
Lysosome-targeting pH indicator based on peri-fused naphthalene monoimide with superior stability for long term live cell imaging†
Astrid
Tannert
 ab,
Javier
Garcia Lopez
cd,
Nikolay
Petkov
e,
Anela
Ivanova
e,
Kalina
Peneva
cd and
Ute
Neugebauer
*abdef
ab,
Javier
Garcia Lopez
cd,
Nikolay
Petkov
e,
Anela
Ivanova
e,
Kalina
Peneva
cd and
Ute
Neugebauer
*abdef
aLeibniz Institute of Photonic Technology, Albert-Einstein-Str. 9, 07745 Jena, Germany. E-mail: ute.neugebauer@leibniz-ipht.de
bCenter for Sepsis Control and Care, Jena University Hospital, Am Klinikum 1, 07747 Jena, Germany
cInstitute of Organic Chemistry and Macromolecular Chemistry, Friedrich Schiller University Jena, Lessingstraße 8, 07743 Jena, Germany
dJena Center of Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
eFaculty of Chemistry and Pharmacy, Sofia University “St. Kliment Ohridski”, 1 James Bourchier Blvd., Sofia, Bulgaria
fInstitute of Physical Chemistry and Abbe School of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
First published on 13th November 2020
Abstract
Lysosomes, the acidic degradation compartments of eukaryotic cells, play an essential role in many physiological processes. Their dysfunction is associated with a number of diseases, which are often related to an altered localization or luminal pH. Thus, the in-depth characterization of lysosomes within the intact eukaryotic cell is of utmost interest. For microscopic evaluation of lysosomal distribution and acidity, a number of labels have been developed, but many showed poor organelle specificity or rapid clearing from lysosomes, rendering them unsuitable for long-term observations. Here, we describe the synthesis and spectroscopic properties of a novel small molecule marker for lysosomes based on naphthalene monoimide with reversible, pH-dependent spectral shifts in both the absorption and the emission spectrum and acidity-associated changes in fluorescence lifetime. The dye can be excited either with single- or two-photon excitation and appears to be very stably associated with lysosomes for several days. We used this chromophore to detect chemically-induced changes of lysosomal pH in HeLa cells by ratiometric and FLIM imaging.
Introduction
Lysosomes are small acidic compartments in eukaryotic cells which play a profound catabolic function being the terminal degradation compartments both for extracellular as well as for intracellular material. Nowadays, we know that they are also involved in a number of other fundamental physiological processes including signaling (especially in signaling processes mediating nutrient sensing, cell metabolism and growth), gene regulation, immunity, cell secretion, plasma membrane repair, cell adhesion and migration;1–5 highlighting the regulatory role of lysosomes.6 Consequently, the list of pathophysiological conditions, in which lysosomal dysfunction plays an important role, has increased rapidly. It is now recognized that lysosomal dysfunction is not only associated with the long-known lysosomal storage disorders (LSD),7 but also contributes in certain types of cancer, autoimmune disorders, neurodegenerative diseases and metabolic disorders, immune-deficiency diseases and pigmentation-bleeding disorders.3–5,8–11 Lysosomes are now seen as very dynamic and heterogeneous organelles, undergoing a number of fusion and fission events and having different morphology, size and luminal pH depending on their current function and position within the cell.3,4,6To understand the involvement of the endo-lysosomal system in different diseases, it is important to monitor alterations of lysosomal pH and localization during various (patho)physiological conditions. First attempts to study lysosomes relied on visualization by immunofluorescence, e.g. by detecting one of the lysosome-associated-membrane proteins (LAMP). These methods are not suited for live-cell imaging and, thus, investigation on the dynamic behaviour of lysosomes and the response to certain stimuli is often not possible. To enable in vivo monitoring of lysosomal localization and to understand possible pH changes in model systems, stable and reliable fluorescent labels are required. These labels should localize specifically to the lysosomes, exhibit no cyto- and phototoxicity and should be ideally stable for several hours to days within the cell for long-term observations. One possibility to achieve lysosomal labelling in living cells is the use of genetically encoded fluorescent marker proteins like variants of the green fluorescent protein (GFP) coupled to LAMP1.12 Variants of GFP may also be used for pH sensing.13 This is an elegant method; however, it requires the transfection of the investigated cellular system with the appropriate plasmids, which is not easy to achieve in certain cell types like neurons or in primary cells. Therefore, small fluorescent molecules which after uptake specifically accumulate in lysosomes are a powerful alternative to fluorescent proteins. In addition to available commercial dyes (like the popular LysoTracker™ series14 for localization and LysoSensor™ series for monitoring changes in lysosomal pH), a number of chromophores for monitoring lysosomal localization and/or pH values have been developed in the last years.15 Pure localization markers often show good lysosomal specificity but lack long term stability in cells. Among the available probes for stable long-term lysosome labelling, there are a number of dextran conjugates, including also commercially available labels which are conjugated to pH-sensitive fluorophores.16–18 Dextrans are internalized by endocytosis and – because of their size – retained in lysosomes.16 Due to this mechanism, however, labelling normally requires incubation for several hours up to days. Among the dyes designed for pH monitoring, there is a huge number of small molecules reported, with only a few of them showing good lysosomal specificity. While the physiological pH of lysosomes is generally between 4.5 and 5,5 pKa values of many reported dyes are well outside that physiological range questioning their suitability for most physiological applications. pH-Sensitive dyes can be divided roughly into sensors with pH-dependent emission intensity at a single wavelength19–32 and ratiometric dyes.33–46 The advantage of the latter is their independence of uneven concentration, excitation power fluctuations or cellular autofluorescence. A literature overview on available lysosomal-targeted ratiometric pH probes and their properties is given in Table 1 and more extensively, including also intensity-based probes, in the ESI,† Table S1. Like for the pure localization marker probes, long term stability in cells for several days has been demonstrated only in few of the published pH-sensitive probes. The available ratiometric pH probes with proofed cellular long term stability generally require labelling times of at least 16 h. Thus, there is a lack of photostable, selective and sensitive pH reporter dyes.
| Type of probe | Spectral range | Readout | Range of pH sensitivity | Labelling time (concentration) | Long term stability in cells | Ref. |
|---|---|---|---|---|---|---|
| NMI-LS | UV-VIS (2P-ex. possible) | Excitation ratio 458 nm/405 nm or emission ratio 467 nm/449 nm or average fluorescence lifetime | 2–6 | 30 min (0.5 μM) | 48 h | This publ. |
| FITC-labeled dextran | UV-VIS | Excitation ratio 495 nm/450 nm or emission ratio 515 nm/610 nm | 4–7 | 16 h–3 d (1 mg ml−1) | Several days | 16–18 |
| Glyco-fluorescein-rhodamine-X-lactam probes | UV-VIS | Emission ratio (with changing excitation) 515 nm/610 nm (pH range 4–5) or 610 nm/515 nm (pH range 5–7.5) | 4–7.5 | 24 h (20 μM) | After 24 h superior retention compared to LysoTracker™ Red, also retention when acidity is lost | 44 |
| Benzimidazole-based probe | UV-VIS | Emission ratio appr. 525 nm/474 nm | 5–7 | 30 min (3 μM) | Not reported | 34 |
| Carbazole-based probes | UV-VIS (2P-ex. possible) | Emission ratio Ich2/Ich1 | 4–5 (probe A) or 4.5–5.5 (probe B) | 30 min (30 μM) | Not reported | 43 |
| Naphthalene-naphthalimide-based probe | UV-VIS (2P-ex. possible) | Emission ratio Ich2/Ich1 | 3–6.5 | 60 min (1 μM) | Not reported | 45 |
| Naphthalimide-rhodamine-based probe | UV-VIS (2P-ex. possible) | Emission ratio Ich2/Ich1 or Ich1/Ich2 | 4–6 | 30 min–6 h (1–10 μM) | Not reported | 41 and 42 |
| Quinolone-based probe | UV-VIS | Emission ratio Ich1/Ich2 (appr. 494 nm/570 nm) | 4–6 | 30 min (2 μM) | Not reported | 35 |
| Naphthalimide-coumarin-based probe | UV-VIS | Emission ratio 530 nm/454 nm (pH range 4–6) or 454 nm/530 nm (pH range 6–8) | 4.5–8 | 10 min (5 μM) | Not reported | 33 |
| Chromenoquinoline-based probe | UV-VIS | Emission ratio Ired/Igreen (appr. 613 nm/560 nm) | 4–6.5 | 15 min (5 μM) | Not reported | 36 |
| Porphyrin-pyranine-based probe | UV-VIS | Emission ratio Ired/Iblue (appr. 660 nm/435 nm) | 2.5–8.5 | 30 min (7.5 μM) | Not reported | 47 |
| Coumarin-rhodamine-based probe | UV-VIS | Emission ratio Ired/Igreen (appr. 605 nm/475 nm) | 4.5–7 | 30 min (3 μM) | Not reported | 40 |
| BODIPY-based probe | UV-VIS | Emission ratio Ich1/Ich2 | 1–4 | 15 min (5 μM) | Not reported | 46 |
| BODIPY-rhodamine-based probes | NIR | Emission ratio Ich2/Ich1 (lower pH range) or Ich1/Ich2 (higher pH range) | 3–6.5 (probe A) or 2.5–7 (probe B) | 30 min (5–15 μM) | Not reported | 39 |
| Hemicyanine-based probe | NIR | Emission ratio Ich1/Ich2 (appr. 670 nm/708 nm) | 4–6 | 5 min (50 nM) | Not reported | 38 |
Organic dyes based on peri-guanidine-fused naphthalene monoimide and perylene monoimide chromophores are biocompatible chromophores with very high photostability. They can be functionalized to selectively target different cellular organelles.48 The preparation of naphthalene and perylene monoimide chromophores functionalized in the peri-positions with two electron donor groups was reported.48 The resulting chromophores possess a strong bathochromic shift of their absorbance and emission maxima in the visible range (>450 nm), excellent chemical and photostability as well as high quantum yield in water.48
Here, we describe a novel peri-fused naphthalene monoimide dye which is a specific lysosomal localization marker that possesses high long-term stability for imaging lysosomal distribution over several days. This dye functions as a sensitive ratiometric pH indicator for monitoring changes in lysosomal pH in the physiologically relevant range of pH 3.5–6. The probe is readily internalized by cells within 20–30 min and can be excited in the blue visible spectrum or using two-photon excitation at 880–900 nm.
