
Understanding the reaction mechanism and performances of 3d transition metal cathodes for all-solid-state fluoride ion batteries†
Datong
Zhang
 a,
Kentaro
Yamamoto
*a,
Aika
Ochi
a,
Yanchang
Wang
a,
Takahiro
Yoshinari
a,
Koji
Nakanishi
a,
Hiroyuki
Nakano
a,
Hidenori
Miki
ab,
Shinji
Nakanishi
b,
Hideki
Iba
b,
Tomoki
Uchiyama
a,
Toshiki
Watanabe
a,
Koji
Amezawa
c and
Yoshiharu
Uchimoto
a
a,
Kentaro
Yamamoto
*a,
Aika
Ochi
a,
Yanchang
Wang
a,
Takahiro
Yoshinari
a,
Koji
Nakanishi
a,
Hiroyuki
Nakano
a,
Hidenori
Miki
ab,
Shinji
Nakanishi
b,
Hideki
Iba
b,
Tomoki
Uchiyama
a,
Toshiki
Watanabe
a,
Koji
Amezawa
c and
Yoshiharu
Uchimoto
a
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida-nihonmatsu-cho, Sakyo, Kyoto 606-8501, Japan. E-mail: yamamoto.kentaro.4e@kyoto-u.ac.jp
bBattery Research Div., Toyota Motor Corporation, 1200 Mishuku, Shizuoka 410-1193, Japan
cIMRAM, Tohoku University, 2-1-1 Katahira, Aoba, Sendai, Miyagi 980-8577, Japan
First published on 8th December 2020
Abstract
Fluoride ion batteries (FIBs) are regarded as promising energy storage devices, and it is important and urgent to develop cathode materials with high energy densities for use in FIBs. However, systematic investigations of 3d transition metal/metal fluorides have been rarely reported thus far because of the restricted reversibility and unfavorable interfacial compatibility of 3d transition metal/metal fluorides with solid-state electrolytes. Herein, 3d transition metals are investigated by utilizing thin-film cells with LaF3 substrates. The highly reversible (de)fluorinations of Cu, Co, and Ni are validated at various temperatures. High capacity utilizations of 79.5%, 100%, and 90.5% are obtained during the initial cycle at 150 °C. By combining results from X-ray absorption spectroscopy (XAS) and electrochemical characterization, the electrochemical behaviors of Cu, Co, and Ni, as well as experimental evidence of the two-phase transition mechanism during the M/MF2 reaction are reported for the first time. This provides new insights required for future cathode designs for use in all-solid-state FIBs.
Introduction
Developing electrochemical energy storage devices is a greatly effective and imperative strategy in developing alternative energy sources to fossil fuels, that have been deemed to cause environmental issues and are possibly close to depletion.1–5 Lithium ion batteries (LIBs), typical representatives of electrochemical energy storage devices, were used worldwide in the last several decades. However, the energy densities of highly commercialized LIBs are difficult to improve which have been limited by their intrinsic characteristics. As a result, there has been a strong desire to design new concepts of beyond-LIBs, such as new systems that employ Na+, K+, Mg2+, Zn2+, Ca2+, Al3+, F−, and Cl− as charge carriers.6–17 Fluoride ion batteries (FIBs) are among these new systems and have been regarded as promising candidates owing to the electrochemical stability of F anions as charge carriers, which is ensured by the high electronegativity of the F element.14,18–25After the successful application of tysonite-type solid solutions La1−xBaxF3−x (0 ≤ x ≤ 0.15) as solid-state electrolytes,14 all-solid-state FIBs utilizing