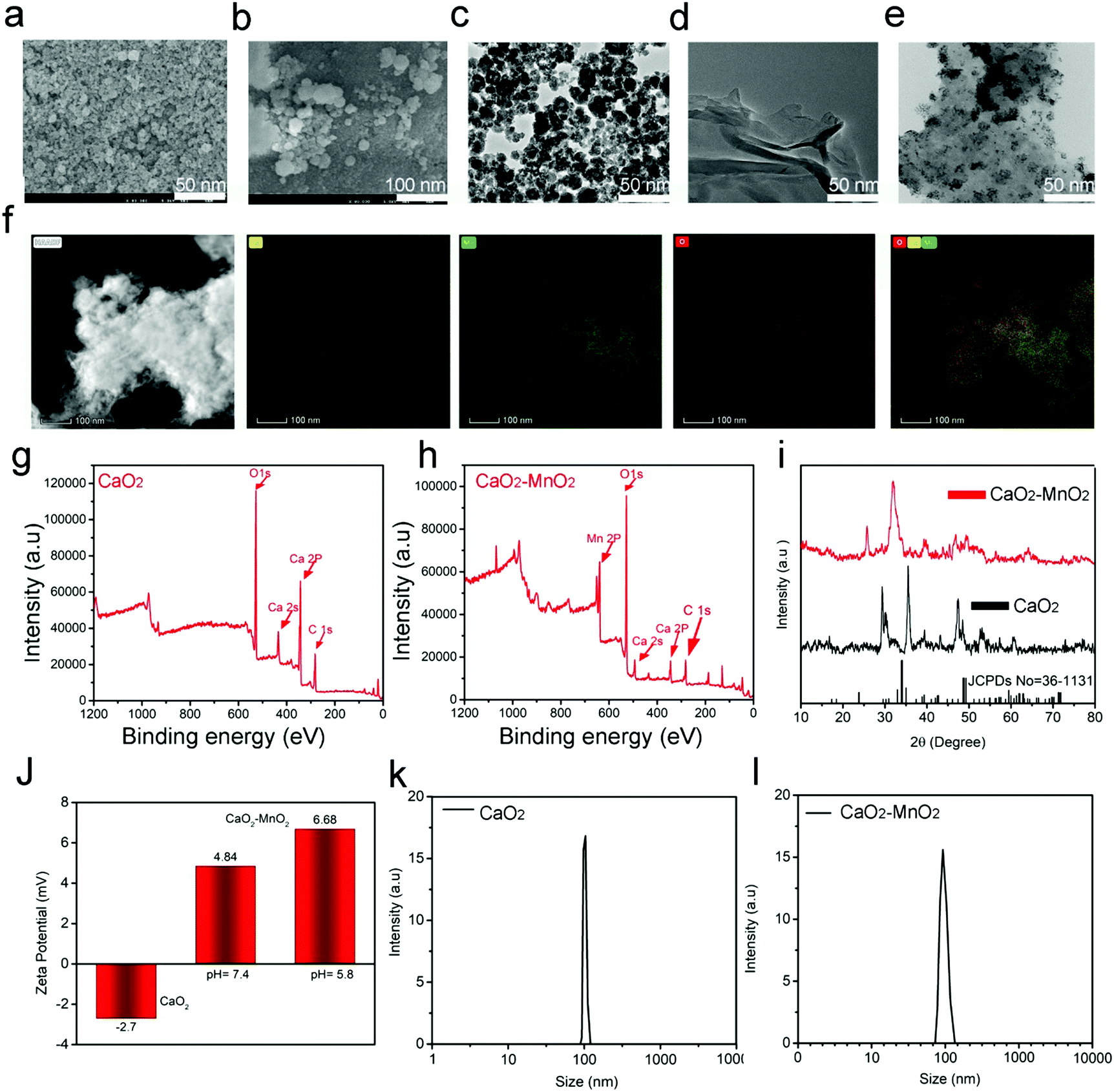 | ||
| Fig. 1 SEM images of (a) CaO2, (b) CaO2–MnO, and the TEM images of (c) CaO2 and (d) CaO2–MnO2 nanosheet. (e) The elemental mapping images of CaO2–MnO2. (f and g) XPS spectra of CaO2 and CaO2–MnO2. (h) XRD spectra of CaO2 and CaO2–MnO2. (I) The zeta potential of CaO2 and CaO2–MnO2. (j and k) Dynamic light scattering (DLS) particle size distributions of CaO2 and CaO2–MnO2. | ||
In order to provide our nanocomposite with multimodal imaging and therapeutic function, core–shell structured NaGdF4:Yb3+,Er3+@NaGdF4:Nd3+,Yb3+ UCNPs with distinctly proficient upconversion luminescence (UCL) were made to perform as a fluorescent probe via our earlier established procedure56 and were covalently conjugated with the CaO2–MnO2 nanocomposite. The UCNPs cores’ average diameter was 10.4 nm and for core–shell nanoparticles, it was 23.1 nm, as revealed by the TEM images (Fig. 2a and b). To enhance the biological compatibility of our nanocomposite, the UCNPs were modified by PEI. As established by the TEM images (Fig. 2c and d), UCNPs–PEI presented identical morphology and is highly monodisperse as compared to the OA-stabilized UCNPs, which possess a normal diameter of about 24.5 nm, which is flawless for biological applications. Using hydrodynamic size distribution, the shell thickness of the core–shell UCNPs was calculated to be about 12.7 nm (Fig. 2e and f). The modification of UCNPs by PEI hardly affected the thickness of the shell, as shown by the hydrodynamic size distribution (Fig. 2g and h). The XRD pattern validated that the UCNPs are in a hexagonal phase (Fig. S1c, ESI†). Excluding an insignificant lessening at 540 nm and 654 nm, the upconversion luminescence spectrum of the UCNPs–PEI continued to be almost the same as that of oleic acid-stabilized UCNPs, confirming that the surface alteration by PEI hardly changed the optical properties of the nanoparticles. The emission spectra of the UCNPs–PEI and CaMn–NUC (Fig. 2i) revealed the emission bands at 520–540 nm and 540–560 nm in the green spectral region, which are attributed to Er3+ transitions from 2H11/2 to 4I15/2 and from 4S3/2 to 4I15/2, respectively. In addition, the emission band in the red spectral region of 640–680 nm is due to the Er3+ transition from 4F9/2 to 4I15/2. Although the emission intensity of CaMn–NUC is to some extent weaker compared to that of the UCNPs–PEI owing to the part absorption by CaO2–MnO2, the held-in reserve emission was hitherto robust and abundant enough to be detected with the naked eye (Fig. S1d, ESI†). In Fig. 2j, the UCL spectra demonstrates the quenching effect of the CaO2–MnO2 nanosheet on the luminescence of UCNPs with and without modification with PEI; as can be seen, there is a negligible quenching of UCL emission in the nanocomposite conjugated with PEI compared to the nanocomposite without modification with PEI. Photoluminescence spectroscopy was used to characterize the energy transfer process from the nanoparticles to the Ce6 photosensitizer. Through the measurement of the UV-vis absorbance of Ce6, it was confirmed that at about 660 nm, the absorption peak matches well with the emission peak of the UCNPs–NUC (Fig. S2a, ESI†). Through the measurement of the temporal behavior of upconversion luminescence, the energy transfer progression was calculated. At 540 and 654 nm, the emission decay curves of the UCNPs–PEI and the CaMn–NUC are shown in Fig. 2k and l. The average decay time at 540 nm diminished to some extent from 123.42 μs to 96.827 μs in the presence of Ce6, which is partially attributed to the feeble absorption of Ce6 at 540 nm. Remarkably, the decay time at 654 nm significantly reduced from 226.212 μs to 175.160 μs.
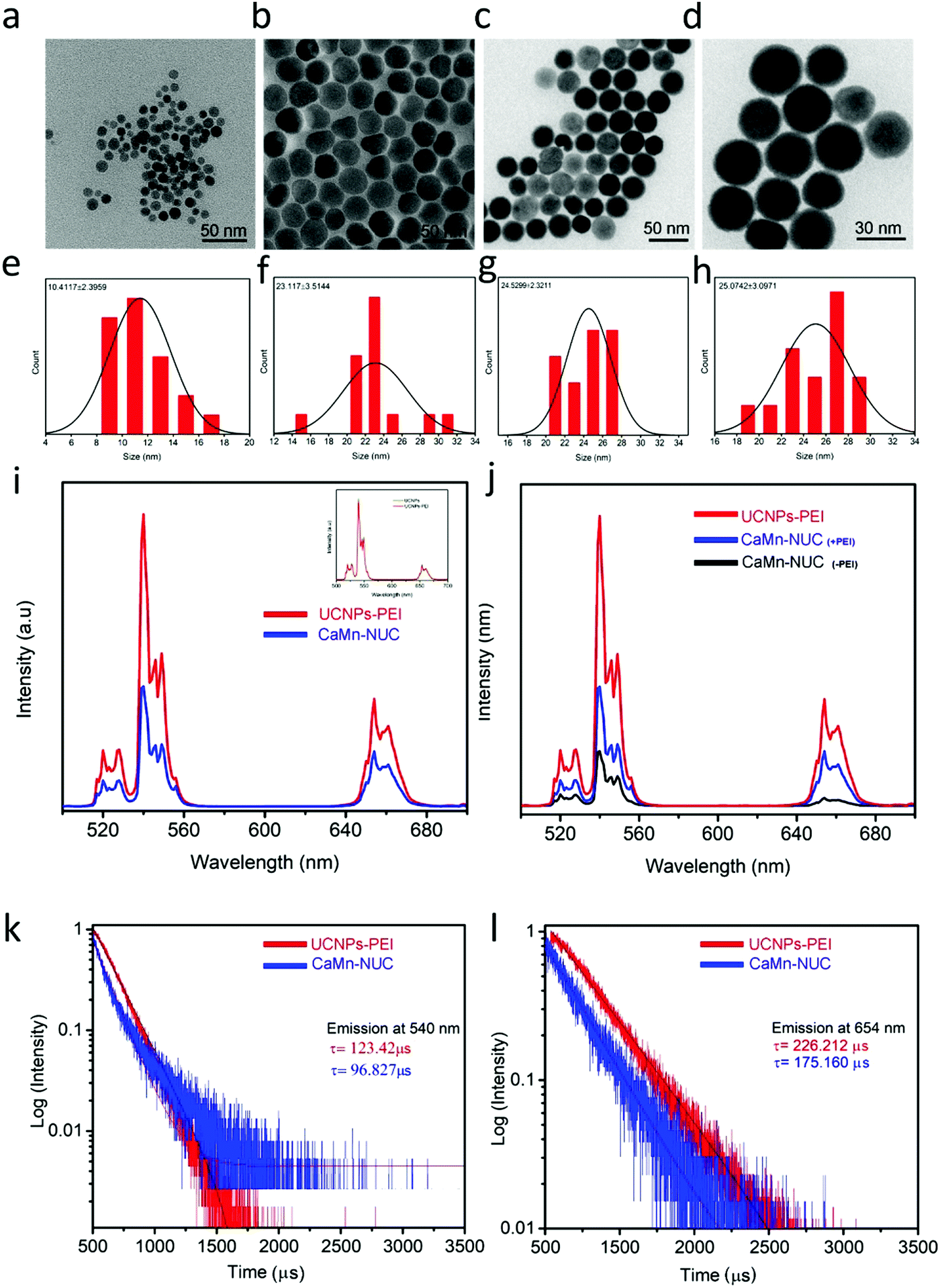 | ||
| Fig. 2 TEM images of (a) the UCNPs core, (b) OA-stabilized core–shell UCNPs, and (c and d) PEI-modified UCNPs. Hydrodynamic size distribution of (e) the UCNPs core, (f) OA-stabilized core–shell UCNPs, and (g and h) PEI-modified UCNPs. (i) Upconversion luminescence spectra of 808 nm NIR excited UCNPs–PEI and CaMn–NUC. (j) Upconversion luminescence spectra of 808 nm NIR excited UCNPs–PEI, CaMn–NUC, and CaMn–NUC without PEI. (k and l) Luminescence decay curves of the emissions at 450 nm and 654 nm of UCNPs–PEI and CaMn–NUC excited by the 808 nm flash. | ||
This outcome was attributed to the robust absorption of Ce6 at about 654 nm, validating the selective and exceedingly efficient energy transfer from the UCNPs to the photosensitizers. The efficiency of quantum yield of light absorption and upconversion emission of CaMn–NUC was found to be 0.89% (Fig. S2b and c, ESI†).