Experimental section
Synthesis
Detailed procedures for the synthesis and characterization of NMI-LS can be found in the ESI.†TD-DFT modeling of the optical properties of NMI-LS
One molecule of NMI-LS (Fig. 1) either in its deprotonated (NMI-LS(+), Fig. S1A, ESI†) or in its protonated (NMI-LS(2+), Fig. S1B, ESI†) form was subjected to 3000 steps of conformational analysis in vacuo in order to determine the most probable conformation of the charged aminoaliphatic tail. Parameters from the force field AMBER9949 were used and the six torsion angles denoted in Fig. S1A (ESI†) were varied. The search was done in the program package Hyperchem 7 [HyperChem 7, Hypercube; Gainesville, FL, USA, 2001]. The geometries of the found stable conformers of the two forms were optimized with ωPBEhPBE/6-31G(d)/PCM(DMSO)50,51 using the Gaussian 16 software.52 The energy minima were verified by frequency analyses. Then, the longest-wavelength electronic absorption transition (LW-AT) of all conformers was calculated with TD-ωPBEhPBE/6-31(d)/PCM(DMSO). Expectedly, the absorption wavelength did not depend markedly on the molecular conformation (Table S2, ESI†) because the transition took place within the naphthalene monoimide fragment (Fig. 1D). Therefore, all remaining computations were performed for the most stable conformers of the two forms only. Since an amino–imino tautomerism is possible in the protonation-prone part of NMI-LS (Fig. S1, ESI†), the geometries of the two tautomers of each form were optimized with ωPBEhPBE/6-31G(d)/PCM(DMSO) and their energies were compared. These two optimized structures (Fig. S2A, ESI†) were used further on for in silico spectral characterization. Their electronic absorption transitions were calculated with TD-ωPBEhPBE/6-31++G(2d,2p)/PCM(DMSO). Then, the geometries of the three lowest-energy singlet excited states (S1 to S3) were optimized with TD-B3LYP-D3/6-31G(d)/PCM(DMSO)53 and the emission transitions thereof were estimated with TD-ωPBEhPBE/6-31++G(2d,2p)/PCM(DMSO). It should be noted that most of the calculations were performed also in implicit water (because a mixture of the two solvents was used in the experimental measurements). There was no qualitative difference in the results and the quantitative deviations were very small. Therefore, only the results in DMSO are shown.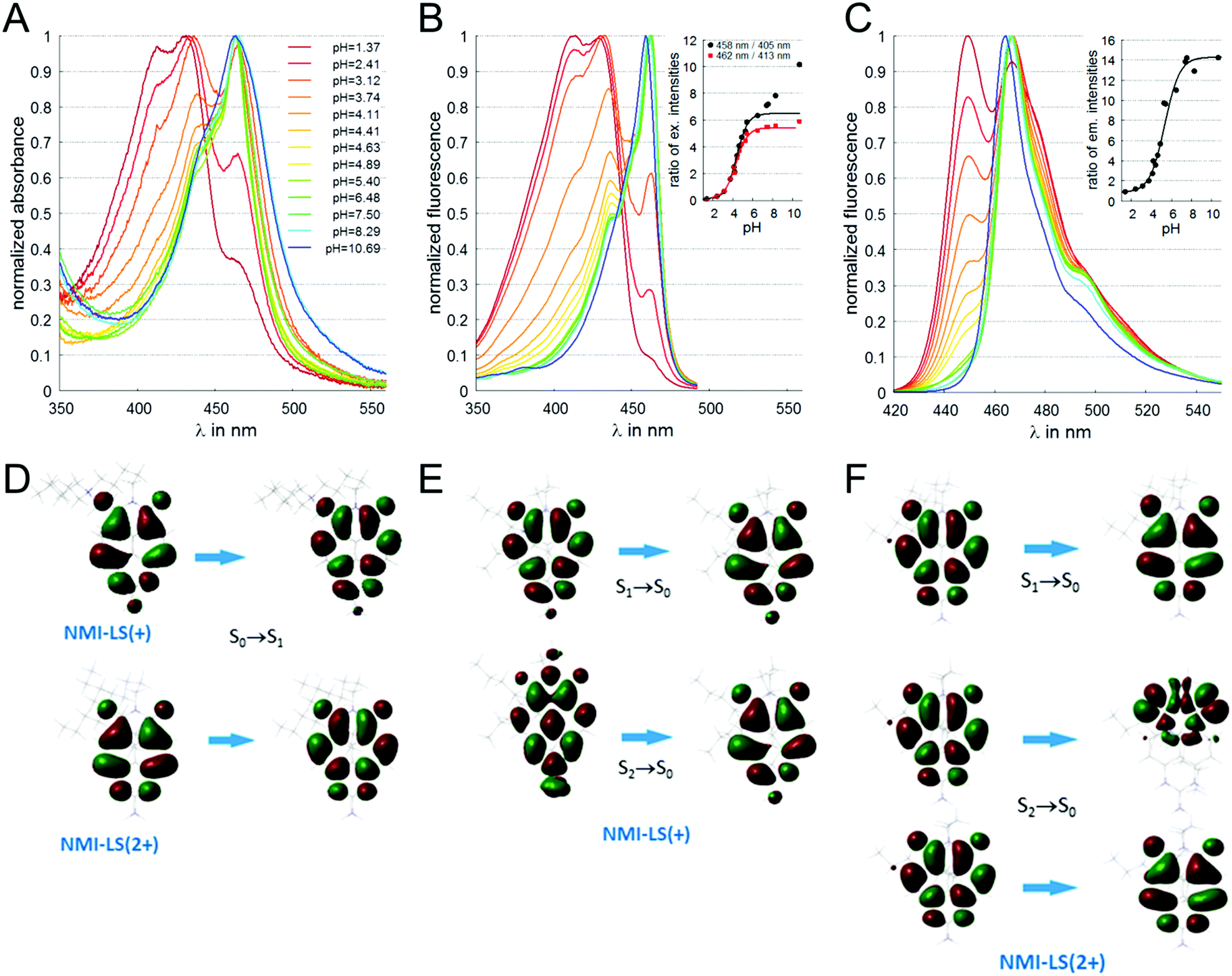 | ||
| Fig. 1 Spectral characterization of NMI-LS (3 μM) in buffers of different pH values. (A) Absorbance spectra of NMI-LS in buffers with varying pH. (B) Fluorescence excitation spectra monitored at λem = 500 nm, inset shows ratios of fluorescence intensities of excitation peaks (red squares 462 nm/413 nm, black circles 458 nm/405 nm, lines indicate fit according to eqn (3)). (C) Fluorescence emission spectra excited at λex = 410 nm, inset depicts ratios of fluorescence intensities of emission peaks 467 nm/449 nm (line indicates fit according to eqn (3)). (D) Molecular orbitals involved in the longest-wavelength absorption transitions of the two forms calculated with TD-DFT. (E) Molecular orbitals involved in the emission transitions of the deprotonated form calculated with TD-DFT. (F) Molecular orbitals involved in the emission transitions of the protonated form calculated with TD-DFT. | ||
Spectral characterization
NMI-LS was dissolved from a 10 mM stock solution in DMSO (Merck, Darmstadt, Germany) to a final concentration of 3 μM in phosphate buffer (PB, 100 mM NaH2PO4/Na2HPO4 (Carl Roth GmbH, Karlsruhe, Germany), 50 mM NaCl (Carl Roth GmbH, Karlsruhe, Germany), pH 6.5–10.7) or McIlvaine buffer, containing NaH2PO4 and citric acid (Carl Roth GmbH, Karlsruhe, Germany)54 supplemented with 50 mM NaCl (MIB, pH 1.4–7.5) of different pH values in the range from pH 1.4–10.7. Absorbance spectra were recorded using a V-760 UV-Visible spectrophotometer (Jasco, Pfungstadt, Germany) in the range from 350–700 nm at 37 °C with 0.2 nm resolution. Fluorescence emission spectra (λex = 410 nm) and fluorescence excitation spectra (λem = 500 nm) were recorded using a FP-8500 fluorescence spectrometer (Jasco, Pfungstadt, Germany) at 37 °C with 1 nm resolution. To record the reversibility of spectral shifts, NMI-LS was dissolved from a 10 mM stock solution in DMSO to a final concentration of 3 μM in deionized H2O and the pH was changed for several times between 7.2 and 4.0 by adding HCl or NaOH. For each cycle, fluorescence excitation and emission spectra were recorded as described above. Time-resolved emission spectra (TRES) were recorded using a FluoTime250 fluorescence lifetime spectrometer (Picoquant, Berlin, Germany) upon excitation at 440 nm in the range from 450–500 nm with a resolution of 5 nm at 37 °C. The instrument response function (IRF) was recorded using Ludox® (Picoquant, Berlin, Germany) after every measurement. Data were fitted to bi- or triexponential model functions using EasyTau 2 software (Picoquant).Calculation of pKa
The pKa value of NMI-LS was obtained from the ratio of excitation (458 nm to 405 nm or 462 nm to 413 nm) or emission (467 nm to 449 nm) at different pH values according to eqn (1):55,56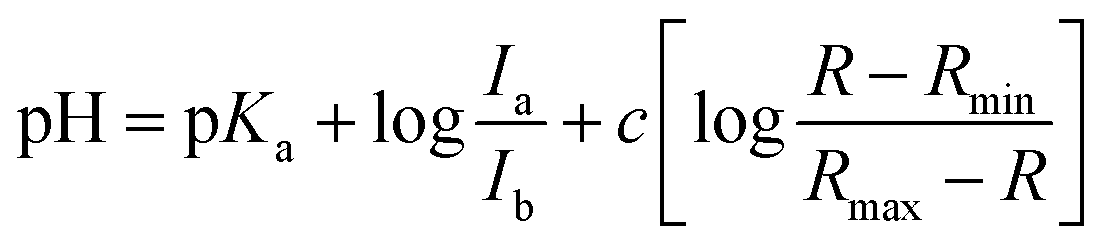 | (1) |
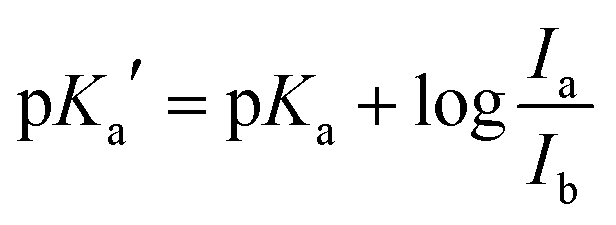 | (2) |
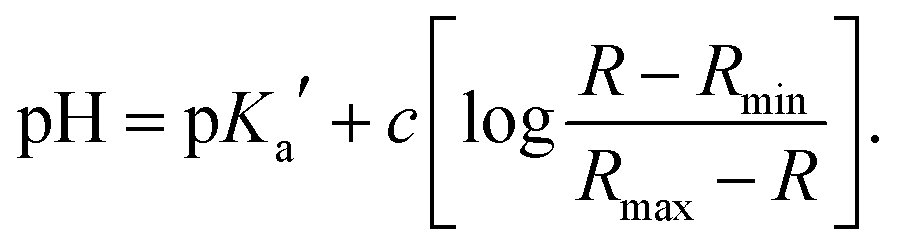 | (3) |
Cultivation and labelling of HeLa cells
HeLa (ATCC CCL-2™, ATCC®, Manassas, Virginia) cells were cultured in very low Endotoxin-DMEM (Merck, Berlin, Germany) supplemented with 10% fetal calf serum (Merck) at 37 °C and 5% CO2. For microscopic observation, cells were trypsinated and seeded on glass bottom Petri dishes or on glass cover slips coated with poly-L-lysin (Merck, Darmstadt, Germany). Cells were labelled by incubation with 100 nM–5 μM NMI-LS and/or 100 nM–5 μM LysoTracker™ Red DND-99 (LTR, ThermoFisher Scientific, Waltham, MA) for 30 min at 37 °C, 5% CO2 in FluoroBrite™ DMEM (ThermoFisher Scientific, Waltham, MA) supplemented with 2 mM L-glutamine (Lonza, Basel, Switzerland) and 10% FCS. For comparison, we also labelled HeLa cells with 500 μg ml−1 of the commercial LysoSensor™ Yellow/Blue dextran (LSYB, ThermoFisher) for 18–24 h at 37 °C, 5% CO2 in complete FluoroBrite™ DMEM (see above). Before imaging, cells were washed twice in phosphate buffered saline (PBS, ThermoFisher Scientific, Waltham, MA) and further incubated in complete FluoroBrite™ DMEM at 37 °C, 5% CO2.Metabolic activity