metal/metal fluoride (M/MFx) compounds as electrodes have attracted broad attention because of their extremely high energy densities. In particular, couples of 3d transition metals and their corresponding metal fluorides (3d-M/MFx), which are lightweight and multivalent, possess huge theoretical capacities (approximately 5–10 times that of common LIB cathode materials) and relatively high potentials (usually 1.5–3.5 V vs. Li/LiF). Therefore, these couples could provide high energy densities of over 2000 W h kg−1 or 5000 W h L−1, as shown in Fig. 1a.25–30
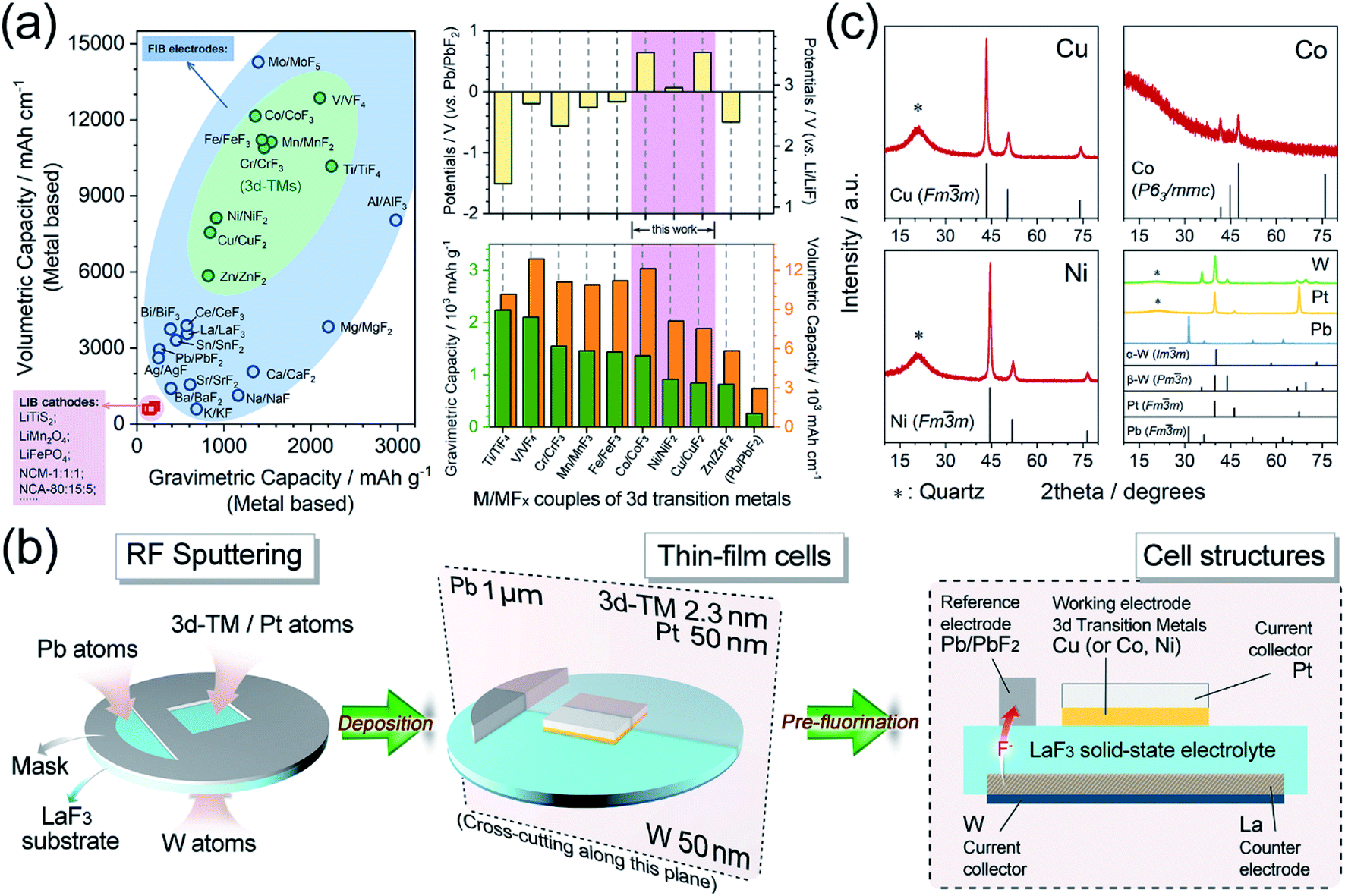 | ||
| Fig. 1 (a) Left: comparison of gravimetric/volumetric energy densities of LIB cathode materials (pink),30 FIB metal/metal fluoride cathode material (blue) and FIB 3d transition metal/metal fluoride cathode materials (green). Right: the theoretical working potentials and gravimetric/volumetric energy densities of 3d transition metal/metal fluoride cathode materials for FIBs. (b) Schematic illustrations of the RF sputtering and pre-fluorination processes. (c) XRD patterns of Cu, Co, Ni, W, Pt, and Pb thin films. | ||
Notwithstanding all these merits in terms of energy densities, there are few reports on the use of 3d-M/MFx as cathode materials for all-solid-state FIBs. In particular, systematic discussions on their typical fluorination process and corresponding phase transitions, based on high battery utilizations, are lacking. Although some pioneering studies on Cu/CuF2 have been reported,26,29,31 most studies have focused more on the optimization of solid-state F− conductors, or on liquid-electrolyte battery systems, without specific perspectives towards the electrode materials of Cu, Zn, Sn, and even Bi.14,17,25,26,32–42 Therefore, it is urgently important to establish a database-type understanding of the basic electrochemical information on the use of 3d-M/MFx as cathodes for all-solid-state FIBs. One main hindrance to such investigations might be the inferior cycling reversibility caused by the enormous volumetric expansion/contraction upon the (de)fluorination process and sluggish ion transport of 3d-M/MFx.26–28,31 Hence, widely used bulk-type cell models are not suitable for evaluating these materials in detail. For example, huge volumetric changes of c.a. 200–300% (Fig. S1†) lead to drastic damage to electrolyte/cathode interfacial contacts.43–48 Therefore, the reaction mechanisms and electrochemical properties of 3d-M/MFx are still unknown and consequently optimization strategies are unclear. Thin-film models are considered powerful tools for evaluating 3d-M/MFx materials, which could be used to control the film thickness to optimize the cathode/electrolyte interfacial compatibility, reduce the internal resistance, and create a nearly ideal reaction environment.31 Herein, we introduced 3d transition metals Cu, Co, and Ni as thin-film cathode materials for all-solid-state FIBs, and for the first time, experimentally explored the two-phase-transition process within the difluorination ranges through X-ray absorption spectroscopy (XAS). To the best of our knowledge, this is the first report to investigate the electrochemical properties of Co and Ni as cathode materials for all-solid-state FIBs.