This result substantiated that our system is highly for avoiding the quenching of UCL with a therapeutic agent, consequently providing the nanoplatform with high fluorescence intensity and a therapeutic effect. The successful conjugation of CaO2–MnO2 and PEI-modified UCNPs were verified by the TEM image, as revealed in Fig. 3a. The UCNPs nanoparticles are loaded on to the CaO2–MnO2 nanosheet. To further support the successful conjugation of UCNPs onto CaO2–MnO2, we carried out the EDS and TEM elemental analysis of CaO2–MnO2–UCNPs, which proves the successful loading of UCNPs onto the nanosheet (Fig. 3b and c).
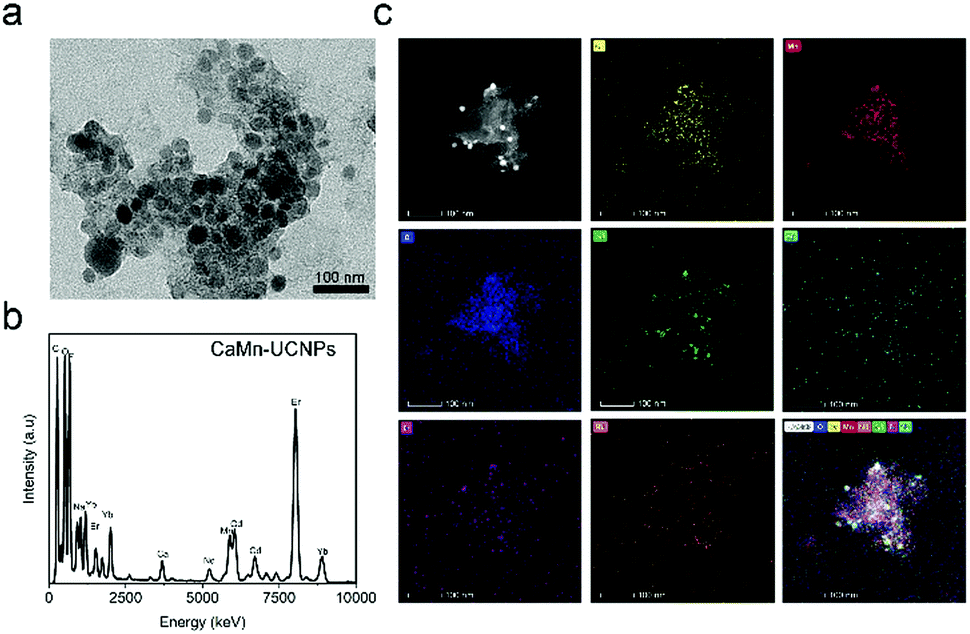 | ||
| Fig. 3 (a) TEM image, (b) EDS spectrum (c), and TEM elemental mapping of CaO2–MnO2–UCNPs. | ||
Moreover, the FTIR analysis of CaMn–NUC confirmed that the peaks at 873 cm−1, 1080 cm−1, and 1490 cm−1 correspond to the presence of the carbonate (CO32−) group, and the peak at 1588 cm−1 is linked to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch even though the peak at 538 cm−1 corresponds to the Mn–O stretching (Fig. S2d, ESI†). In addition, the peak at 3461 cm−1 corresponds to the stretching mode of O–H. The peaks for C–H stretching in oleic acid at 2920 and 2850 cm−1 in OA–UCNPs vanished after hydrophilic modification. The peaks at 743 and 1234 cm−1 in UCNPs–PEI were for C–N stretching and C–O–C stretching, respectively, settling the efficacious surface amendment with PEI. The FTIR analysis of CaMn–NUC exhibited that the peak at 1637 cm−1 was linked with the CO stretching vibration from the amide group and the 1736 cm−1 peak was from the unreacted carboxylate groups in Ce6 since the molecule contains three carboxylate groups. The band at 1622 cm−1 for the DOX-loaded CaO2–MnO2–UCNPs might perhaps be owing to the overlapping of the bands of pure DOX at 1638 cm−1 and UNCPs peak at 1637 cm−1, demonstrating the DOX drug alliance with the UNCPs. The carboxylate band at 1338 cm−1 for DOX does not somewhat indicate a substantial red shift, signifying the disappearance of chemical bonding amongst the surface calcium ions of CaO2–MnO2 and the carboxylate radical of DOX. Hence, the process of drug loading seems to be mainly through electrostatic communication amongst the positive calcium ions on the MnO2 surface and the negative carboxylate radical on DOX.
C stretch even though the peak at 538 cm−1 corresponds to the Mn–O stretching (Fig. S2d, ESI†). In addition, the peak at 3461 cm−1 corresponds to the stretching mode of O–H. The peaks for C–H stretching in oleic acid at 2920 and 2850 cm−1 in OA–UCNPs vanished after hydrophilic modification. The peaks at 743 and 1234 cm−1 in UCNPs–PEI were for C–N stretching and C–O–C stretching, respectively, settling the efficacious surface amendment with PEI. The FTIR analysis of CaMn–NUC exhibited that the peak at 1637 cm−1 was linked with the CO stretching vibration from the amide group and the 1736 cm−1 peak was from the unreacted carboxylate groups in Ce6 since the molecule contains three carboxylate groups. The band at 1622 cm−1 for the DOX-loaded CaO2–MnO2–UCNPs might perhaps be owing to the overlapping of the bands of pure DOX at 1638 cm−1 and UNCPs peak at 1637 cm−1, demonstrating the DOX drug alliance with the UNCPs. The carboxylate band at 1338 cm−1 for DOX does not somewhat indicate a substantial red shift, signifying the disappearance of chemical bonding amongst the surface calcium ions of CaO2–MnO2 and the carboxylate radical of DOX. Hence, the process of drug loading seems to be mainly through electrostatic communication amongst the positive calcium ions on the MnO2 surface and the negative carboxylate radical on DOX.
The CaMn–NUC nanocomposite shows a typical type IV isotherm, thus confirming the mesoporous structure of our nanocomposite and possesses a relatively higher Brunauer–Emmett–Teller (BET) surface area of 307.23 m2 g−1 (Fig. S3a, ESI†).
Since CaO2 produces H2O2 under acidic conditions, the generation of H2O2 was determined according to the described protocol57 under different pH conditions in different physiological media such as H2O, PBS, and FBS, as shown in Fig. S3b (ESI†). As we know, MnO2 nanoparticles have been regarded as the agents to quicken the oxygenation. Under acidic pH conditions, the catalytic pathway of MnO2 decomposition of H2O2 is shown in eqn (1). Afterwards, the JPBJ-608 Shanghai REX instrument acts as an oxygen probe as in vitro O2 progression testing from H2O2 was carried out on CaO2, MnO2, and the CaMn–NUC composite. A substantial amount of oxygen production by CaMn–NUC was accomplished in the H2O2 (100 μM) solution compared to that of H2O2, MnO2, and CaMn–NUC, respectively, which is indicative of MnO2 playing the role of a catalyst to activate the oxygenation in the presence of H2O2 (Fig. S3c, ESI†). In addition, all through the procedure for the disintegration of H2O2 in an acidic environment, as portrayed in eqn (1), Mn2+ liberated from MnO2 acts as a contract agent for T1-weighted MR imaging.
Excited by the results obtained from the photo-properties of CaMn–NUC, the singlet oxygen (1O2) generation of CaMn–NUC was explored by the photoluminescence spectrum. At first, the photo-stability and UV-visible spectroscopy of CaMn–NUC should be discussed. As shown in Fig. S3d (ESI†), CaMn showed excellent photo-stability after 10 min of laser irradiation (808 nm laser, 0.5 W cm−2). In order to achieve a multimodal therapeutic efficiency, Ce6, having a higher optical absorption coefficient in the 600–800 nm wavelength range, was employed as a photosensitizer, which was loaded on to the CaO2–MnO2 surface for PDT via the mixing solution of Ce6/DMSO with the CaMn–UCNPs solution for a period of 12 h. As we know, ROS may well prompt damage to the mitochondria as well as DNA, inducing an increase in the death of the cancer cells. ROS damages the cellular functions through numerous reaction mechanisms, ultimately leading to cell death. A catalyst nanocomposite possessing catalytic functions was revealed in this study, which may perhaps provide an additional reaction pathway for ROS, which can produce harmless/beneficial molecules such as H2O and O2.
| O2− or H2O2 + cell → damage | (3) |
| O2− or H2O2 → O2 + H2O | (4) |
The capability of the nanocomposite to produce ROS is an imperative trait, which governs the PDT efficiency. Dichlorofluorescein (DFC), which is converted from non-fluorescent DCFH-DA oxidized by means of ROS, was employed for observing the extracellular ROS. The comparison of the ROS generation competence of CaMn–NUC dissolved in PBS at pH 5.8 as well as pH 7.4 upon irradiation through an 808 nm NIR laser in dark was carried out. The quantity of ROS generated via the combination of DCFH-DA with CaMn–NUC at pH 7.4 and pH 5.8 through variable irradiation time symbolizes the fluorescence intensity, as shown in Fig. 4a and b. It is apparent that in both pH situations, the fluorescent intensities of CaMn–NUC were increased; nevertheless, there is an extraordinarily greater upsurge in the intensity of ROS production at pH 5.8, therefore establishing our approach towards scheming a pH-receptive nanocomposite intendedly designed for competent PDT at pH = 5.8. Using eqn (4), the singlet oxygen yield of the nanocomposites was calculated.