HeLa cells were seeded into a 96 well plate (μCLEAR®, Greiner Bio-One GmbH, Frickenhausen, Germany) at a density of 4 × 103 cells per well and incubated without (control) or with 500 nM or 5 μM of NMI-LS in culture medium for 48 h at 37 °C, 5% CO2. To assess metabolic activity, 10 μl alamarBlue™ (ThermoFisher Scientific, Waltham, MA) were added per well and further incubated for 2 h at 37 °C, 5% CO2 followed by reading resazurin fluorescence (λex = 550 nm, λem = 600 nm) with a microplate reader Spark® 20M (Tecan, Männedorf, Switzerland).Confocal laser scanning microscopy and ratiometric excitation and emission imaging
Confocal laser scanning microscopy was performed using a CLSM 780 Meta (Zeiss, Jena, Germany). NMI-LS was excited at 405 nm, 458 nm or 488 nm (single-photon excitation) or at 880–900 nm (two-photon excitation), LTR was excited at 561 nm. All images were acquired using a 63× oil immersion objective (NA 1.4) at 37 °C, 5% CO2. Photostability of NMI-LS was tested by repeated illumination every 30 s over a time period of 60 min at 458 nm and 488 nm for single-photon excitation and at 900 nm for two-photon excitation. The laser intensity was adjusted for each excitation wavelength to give high quality fluorescence images. The power of the laser was measured before it entered the objective yielding appr. 0.13 μW, 10 μW, and 17.8 mW for excitation at 458 nm, 488 nm, and 900 nm, respectively. To record changes in the excitation ratio of NMI-LS, the excitation wavelength was changed line-wise during scan between 405 nm and 458 nm, fluorescence emission was detected in the range of 464–544 nm. Images were acquired every 10–20 s and after the acquisition of the first 20–30 images, 10 mM NH4Cl (Carl Roth GmbH, Karlsruhe, Germany) was added to increase lysosomal pH. To record changes in the emission ratio, NMI-LS-labelled HeLa cells were excited using two-photon excitation (880 nm) and emission was recorded in two channels (410–455 nm and 466–518 nm). Images were acquired every 30 s and after the acquisition of the first 10 images, 10 mM NH4Cl was added.Fluorescence lifetime imaging
Fluorescence lifetime imaging was performed either with two-photon excitation at 900 nm or with single-photon excitation at 440 nm. In both cases a 63× oil immersion objective (NA 1.4) was used and the sample was constantly held at 37 °C, 5% CO2. In the first approach, a CLSM 780 Meta (Zeiss, Jena, Germany) and an attached FLIM module (Becker & Hickl, Berlin, Germany) was used. Two-photon excitation was achieved at 900 nm, 80 MHz, data were collected using a 470/60 BP filter and fitted using SPCImage software (Becker & Hickl) to an incomplete biexponential decay model. In the second approach we used a CLSM 980 (Zeiss, Jena, Germany) with an attached rapidFLIM™ module (Picoquant, Berlin, Germany). Pulsed excitation was achieved by a 440 nm diode laser (Picoquant) operated at 20 MHz. Fluorescence was filtered using a 482/35 BP filter. Data were collected with a PMA Hybrid and the MultiHarp15. To record changes in lysosomal pH, FLIM images were recorded every 2 min and after 10 min 10 mM ammonium chloride was added to increase lysosomal pH. Images were binned 2 × 2 before fitting to a global triexponential model function including reconvolution with a calculated instrument response function using the SymphoTime software (Picoquant).Image analysis
Confocal images were analyzed using Fiji ImageJ 1.52p.57 Colocalization analysis was performed using the coloc2 plugin of Fiji. Images were background-corrected, and regions were defined in which cells were located. To analyze long term stability of fluorescence in labelled cells, areas covered with cells were defined and the mean fluorescence intensity in these areas (corrected by the mean background fluorescence intensity) was calculated and related to the mean fluorescence intensity of the first image acquired (approximately 1.5 h after labelling). The ratiometric analysis for both excitation and emission ratiometric imaging was performed using the ratioplus plugin. To avoid background noise during ratio calculation, the threshold for ratio calculation was set well above the background so that only particles with detectable fluorescence emission in both channels were included into ratio calculation. The images were pseudo-colored according to the calculated ratio of ch2/ch1. Please note that this ratio is dependent on the image acquisition parameters set for each channel (i.e. laser power, detector gain). Therefore, only changes in lysosomal pH or differences between individual lysosomes and no absolute pH values have been detected.Results and discussion
Design and synthesis
NMI-LS was designed with a large alkyl/hydrophobic system to facilitate passing through the cell and organelle membranes. The naphthalene dye was functionalized with a guanidine group (at the peri position) to improve cell penetration and water solubility. It further introduces the pH-responsive moiety into the molecule. At low pH (e.g. in the lysosomes) this guanidine group gets protonated and this second positive charge at the guanidinium form of the dye prevents it from going out through the lysosomal membrane. The mechanism of lysosomal specificity is similar to a previously reported Zn ion probe.66The preparation of the positively charged naphthalene monoimide dye NMI-LS was achieved in one step from N-(6-bromohexyl)-guanidine derivative of naphthalene monoimide (Scheme 1). Briefly, the synthesis of the naphthalene dye starts from 4,5-dibromo-1,8-naphthalic anhydride which is reacted with 6-amino-1-hexanol, where subsequent reaction with guanidine leads to N-(6-hydroxyhexyl)-guanidine derivative (Scheme 1).