Experimental
Preparations for the thin-film cell
The thin-film preparation procedure is schematically presented in Fig. 1b. Typically, the thin-film cells were prepared via a radio-frequency magnetron sputtering (RF Sputtering, Eiko, Japan) method by depositing all the necessary metals onto a single-crystal LaF3 substrate (used as a solid-state electrolyte). The parameters and details of the RF sputtering are described in Table S1.† The as-deposited thin-film cells were assembled into battery vessels in an argon-filled glovebox (Miwa, Japan). A Pb/PbF2 reference electrode, which could offer stable reference potentials (Fig. S2†), was constructed by the pre-fluorination step after the thicknesses of all the metals had reached their design values. The pre-fluorination was achieved by connecting the as-deposited Pb and W current collector with a current of 8.3 μA for 300 s at 150 °C. As a result, pure Pb and LaF3 were fluorinated/defluorinated to Pb/PbF2 and La/LaF3, which were employed as the reference and counter electrodes, respectively. The pre-fluorination and galvanostatic tests were processed using a PARSTAT MC2000 battery testing system (Princeton Applied Research, USA). The smoothness and thickness of the as-deposited thin films were also examined by atomic force microscopy (Fig. S3†) and transmission electron microscopy (TEM, Fig. S4†). Since it is difficult to observe ultra-thin films with thicknesses of ∼2.3 nm, TEM images were acquired from thicker films (c.a. 60 nm).The metal deposition rates were determined via a Dektak-8 Stylus Profiler (Veeco, USA) by preparing films of various design thicknesses and then fitting a linear relationship (y = kx) of the actual thickness versus the deposition time, as shown in Fig. S5.† Upon preparing the cell, the deposition rates were measured in real-time using the quartz crystal microbalance (QCM) inside the RF sputtering chamber.
The surface of the single-crystal LaF3 substrate was treated by reverse sputtering under argon (2.0 Pa) to remove most of the surficial oxidized layers. Then, the surface was annealed in a NF3 atmosphere (3.6 Pa) at 300 °C for 1 hour to repair the defective lattice (which included remnants of LaOF and La2O3 species) based on the following reactions.
 | (1) |
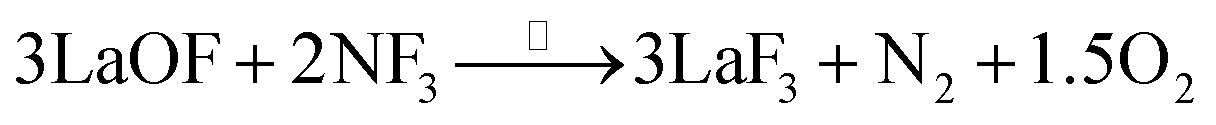 | (2) |
Characterization and electrochemical tests
The X-ray diffraction (XRD) patterns were obtained using an X-ray diffractometer (Rigaku Ultima IV, Japan) with Cu-Kα radiation (λ = 1.54056 Å). X-ray adsorption near-edge spectroscopy (XANES) was carried out at BL37XU beamline at the SPring-8 synchrotron radiation facilities (Hyogo, Japan). The signals were collected in partial fluorescence yield (PFY) mode by using a 19-element solid-state detector.The ionic conductivity of LaF3 was measured by electrochemical impedance spectroscopy (EIS) using a ModuLab XM ECS electrochemical test system (Solartron analytical, UK) at a frequency in the range of 106 to 10−1 Hz and an amplitude voltage of 50 mV at temperatures of 15, 50, 80, 100, 120, and 150 °C. The activation energy (Ea) was calculated using the Arrhenius equation:35,49
 | (3) |
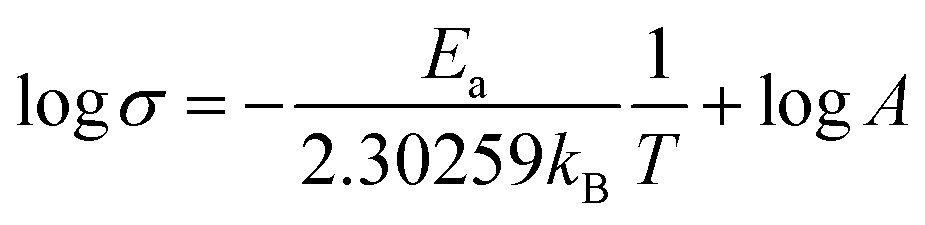 | (4) |
Therefore, the Ea can be determined by linear fitting of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σ vs. T−1, as depicted in Fig. S6.†
σ vs. T−1, as depicted in Fig. S6.†
Results and discussion
Structural analysis of the as-deposited metals
The XRD of the thin films was accomplished by depositing the metals onto quartz substrates instead of LaF3 substrates to avoid the disturbance of the strong LaF3 diffraction peaks, as demonstrated in Fig. 1c. The diffraction peaks of Cu and Ni were both well indexed to a face-centered cubic (fcc) structure (JCPDS#04-0836 and #04-0850, respectively) with a space group of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The metallic cobalt exhibited rough diffraction with a P63/mmc hexagonal close packed (hcp) structure (JCPDS#05-0727). Additionally, the Pt and Pb also corresponded well to the cubic structure (JCPDS#04-0802 and #04-0686) and W to α + β multiphases (JCPDS#04-0806 and #47-1319), respectively.
m. The metallic cobalt exhibited rough diffraction with a P63/mmc hexagonal close packed (hcp) structure (JCPDS#05-0727). Additionally, the Pt and Pb also corresponded well to the cubic structure (JCPDS#04-0802 and #04-0686) and W to α + β multiphases (JCPDS#04-0806 and #47-1319), respectively.