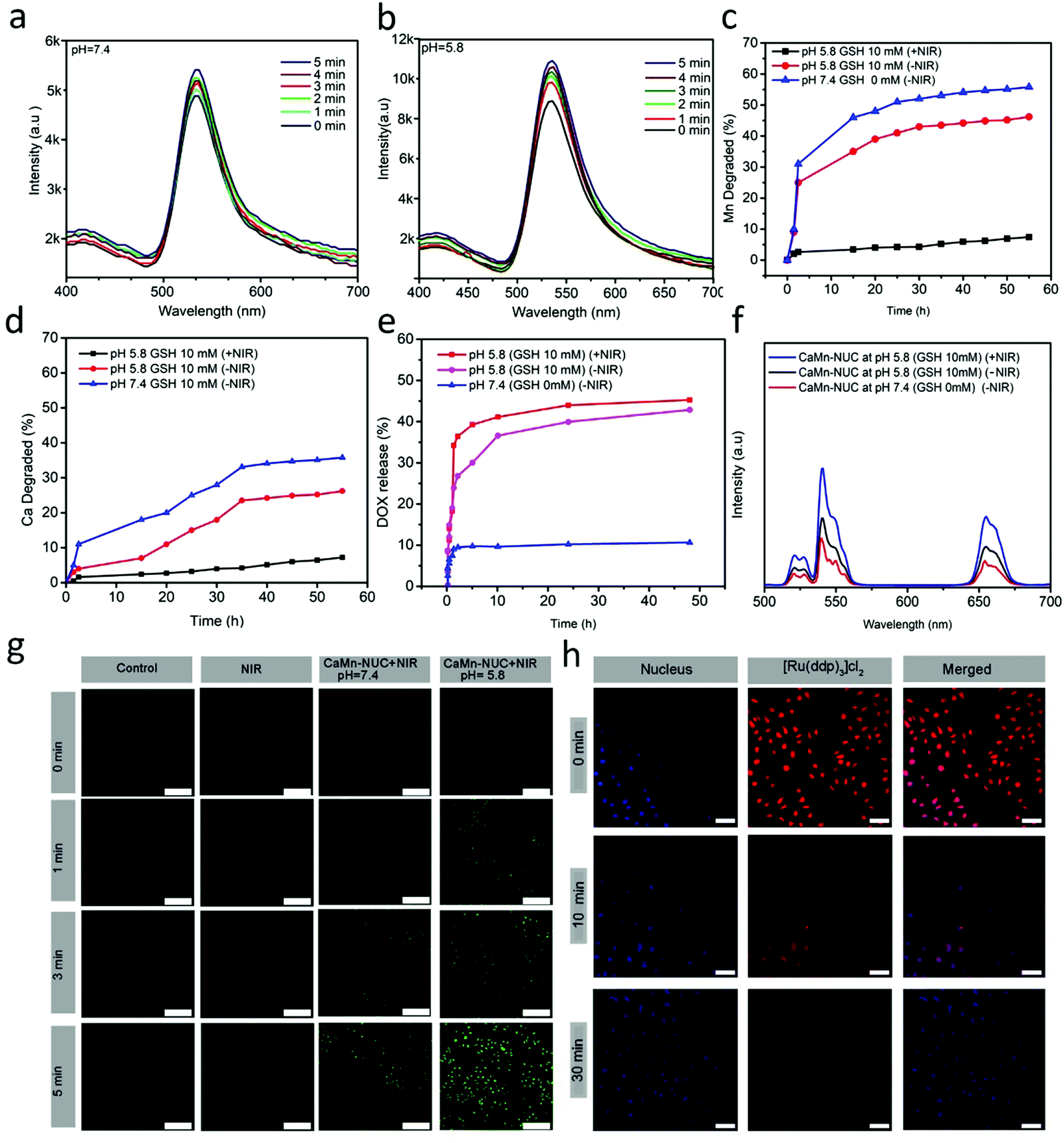 | ||
| Fig. 4 (a and b) Photoluminescence intensities of DCFH-DA-containing solution of the CaMn–NUC nanocomposite at 525 nm under 808 nm NIR irradiation as a function of time at pH 7.4 and pH 5.8. (c and d) Degraded Mn and Ca concentrations in PBS (pH 7.4, GSH 0 mM and pH 5.8, GSH 10 mM) without and with NIR laser irradiation. (e) DOX release profiles from CaMn–NUC in PBS without and with NIR laser irradiation. (f) Upconversion emission profiles of CaMn–NUC as a function of release time in PBS. (g) In vitro ROS generation in HeLa cells under different pH conditions. (h) In vitro hypoxia study on O2 production in vitro: the HeLa cells were stained with DAPI and [Ru(dpp)3]Cl2 as the nucleus and dissolved O2 probes and then treated with CaMn–NUC nanocomposites at different incubation times. All scale bars are 50 μm. | ||
As is known, elevated level of GSH in TME is another hurdle in the efficient application of PDT since it is known that the Mn–O bonds are responsive to reduction, besides a minor acidic environment. Consequently, we foresee the idea that the proposed Mn–O bonds in the nanocomposite might disintegrate in a condition identical to the tumor, thus accelerating the nanocomposite biodegradation. The extracellular pH in TME is mostly in the range of 7.4–6.5, which is mostly reliant on the stage besides the type of the tumor, whereas that of the intracellular initial endosome and lysome touched 6.2–5.0. In addition, the intracellular microenvironment has a reductive nature with a GSH content that is at least 4-fold more than that in a normal tissue. In order to model the TME, for comprehensive biodegradation assays, the PBS buffer solution with pH 5.5 and 10 mM GSH was employed. To model the normal body, PBS buffer with pH 7.4 and 0 mM GSH was used for comparison. CaMn–NUC was dissolved in numerous PBS solutions and ICP tests were employed to examine the progress of degradation. In comparison with that in neutral and non-reductive PBS, Mn release is noticeably boosted in a slightly acidic as well as reductive condition (Fig. 4c). In addition, rapid Mn discharge from CaMn–NUC also led to accelerated Ca discharge (Fig. 4d). Throughout the degradation time of 4 h to 8 h, the 808 nm laser irradiation hastened the Mn and Ca release. Thus, we can conclude that the acidity and reducibility of the tumor might prompt the Mn–O bond rupture and initialize the degradation of the nanocomposite. Importantly, the NIR photon an accelerating effect over the nanocomposite degradation rate. The release profiles of Ca/Mn specify that the disintegration of Mn–O bonds in acidic conditions encourages the rapid rupture of the CaMn–NUC framework afterwards and the NIR irradiation functions as a promoter for the breakdown nanocomposite. Convinced by the excellent biodegradable performance of our nanocomposite, the DOX drug release profile of CaMn–NUC was studied. There is a fastened drug release from PBS in a mildly acidic and reductive environment. Furthermore, DOX release shows greater efficiency in a simultaneously acidic as well as reductive environment upon irradiation with the NIR laser, hence leading to the fast dilapidation of the nanocomposite, as shown in Fig. 4e. The CaMn–NUC upconversion emission spectra in various solutions of PBS deprived of and by means of NIR irradiation against the release time are revealed in Fig. 4f. Due to the fluorescence resonance energy transfer (FRET) amongst UCNPs and DOX, there is a great inhibition of green-emissive peaks of the UCNPs. It is believed that more than 10 nm distance between the donor and the acceptor will result in the FRET effect to vanish. Accordingly, with heightened DOX release, there is an increased intensity of emission of the green peaks compared to the red emissions and the augmentation of combined intensity is primarily triggered by abridged quenching effect from the MnO2 nanosheet. Notably, for 1 to 4 h without NIR irradiation, emission retrieval, particularly in the green range, is higher than that during 4 to 6 h with NIR irradiation, hence validating that the NIR laser might augment nanocomposite disintegration, and thus, the DOX release.
Besides, we employed HeLa cells to study the ROS generation in vitro. At two different pH values, the cancerous HeLa cells were incubated with CaMn–NUC for 24 h, followed by irradiation with an 808 nm NIR irradiation for 0–5 min. There is an insignificant ROS production detected with CLSM in HeLa cells in the control group and cells in groups treated with 808 nm NIR irradiation for different time intervals, as shown in Fig. 4g. A substantial quantity of ROS production, as the intensity of green color, was amplified in the cells in the group with pH 7.4. Nevertheless, there was large amount of ROS generation in the cell groups with pH value 5.8, given that there was an exponential increase in the intensity of the green color, as revealed in Fig. 4g. Therefore, concluding from the above-mentioned outcomes, we are certain that our nanocomposite may perform well as an efficient PDT therapeutic agent.
As we know, another TME characteristic that drastically affects the efficiency of PDT is tumor hypoxia. To authenticate the efficiency of our nanocomposite to overcome the TME hypoxia by in situ oxygen generation in vitro, we employed bis(triphenylphosphine)ruthenium(II)dicarbonylchloride ([Ru(dpp)3]Cl2) (working as the O2 probe) to assess the in vitro O2 generation. The O2 produced can oxidize the fluorescent [Ru(dpp)3]Cl2. As revealed in Fig. 4h, post 30 min incubation with CaMn–NUC nanocomposites, the fluorescence intensity of [Ru(dpp)3]Cl2 was very feeble, thus demonstrating that the nanocomposites may perhaps produce O2in vitro, thereby dismissing tumor hypoxia in addition to augmenting the efficiency of PDT. Thus, it is evident from the above studies that our nanocomposite is a very efficient candidate for photo-chemodynamic therapy by overcoming the tumor microenvironment GSH and hypoxia, and elevated the level of singlet oxygen generation.
To further explore the antitumor effect of CaMn–NUC in vitro, the cellular uptake and in vitro cytotoxicity performance of the nanocomposites might be measured by the cell experiments. The cell viability of CaMn–NUC should be confirmed to ensure its negligible damage to the normal cells. As revealed in Fig. S4a and b (ESI†) the L929 fibroblast cells were applied for evaluating the biocompatibility of CaMn–NUC. Then, the cells were treated with various concentrations of CaMn–UCNPs–Ce6 and CaMn–NUC (15.6, 31.2, 62.5, 125, 250, and 500 μg mL−1). The cells treated with CaMn–UCNPs–Ce6 displayed a high rate of cell viability (>85%) at the highest concentration of 500 μg mL−1 after 24 h, while the cells treated with CaMn–NUC displayed a cell viability of (>80%) at the highest concentration of 500 μg mL−1 after 24 h, suggesting that CaMn–NUC has more toxicity towards the normal cells than the CaMn–UCNPs–Ce6 nanocomposites due to the presence of DOX. Nevertheless, the cell viability (>80%) at the highest concentration of 500 μg mL−1 after 24 h suggests that our nanocomposite had low toxicity for normal cells. As is known, CaO2 generates H2O2 and H2O, which can be dangerous to normal cells; thus, we evaluated the cell viability of CaO2 at different pH values of 7.4 and 5.8. The cell viability at pH 7.4 was found to be well above 90%, thus suggesting that CaO2 does not show any significant toxicity at normal physiological conditions. The cell viability of cells treated with CaO2 at pH 5.8 was found to be 81%, which suggests the production of H2O2 under acidic tumor conditions, as shown in Fig. 4c. HeLa cells were incubated with CaO2 at pH 7.4 and 5.8 to confirm the cytotoxicity of CaO2 by calcein AM and propidium iodide (PI) double-staining method. As shown in Fig. S4d (ESI†), we can see that there is no obvious cancer cell killing at pH 7.4, while a minor cell toxicity is noticed at pH 5.8.
Through the incubation of HeLa cells with CaMn–NUC for 0.5, 1, and 3 h at 37 °C, the cell uptake conduct of the sample was investigated and the respective CLSM images were unveiled in Fig. 5a. To stain the cell nuclei, DAPI, which can radiate blue emission, was employed and the DOX loaded in the nanosystem emits red fluorescence upon 488 nm laser excitation. Afterwards, the merged images from the above two channels were shown. As we can see, there is a weak red emission in the first 0.5 h, demonstrating that only a small quantity of the nanocomposite is swallowed by the cells. Once the incubation time was extended, we can notice that the red signal turns out to be robust, suggesting that more nanocomposites were localized in the cells. Thus, we can confirm from these results that our nanocomposite was proficiently internalized by the HeLa cells. As unveiled in Fig. 5b and c, the cytotoxicities of different samples were detected by employing the standard methyl thiazolyl tetrazolium (MTT) assay. The HeLa cells were incubated with non-NIR treatment groups as follows: Control, CaO2–MnO2, CaO2–MnO2–UCNPs, and CaMn–UCNPs–Ce6. The concentrations of CaO2–MnO2, CaO2–MnO2–UCNPs, and CaMn–UCNPs–Ce6 were set as 15.63, 31.5, 62.5, 125, 250, 500, and 1000 μg mL−1. The cells treated with CaO2–MnO2, CaO2–MnO2–UCNPs, and CaMn–UCNPs–Ce6 without NIR irradiation showed high cell viability of about 75%, implying little damage to the HeLa cells. In contrast to the groups treated with pure CaO2–MnO2/DOX and NIR irradiation, the highest concentration of (1000 μg mL−1) exhibited an inhibition over the proliferation of HeLa cells with a killing efficiency that could reach 31%. Another group treated with NIR irradiation and CaMn–UCNPs/DOX at its highest concentration exhibited an inhibition of the HeLa cells with the killing efficiency reaching about 24%. Though the survival rate of the CaMn–UCNPs/DOX + NIR treated group was much lower than that of the groups treated with only NIR and CaO2–MnO2/DOX + NIR, the killing efficiency was still below our expectations. Therefore, the cells that were incubated with CaMn–NUC + NIR exhibited the highest killing efficiency, which was contributed to the synergistic therapeutic effect of mitochondrial therapy and chemo-photodynamic therapy. The synergistic treatment of CaMn–NUC was also ensured by calcein AM and propidium iodide (PI) double-staining method in Fig. 5d, in which the live cells were stained by green color and the dead cells were red. For groups treated with pure NIR or CaO2–MnO2/DOX + NIR, there was no obvious damage to the HeLa cells. Then, the CaMn–UCNPs/DOX + NIR treated group exhibited a small number of dead cells. Almost 50% of the HeLa cells were dead when treated with CaMn–UCNPs/DOX + NIR. Obviously, when the cells were incubated with CaMn–NUC + NIR, all the cells were killed. The result from the AM-PI method was matched with the cytotoxicity experiment of CaMn–NUC.