 | ||
| Scheme 1 Synthesis of naphthalene monoimide dye – NMI-LS. | ||
In the following step, the N-(6-hydroxyhexyl)-guanidine derivative is reacted with phosphorous tribromide to introduce a bromine functionality, which is subsequently reacted with tributylamine in 1,2-dichlorobenzene. The facile preparation of the naphthalene chromophore enables the attachment of functional groups for targeting subcellular structures, such as in this case, the lysosomes. We thus obtained the final chromophore in a few simple steps in high yield starting from cheap and easily available chemicals. NMI-LS possesses excellent solubility in water, high molar absorptivity and a large bathochromic shift of both the absorbance and emission properties compared to naphthalene monoimide dyes like 2.
Spectral characterization of pH-dependent properties
Absorbance spectra of NMI-LS dissolved in buffer solutions in the pH range of pH 1.4 and 10.7 are depicted in Fig. 1A. A clear hypsochromic shift was observed upon acidification. A similar shift was visible in the fluorescence excitation spectra (Fig. 1B). In both spectral series, the main peak at neutral pH is found at 462 nm. A second peak at 433 nm becomes more pronounced upon acidification, and a third peak appeared at very low pH (pH < 4) at about 413 nm. The position of the different maxima in absorbance and the calculated molar extinction coefficients at the different pH values are given in ESI,† Table S3. The fluorescence emission was also hypsochromically shifted upon acidification of the solvent (Fig. 1C) with two emission peaks at 449 nm (more pronounced in the acidic form) and 467 nm (main band at neutral pH). To interpret the spectral shifts of the absorbance and emission maxima, we performed DFT calculations of the two possible tautomers of the dye (see ESI,† Fig. S1). It turned out that the amino-tautomer of the deprotonated form was more stable by 5.5 kcal mol−1, while the imino-one was preferred by 21.2 kcal mol−1 for the protonated molecule. The DFT results are visualized in Fig. 1D–F, ESI,† Fig. S2 and summarized in Table 2. The calculations assign the experimentally observed hypsochromic shift of ca. 30 nm upon protonation to a π → π* transition which is expectedly delocalized over the naphthalene scaffold of the molecule (Fig. 1D). The origin of the hypsochromic shift in the protonated form is traced back to local electron density redistribution upon protonation (illustrated in Fig. S2B, ESI†). This results in a weaker π-conjugation of the protonated part to the polycyclic system as compared to the deprotonated form, yielding a blue shift in the spectra. The computed emission transitions also provide an explanation of the experimentally observed features. In the deprotonated form, there is only one dominant transition at 469 nm, which has the same nature (Fig. 1E) as the absorption one described above. The emission from S2 is less intense because of symmetry restrictions (perpendicular nodal planes in the two orbitals, Fig. 1E), while that from S3 is in the UV range and is forbidden. The picture of the excited states changes upon protonation: In NMI-LS (2+), S1 and S2 (Fig. 1F) become practically degenerate at 450 nm and the fluorescence from both of them is allowed, resulting in the more intense transition observed at this wavelength at lower pH. There is also another transition in the visible range but its much smaller oscillator strength explains why it could be recorded only at the most acidic pH values (where the protonated form is highly predominant). Overall, the measured absorption and emission maxima are in good agreement with the calculated transitions.| NMI-LS(+) | NMI-LS(2+) | |||||
|---|---|---|---|---|---|---|
| Absorption | S0 → S1 | S0 → S2 | S0 → S1 | S0 → S2 | ||
| λ | 449 | 394 | 420 | 418 | ||
| f | 0.323 | 0.019 | 0.264 | 0.004 | ||
| Emission | S1 → S0 | S2 → S0 | S3 → S0 | S1 → S0 | S2 → S0 | S3 → S0 |
| λ | 469 | 444 | 388 | 450 | 449 | 406 |
| f | 0.254 | 0.054 | 0.000 | 0.139 | 0.205 | 0.017 |
To further characterize the pH-dependent changes, we calculated the ratio between the experimental excitation intensities at 462 nm and 413 nm, or (in accordance to the available laser lines for confocal imaging) 458 nm and 405 nm (Fig. 1B inset) as well as the ratio of the emission intensities at 467 and 449 nm (Fig. 1C inset). These ratios show an almost linear dependency on the pH value in the range of 4–5, which is the physiologically relevant pH range of lysosomes.5,58 The apparent pKa′ values were determined by linear regression of the intensity ratio in the pH range of 1.5–7.5 using eqn (3). We did not include higher pH values to avoid the influence of a second deprotonation step which is characterized by a slight bathochromic shift of the 467 nm excitation maximum in the basic pH region (pH > 8). The apparent pKa′ values for the excitation ratios 462 nm/413 nm and 458 nm/405 nm were 4.33 and 4.39, respectively, whereas for the emission ratio 467 nm/449 nm we calculated a pKa′ of 5.33. From the intensities measured at maximal protonation and deprotonation (Ia/Ib) the true pKa of NMI-LS value was estimated to be about 3.2 (see ESI,† Table S4). This value is below the physiological value of most lysosomal associated processes, however when using the ratios suggested above, pH values around the apparent pKa′ rather than the true pKa values will be linearly dependent on these ratios. Please note that much lower pH values will give linear dependency on the reciprocal ratios, e.g. 413 nm/462 nm for excitation ratiometric imaging or 449 nm/467 nm.
Spectral properties of the fluorophore remained constant after repeatedly changing the pH value between 4.0 and 7.2 as indicated in ESI,† Fig. S3 for the excitation ratios 462 nm/413 nm and the emission ratios of 467 nm/449 nm. These spectral changes indicate that NMI-LS is very well suited to be used as a pH indicator.
