
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shear-induced alignment of block copolymer worms in mineral oil†
Matthew J.
Derry‡
 *,
Oleksandr O.
Mykhaylyk
and
Steven P.
Armes
*,
Oleksandr O.
Mykhaylyk
and
Steven P.
Armes
Department of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, S3 7HF, UK. E-mail: m.derry@aston.ac.uk
First published on 15th September 2021
Abstract
Poly(stearyl methacrylate)–poly(benzyl methacrylate) [PSMA–PBzMA] diblock copolymer worms were synthesized directly in mineral oil via reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization at 90 °C. Free-standing gels were obtained from this polymerization-induced self-assembly (PISA) formulation when targeting PSMA13–PBzMA65 dispersions at 5% w/w to 20% w/w copolymer concentration. Gel permeation chromatography (GPC) studies indicated that almost identical copolymer chains were obtained in all cases, while transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS) studies confirmed that highly anisotropic worms were formed with mean cross-sectional diameters of 11.9–13.1 nm. These worms undergo a thermoreversible worm-to-sphere transition on heating up to 150 °C. Rheological studies were conducted to characterize the shear rate- and concentration-dependent behaviour caused by this change in copolymer morphology, where the initial shear-thinning worm gels form spheres (i.e. a Newtonian fluid) on heating up to 150 °C. Complementary shear-induced polarized light imaging (SIPLI) experiments confirmed the formation of aligned linear worms under applied shear between 80 °C and 110 °C, with high-viscosity dispersions of branched worms being obtained at 20–60 °C and low-viscosity spheres being produced at 150 °C. This study informs the use of such block copolymer worms as rheology modifiers for non-polar oils, which is of potential interest for the automotive industry.
Introduction
Block copolymer worms have been known for several decades.1–3 Traditionally, they have been prepared using a post-polymerization processing protocol via either a solvent switch4,5 or by direct dissolution.1 Their highly anisotropic morphology leads to relatively high solution viscosity.1,6 Indeed, block copolymer worms have been recently evaluated as thickeners for silicone oils.7 Above a certain critical copolymer concentration, block copolymer worms form free-standing gels.8 In the case of aqueous worm gels, this has led to various potential biomedical applications as 3D matrices for cell culture,9–11 cryopreservation of red blood cells,12 or the long-term storage of human stem cells.13 Alternatively, Discher and co-workers examined degradable block copolymer worms in the context of drug delivery applications.14–16Over the past decade or so, polymerization-induced self-assembly (PISA) has become widely recognized as a powerful platform technology for the rational synthesis of block copolymer nano-objects in the form of concentrated colloidal dispersions.17–21 PISA has proved to be particularly useful for the identification of suitable diblock copolymer compositions and synthesis conditions that enable the formation of purely linear worms in various solvents.22,23 More specifically, various diblock copolymer worm gels have been prepared via PISA syntheses conducted in water,17 lower alcohols18,19 and n-alkanes18 using reversible addition–fragmentation chain transfer (RAFT)-mediated polymerization.24–26
For example, we reported the synthesis of poly(stearyl methacrylate)–poly(benzyl methacrylate) [PSMA–PBzMA] diblock copolymer spheres, worms or vesicles directly in mineral oil via RAFT dispersion polymerization. The construction of a pseudo-phase diagram for this PISA formulation indicated a very narrow worm phase.27 Nevertheless, a pure worm morphology could be accessed at copolymer concentrations ranging from 5.0 to 20% w/w when targeting PSMA13–PBzMA65. Similarly, poly(lauryl methacrylate)–poly(benzyl methacrylate) [PLMA–PBzMA] worms can be prepared in n-heptane,28n-dodecane,23 mineral oil29 or a poly(α-olefin).29 Interestingly, a thermoreversible worm-to-sphere transition is observed on heating such dispersions. This is caused by surface plasticization of the core-forming PBzMA block by ingress of hot solvent. This leads to a subtle change in the relative volume fractions of the solvophobic and solvophilic blocks and thus the preferred diblock copolymer morphology.23,30,31 Although it has been demonstrated that these PSMA–PBzMA worm-like nanoparticles can act as a thickener for mineral oil,30,31 their shear-dependent behavior has not been investigated. It is well-known that such block copolymer worms become aligned when subjected to an applied shear,32 and in the case of worm gels this typically leads to shear-induced flow.33–35 Such worm alignment can be studied by combining scattering techniques such as small-angle X-ray scattering (SAXS) with rheological experiments (in particular, using a Couette shear cell).33,34