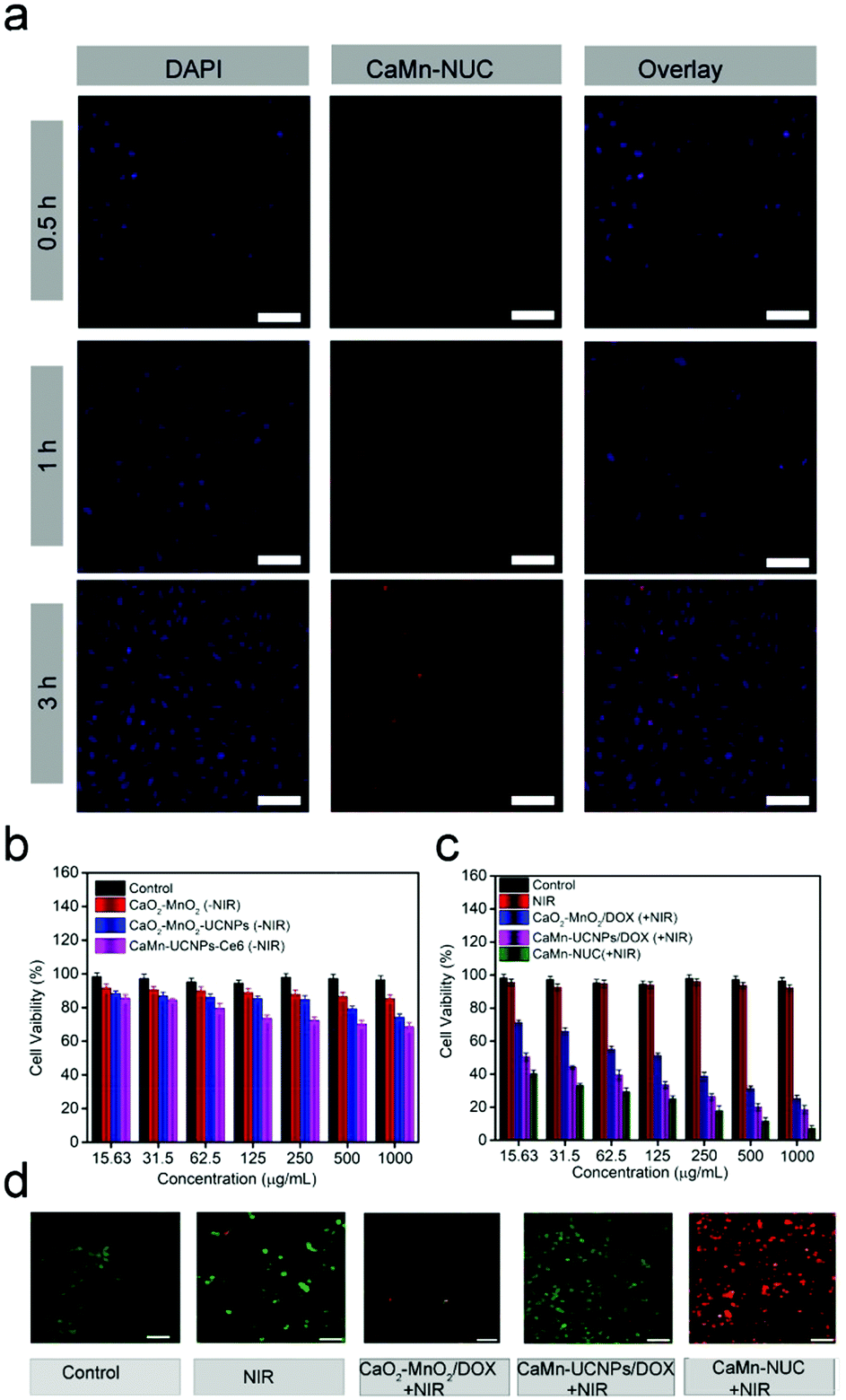 | ||
| Fig. 5 (a) CLSM images of HeLa cells incubated with CaMn–NUC for 0.5, 1, and 3 h. Scale bar: 75 μm. (b) In vitro cytotoxicity of Control, CaO2–MnO2, CaO2–MnO2–UCNPs, and CaMn–UCNPs–Ce6 without NIR. (c) In vitro cytotoxicity of Control, NIR, CaO2–MnO2/DOX, CaMn–UCNPs/DOX, and CaMn–NUC with NIR. Statistical analysis was performed using Student's two-tailed t test (*P < 0.05). (d) CLSM images of HeLa cancer cells after various treatments, dyed with AM/PI. Scale bar: 50 μm. | ||
Mitochondria is a crucial cellular organelle; besides, it is responsible for the bulk of the energy supply of the cells. Also, it has a decisive role in cell-programmed death, for instance, apoptosis. Likewise, as is known that malignant tumor cells are very active and rapidly proliferating cells, therefore, they require supplementary mitochondria compared to the normal cells.58 Hence, if the cancer cell mitochondria are damaged, it will inhibit the division of cancer cells. Thus, to confirm the mitochondrial uptake of the CaMn–NUC nanocomposite in the HeLa cells, we used CLSM and a commercially accessible red-selective Mito-tracker. Mito-tracker dyes the mitochondria red, which can be then examined using CLSM. In the meantime, FITC will make the CaMn–NUC nanocomposites show green fluorescence. As revealed in Fig. 6a, the green fluorescence imaging of the CaMn–NUC–FITC nanocomposite agrees with the red fluorescence imaging of the Mito-Tracker in the time period of 0.5, 1.0, and 1.5 h. This occurrence proposes that the CaMn–NUC nanocomposite can enter the mitochondria. Besides, the mitochondria became more and more green with more time, suggesting that the materials kept in flowing into the mitochondria, as shown by the pictures in the second and fourth columns. Thus, we can conclude from the images (Fig. 6a) that the CaMn–NUC nanocomposites kept entering the mitochondria for mitochondria-assisted cancer cell therapy. The mechanism of mitochondrial uptake and mitochondria-assisted cancer therapy is shown in Fig. 6b.
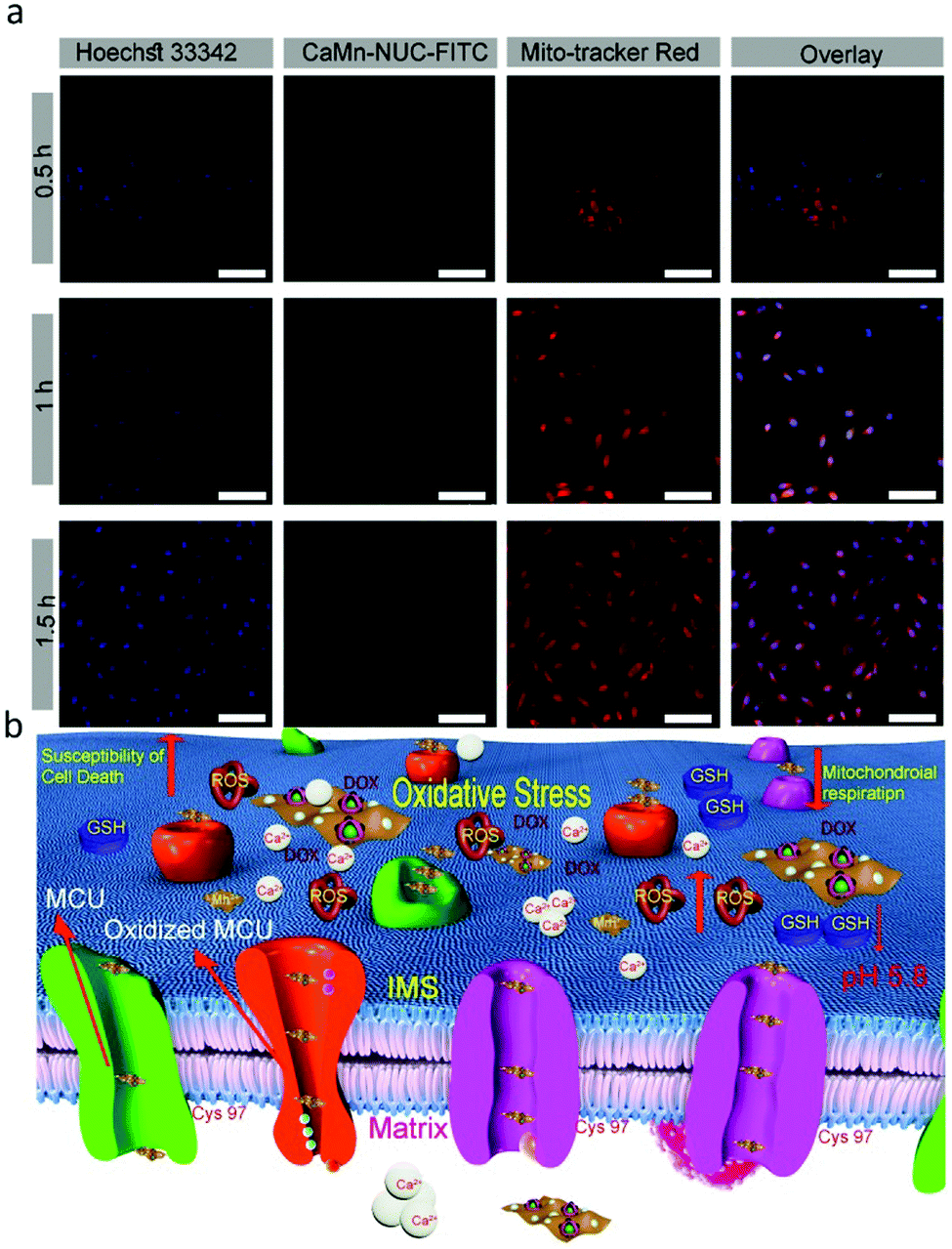 | ||
| Fig. 6 (a) CLSM images of mitochondrial uptake of CaMn–NUC by HeLa cells incubated with CaMn–NUC–FITC for 0.5, 1, and 3 h. Scale bar: 75 μm. (b) the schematic illustration of mitochondrial uptake of CaMn–NUC and the ensuing cancer cell death. | ||
The multimodal imaging properties of our nanocomposite were investigated by MR, CT, and UCL imaging after its encouraging and outstanding antitumor operations in vitro. We know from the preceding studies that positive augmentation of T1 MR imaging signal is shown by Mn2+ and Gd3+ ions. Thus, for a superior pH-responsive MR imaging result, we proposed the idea of grouping of CaO2–MnO2 and NaGdF4-based UCNPs. In this, we firstly explored the T1-weighted MR imaging outcome of the CaMn–NUC nanocomposite dispersed in the PSB solvent at the pH values of 7.4 and 5.8. We found that at two dissimilar pH values, the relaxation r1(1/T1) signal displayed a linear intensification against the overall concentration of Mn2+ and Gd3+, which changed from 0 to 0.16 mM (Fig. 7a). At pH = 5.8, the relaxation rate (r1 value) was found to be 10.903 mM−1 s−1, which is an extraordinarily better value compared to the relaxation rate (r1 value) of 4.2012 mM−1 s−1 at pH = 7.4. Thus, it confirms that CaMn–NUC is able to produce MR contrast on a transverse photon relaxation-time-weighted sequence to efficaciously curtail the T1 relaxation time. There was a concentration-dependent whitening effect of the pH-responsive in vitro T1-weighted images (Fig. 7b). Herein, we perceive that due to the disintegration of MnO2 into Mn2+ ions at pH = 5.8, the concentration-reliant whitening effect at pH 5.8 was greater compared to that at pH = 7.4. The in vivo T1-weighted MR imaging studies on tumor-bearing mice were carried out post the achievement of the encouraging in vitro results. As shown in Fig. 7c, we can see a strong T1-MR signal attenuation effect for the mouse, which has been injected in the sample in contrast to the mouse deprived of sample injection, signifying the extraordinary capability of CaMn–NUC as a T1 MR imaging contract agent.