Time-resolved fluorescence of NMI-LS at different pH values
Time-resolved emission spectra (TRES) of NMI-LS were recorded in buffers of different pH values upon excitation at 440 nm. Decay curves were fitted to biexponential model functions. As indicated in ESI,† Fig. S4, the average fluorescence lifetime increases at all monitored wavelengths with increasing pH. At lower emission wavelength, this effect is more pronounced and is better observable in the physiologically relevant pH range. This finding is not unexpected since the shorter fluorescence lifetime of NMI-LS seems to originate from the protonated form which has also the major contribution to the shorter wavelength emission peak. We suggest that for FLIM of NMI-LS in live cells the emission wavelengths should cover the range of 450–470 nm for most relevant physiological conclusions.NMI-LS is stable at longer incubation times
To investigate whether NMI-LS might be degraded during longer incubation periods at 37 °C, we measured fluorescence excitation and emission spectra before and 24 h or 48 h after incubation at 37 °C in buffers of pH 4.4 or 7.5. As shown in ESI,† Fig. S5, major features of the spectra remain unchanged over time. We conclude that NMI-LS is stable for long term observation in living cells for at least up to 2 days. This stability is in agreement with expectations from the chemical structure: NMI-LS has an imide group fused to an aromatic system generating a six-member flat ring where all carbons are sp2 hybridized which makes them exceptionally chemically stable. NMI-LS has both carbonyls facing at each other, preventing the ring-opening hydrolysis of the imide. Naphthalimides are not sensitive to acidic conditions and were reported to be present in aggressive basic/acidic reaction mixtures without compromising the reaction yield.59 Additionally, the NMI-LS has a push–pull system with a strong electron-donating group – the guanidine – and a strong electron-withdrawing group, the imide. This system stabilizes the imide considerably by reducing the electron deficiency of the carbonyl groups of the imide.NMI-LS accumulates in lysosomes
To analyze the subcellular distribution of NMI-LS, HeLa cells were incubated with the dye and additionally stained with commercial LysoTracker™ Red DND-99 (LTR), which accumulates in lysosomes. Dyes of the LysoTracker™ family are among the most frequently used chromophores to stain lysosomes and they show good lysosomal staining after short incubation times (usually <30 min). Using confocal laser scanning microscopy (CLSM), NMI-LS was excited with either 458 nm (Fig. 2A) or 488 nm (Fig. 2B), and detected emission is shown in green pseudocolor. Simultaneously, LTR was excited at 561 nm, where the detected emission is shown in red pseudocolor. As indicated by the yellow pseudocolor, NMI-LS largely colocalizes with LTR proving its lysosomal accumulation. Colocalization was further visualized by scatter plots of the pixel intensities in both channels (Fig. 2) and calculation of the Pearson coefficient which was for all analyzed fields of view in the range of 0.75–0.9 indicating good correlation. To investigate whether NMI-LS can also be excited by two-photon excitation, we excited labelled HeLa cells with 880 or 900 nm. Both excitation wavelengths permitted imaging of the punctuate lysosomal pattern with high emission rate (see ESI,† Fig. S6 for an example imaged using 900 nm excitation). Therefore, the probe might as well be used for deep tissue imaging of lysosomes by two-photon excitation. | ||
| Fig. 2 Colocalization of NMI-LS (green) and LTR (red) in HeLa cells. Cells were labelled with NMI-LS (500 nM) and LTR (200 nM). (A) Dual excitation scan with 458/561 nm, emission was detected from 460–540 nm (green pseudo-color channel) and from 570–700 nm (red pseudo-color channel). (B) Dual excitation scan with 488/561 nm, emission was detected from 491–540 nm (green pseudo-color channel) and from 570–700 nm (red pseudo-color channel). The third panel in each row shows the overlay of the green and red channel with yellow color indicating colocalization, the fourth panel in each row is an enlarged visualization of the area indicated by a square in the third panel. A scatter plot of pixel intensities is shown in the fifth panel of each row indicating colocalization. | ||
NMI-LS is stable in living cells for several days
To evaluate the potential of NMI-LS for long-term observation of lysosomal processes, we labeled HeLa cells with 500 nM of NMI-LS and for comparison with 500 nM of the commercial LTR. After labeling, the cells were incubated in the incubation chamber of a CLSM at 37 °C, 5% CO2 and images were acquired at different defined positions every few hours over a time period of 48 h (Fig. 3A). NMI-LS was either excited at 458 nm or at 488 nm. The mean fluorescence intensity of the areas covered by cells was then quantified for each time point and position and the fluorescence intensity compared to the initially acquired intensity value (Fig. 3B). The observed decrease in fluorescence intensity over time corresponds to the clearing or degradation of the dye from the cells. After 48 h, fluorescence from the LTR was hardly detectable, while NMI-LS was still showing bright fluorescence (Fig. 3A). After 48 h, the percentage of remaining fluorescence intensity was significantly higher in NMI-LS labeled cells – both excited at 458 nm or 488 nm – compared to LTR. Therefore, the stability of NMI-LS in living cells and its suitability for long-term tracking is superior compared to the commercially available dyes. To assess if NMI-LS has any cytotoxic effects on living cells, HeLa cells were incubated for 48 h with 500 nM or 5 μM NMI-LS. Metabolic activity was determined afterwards using the AlamarBlue™ assay and as shown in Fig. 3C, no reduction in metabolic activity, and thus, no cytotoxicity was detectable. Photostability was assessed by repeated illumination over a time period of 60 min at different single- or two-photon excitation wavelengths (see Fig. 4). Total fluorescence intensity of the background-corrected images was quantified showing after 1 h of time lapse measurement still 80–93% of the initial fluorescence intensity. Bleaching was lowest using the two-photon excitation at 900 nm. When using single photon excitation, excitation at 488 nm, which is well outside the excitation optimum, resulted in less bleaching than excitation at 458 nm, even though the laser power required for optimal excitation at 488 nm was two orders of magnitude higher than at 458 nm. We conclude that NMI-LS is quite photostable, especially when excited at 488 nm or using two-photon excitation.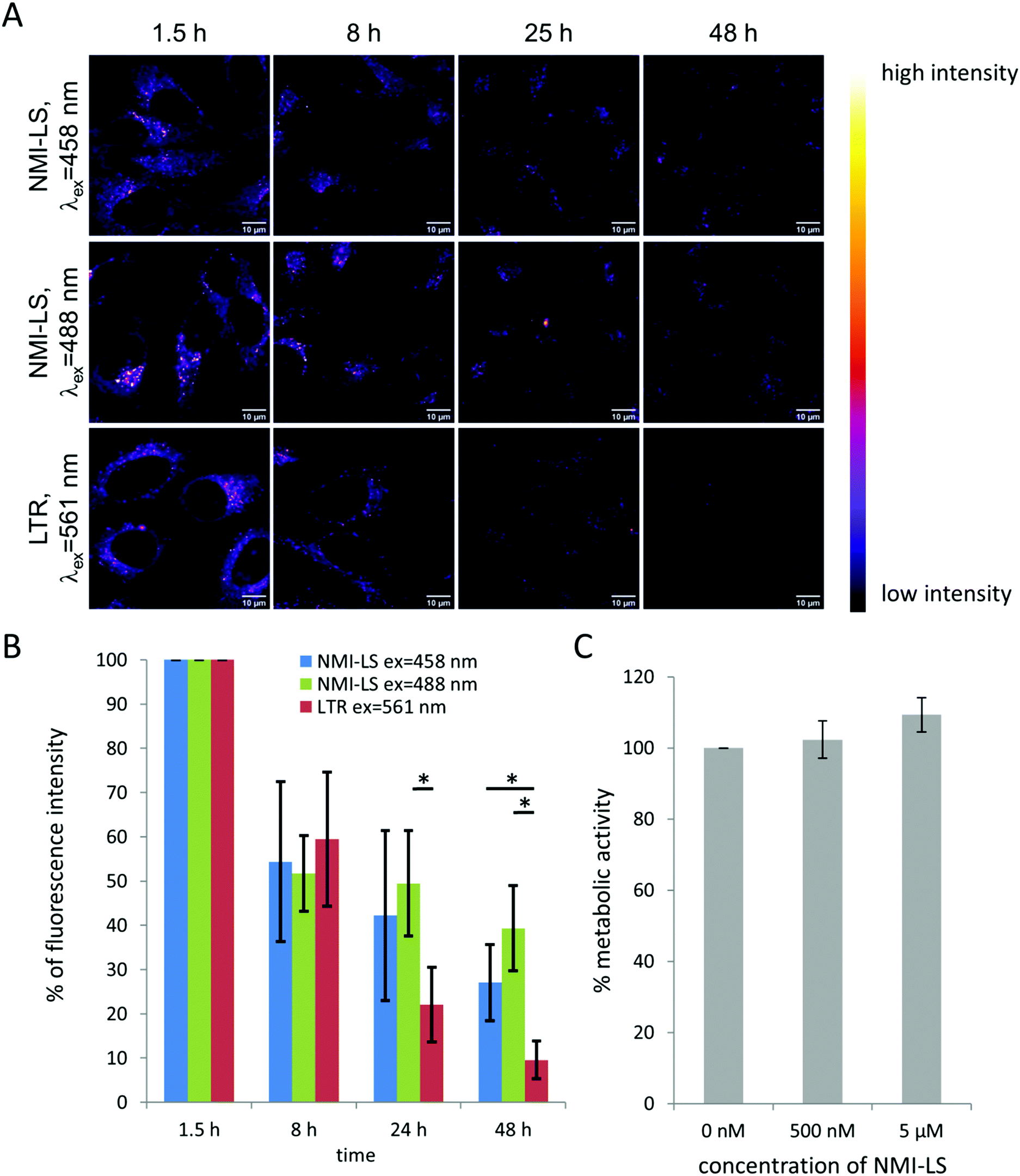 | ||