Electrochemical properties
To acquire the ideal ionic conductivity of LaF3, the galvanostatic charge/discharge tests were mainly carried out at 150 °C. The ionic conductivities (σ) of LaF3 at various temperatures and its activation energy (Ea) were measured by EIS and calculated using the Arrhenius equation, respectively, as described in the Experimental section. As shown in Fig. S6,† the σ(LaF3) at 150 °C was approximately 1.1 × 10−3 S cm−1. This was adequate for the smooth electrochemical reactions in the all-solid-state cells. Meanwhile, the calculated Ea was 0.527 eV (the reference value was 0.43–0.46 eV (ref. 50 and 51)). The Pb/PbF2 reference electrode also provided stable reference potentials of c.a. 2.5 V (theoretically 2.41 V). As shown in Fig. S2,† the increase in the reference potential from 0 to 75 s was attributed to the generation and growth of La grains from pure LaF3, and PbF2 grains from pure Pb, i.e., the reference voltages changed from Pb vs. LaF3 (0 s) to Pb/PbF2vs. La/LaF3 (75 s). The Cu, Co, and Ni metals exhibited high (de)fluorination reversibility upon cycling as shown in Fig. 2. The initial reversible capacity of Cu, Co, and Ni at 150 °C was 670.5, 1366.6, and 826.9 mA h g−1, which were 79.5%, 100%, and 90.5% of the theoretical values of Cu/CuF2, Co/CoF3, and Ni/NiF2, respectively. Notice that all three metals possessed flat single charge/discharge plateaus, which was indicative of the possibility of a phase-transition mechanism upon cycling. The deteriorations in the average working potentials of Cu, Co, and Ni were inappreciable as presented in Fig. S7,† with potential changes of 0.047/−0.018 V, 0.001/−0.026 V, and 0.018/−0.012 V during charge/discharge processes after 20 cycles, respectively. In addition, the reversible (de)fluorination of Cu, Co, and Ni at mild temperatures was also verified. Although the σ(LaF3) dropped to 2.1 × 10−5 S cm−1 (60 °C) and 2.4 × 10−6 S cm−1 (25 °C), the reversible M/MFx conversion was still achieved at mild temperatures. As shown in Fig. 2, the 3d-TM cathode materials at 25 °C exhibited considerable capacities compared with typical LIB cathodes, while the polarization became exceedingly large owing to the very limited σ(LaF3) value. But the potential for wider application of 3d-TMs as FIB cathodes in the future was also revealed. | ||
| Fig. 2 Galvanostatic charge/discharge profiles at 0.1C of Cu (left column), Co (middle column), and Ni (right column) at 150 (upper), 60 (middle) and 25 °C (lower). The vertical dashed lines refer to the theoretical capacity of x = 2.0 in MFx (3.0 for CoFx additionally). | ||
X-ray absorption spectroscopy and the reaction mechanism
As mentioned above, a two-phase-transition mechanism was thought to be responsible for the reaction based on the electrochemical behaviors. XANES was conducted to afford firmer experimental evidence. The ex situ XANES samples were prepared by controlling the state of charge (SOC) into a certain F content in the MFx. It must be emphasized that the MFx formulas used in this work do not represent the homogeneous solid solutions, but only the intermediate products of (1 − 0.5x)M·0.5xMF2 (0 ≤ x ≤ 2.0) with a nominal F content of x according to the two-phase-transition mechanism. The Cu K-edge, Co K-edge, and Ni K-edge spectra are shown in Fig. 3a–d. The spectra of all the pristine metals (CuF0, CoF0, and NiF0) were consistent with the reference (Ref.) spectra of Cu, Co, and Ni. The tendencies of the spectra to transfer from Ref.