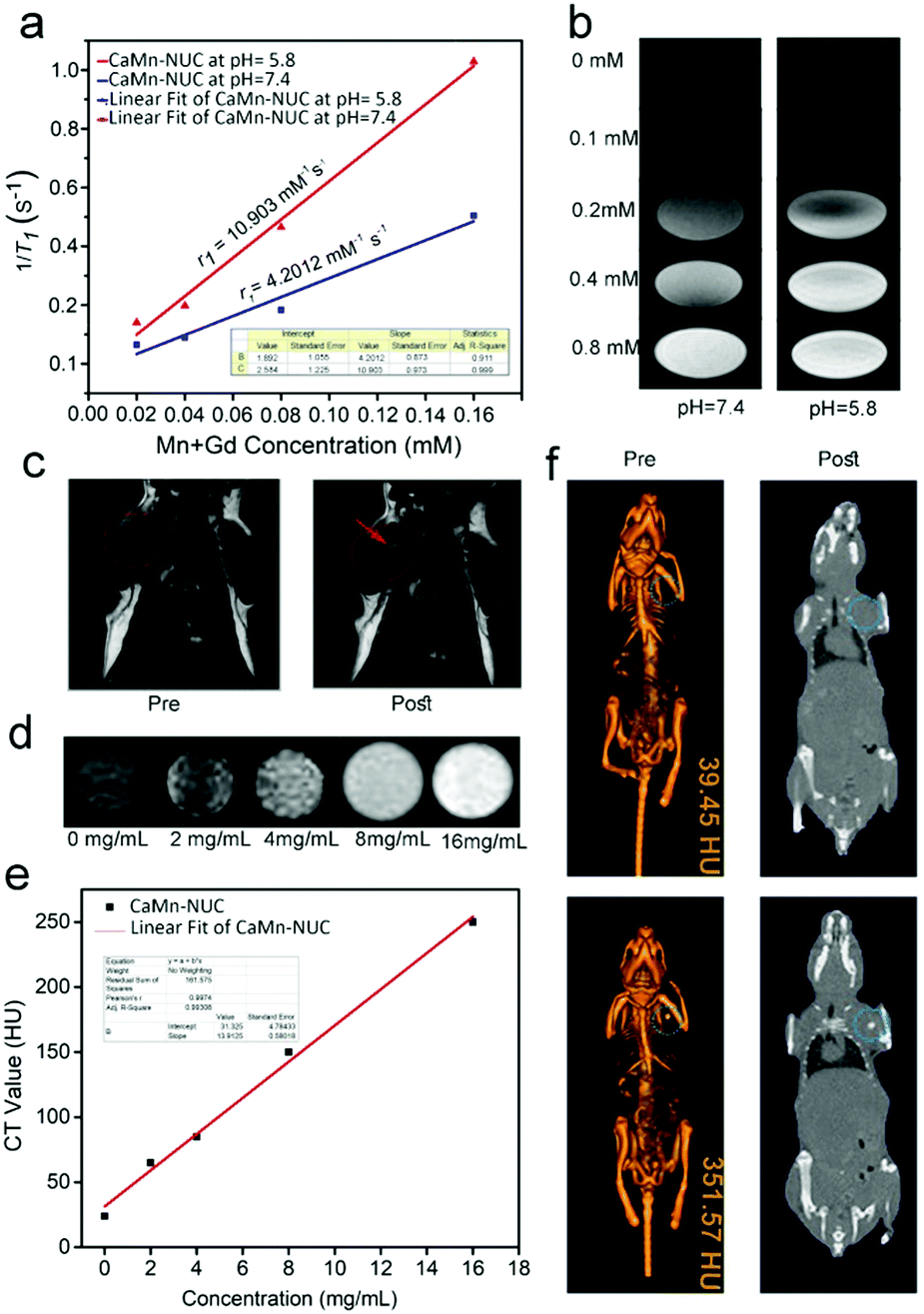 | ||
| Fig. 7 (a) r1 relaxation rate versus total concentration of Mn and Gd. (b) In vitro T1-weighted MR images of CaMn–NUC. (c) In vivo T1-weighted MR images of tumor-bearing mice pre-treatment and post treatment. (d) In vitro CT images with different concentrations of CaMn–NUC, when incubated in PBS with different pH values of 5.8 and 7.4 (e) CT values of aqueous CaMn–NUC versus concentration of CaMn–NUC. (f) In vivo CT images of tumor-bearing mice treated with CaMn–NUC and untreated mouse as the control. | ||
As is commonly recognized, the CT imaging technology bids the essentials of 3D assemblies with deep tissue penetration. Furthermore, nanomaterials with lanthanide doping were extensively studied for X-ray attenuation for high atomic number lanthanide elements. In this study, the in vitro and in vivo CT imaging properties of the CaMn–NUC nanocomposite were studied. As revealed in Fig. 7d, X-ray attenuation rises phenomenally when the concentrations of CaMn–NUC were increased. Furthermore, the CT values show a linear dependence on the sample concentration, as portrayed in Fig. 7e; therefore, a slope value of 13.9125 was calculated. We carried out the in vivo CT studies on tumor-bearing mice to determine the in vivo CT imaging potential of CaMn–NUC after being encouraged by the results of the in vitro studies. A small animal CT imaging device was used to analyze the imaging outcome of tumor-bearing mice injected with and without CaMn–NUC. The tumor site starved of injection displayed a lesser CT value (39.45 HU) compared with the CT value (351.57 HU) of the tumor site with injection (Fig. 7f). Such outcomes indicate that CaMn–NUC may well be used as an efficient CT contrast agent intended for multimodal imaging. Encouraged by the above imaging properties of CaMn–NUC, we conducted in vivo UCL imaging on a U14-tumor-bearing Kunming female mouse. The pre- and post-intra tumor injection UCL imaging results are shown in Fig. S4c (ESI†), which demonstrate that the mice treated with CaMn–NUC showed no luminescence when the laser was turned off, while the mice treated with CaMn–NUC and irradiated by a 808 nm laser at a power density of 0.5 W cm−2 displayed a strong upconversion luminescence, which is clear to the naked eye.
Then, the outstanding antitumor performance of CaMn–NUC was further explored by the in vivo therapeutic experiment. The therapeutic effect of CaMn–NUC was derived from the combination of highly toxic 1O2, elevated levels of oxygen generation, and damage to the tumor mitochondrial DNA by Ca2+ uptake and the chemotherapeutic effect of DOX. In the specific procedure of antitumor behavior, CaMn–NUC could be degraded under acidic TME and then Ca2+, Mn2+, DOX, and UNCPs–Ce6 were swallowed into the tumor cells. 1O2 was generated from UNCPs–Ce6 with 808 nm laser irradiation using the energy transfer process from the T1 state of the excited photosensitizer to the triplet oxygen (3O2). Therefore, the synergistic therapeutic effect of highly toxic 1O2, Ca2+, and DOX could easily eliminate the tumor cells by destroying the DNA of the tumor mitochondria. In order to confirm the antitumor efficiency of CaMn–NUC, it was evaluated in the model of U14-tumor-bearing Kunming female mice. At first, the tumor-bearing mice were stochastically categorized into group A and B. The mice in group A were further divided randomly into five groups: control group (group I), injection of normal saline with 808 nm laser irradiation (group II), injection of CaO2–MnO2/DOX with 808 nm laser irradiation (group III), injection of CaMn–UCNPs/DOX with 808 nm laser irradiation (group IV), and injection of CaMn–NUC with 808 nm laser irradiation (group V). As required by the four groups (groups II–V) needed, the light irradiation time for all was set as 10 min. After 2 h intravenous injection, groups II–V were irradiated by an 808 nm laser (0.5 W cm−2). Fig. 8a shows the schematic illustration of the progress of animal experimentation. There was a negligible effect on the relative tumor volume and tumor weight in the groups treated with saline only and NIR, revealing that laser irradiation could not mitigate tumor growth, while the tumor treated with CaO2–MnO2/DOX and NIR irradiation displayed slight tumor inhibition due to DOX. In contrast, the group IV exhibited clearly better tumor treatment efficacy because the released DOX, Ca2+, and production of H2O2in situ in acidic TME which in turn reduce the hypoxia in the TME. Fortunately, the tumor of group V was completely inhibited after the intravenous injection of CaMn–NUC under 808 nm laser irradiation for 10 min. Compared with the treatment effect of the other four groups, group V exhibited a remarkable curative effect owing to the synergistic therapeutic efficacy of PDT due to the presence of photosensitizer Ce6, which resulted in the production of singlet oxygen species due to reduced hypoxia in TME, which ensured mitochondrial DNA damage and chemotherapeutic effect of DOX, as shown in Fig. 8b and c, after two weeks of treatment. Digital photographs of the tumor were taken at the same time and it is evident from the photographs in Fig. 8d that there has been a huge tumor inhibition effect achieved in group V due to the synergetic chemo-photodynamic therapeutic effect of CaMn–NUC. The body weight of different groups showed little differences (Fig. 8e), indicating that the treatment did not pose any threat to the normal cells. The average life span and survival rates showed that the mice treated with the CaMn–NUC nanocomposite and irradiated with 808 nm laser survived for over 35 days, as shown in Fig. 8f and g. Tumor tissue destruction was confirmed by H&E staining, which clearly shows that the tumor tissues in the groups I and II are intact without any damage, while groups III and IV display a slight damage to the tumor tissue. The tumor tissue in the group V displays significant damage to the tissue morphology, as shown in Fig. 8h.