Above a certain critical shear rate, worm gels display shear-thinning behavior owing to alignment of these anisotropic particles in the direction of the flow.11,22 This relationship between viscosity and shear alignment can be assessed using an opto-rheological technique known as shear-induced polarized light imaging (SIPLI).36–39 SIPLI uses crossed polarizers to monitor the interaction of a sample with linearly polarized white light, with the presence of a Maltese cross motif in the resulting polarized light images (PLIs) indicating sample birefringence owing to structural morphology alignment. Originally, this technique was developed to monitor the shear-induced crystallization of thermoplastic polymers.36,37 In this case, birefringence occurs as a result of the alignment of stretched polymer chains at the onset of shear flow, followed by nucleation of oriented fibrils formed by the polymer chains. More recently, SIPLI has been used to examine the shear alignment of native silk proteins,38 diblock copolymer lamellae,39,40 worm-like micelles comprising small molecule gelators,41 and, of particular relevance to the present work, poly(glycerol monomethacrylate)-poly(2-hydroxypropyl methacrylate) worms dispersed in aqueous media.22
Herein, the rheological properties of PSMA13–PBzMAx diblock copolymer worm gels prepared in mineral oil are assessed over a range of copolymer concentrations. This enables important physical properties such as the critical gelation temperature (CGT) and the critical gelation concentration (CGC) to be determined. Moreover, the shear-induced alignment of these worm gels is monitored using the SIPLI technique as a function of temperature, shear rate and copolymer concentration.
Experimental
Materials
Stearyl methacrylate (SMA) and benzyl methacrylate (BzMA) were each purchased from Sigma-Aldrich (UK) and passed in turn through a basic alumina column to remove inhibitor prior to use. tert-Butyl peroxy-2-ethylhexanoate (T21s) initiator was purchased from AkzoNobel (The Netherlands). Cumyl dithiobenzoate (CDB), CDCl3, and all other reagents were purchased from Sigma-Aldrich (UK) and were used as received, unless otherwise noted. THF, n-heptane and toluene were purchased from Fisher Scientific (UK), CD2Cl2 was purchased from Goss Scientific (UK) and industrial-grade mineral oil was provided by The Lubrizol Corporation (UK). All solvents were used as received.Synthesis of the poly(stearyl methacrylate) precursor
The synthesis of the PSMA13 precursor used in this work, via RAFT solution polymerization in toluene using CDB at 70 °C, has been described previously.27 THF gel permeation chromatography (GPC) studies indicated an Mn of 5700 g mol−1 and an Mw/Mn of 1.17, while the mean degree of polymerization (DP) was determined to be 13 via end-group analysis using 1H NMR spectroscopy.Synthesis of poly(stearyl methacrylate)–poly(benzyl methacrylate) diblock copolymer worms
The PISA synthesis of the PSMA13–PBzMAx diblock copolymer worms used in this study has been described previously.27 Specifically, a typical RAFT dispersion polymerization synthesis of PSMA13–PBzMA65 worms in mineral oil targeting 20% w/w solids was conducted as follows: a 10 mL round-bottomed flask was charged with benzyl methacrylate (BzMA; 0.368 g; 2.09 mmol), T21s initiator (1.39 mg; 6.42 μmol; dissolved at 10.0% v/v in mineral oil), the PSMA13 precursor (0.15 g; 32.1 μmol; PSMA13/T21s molar ratio = 5.0) and mineral oil (2.07 g). The reaction mixture was purged with nitrogen gas for 30 min and the deoxygenated solution was then placed in a pre-heated oil bath at 90 °C for 5 h (final BzMA conversion = 98%; Mn = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1, Mw/Mn = 1.15).
700 g mol−1, Mw/Mn = 1.15).
Gel permeation chromatography
Molecular weight distributions (MWDs) were assessed by GPC using THF eluent. The GPC instrument was equipped with two 5 μm (30 cm) Mixed C columns and a WellChrom K-2301 refractive index detector operating at 950 ± 30 nm. The mobile phase contained 2.0% v/v triethylamine and 0.05% w/v butylhydroxytoluene (BHT) with a toluene flow rate marker and the flow rate was fixed at 1.0 mL min−1. A series of ten near-monodisperse poly(methyl methacrylate) standards (Mp values ranging from 645 to 2480000 g mol−1) were used for calibration.