-M toward Ref.-MF2 were exhibited upon gradual fluorination. The isosbestic points can be observed in Fig. 3a–d which are located at c.a. 8990, 7717, and 8342 eV for the Cu K-edge, Co K-edge, and Ni K-edge, respectively. The occurrence of the isosbestic points usually indicates the direct transition from phase A toward phase B;52–55 consequently we linearly fitted the M and MF2 (M = Cu, Co and Ni) phase amounts versus the F content, as shown in Fig. 3e–g. According to the results from the fitted lines, the phase fraction changes of M and MF2 were linear in the range of 0 ≤ x ≤ 2.0 in the MFx, which implied the two-phase-transition nature of the M/MF2 reactions. A recent report by Haruyama J. et al.56 also concluded the same, in which the authors discussed the thermodynamically favored reaction patterns (i.e., solid-solution or two-phase-coexistent) of various M/MFx combinations via density functional theory calculations. They also discussed both models and concluded that the solid-solution model was unreasonable, because the F vacancy would be uniformly distributed and equally introduce electron dopants, whereas the large forbidden band gap made these doped electrons unstable.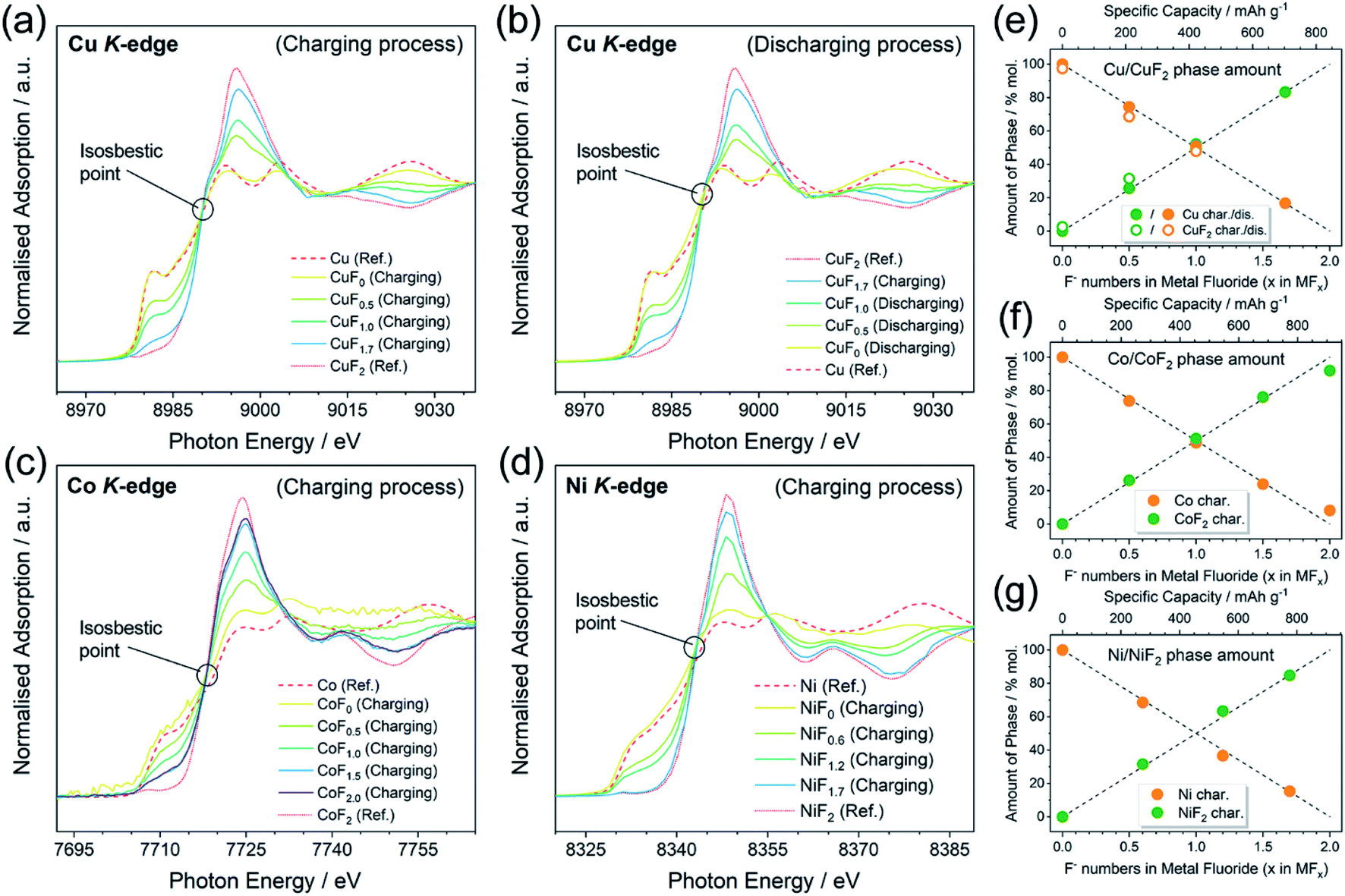 | ||
| Fig. 3 Ex situ X-ray absorption near-edge spectra of (a) Cu K-edge upon initial charging, (b) Cu K-edge upon initial discharging, (c) Co K-edge upon initial charging with Co/CoF2 range and (d) Ni K-edge upon initial charging, and the corresponding linear fittings of the phase amounts of (e) Cu/CuF2, (f) Co/CoF2, and (g) Ni/NiF2. The dashed lines in (e), (f), and (g) are assistant lines that connect (0, 0) with (2.0, 100%), and (0, 100%) with (2.0, 0), which represent the ideal linear changes of the phase amounts. | ||
The galvanostatic intermittent titration open circuit potentials (OCPs) at various SOC were also analyzed and are shown in Fig. 4a–c. The OCPs of CuFx, CoFx, and NiFx almost remained stable in the range of 0 ≤ x ≤ 2.0 except at the beginning of the charging. This inferred the phase-transition mechanism because the Gibbs free energy of formation of the M and MF2 phases remained unchanged during the (de)fluorination process within the phase-transition dominations.57 In particular, the Co with a higher metallic character showed a different mechanism in which the OCPs gradually increased upon further charging in the range of 2.0 ≤ x ≤ 3.0 (Fig. 4b). This was inconsistent with the two-phase transition characteristics. Meanwhile, the XANES patterns of the CoF2.5 and CoF3.0 transferred into higher oxidation states but were still similar to Ref.-CoF2 spectra (Fig. 4d). For the whole Co/CoF3 fluorination process, the reaction mechanisms in the initial and later stages (i.e., 0 ≤ x ≤ 2.0 and 2.0 ≤ x ≤ 3.0) were completely distinct. The mechanism of CoF2/CoF3 has not been clearly elucidated; however a possible reason for the abnormal XANES spectra is that the XANES data were collected through ex situ approaches which did not adequately shield the chemically unstable samples such as CoF3. Although CoF3 can exist independently, it is decomposed into CoF2 by traces of moisture during sample storage or transfer. Considering the ultra-thin thickness employed in this work, small current leakage might also accelerate this decomposition. These facts point to some critical requirements for in situ measurements of thin-film systems.