| Fig. 3 Long-term fluorescence stability and cytotoxicity assay of NMI-LS compared to LTR in HeLa cells. (A) Fluorescence intensity after different time points of incubation depicted in pseudo-color. (B) Quantification of the mean fluorescence intensity in background-corrected images after different time points of incubation with NMI LS or LTR; *p ≤ 0.01; n = 9 fields of view (FOV) measured on 3 biological replicates; data represent mean ± standard deviation related to the value of the first acquired image which was taken approx. 1.5 h after the start of labelling. (C) Metabolic activity assessed by Alamar Blue™ assay after incubating HeLa cells for 2 days with NMI-LS. Error bars indicate mean ± standard deviation of 3 biological replicates, each containing at least 14 technical replicates. | ||
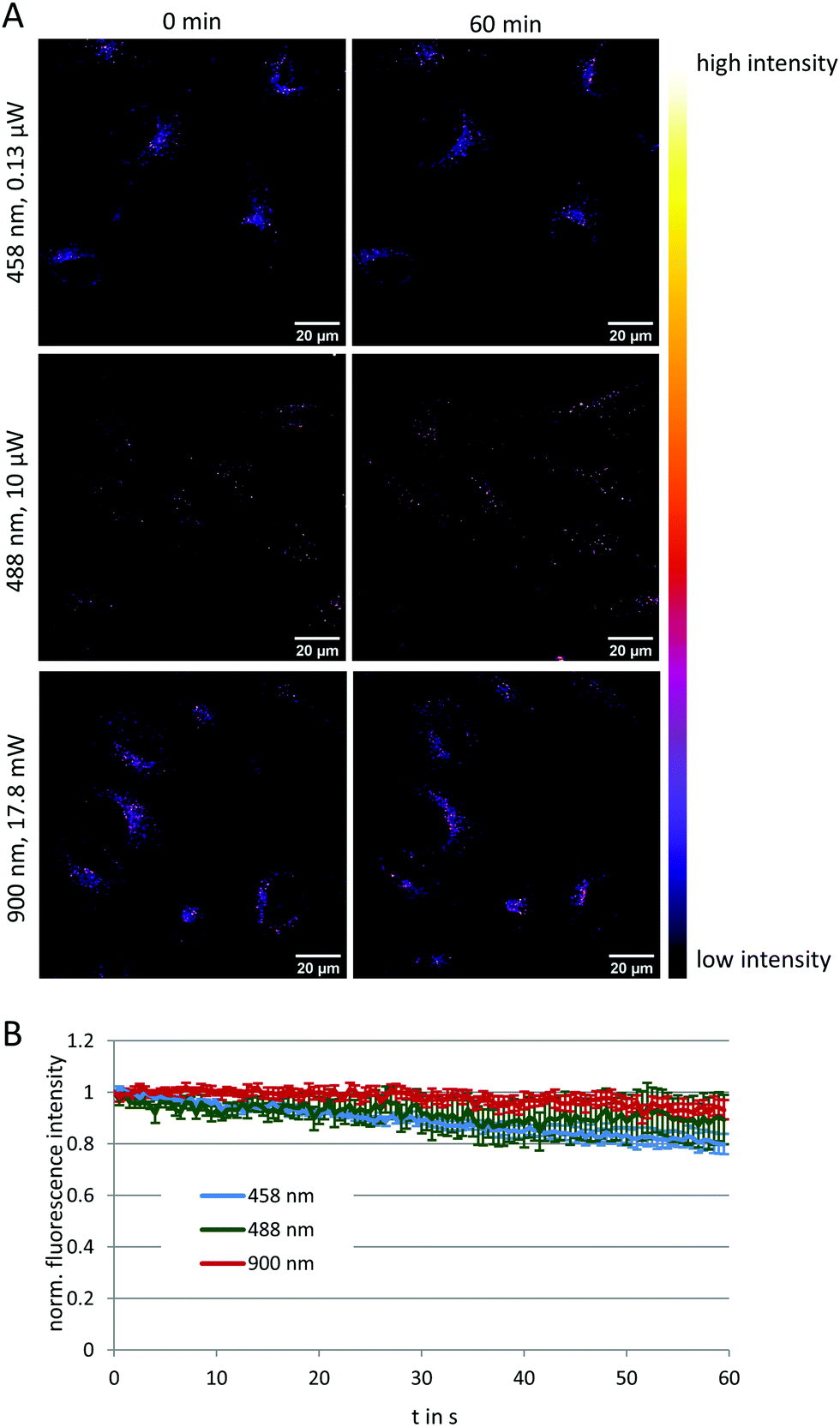 | ||
| Fig. 4 Photostability of NMI-LS under illumination with different excitation wavelengths (458 nm and 488 nm for single-photon excitation, 900 nm for two-photon excitation). Time lapse images (1024 × 1024 pixels) were acquired in 3 FOV for 60 min every 30 s with a pixel dwell time of 1.58 μs with the indicated laser power. (A) The first and last image of one representative time lapse is shown in intensity-based pseudocolor. (B) Quantification of fluorescence intensity during the acquisition time. Data represent mean ± standard deviation background-corrected fluorescence intensity normalised to the first acquired image of minimal 8 FOV from total three biological replicates. | ||
NMI-LS can record pH variations of lysosomes in living cells
A vast number of diseases have been associated with lysosomal pH variations. These include defects in acidification as assumed in a number of autoimmune diseases like Lupus or Sjögrens syndrome10 and neurodegenerative disorders.60 In addition, age-related complications have been attributed to reduced lysosomal acidification efficiency.60 Detecting luminal pH in lysosomes may, thus, help to establish disease models and to test therapeutic strategies to restore physiological pH values.61 Moreover, it has been demonstrated that the luminal pH is dependent on the localization of the lysosomes within the cell.62 Correlating localization with pH and the distinct functions of lysosomes over time will certainly help to better understand the numerous complex processes in physiology and pathophysiology.To explore the ability of NMI-LS to record lysosomal pH variations, we performed ratiometric excitation imaging using the 405 nm and 458 nm laser lines as excitation sources. An increased cellular pH should result in an increased fluorescence when excited with 458 nm compared to 405 nm. Indeed, upon addition of 10 mM ammonium chloride, a compound known to increase lysosomal pH,17,63 we observed an increase in the ratio of fluorescence intensities recorded upon 458 nm to 405 nm excitation in HeLa cells labeled with 500 nM NMI-LS (Fig. 5A). Please note that the intensity values for each channel are arbitrary units and are dependent on the excitation power as well as on the gain of the detector. These values were optimized for the chosen labeled sample. Therefore, also the obtained ratio is dependent on these settings and can vary when image acquisition parameters are modified.
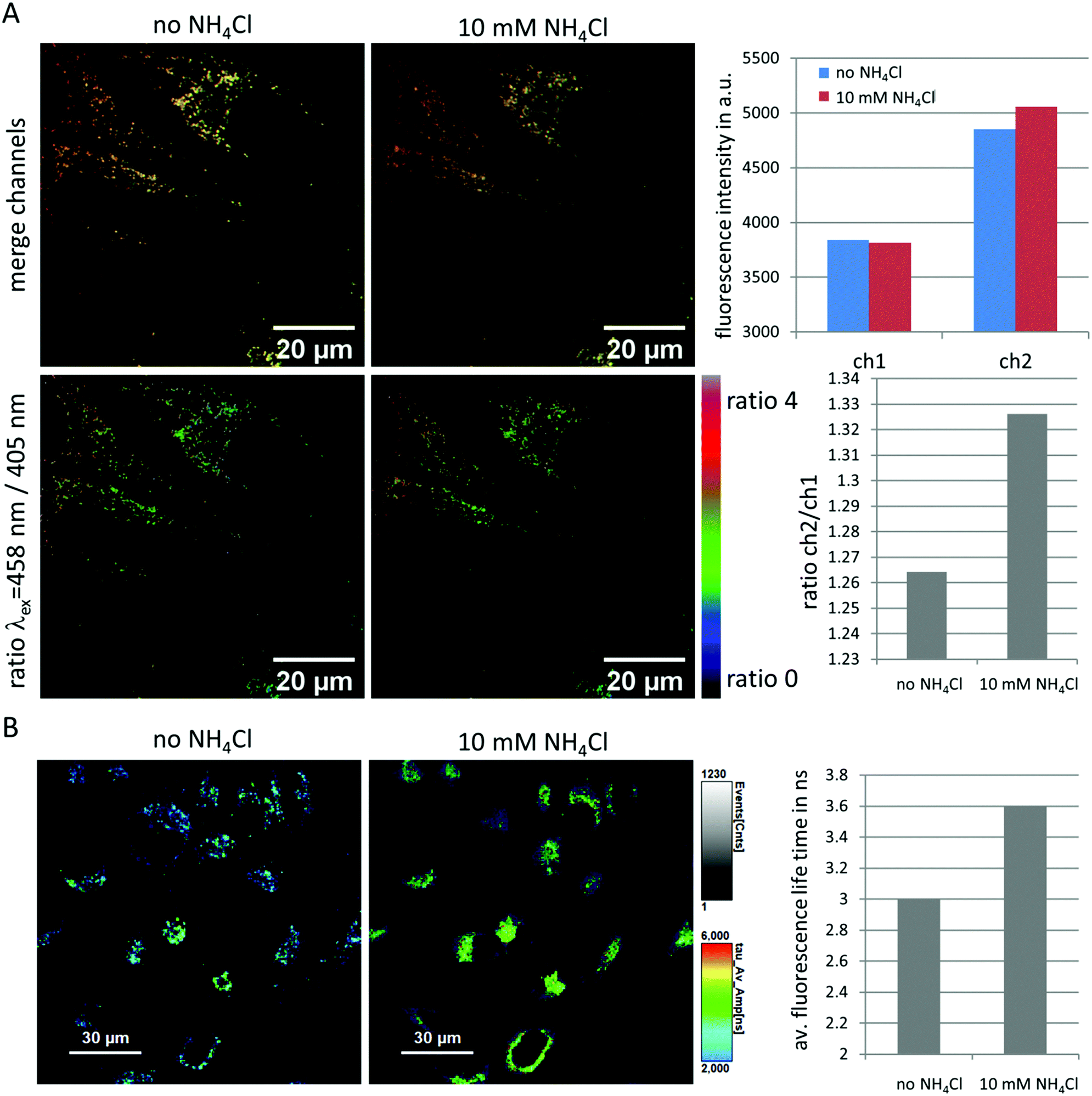 | ||
| Fig. 5 Feasibility of NMI-LS as pH indicator in living HeLa cells. (A) Ratiometric excitation imaging before and 2 min after the addition of 10 mM ammonium chloride. The upper panels show a merged image of the channel excited with 405 nm (green) and 458 nm (red), the lower panels the pseudo-colored ratio between 458 nm excitation and 405 nm excitation channel. An increase in the pH is visualized by an increase in this ratio. Mean fluorescence intensity before and after addition of ammonium chloride was assessed from areas covered by the three cells in the FOV and is shown in the upper bar plot. The ratio of ch2/ch1 in the second bar plot was calculated from this mean cellular fluorescence intensity. (B) FLIM image before and 2 min after addition of 10 mM ammonium chloride (λex = 440 nm, 20 MHz, λem = 465–500 nm), triexponential fit; τAv = a1τ1 + a2τ2 + a3τ3. The average fluorescence life time of the cells in the image is shown in the bar plot. A time lapse Video is available in ESI.† | ||
Similarly, ratiometric imaging of two emission channels (465–518 nm/410–455 nm) yielded an increase of this ratio after addition of 10 mM ammonium chloride (see ESI,† Fig. S7). However, due to the narrow band of the chosen emission channels, less light can be detected, so in our hands ratiometric excitation imaging proved more reliable in monitoring a chemically induced increase in lysosomal pH. Increased dye concentration during the labelling procedure will result in brighter cells but also in more unspecific staining and artifacts during ratiometric imaging, probably due to quenching/unquenching processes (data not shown). We therefore recommend to use low dye concentrations (0.1–0.5 μM).