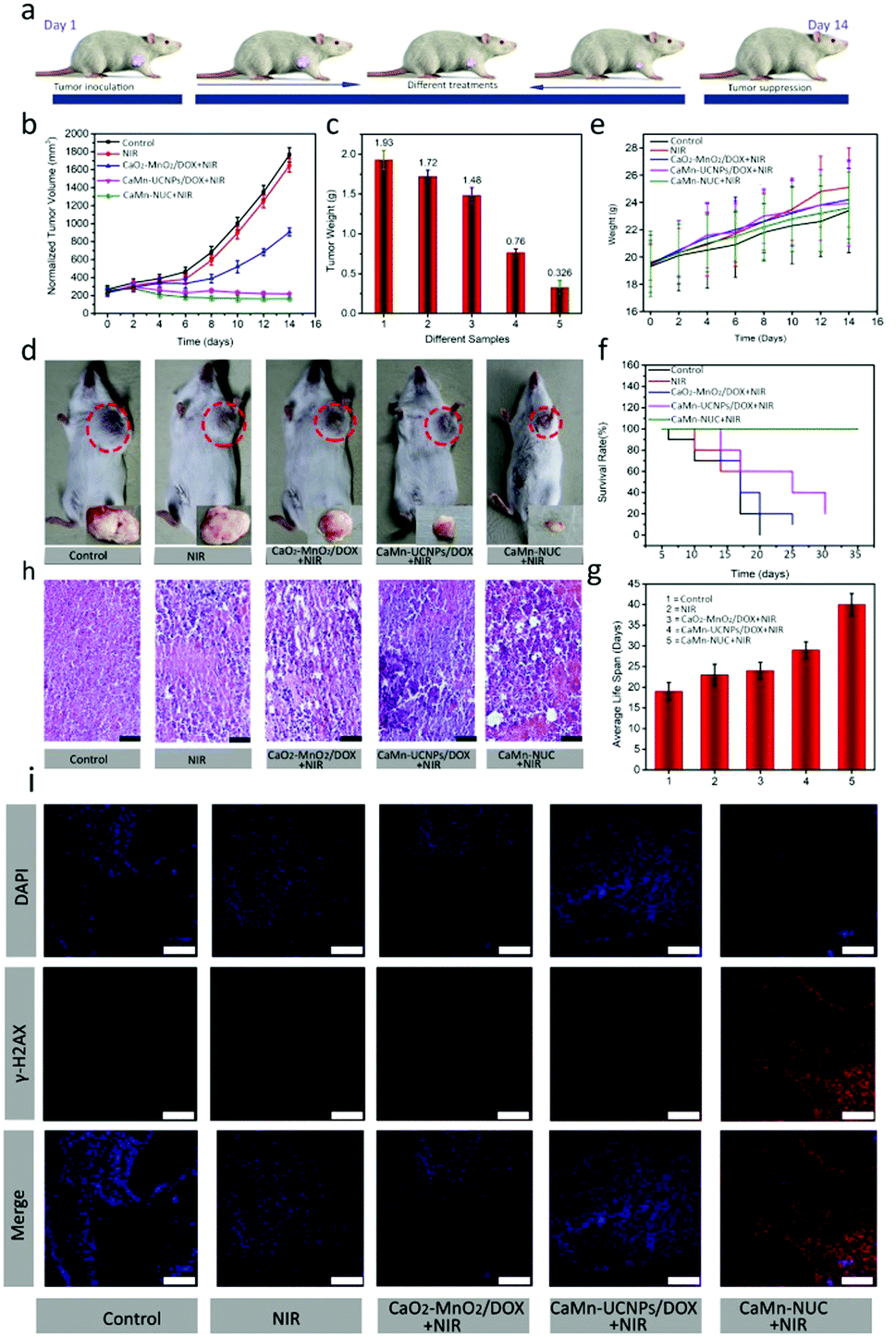 | ||
| Fig. 8 (a) Schematic representation of the animal experiment progress from day 1 to day 14 of the treatment. (b and c) Variation in the normalized tumor volume and tumor weight of mice with different treatment groups. (d) Digital photographs of mice and dissected tumors of different treatment groups. (e) The variation in the body weight in different treatment groups. (f and g) Survival rate and average life span of mice in different treatment groups. (h) H&E stained images of tumor tissues from mice after different treatments on the 14th day (scale bar: 100 μm). (i) Immunofluorescence staining by the nucleus (DAPI: blue) and the marker (γ-H2AX: red) of double-strand DNA breaks for tumor slices in different groups. These images were acquired by CLSM at the scale bar of 50 μm. Statistical analysis was performed using Student's two-tailed t test. *p < 0.05. | ||
In addition, we investigated the ability of the CaMn–NUC composite towards in vivo DNA damage. In this experiment, an immunofluorescence assay by staining γ-H2AX was performed on 5 μm frozen sections of each tumor tissue slices. The cell nuclei and γ-H2AX areas in the tumor slices were stained with DAPI (blue) and goat anti-rabbit IgG/Cy5-conjugated antibody (red), respectively. The red fluorescence emission of goat anti-rabbit IgG/Cy5-conjugated antibody will be enhanced if a greater number of DNA double strands are damaged. In the meantime, the DAPI area will dwindle in the fluorescence imaging as they were damaged. As can be seen in groups I & II, no or negligible γ-H2AX expression levels were observed, which indicates no damage to the DNA of the tumor tissue, while an apparent change was observed in groups III and IV, demonstrating that CaO2–MnO2/DOX + NIR and CaMn–UCNPs/DOX + NIR displayed therapeutic effect by damaging the tumor DNA as there is significant expression of the γ-H2AX levels. As can be seen for group V treated with CaMn–NUC + NIR, the blue area of the cell nuclei was completely shrunk, while the red area of γ-H2AX was broadened and brightened, which indicated severe damage to the DNA double strand in group V, as shown in Fig. 8i. Thus, the γ-H2AX immunofluorescence assay further verifies the synergetic chemo-photodynamic efficiency of our nanocomposite for inhibition.
In Fig. 9, the H&E stained images of the main organs including spleen, lung, heart, liver, and kidney extracted from groups I to V presented that there were no obvious injury and necrosis.
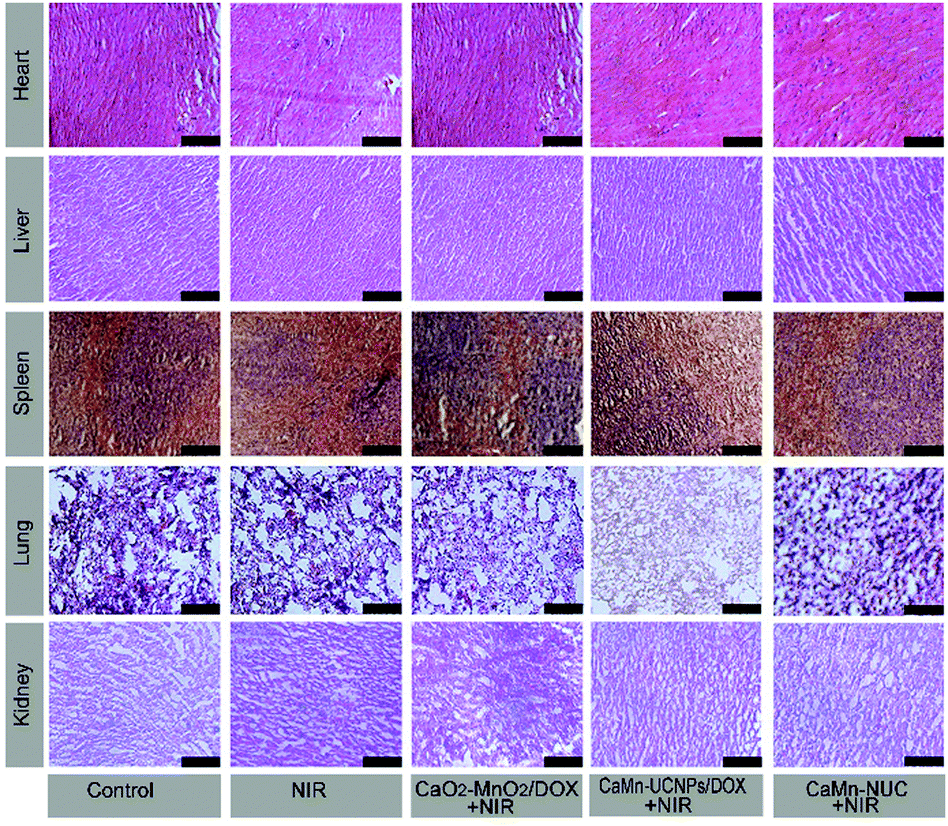 | ||
| Fig. 9 Representative histological H&E stained tissue sections from mice to monitor the histological change in the heart, liver, spleen, lung, and kidney excised from different groups after 14 days of treatment. All the images share the same scale bar of 100 μm. | ||
Conclusions
In summary, we have designed a multifunctional CaMn–NUC nanoplatform that possesses excellent performance of TME-response, targeting ability, and the synergistic therapeutic effect of PDT and chemotherapy. UCNPs–Ce6/DOX is incorporated onto the CaO2–MnO2 nanosheet by covalent conjugation and the chemotherapeutic drug DOX can be effectively loaded. Fascinatingly, minute fluorescent CaMn–NUC can be decomposed in the acidic environment (pH = 5.5–6.5), which then releases strong fluorescent UCNPs–Ce6, Mn2+, Ca2+ and the anticancer drug DOX. In particular, the PDT performance of UCNPs–Ce6 can be recovered when CaMn–NUC is mixed with the acidic solution such as TME, which is confirmed by the intracellular or extracellular 1O2 generation test. In addition, CaMn–NUC epitomizes higher selectivity towards the tumor cells than the normal cells. Therefore, the kind of stimuli-responsive “all-in-one” nanoplatform that combines targeting ability, TME-response, PDT, chemotherapy, and guided imaging will be an excellent therapeutic agent for mitochondria-enhanced tumor treatment.Experimental section
Reagents and materials
In this research work, all the chemical reagents employed were of analytical grade without further purification, such as Gd2O3, Er2O3, Nd2O3 (99.99%), Yb2O3 (99.99% purity), and concentrated HCl. NH4F, Ca(OH)2, KMnO4, and NaOH were purchased from Sino Pharm Chemical Reagent Co., Ltd; sodium trifluoroacetate (CF3COONa) and trifluoroacetic acid (CF3COOH) were purchased from Beijing Chemical Regent Co., Ltd; oleic acid (OA) and 1-octadecene (ODE) were purchased from Sigma-Aldrich. Co. LLC; N,N-dimethylformamide (DMF) was purchased from Tianjin Kermel Chemical Co., Ltd; poly(maleic anhydride-alt-1-octadecene) (C18PMH-PEG5000), propidiumiodide (PI),3-(4,5-dimethyl-2 thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), Calcein AM, and 2,7-dichlorofluorescein diacetate (DCFH-DA) were procured from Sigma-Aldrich Co. LLC.Synthesis of CaO2–MnO2
6 g of PVP was dispersed in 12 mL deionized water via ultrasonication in an ultrasonic bath until it dissolved completely. At that, 0.6 g of Ca(OH)2 was supplemented into the above solution, followed by stirring for further 20 min. With repeated stirring, 6.3 mL of H2O2 was supplemented dropwise in the above solution. Stable CaO2 nanoparticles were formed after reaction, which was continued for about 15 min. Lastly, the final product was separated through centrifugation, followed by washing with deionized water as well as ethanol absolute and followed by vacuum drying at 38 °C in a vacuum oven.The synthetic procedure for CaO2–MnO2 is as follows. Through ultrasonication, 1.44 mg of the as-prepared CaO2 was dissolved in 3 mL of phosphate buffer saline (PBS) (pH 8.5). Afterwards, 100 μL morpholinoethanesulphonic acid (MES, 100 mM) was supplied to the above solution with continuous ultrasonication. The solution was sonicated for 30 min after the addition of 100 μL KMnO4 (10 mM). Lastly, the as-obtained products were washed with PBS (pH 8.5) three times and put it in drying oven for further use.