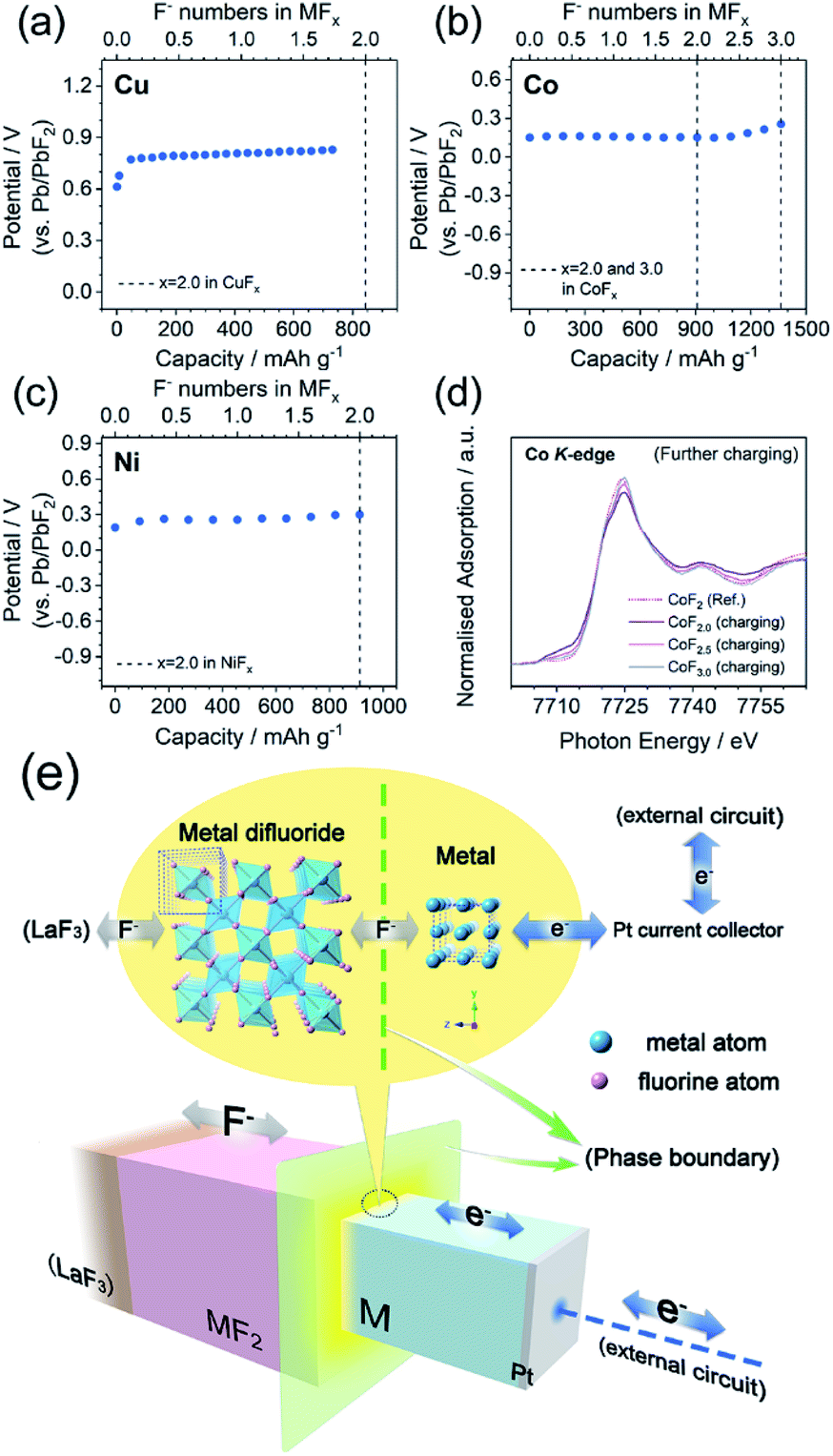 | ||
| Fig. 4 (a–c) Galvanostatic intermittent titration open circuit potentials (OCPs) upon initial charging of Cu, Co, and Ni. The vertical dashed lines refer to the theoretical capacities of x = 2.0 or 3.0 in the MFx formula. (d) Ex situ XANES of the Co K-edge upon initial charging with the CoF2/CoF3 range. (e) Schematic illustration of the two-phase transition mechanism during the M/MF2 reaction. | ||
Discussion and prospection
A typical charge/mass transfer procedure of M/MF2 phase transitions (the schematic description is shown in Fig. 4e) can be summarized as the following categories; (i) the interphase F− transport, i.e., the F− diffuses across the M/MFx phase boundaries, (ii) the internal F− transport, i.e., F− inside the MFx bulks, and (iii) the electron transfer toward the current collectors via the M bulks. The resistance from the electrode was deemed to be the macroscale rate-determining step; however more microscale contributions from the internal and interphase components (i or ii) could not be identified.56 Primary evaluations towards kinetic factors were given by the results of the film-thickness-dependent electrochemical properties of Cu as demonstrated in Fig. S8.† Thin films of Cu with various thicknesses of 2.3 (this work), 10 and 50 nm were prepared and galvanostatically tested at 150 °C. The samples with 2.3 and 10 nm thicknesses almost delivered capacities near theoretical values, whereas the 50 nm sample only delivered an initial charge capacity of ∼170 mA h g−1 which was c.a. 20% of the theoretical value, corresponding to a diffusion depth of c.a. 10 nm. The specific rate-determining step, factors effecting the (de)fluorination kinetics, and CoF2/CoF3 reaction mechanisms will be researched in detail in future studies, and are beyond the discussions of this work.Conclusions
The 3d transition metals Cu, Co, and Ni were investigated as reliable cathode materials for all-solid-state FIBs using thin-film cell models. The electrochemical properties with buffered polarizations and high reversible capacities were determined and analyzed in the ideal reaction environments created using the thin-film cell models. Based on the thorough fluorination, a possibility was implied that the fluorination of 3d transition metals proceeds via two-phase transition by ex situ XAS analyses and electrochemical assessments. The electrochemical properties of Co and Ni for use in FIBs were also reported for the first time. From a wider perspective, the overall performance of the all-solid-state FIB depended on various aspects; therefore deeper understanding and knowledge are required to design the FIB. Thus, besides the insights acquired in this study, more investigations are required to achieve both high reversibility and high energy density in FIBs.Author contributions