For comparison we also tested a commercial lysosomal sensor probe. We chose LysoSensor™ Yellow/Blue dextran (LSYB) from ThermoFisher, since this is one of the two ratiometric dyes available from this provider. We chose the dextran version because this dye will be also retained for longer times in the lysosomes. The other ratiometric dye LysoSensor™ Yellow/Blue DND-160 requires very minute labelling times and will induce pH changes to the lysosomes upon longer incubation times according to the manufacturers data sheet. The chosen commercial LSYB has a pKa of 3.9 according to the data sheet which is in a similar range as that of NMI-LS. The manufacturer suggests ratiometric emission monitoring upon excitation at 360 nm. Upon neutralization the color should change from yellow to blue. Since we do not possess UV excitation with our confocal systems, we performed ratiometric emission imaging upon two photon excitation at 730 nm. Emission was detected in two channels (ch1 = 419–499 nm and ch2 = 517–624 nm). Fig. S8A (ESI†) shows the merged images of ch1 (pseudo-colored in green) and ch2 (pseudo-colored in red) as well as the ratio of ch1/ch2 in intensity-based pseudo-color. Upon raise of lysosomal pH by addition of 10 mM ammonium chloride, we observed a strong decrease of the already weak fluorescence intensity. Indeed the change of lysosomal pH could be detected by an increase of the ratio of ch1/ch2, since the fluorescence in ch2 was fading stronger. We also noticed a rather higher bleaching of the commercial LSYB compared to NMI-LS using two photon excitation at similar laser power (see ESI,† Fig. S8B). In conclusion, both sensors can be excited using two-photon excitation, but our new sensor offers slightly more pH-stable and photo-stable detection of lysosomal pH changes, while the required labelling time is orders of magnitude lower than with the commercial probe designed for long term studies.
Since also the fluorescence life time of NMI-LS proofed as sensitive indicator for pH (see above and ESI,† Fig. S4), we performed fluorescence lifetime imaging (FLIM) before and after addition of 10 mM ammonium chloride to NMI-LS labeled HeLa cells. RapidFLIM images revealed a gradual increase in the average fluorescence life time after ammonium chloride addition (Fig. 5B and Video in ESI†) which was not detected before compound addition. This increase in fluorescence life time is expected when the pH rises (see above). Please note that the fluorescence life time of probes is highly dependent on various environmental factors like hydrophobicity and ion concentrations. This is probably the reason for the lower average fluorescence life times detected in a cellular environment compared to that measured in simple buffer systems (ESI;† Fig. S4).
Additionally, FLIM of NMI-LS-labelled HeLa cells was carried out using two-photon excitation at 900 nm. FLIM data were fitted to biexponential model functions and the resulting detailed average fluorescence lifetime is shown in pseudocolor in ESI,† Fig. S9. It is clearly visible that the average fluorescence lifetime shows variation between different lysosomes indicating variations in lysosomal pH values. pH variations of lysosomes in one cell are known and have been reported in the literature.62
The results demonstrate that NMI-LS is a suitable marker for live-cell imaging of lysosomes and tracking of real-time lysosomal pH variations within a living cell as was shown here, after addition of chemical stimuli. The physicochemical properties of the NMI-LS dye enable different readout options, such as either ratiometric imaging of the spectral changes of NMI-LS upon acidification/neutralization or FLIM utilizing the pH-dependence of NMI-LS fluorescence lifetime.
Conclusions
Here we report the synthesis and spectroscopic characterization of the peri-fused naphthalene monoimide that can serve as a stable, non-toxic lysosomal marker and read-out dye for long term observation of lysosomal localization as well as the detection of lysosomal pH variations using ratiometric imaging or FLIM in living cells.Following the time-course of uptake and lysosomal association of intra- and extracellular material in live cell imaging is of crucial interest to investigate several pathophysiological conditions and to develop and assess the effectiveness of possible therapeutic interventions. The lysosomes may accumulate non-digestible material, for instance, in amyloid-associated disease10 or may house certain intracellular pathogens.64,65 Likewise, newly developed drug formulations for different diseases may pass through the lysosomes and eventually be released from or degraded by this organelle. Stable ratiometric dyes with reasonable loading times for long term tracking of lysosomes and their pH variations in living cells have not been reported so far. NMI-LS may, thus, become a useful tool for further analysis and understanding of the many lysosomal-related biological processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 316213987 – SFB 1278 (projects Z01 and B03) and the Core Facility Jena Biophotonic and Imaging Laboratory (JBIL, FKZ: PO 563/29-1, BA 1601/10-1), the BMBF via the Integrated Research and Treatment Center “Center for Sepsis Control and Care” (CSCC, FKZ 01EO1502) and the Leibniz Association via the Leibniz ScienceCampus InfectoOptics (W8/2018) is highly acknowledged. Furthermore, the project was supported by the Free State of Thuringia (2016FGI0010 and 2017FGI0026) with co-financing from the European Union within the European Regional Development Fund (EFRE) and by the Thüringer Innovationszentrum für Medizintechnik-Lösungen (ThIMEDOP) (FKZ IZN 2018 0002).Notes and references
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Nat. Rev. Mol. Cell Biol., 2013, 14, 283–296 CrossRef CAS.
- H. X. Xu and D. J. Ren, Annu. Rev. Physiol., 2015, 77, 57–80 CrossRef CAS.
- A. Ballabio and J. S. Bonifacino, Nat. Rev. Mol. Cell Biol., 2020, 21, 110–118, DOI:10.1038/s41580-019-0185-4.
- R. E. Lawrence and R. Zoncu, Nat. Cell Biol., 2019, 21, 133–142 CrossRef CAS.
- J. E. Oyarzun, J. Lagos, M. C. Vazquez, C. Valls, C. De la Fuente, M. I. Yuseff, A. R. Alvarez and S. Zanlungo, Biochim. Biophys. Acta, Mol. Basis Dis., 2019, 1865, 1076–1087 CrossRef CAS.
- G. T. Saffi and R. J. Botelho, Trends Cell Biol., 2019, 29, 635–646 CrossRef CAS.
- E. J. Parkinson-Lawrence, T. Shandala, M. Prodoehl, R. Plew, G. N. Borlace and D. A. Brooks, Physiology, 2010, 25, 102–115 CrossRef CAS.
- E. Van Meel and J. Klumperman, Histochem. Cell Biol., 2008, 129, 253–266 CrossRef.
- C. Fennelly and R. K. Amaravadi, Methods Mol. Biol., 2017, 1594, 293–308 CrossRef CAS.
- S. R. Bonam, F. J. Wang and S. Muller, Nat. Rev. Drug Discovery, 2019, 18, 923–948 CrossRef CAS.
- S. M. Davidson and M. G. Vander Heiden, Annu. Rev. Pharmacol., 2017, 57, 481–507 CrossRef CAS.
- S. Ishii, A. Matsuura and E. Itakura, Sci. Rep., 2019, 9(1), 11635, DOI:10.1038/s41598-019-48131-2.
- S. Burgstaller, H. Bischof, T. Gensch, S. Stryeck, B. Gottschalk, J. Ramadani-Muja, E. Eroglu, R. Rost, S. Balfanz, A. Baumann, M. Waldeck-Weiermair, J. C. Hay, T. Madl, W. F. Graier and R. Malli, ACS Sens., 2019, 4, 883–891 CrossRef CAS.
- A. Pierzynska-Mach, P. A. Janowski and J. W. Dobrucki, Cytometry, Part A, 2014, 85, 729–737 CrossRef.