1H NMR spectroscopy
1H NMR spectra were recorded in either CD2Cl2 or CDCl3 using a Bruker AV1-400 or AV1-250 MHz spectrometer. Typically, sixty-four scans were averaged per spectrum.Transmission electron microscopy
Transmission electron microscopy (TEM) studies were conducted using a Philips CM 100 instrument operating at 100 kV and equipped with a Gatan 1k CCD camera. Diluted block copolymer solutions (0.10% w/w) were placed on carbon-coated copper grids and exposed to ruthenium(VIII) oxide vapor for 7 min at 20 °C prior to analysis.42 This heavy metal compound acted as a positive stain for the core-forming PBzMA block to enhance electron contrast. The ruthenium(VIII) oxide was prepared as follows: ruthenium(IV) oxide (0.30 g) was added to water (50 g) to form a black slurry; addition of sodium periodate (2.0 g) with stirring produced a yellow solution of ruthenium(VIII) oxide within 1 min.Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) patterns were recorded at 20 °C using a synchrotron source (Diamond Light Source, station I22, Didcot, UK). A monochromatic X-ray radiation (wavelength λ = 0.1001 nm) and a 2D Pilatus 2M detector (Dectris, Switzerland) were used for the experiments. The SAXS instrument was configured to cover the q range from 0.015 nm−1 to 1.5 nm−1, where q = 4πsinθ/λ is the modulus of the scattering vector and θ is one half of the scattering angle. A 2.0 mm round glass capillary was used as a sample holder. X-ray scattering data were reduced (integration and calibration) using Dawn software version 1.7.143 and were further analyzed (background subtraction and data modelling) using the Irena SAS macro for Igor Pro.44 SAXS experiments were conducted on 1.0% w/w dispersions of PSMA13–PBzMAx nano-objects in mineral oil.
Oscillatory rheology studies
A TA Instruments AR-G2 rheometer equipped with a variable temperature Peltier plate and a 40 mm 2° aluminum cone was used for all nanoparticle dispersion measurements. The storage (G′) and loss (G′′) moduli were determined as a function of (i) angular frequency (from 0.1 to 100 rad s−1 at 20 °C with a fixed strain of 1.0%), and (ii) temperature (from 20 to 90 °C at 5 °C intervals with a fixed strain of 1.0% and an angular frequency of 10 rad s−1). During thermal cycles, 5 min was allowed for thermal equilibration prior to each measurement. A TA Instruments Discovery HR-3 rheometer equipped with a variable temperature Peltier plate and a 60 mm 2° aluminum cone was used to acquire the viscosity-temperature profile for mineral oil on heating from 20 °C to 147 °C at 5 °C min−1.Shear-induced polarized light imaging
A mechano-optical rheometer (Anton Paar Physica MCR301 with SIPLI attachment) was used for all rotational shear alignment experiments. These measurements were performed using a plate–plate geometry composed of a 25 mm polished steel (top rotating) plate and a fused quartz (bottom fixed) plate connected to a variable temperature Peltier system. The gap between plates was set at 1 mm and an additional Peltier hood was used to control the sample temperature. The dispersion viscosity was determined as a function of temperature at a heating/cooling rate of 2.0 °C min−1 and a constant angular speed of 0.08 rad s−1, 0.8 rad s−1 or 8.0 rad s−1, which provides a maximum (sample edge) shear rate of 1, 10 or 100 s−1, respectively. An Edmund Optics 150 W MI-150 high intensity fiber-optic white light source was used for sample illumination. The polarizer and analyzer axes were crossed at 90° to obtain PLIs), which were recorded using a color CCD camera (Lumenera Lu165c).Results and discussion
The PSMA13 precursor was chain-extended via RAFT dispersion polymerization of BzMA at 90 °C targeting PSMA13–PBzMA65 worms (see Scheme 1). Highly anisotropic worms were obtained over a range of copolymer concentrations (from 5.0 to 20% w/w solids) and analyzed using 1H NMR spectroscopy, GPC, TEM and SAXS (see Table 1 and Fig. 1, 2). | ||
| Scheme 1 Synthesis of targeted poly(stearyl methacrylate)13–poly(benzyl methacrylate)65 (PSMA13–PBzMA65) worms via RAFT dispersion polymerization of BzMA in mineral oil at 90 °C. Final copolymer concentrations ranged from 5.0 to 20% w/w solids. | ||