D. Zhang, K. Yamamoto, and A. Ochi conducted the experimental work. D. Zhang, and T. Yoshinari analyzed the electrochemical data. Y. Wang, K. Yamamoto, T. Uchiyama, T. Watanabe, and K. Nakanishi carried out the synchrotron X-ray absorption spectroscopy. H. Nakano, H. Miki, S. Nakanishi, H. Iba and K. Amezawa prepared the measurement environment. D. Zhang and K. Yamamoto wrote the manuscript. The ideas and experiments were conceived, planned, and analyzed by all co-authors under the supervision of K. Yamamoto and Y. Uchimoto. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST-Mirai Program Grant Number JPMJMI18E2, Japan. Synchrotron radiation experiments were performed at beamline BL37XU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal number 2016B1021, 2016B1517, 2017A1456, 2017B1038, 2018B1029, 2018B1616, and 2019A1020). Acknowledgements are also due to China Scholarship Council (CSC No. 201806370208) and Kyoto University for their financial supports to D. Zhang.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- C. Grey and J. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- C. Delmas, J. J. Braconnier, C. Fouassier and P. Hagenmuller, Solid State Ionics, 1981, 3–4, 165–169 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed. Engl., 2012, 51, 933–935 CrossRef CAS.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 RSC.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 325–328 CrossRef PubMed.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- P. Gao, M. A. Reddy, X. Mu, T. Diemant, L. Zhang, Z. Zhao-Karger, V. S. Chakravadhanula, O. Clemens, R. J. Behm and M. Fichtner, Angew. Chem., Int. Ed. Engl., 2016, 55, 4285–4290 CrossRef CAS PubMed.
- X. Zhao, Z. Zhao-Karger, D. Wang and M. Fichtner, Angew. Chem., Int. Ed. Engl., 2013, 52, 13621–13624 CrossRef CAS PubMed.
- M. Anji Reddy and M. Fichtner, J. Mater. Chem., 2011, 21, 17059–17062 RSC.
- A. Ponrouch, C. Frontera, F. Barde and M. R. Palacin, Nat. Mater., 2016, 15, 169–172 CrossRef CAS PubMed.
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H. M. Cheng, Nat. Chem., 2018, 10, 667–672 CrossRef CAS PubMed.
- V. K. Davis, C. M. Bates, K. Omichi, B. M. Savoie, N. Momcilovic, Q. Xu, W. J. Wolf, M. A. Webb, K. J. Billings, N. H. Chou, S. Alayoglu, R. K. McKenney, I. M. Darolles, N. G. Nair, A. Hightower, D. Rosenberg, M. Ahmed, C. J. Brooks, T. F. Miller III, R. H. Grubbs and S. C. Jones, Science, 2018, 362, 1144–1148 CrossRef CAS PubMed.
- J. H. Kennedy and J. C. Hunter, J. Electrochem. Soc., 1976, 123, 10–14 CrossRef CAS.
- J. Schoonman, J. Electrochem. Soc., 1976, 123, 1772–1775 CrossRef CAS.
- Y. Danto, G. Poujade, J. Pistre, C. Lucat and J. Salardenne, Thin Solid Films, 1978, 55, 347–354 CrossRef CAS.
- J. Schoonman, K. E. D. Wapenaar, G. Oversluizen and G. J. Dirksen, J. Electrochem. Soc., 1979, 126, 709–713 CrossRef CAS.
- P. Hagenmuller, J.-M. Réau, C. Lucat, S. Matar and G. Villeneuve, Solid State Ionics, 1981, 3, 341–345 CrossRef.
- J. Schoonman and A. Wolfert, Solid State Ionics, 1981, 3, 373–379 CrossRef.
- I. Kosacki, Appl. Phys. A, 1989, 49, 413–424 CrossRef.
- K.-i. Okazaki, Y. Uchimoto, T. Abe and Z. Ogumi, ACS Energy Lett., 2017, 2, 1460–1464 CrossRef CAS.
- D. T. Thieu, M. H. Fawey, H. Bhatia, T. Diemant, V. S. K. Chakravadhanula, R. J. Behm, C. Kübel and M. Fichtner, Adv. Funct. Mater., 2017, 27, 1701051 CrossRef.
- M. A. Reddy and M. Fichtner, in Advanced Fluoride-Based Materials for Energy Conversion, ed. T. Nakajima and H. Groult, Elsevier, 2015, pp. 51–76, DOI:10.1016/b978-0-12-800679-5.00003-8.