- J. T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X. Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC.
- I. Eriksson, K. Öllinger and H. Appelqvist, in Lysosomes: Methods and Protocols, ed. K. Öllinger and H. Appelqvist, Springer, New York, New York, NY, 2017, pp. 179–189 DOI:10.1007/978-1-4939-6934-0_11.
- S. Ohkuma and B. Poole, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3327–3331 CrossRef CAS.
- M. J. Geisow, Exp. Cell Res., 1984, 150, 29–35 CrossRef CAS.
- L. Fan, M. Nan, J. Y. Ge, X. D. Wang, B. Lin, W. J. Zhang, S. M. Shuang and C. Dong, New J. Chem., 2018, 42, 13479–13485 RSC.
- D. Lee, K. M. K. Swamy, J. Hong, S. Lee and J. Yoon, Sens. Actuators, B, 2018, 266, 416–421 CrossRef CAS.
- B. Li, G. B. Ge, L. L. Wen, Y. Yuan, R. Zhang, X. J. Peng, F. Y. Liu and S. G. Sun, Dyes Pigm., 2017, 139, 318–325 CrossRef CAS.
- H. S. Lv, S. Y. Huang, Y. Xu, X. Dai, J. Y. Miao and B. X. Zhao, Bioorg. Med. Chem. Lett., 2014, 24, 535–538 CrossRef CAS.
- G. L. Niu, P. P. Zhang, W. M. Liu, M. Q. Wang, H. Y. Zhang, J. S. Wu, L. P. Zhang and P. F. Wang, Anal. Chem., 2017, 89, 1922–1929 CrossRef CAS.
- X. J. Cao, L. N. Chen, X. Zhang, J. T. Liu, M. Y. Chen, Q. R. Wu, J. Y. Miao and B. X. Zhao, Anal. Chim. Acta, 2016, 920, 86–93 CrossRef CAS.
- X. Chen, Y. Bi, T. Wang, P. Li, X. Yan, S. Hou, C. E. Bammert, J. Ju, K. M. Gibson, W. J. Pavan and L. Bi, Sci. Rep., 2015, 5, 9004 CrossRef CAS.
- Y. X. Fu, J. J. Zhang, H. Wang, J. L. Chen, P. Zhao, G. R. Chen and X. P. He, Dyes Pigm., 2016, 133, 372–379 CrossRef CAS.
- D. D. He, W. Liu, R. Sun, C. Fan, Y. J. Xu and J. F. Ge, Anal. Chem., 2015, 87, 1499–1502 CrossRef CAS.
- G. K. Vegesna, J. Janjanam, J. H. Bi, F. T. Luo, J. T. Zhang, C. Olds, A. Tiwari and H. Y. Liu, J. Mater. Chem. B, 2014, 2, 4500–4508 RSC.
- C. Wang, B. L. Dong, X. Q. Kong, N. Zhang, W. H. Song and W. Y. Lin, Luminescence, 2018, 33, 1275–1280 CrossRef CAS.
- K. K. Yu, K. Li, J. T. Hou, H. H. Qin, Y. M. Xie, C. H. Qian and X. Q. Yu, RSC Adv., 2014, 4, 33975–33980 RSC.
- J. T. Zhang, M. Yang, C. Li, N. Dorh, F. Xie, F. T. Luo, A. Tiwari and H. Y. Liu, J. Mater. Chem. B, 2015, 3, 2173–2184 RSC.
- H. Zhu, J. L. Fan, Q. L. Xu, H. L. Li, J. Y. Wang, P. Gao and X. J. Peng, Chem. Commun., 2012, 48, 11766–11768 RSC.
- B. L. Dong, X. Z. Song, C. Wang, X. Q. Kong, Y. H. Tang and W. Y. Lin, Anal. Chem., 2016, 88, 4085–4091 CrossRef CAS.
- H. J. Kim, C. H. Heo and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 17969–17977 CrossRef CAS.
- G. Li, D. Zhu, L. Xue and H. Jiang, Org. Lett., 2013, 15, 5020–5023 CrossRef CAS.
- X. J. Liu, Y. A. Su, H. H. Tian, L. Yang, H. Y. Zhang, X. Z. Song and J. W. Foley, Anal. Chem., 2017, 89, 7038–7045 CrossRef CAS.
- D. G. Smith, B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2012, 48, 8520–8522 RSC.
- Q. Q. Wan, S. M. Chen, W. Shi, L. H. Li and H. M. Ma, Angew. Chem., Int. Ed., 2014, 53, 10916–10920 CrossRef CAS.
- S. Xia, M. X. Fang, J. B. Wang, J. H. Bi, W. Mazi, Y. B. Zhang, R. L. Luck and H. Y. Liu, Sens. Actuators, B, 2019, 294, 1–13 CrossRef CAS.
- Z. W. Xue, H. Zhao, J. Liu, J. H. Han and S. F. Han, ACS Sens., 2017, 2, 436–442 CrossRef CAS.
- G. Q. Yuan, H. Y. Ding and L. Y. Zhou, Spectrochim. Acta, Part A, 2020, 224, 117397, DOI:10.1016/j.saa.2019.117397.
- X. F. Zhang, T. Zhang, S. L. Shen, J. Y. Miao and B. X. Zhao, J. Mater. Chem. B, 2015, 3, 3260–3266 RSC.
- T. Zhang, D. Xu, C. Y. Poon, X. Wang, F. Bolze, H. W. Li and M. S. Wong, Talanta, 2019, 202, 34–41 CrossRef CAS.
- E. Zhang, S. Wang, X. Su and S. Han, Analyst, 2020, 145(5), 1319–1327, 10.1039/c9an02441g.
- X. J. Zhao, C. Wang, G. Q. Yuan, H. Y. Ding, L. Y. Zhou, X. G. Liu and Q. L. Lin, Sens. Actuators, B, 2019, 290, 79–86 CrossRef CAS.
- M. L. Zhu, P. P. Xing, Y. B. Zhou, L. Gong, J. H. Zhang, D. D. Qi, Y. Z. Bian, H. W. Du and J. Z. Jiang, J. Mater. Chem. B, 2018, 6, 4422–4426 RSC.
- J. H. Zhang, M. L. Zhu, J. W. Cui, C. M. Wang, Z. W. Zhou, T. H. Wang, L. Gong, C. R. Su, D. D. Qi, Y. Z. Bian, H. W. Du and J. Z. Jiang, J. Photochem. Photobiol., A, 2020, 396, 112524, DOI:10.1016/j.jphotochem.2020.112524.
- S. Kaloyanova, Y. Zagranyarski, S. Ritz, M. Hanulova, K. Koynov, A. Vonderheit, K. Mullen and K. Peneva, J. Am. Chem. Soc., 2016, 138, 2881–2884 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- T. M. Henderson, A. F. Izmaylov, G. Scalmani and G. E. Scuseria, J. Chem. Phys., 2009, 131, 044108 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian Inc., Wallingford, USA, 2016 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- T. C. McIlvaine, J. Biol. Chem., 1921, 49, 183–186 CAS.
- J. E. Whitaker, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1991, 194, 330–344 CrossRef CAS.
- T. Myochin, K. Kiyose, K. Hanaoka, H. Kojima, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 3401–3409 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS.
- F. Wang, R. Gomez-Sintes and P. Boya, Traffic, 2018, 19, 918–931 CrossRef CAS.
- A. Wu, Y. Xu and X. Qian, Bioorg. Med. Chem., 2009, 17, 592–599 CrossRef CAS.
- D. J. Colacurcio and R. A. Nixon, Ageing Res. Rev., 2016, 32, 75–88 CrossRef CAS.
- J. H. Lee, M. K. McBrayer, D. M. Wolfe, L. J. Haslett, A. Kumar, Y. Sato, P. P. Y. Lie, P. Mohan, E. E. Coffey, U. Kompella, C. H. Mitchell, E. Lloyd-Evans and R. A. Nixon, Cell Rep., 2015, 12, 1430–1444 CrossRef CAS.
- D. E. Johnson, P. Ostrowski, V. Jaumouille and S. Grinstein, J. Cell Biol., 2016, 212, 677–692 CrossRef CAS.
- V. Ambriz-Avina, J. A. Contreras-Garduno and M. Pedraza-Reyes, Biomed. Res. Int., 2014, 461941, DOI:10.1155/2014/461941.
- T. O. Omotade and C. R. Roy, Microbiol. Spectrum, 2019, 7(2) DOI:10.1128/microbiolspec.BAI-0022-2019.
- D. E. Voth and R. A. Heinzen, Cell. Microbiol., 2007, 9, 829–840 CrossRef CAS.
- H. J. Lee, C. W. Cho, H. Seo, S. Singha, Y. W. Jun, K. H. Lee, Y. Jung, K. T. Kim, S. Park, S. C. Bae and K. H. Ahn, Chem. Comm., 2016, 52, 124–127 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure, NMR characterization, supplementary tables and figures, time lapse video. See DOI: 10.1039/d0tb02208j |
| This journal is © The Royal Society of Chemistry 2021 |