Synthesis of OA-stabilized NaGdF4:Yb,Er core nanoparticles
For the synthesis of OA-capped NaGdF4:20%Yb,2%Er core nanoparticles, the synthetic procedure is as follows. Firstly, 1.0 mmol of RECl3 (RE = 0.78Gd + 0.2Yb + 0.02Er), 6 mL OA, and 15 mL ODE were mixed in a three-neck glass flask. Under vacuum, the solution was subjected to heating to reach 156 °C so as to obtain a homogeneous solution, which was followed by cooling the mixture down to room temperature. To this homogenous reaction mixture, 10 mL methanol solution comprising of NaOH (2.5 mmol, 0.100 g) and NH4F (4 mmol, 0.148 g) was slowly added, which was stirred for an additional 30 min. Afterwards, in order to remove methanol from the reaction mixture, it was heated gradually, degassed for 10 min at 110 °C, and heated to 300 °C for 90 minutes under N2 protection. After 90 min, the solution was cooled down slowly to room temperature, which was followed by removing the final products by centrifugation in ethanol and cyclohexane. In 5 mL cyclohexane, the obtained NaGdF4:Yb,Er nanoparticles were dispersed for further testing.Production of OA-stabilized NaGdF4:Yb,Er@NaGdF4:Nd,Yb core–shell nanoparticles (named as UCNPs)
Briefly, in a three-neck reaction vessel, the cyclohexane solution containing NaGdF4:20%Yb,2%Er core nanoparticles was supplied, followed by the addition of 0.3 mmol of Gd(CF3COO)3, 0.1 mmol of Nd(CF3COO)3, 0.1 mmol of Yb(CF3COO)3, 1 mmol of CF3COONa, 15 mL of OA, and 15 mL of ODE to the reacting solution. With strong magnetic stirring for about 60 min under a vacuum environment, the reacting solution was heated to 120 °C, which was followed by N2 flushing. Afterwards, the temperature of the reaction mixture was increased to 310 °C, which was then set aside undisturbed for 60 min with N2 protection. Lastly, for further investigation, the obtained NaGdF4:20%Yb,2%Er@NaGdF4:10%Nd,10%Yb core–shell nanoparticles were preserved in cyclohexane.Preparation of CaO2–MnO2–UCNPs and CaO2–MnO2–UCNPs–Ce6
CaO2–MnO2 was mixed with 1 mL aqueous solution of 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide (EDC) (0.03 mM) under ultra-sonication for 30 min at room temperature. After that, in a dropwise manner, 2 mL of UCNP–PEI (0.5 mg mL−1) solution was added into the above solution, followed by stirring strongly for about 24 h. The as-attained CaO2–MnO2–UCNPs nanoparticles were ultra-centrifugated, followed by rinsing with the deionized water several times.Intended for additional usage as a supply solution, Ce6 was dissolved in dimethyl sulphoxide (DMSO) at 10 mM. To obtain CaO2–MnO2–UCNPs–Ce6, 5 mL CaO2–MnO2–UCNPs (1.0 mg mL−1) was made to react with 200 μL Ce6 (10 mM) in 2 mL of PBS solvent and then magnetically stirred for about 12 h at room temperature. To remove the excess Ce6, CaO2–MnO2−UCNPs–Ce6 was left to dialyse in PBS using a 10 kDa dialysis membrane for 24 h until the filtrate left no greenish color.
Synthesis of CaO2–MnO2–UCNPs–Ce6/DOX (CaMn–NUC)
At first, DOX (2 mg) was supplemented into the CaO2–MnO2–UCNPs–Ce6 solution (10 mL, 0.3 mg mL−1), followed by stirring in the dark overnight at room temperature. Then, CaMn–NUC was obtained by ultracentrifugation, followed by washing with deionized water. The DOX loading content was calculated from the UV-vis absorption spectra of the supernatant solution. The loading rate value of DOX was calculated as per the subsequent formula: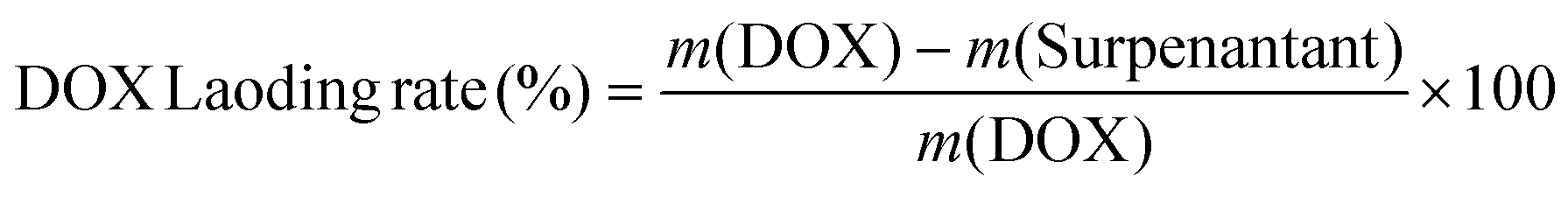 | (5) |
Characterization
The SEM images were recorded using a JSM-6700F (JEOL) instrument. The TEM and HR-TEM images were recorded on a JEM-2100F (JEOL). Digital Instruments Nanoscope IIIA was used to obtain the AFM images. A Rigaku D/max-TTR-III diffractometer was employed for carrying out the X-ray diffraction (XRD) studies of the samples. The X-ray photoelectron spectroscopy (XPS) of the samples was carried out by a Thermo ESCALAB 250 spectrometer with monochromatic Al Kα X-rays as the source (15 kV, 150 W). The UV-vis spectra of different samples were recorded on a UV-visible spectrophotometer (SHIMADZU UV-2550). An Edinburgh FLS 980 device, with an excitation wavelength irradiation source of 808 nm, was used for recording the fluorescence spectra of different samples. Leica SP8 was used for confocal laser scanning microscopy (CLSM).In vitro 1O2 generation test
By using DCFH-DA, the 1O2 generation capacity of CaMn–NUC was evaluated in vitro51–55 since the non-fluorescent DCFH would generate fluorescent DCF by the oxidation of reactive oxygen species (ROS), such as 1O2, ˙OH, and O2−. Then, for the 1O2 production experiment in vitro, we used HeLa cells as the model. At first, the HeLa cells were separated in groups of three and then cultivated in a well-plate for 24 h. Next, 3 mL of CaMn–NUC solution (500 g mL−1) was supplied into the three groups of cells on an average and the samples were respectively mixed in acidic PBS solution (pH = 5.5) for various times (0, 0.5 h, and 1 h). At this point, the cells were continually incubating for 4 h. Subsequently, 3 mL PBS solution mixed with 60 μM DCFH-DA was equally injected into the plate. After 15 min, the plate was exposed for 10 min to NIR laser (808 nm, 0.5 W cm−2) and the fluorescence images were recorded by CLSM.In this experiment, 0.5 μL of DCFH-DA dissolved in DMSO was added to 1.5 mL CaMn–NUC solution, followed by irradiation with a laser light (400–800 nm) having a power density of 0.5 mW cm−2. The absorption maxima of DCFH-DA and CaMn–NUC was adjusted to ∼0.2 D in order to eradicate the inner-filter effect. Through the comparison of the DCFH-DA-reduced absorption area (500 to 700 nm integration area), the yield of singlet oxygen of the nanocomposites was evaluated. The quantum yield of singlet oxygen sensitization through photoexcited Ce6 was calculated by using eqn (4).
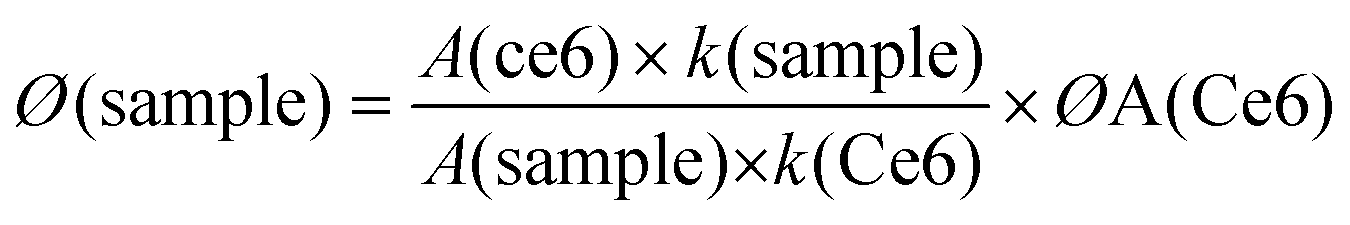 | (6) |
| Sample | A(Ce6) | ϕ(Ce6) | A(sample) | ϕ(sample) |
|---|---|---|---|---|
| Ce6 | 0.850 | 0.52 | — | — |
| CaMn–NUC at pH 5.8 | 0.850 | 0.52 | 2.153 | 0.22 |
| CaMn–NUC at pH 7.4 | 0.850 | 0.52 | 4.65 | 0.68 |
Cell viability of CaMn–NUC
The MTT technique was employed for confirming the CaMn–NUC cell viability. L929 fibroblast cells were employed as a cell type to determine the cell viability. In brief, L929 cells were incubated for 24 h at 37 °C in 5% CO2 in a 96-well plate (6000–7000 per well). At various concentrations of (15.6, 31.2, 62.5, 125, 250, and 500 μg mL−1), CaMn–NUC solutions were supplied into the cells that were continually cultivated for 24 h. Afterwards, 15 μL of MTT solution (5 mg mL−1) was supplied individually into every well and the treated cells were incubated at 37 °C for an additional 3 h. Lastly, into each well, 120 μL of DMSO was supplemented and a 490 nm microplate reader was used for measuring the cell viability data.In vitro cytotoxicity of CaMn–NUC
The in vitro cytotoxicity of the samples may well be established through MTT methods. At first, we cultivated the HeLa cells in a 96-well plate with a density of 7000 per well, which was placed into an incubator (37 °C, 5% CO2) for a period of 24 h. Next, CaO2–MnO2/DOX, CaMn–UCNPs/DOX, and CaMn–NUC with various concentrations of 0, 15.6, 31.2, 62.5, 125, 250, and 500 μg mL−1 were injected into the 96-well plate. Then, the cells of the 96-well plate were treated as follows: Control, NIR, CaO2–MnO2/DOX + NIR, CaMn–UCNPs/DOX + NIR, and CaMn–NUC + NIR (NIR laser: 808 nm, 0.5 W cm−2). Before NIR laser irradiation, each sample was already supplied and rested to incubate for about 4 h for completing the cellular uptake. Afterwards, the treatment of cells was done in the same way as in the MTT methods as per the cell viability assay on L929 cells.In vitro cytotoxicity via live/dead detection
By calcein AM/propidium iodide labelling methods, the in vitro cytotoxicity of CaMn–NUC may well be additionally discovered. Calcein AM with green color was employed to dye the living cells and PI with red color could be employed to dye the dead cells. At first, HeLa cells were incubated for 24 h in a 6-well plate to form a monolayer cell in the plate. Next, the cells of the plate were individually subjected to various treatments such as Control, NIR, CaO2–MnO2/DOX + NIR, CaMn–UCNPs/DOX + NIR, and CaMn–NUC (NIR laser: 808 nm, 0.5 W cm−2). Afterwards, AM/PI solution was supplied to the 6-well plate and the cells were further incubated at 37 °C for a period of 30 min. In the end, CLSM was used to observe the images.In vitro cellular uptake test
The CaMn–NUC cellular uptake procedure was as follows: To begin with, HeLa cells in a 6-well plate were cultivated to render a monolayer cell. Then, a (500 μg mL−1) CaMn–NUC solution was added into each plate and the cells of the plate were further incubated for various time periods (0.5, 1, and 3 h). After incubation, the cells were rinsed with PBS quite a few times, followed by staining of DAPI (25 μg mL−1) for a time period of 5 min so that the dye may stain the nuclei of the cells well. Finally, 2.5% glutaraldehyde (1 mL) was used to fix the cells for about 10 min, followed by CLSM observation.In vivo anticancer treatment performance
All the Female Kunming mice were purchased from Second Affiliated Hospital, Harbin Medical University. All the animal experiments were conducted in compliance with the specifications of The National Regulation of China for Care and Use of Laboratory Animals and were approved by Harbin Medical University (SYDW 2019-82). Initially, the mice where categorized into group A and B. We rooted the U14 cells in the left axilla in each mice; after this, mice in group A were indiscriminately separated into groups of five: control group (group I), NIR irradiated group (group II), pure CaO2–MnO2/DOX group with NIR irradiation (group III), CaMn–UCNPs/DOX with NIR irradiation group (group IV), and CaMn–NUC with NIR irradiation group (group V). 0.5 W cm−2 was the power density of the 808 nm laser that was used. Before intravenous injection of the mice, each sample was dissolved in normal saline. Through intravenous injection, these samples were inoculated in each mouse (500 μg mL−1, 100 μL) apart from that of group I and II mice, which were administered with 100 μL of regular saline as soon as the tumor location of the mice increased up to 5 mm3. After 2 h of the dose, the tumor spots of groups II, IV, and V were subjected to 808 nm laser for a period of 10 min. Subsequently, the tumor sizes of groups I–V were gauged through a caliper every 2nd day; in addition, the tumor volume of each group was calculated as per the following formula:| V = L × W2/2 | (7) |
Histology examination
Post 14 days of treatment, various vital organs such as heart, liver, spleen, lung, and kidney of all the groups were studied through histological analysis so as to ensure that the various treatments had little damage on the organs. Firstly, from each group, the organs were extracted, followed by dehydration in buffered formalin with various ethanol solution concentrations as well as xylene. Following this, the dehydrated organs were put on a 3 mm × 5 mm slice after treatment in liquid paraffin. Lastly, the slice of each organs was H&E stained and then, Leica TCS SP8 was used to obtain the images.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Notes Financial support from the National Natural Science Foundation of China (NSFC 51772059, 51972075, 51972076 and 51602072), Natural Science Foundation of Shandong Province (ZR2019ZD29), Natural Science Foundation of Heilongjiang Province (YQ2019E016), Postdoctoral Scientific Research Developmental Fund (LBH-Q18034), and the Fundamental Research funds for the Central Universities are greatly acknowledged.Notes and references
- D. M. Gilkes, G. L. Semenza and D. Wirtz, Nat. Rev. Cancer, 2014, 14, 430–439 CrossRef CAS.