- F. Gschwind, G. Rodriguez-Garcia, D. J. S. Sandbeck, A. Gross, M. Weil, M. Fichtner and N. Hörmann, J. Fluorine Chem., 2016, 182, 76–90 CrossRef CAS.
- M. H. Fawey, V. S. K. Chakravadhanula, A. R. Munnangi, C. Rongeat, H. Hahn, M. Fichtner and C. Kübel, J. Power Sources, 2020, 466, 228283 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- L. Zhang, M. A. Reddy, P. Gao, T. Diemant, R. Jürgen Behm and M. Fichtner, J. Solid State Electrochem., 2016, 21, 1243–1251 CrossRef.
- C. Rongeat, M. A. Reddy, R. Witter and M. Fichtner, J. Phys. Chem. C, 2013, 117, 4943–4950 CrossRef CAS.
- C. Rongeat, M. Anji Reddy, T. Diemant, R. J. Behm and M. Fichtner, J. Mater. Chem. A, 2014, 2, 20861–20872 RSC.
- L. Zhang, M. A. Reddy and M. Fichtner, J. Solid State Electrochem., 2017, 22, 997–1006 CrossRef.
- I. Mohammad, J. Chable, R. Witter, M. Fichtner and M. A. Reddy, ACS Appl. Mater. Interfaces, 2018, 10, 17249–17256 CrossRef CAS PubMed.
- A. Grenier, A.-G. Porras-Gutierrez, H. Groult, K. A. Beyer, O. J. Borkiewicz, K. W. Chapman and D. Dambournet, J. Mater. Chem. A, 2017, 5, 15700–15705 RSC.
- A. Grenier, A. G. Porras-Gutierrez, M. Body, C. Legein, F. Chrétien, E. Raymundo-Piñero, M. Dollé, H. Groult and D. Dambournet, J. Phys. Chem. C, 2017, 121, 24962–24970 CrossRef CAS.
- I. Mohammad, R. Witter, M. Fichtner and M. A. Reddy, ACS Appl. Energy Mater., 2019, 2, 1553–1562 CrossRef CAS.
- X. Hou, Z. Zhang, K. Shen, S. Cheng, Q. He, Y. Shi, D. Y. W. Yu, C.-y. Su, L.-J. Li and F. Chen, J. Electrochem. Soc., 2019, 166, A2419–A2424 CrossRef CAS.
- Z. Zhang, X. Hu, Y. Zhou, S. Wang, L. Yao, H. Pan, C.-Y. Su, F. Chen and X. Hou, J. Mater. Chem. A, 2018, 6, 8244–8250 RSC.
- T. Yamanaka, H. Nakamoto, T. Abe, K. Nishio and Z. Ogumi, ACS Appl. Energy Mater., 2019, 2, 3092–3097 CrossRef CAS.
- A. Grenier, A. G. Porras Gutierrez, H. Groult and D. Dambournet, J. Fluorine Chem., 2016, 191, 23–28 CrossRef CAS.
- E. A. Owen and E. L. Yates, Philos. Mag., 1933, 15, 472–488 CAS.
- P. Fischer, W. Hälg, D. Schwarzenbach and H. Gamsjäger, J. Phys. Chem. Solids, 1974, 35, 1683–1689 CrossRef CAS.
- J. Zemann, Acta Crystallogr., 1965, 18, 139 CrossRef.
- J. E. Jorgensen and R. I. Smith, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 987–992 CrossRef CAS PubMed.
- M. M. R. Costa, J. A. Paixão, M. J. M. de Almeida and L. C. R. Andrade, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 591–599 CrossRef.
- M. A. Hepworth, K. H. Jack, R. D. Peacock and G. J. Westland, Acta Crystallogr., 1957, 10, 63–69 CrossRef CAS.
- P. Molaiyan and R. Witter, Mater. Des. Process. Commun., 2019, 1, e44 CAS.
- C. Rongeat, M. A. Reddy, R. Witter and M. Fichtner, ACS Appl. Mater. Interfaces, 2014, 6, 2103–2110 CrossRef CAS.
- A. Roos, F. Vandepol, R. Keim and J. Schoonman, Solid State Ionics, 1984, 13, 191–203 CrossRef CAS.
- Y. Orikasa, T. Maeda, Y. Koyama, H. Murayama, K. Fukuda, H. Tanida, H. Arai, E. Matsubara, Y. Uchimoto and Z. Ogumi, Chem. Mater., 2013, 25, 1032–1039 CrossRef CAS.
- G. Jain, M. Balasubramanian and J. J. Xu, Chem. Mater., 2006, 18, 423–434 CrossRef CAS.
- G. R. Jain, J. S. Yang, M. Balasubramanian and J. J. Xu, Chem. Mater., 2005, 17, 3850–3860 CrossRef CAS.
- X. Q. Wang, J. C. Hanson, A. I. Frenkel, J. Y. Kim and J. A. Rodriguez, J. Phys. Chem. B, 2004, 108, 13667–13673 CrossRef CAS.
- J. Haruyama, K. I. Okazaki, Y. Morita, H. Nakamoto, E. Matsubara, T. Ikeshoji and M. Otani, ACS Appl. Mater. Interfaces, 2020, 12, 428–435 CrossRef CAS PubMed.
- C.-X. Zu and H. Li, Energy Environ. Sci., 2011, 4, 2614–2624 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta08824b |
| This journal is © The Royal Society of Chemistry 2021 |