- G. L. Semenza, Oxygen sensing, homeostasis, and disease, N. Engl. J. Med., 2011, 6, 537–547 CrossRef.
- O. Trédan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 19, 1441–1454 CrossRef.
- T. Kietzmann and A. Görlach, Semin. Cell Dev. Biol., 2005, 16, 474–478 CrossRef CAS.
- C. Gorrini, I. S. Harris and T. W. Mak, Nat. Rev. Drug Discovery, 2013, 12, 931–947 CrossRef CAS.
- W. Li and A. N. Kong, Mol. Carcinog., 2009, 48, 91–104 CrossRef CAS.
- P. Prasad, C. R. Gordijo, A. Z. Abbasi, A. Maeda, A. Ip, M. Rauth, R. S. Dacosta and X. Y. Wu, ACS Nano, 2013, 8, 3202–3212 CrossRef.
- L. Tong, C. C. Chuang, S. Wu and L. Zuo, Cancer Lett., 2015, 367, 18–25 CrossRef CAS.
- S. Doublier, D. C. Belisario, M. Polimeni, L. Annaratone, C. Riganti, E. Allia, D. Ghigo, A. Bosia and A. Sapino, BMC Cancer, 2012, 12, 1–15 CrossRef.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 1–8 CrossRef.
- H. E. Barker, J. T. E. Paget, A. A. Khan and K. J. Harrington, Nat. Rev. Cancer, 2015, 15, 409–425 CrossRef CAS.
- Z. Z. Yu, Q. Q. Sun, W. Pan, N. Li and B. Tang, ACS Nano, 2015, 1, 11064–11074 CrossRef.
- M. Olivo, R. Bhuvaneswari, S. S. Lucky, N. Dendukuri and P. S. Thong, Pharmaceuticals, 2010, 3, 1507–1529 CrossRef.
- C. M. Moore, D. Pendse and M. Emberton, Nat. Clin. Pract. Urol., 2009, 6, 18–30 CrossRef CAS.
- D. Zhang, M. Wu, Y. Y. Zeng, L. J. Wu, Q. T. Wang, X. Han, X. L. Liu and J. F. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 8176–8187 CrossRef CAS.
- B. Oleg, A. Sergii, V. G. Liudmyla, V. N. Viktoria, V. N. Tetiana, V. S. Olha, V. S. Dmytro, I. D. Olga, I. O. Liudmyla, L. Volodymyr, S. U. Anne and V. K. Igor, Angew. Chem., Int. Ed., 2016, 55, 5493–5496 CrossRef.
- N. Solban, I. Rizvi and T. Hasan, Lasers Surg. Med., 2006, 38, 522–523 CrossRef.
- W. Fan, W. Bu and J. Shi, Adv. Mater., 2016, 28, 3987–4011 CrossRef CAS.
- L. Xia, X. Kong, X. Liu, L. Tu, Y. Zhang, Y. Chang, K. Liu, D. Shen, H. Zhao and H. Zhang, Biomaterials, 2014, 35, 4146–4156 CrossRef CAS.
- P. Vaupel, O. Thews and M. Hoeckel, Med. Oncol., 2001, 18, 243–259 CrossRef CAS.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- J. Yuan, Y. Cen, X. J. Kong, S. Wu, C.-L. W. Liu, R. Q. Yu and X. Chu, ACS Appl. Mater. Interfaces, 2015, 7, 10548–10555 CrossRef CAS.
- X. J. Song, L. Z. Feng, C. Liang, K. Yang and Z. Liu, Nano Lett., 2016, 16, 6145–6153 CrossRef CAS.
- P. Prasad, C. R. Gordijo, A. Z. Abbasi, A. Maeda, A. Ip, A. M. Rauth, R. S. DaCosta and X. Y. Wu, ACS Nano, 2014, 8, 3202–3212 CrossRef CAS.
- R. Deng, X. Xie, M. Vendrell, Y. T. Chang and X. Liu, J. Am. Chem. Soc., 2011, 133, 20168–20171 CrossRef CAS.
- H. H. Fan, G. B. Yan, Z. L. Zhao, X. X. Hu, W. H. Zhang, H. Liu, X. Y. Fu, T. Fu, X. B. Zhang and W. H. Tan, Angew. Chem., Int. Ed., 2016, 55, 5477–5482 CrossRef CAS.
- Y. Kuang, K. Balakrishnan, V. Gandhi and X. Peng, J. Am. Chem. Soc., 2011, 133, 19278–19281 CrossRef CAS.
- S. J. Liu, B. Jiang, G. Q. Huang and X. G. Li, Water Res., 2006, 40, 3401–3408 CrossRef CAS.
- C. M. Kao, S. C. Chen, J. Y. Wang, Y. L. Chen and S. Z. Lee, Water Res., 2003, 37, 27–38 CrossRef CAS.
- Q. Jia, J. Ge, W. Liu, X. Zheng, M. Wang, H. Zhang and P. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 21124–21132 CrossRef CAS.
- Z. Dong, S. Shanmughapriya, D. Tomar, N. Siddiqu, S. Lynch, N. Nemani, S. L. Breves, X. Zhang, A. Tripathi, P. Palaniappan, M. F. Riitano, A. M. Worth, A. Seelam, E. Carvalho, R. Subbiah, F. Jaña, J. Soboloff, Y. Peng, J. Y. Cheung, S. K. Joseph, J. Caplan, S. Rajan, P. B. Stathopulos and M. Madesh, Mol. Cell, 2017, 65, 1014–1028 CrossRef CAS.
- J. M. Baughman, F. Perocchi, H. S. Girgis, M. Plovanich, C. A. Belcher-Timme, Y. Sancak, X. R. Bao, L. Strittmatter, O. Goldberger, R. L. Bogorad, V. Koteliansky and V. K. Mootha, Nature, 2011, 476, 341–350 CrossRef CAS.
- D. De Stefani, R. Rizzuto and T. Pozzan, Annu. Rev. Biochem., 2016, 85, 161–192 CrossRef CAS.
- C. V. Logan, G. Szabadkai, J. A. Sharpe, D. A. Parry, S. Torelli, A. M. Childs, M. Kriek, R. Phadke, C. A. Johnson, N. Y. Roberts, D. T. Bonthron, K. A. Pysden, T. Whyte, I. Munteanu, A. R. Foley, G. Wheway, K. Szymanska, S. Natarajan, Z. A. Abdelhamed, J. E. Morgan, H. Roper, G. W. Santen, E. H. Niks, W. L. van der. Pol, D. Lindhout, A. Raffaello, D. De Stefani, J. T. den Dunnen, Y. Sun, I. Ginjaar, C. A. Sewry, M. Hurles and R. Rizzuto, Nat. Genet., 2014, 46, 188–193 CrossRef CAS.
- F. Perocchi, V. M. Gohil, H. S. Girgis, X. R. Bao, J. E. McCombs, A. E. Palmer and V. K. Mootha, Nature, 2010, 467, 291–296 CrossRef CAS.
- N. Demaurex, D. Poburko and M. Frieden, Biochim. Biophys. Acta., 2009, 1787, 1383–1394 CrossRef CAS.
- D. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 375–380 CrossRef.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef.
- A. Gulzar, J. Xu, D. Yang, L. Xu, F. He, S. Gai and P. Yang, Dalton Trans., 2018, 47, 3931–3939 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS.
- Y. Wang, S. Song, S. Zhang and H. Zhang, Nano Today, 2019, 25, 38–67 CrossRef CAS.
- W. Fan, W. Bu and J. Shi, Adv. Mater., 2016, 28, 3987–4011 CrossRef CAS.
- Z. Yin, D. Chen, J. Zou, J. Shao, H. Tang, H. Xu, W. Si and X. Dong, ChemistrySelect, 2018, 3, 4366–4373 CrossRef CAS.
- H. Dong, S. Du, X. Zheng, G. M. Lyu, L. D. Sun, L. D. Li, P. Z. Zhang, C. Zhang and C. H. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS.
- L. Sun, R. Wei, J. Feng and H. Zhang, Coord. Chem. Rev., 2019, 346, 10–32 Search PubMed.
- A. Gulzar, J. Xu, P. Yang, F. He and L. Xu, Nanoscale, 2017, 9, 12248–12282 RSC.
- A. Gulzar, W. Zhao, F. He, D. Yang, F. Zhang, S. Gai and P. Yang., Inorg. Chem., 2020, 59, 4909–4923 CrossRef CAS.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS.
- J. Shen, G. Chen, A.-M. Vu, W. Fan, O. S. Bilsel, C.-C. Chang and G. Han, Adv. Opt. Mater., 2013, 1, 644–650 CrossRef.
- D. Zhang, M. Wu, Z. Cai, N. Liao, K. Ke, H. Liu, M. Li, G. Liu, H. Yang, X. Liu and J. Liu, Adv. Sci., 2018, 5, 1700648 CrossRef.
- W. Chen, J. Ouyang, H. Liu, M. Chen, K. Zeng, J. Sheng, Z. Liu, Y. Han, L. Wang, J. Li, L. Deng, Y. N. Liu and S. Guo, Adv. Mater., 2017, 29, 1603864 CrossRef.
- S. Lan, Z. Lin, D. Zhang, Y. Zeng and X. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 9804–9813 CrossRef CAS.
- Y. Xu, M. Zhao, L. Zou, L. Wu, M. Xie, T. Yang, S. Liu, W. Huang and Q. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 44324–44335 CrossRef CAS.
- L. Liu, J. Wang, Q. You, Q. Sun, Y. Song, Y. Wang, Y. Cheng, S. Wang, F. Tan and N. Li, J. Mater. Chem. B, 2018, 6, 4239–4250 RSC.
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. A. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS.
- H. Wang, Y. Zhao, T. Li, Y. Wang and C. Qin, Chem. Eng. J., 2016, 303, 450–457 CrossRef CAS.
- J. S. Modica-Napolitano and V. Weissig, Int. J. Mol. Sci., 2015, 16, 17394 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: AFM and XRD of CaO2–MnO2 and UCNPs. UV-vis of CaMn, FT-IR of CaMn–NUC, BET and in vitro O2 evolution of CaMn–NUC, cell viability of L929. See DOI: 10.1039/d0tb01967d |
| This journal is © The Royal Society of Chemistry 2021 |