| Copolymer conc. (% w/w) | BzMA conversiona (%) | Actual copolymer compositiona | M n (g mol−1) | M w/Mnb | Copolymer morphologycd | D wc ± σDwcd (nm) |
|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by THF GPC against poly(methyl methacrylate) standards. c Assigned by TEM. d Determined by fitting representative SAXS patterns (obtained for 1.0% w/w copolymer dispersions in mineral oil) to an established worm-like micelle model.48 | ||||||
| — | — | PSMA13 | 4900 | 1.17 | — | — |
| 5.0 | 98 | PSMA13–PBzMA64 | 13300 |
1.19 | Worms | 13.0 ± 1.4 |
| 7.5 | 96 | PSMA13–PBzMA62 | 12200 |
1.19 | Worms | 13.1 ± 1.4 |
| 10 | 97 | PSMA13–PBzMA63 | 13100 |
1.17 | Worms | 12.0 ± 1.2 |
| 15 | 98 | PSMA13–PBzMA64 | 12800 |
1.16 | Worms | 12.6 ± 1.4 |
| 20 | 98 | PSMA13–PBzMA64 | 12700 |
1.15 | Worms | 11.9 ± 1.3 |
 | ||
| Fig. 1 Representative transmission electron micrograph recorded for a 0.10% w/w dispersion of PSMA13–PBzMA63 worms prepared in mineral oil at 10% w/w solids. | ||
 | ||
| Fig. 2 Representative SAXS patterns recorded at 20 °C for 1.0% w/w dispersions of PSMA13–PBzMAx worms prepared in mineral oil at 5.0% w/w (black symbols), 7.5% w/w (red symbols), 10% w/w (blue symbols), 15% w/w (green symbols) or 20% w/w (orange symbols). The white lines for each SAXS pattern indicate the data fit obtained when using an established worm-like micelle model (eqn (S1)–(S8), ESI†).48 Each pattern and its corresponding data fit is offset by the stated arbitrary factor for the sake of clarity. | ||
GPC analysis confirms the synthesis of the targeted PSMA13–PBzMA65 diblock copolymers with very similar identical molecular weight distributions being obtained for each copolymer concentration (see Fig. S1, ESI†). Moreover, comparison to the GPC curve recorded for the PSMA13 precursor indicates that a relatively high blocking efficiency was achieved in each case. TEM studies confirm that the target PSMA13–PBzMA65 diblock copolymers form a highly anisotropic worm-like morphology, see Fig. 1.
SAXS analysis was also conducted to confirm the worm morphology (Fig. 2). This is because TEM only enables the copolymer morphology to be assessed for a few hundred nanoparticles after removing all traces of solvent under ultrahigh vacuum. In contrast, SAXS is averaged over millions of nanoparticles within the ‘wet’ dispersion, thus providing a more statistically robust assessment. All SAXS patterns exhibited at low q a scattered intensity gradient of approximately −1, which is consistent with a highly anisotropic worm morphology.23,45,46 Moreover, the local minimum observed at q ∼ 0.5 nm−1 in each case indicated a mean worm core diameter (Dwc) ranging between 11.9 and 13.1 nm (see Table 1). However, the SAXS patterns recorded for worms prepared at 15 and 20% w/w deviate from the worm-like micelle model at low q. This suggests the presence of branched worms formed when higher copolymer concentrations are targeted during synthesis,22 which is important when considering the shear alignment of such anisotropic nanoparticles.
These SAXS patterns were recorded over a relatively limited q range of ∼0.015–1.5 nm−1. Thus, scattering angles required for measuring the mean worm contour length (Lw), which is typically indicated by a subtle feature at q < 0.01 nm−1, were not resolved in the experiment. Furthermore, if sufficient worm branching exists this Lw feature may not be observed.47 For PLMA16–PBzMA37 worms prepared in n-dodecane via RAFT dispersion polymerization,23Lw was found to be 591 nm from SAXS patterns recorded over a wider q range (0.005 nm−1 < q < 1.6 nm−1), with this analysis requiring the combination of data collected at both short (for 0.08 nm−1 < q < 1.6 nm−1) and long (for 0.005 nm−1 < q < 0.12 nm−1) sample-to-detector distances.49 In principle, a population of branched worms within worm dispersions prepared at 10.0% w/w solids or below (Fig. 2) may not be experimentally observable owing to the limited q range used for the SAXS experiments reported herein.
Rheological properties of PSMA13–PBzMAx worm gels
Prior studies of the PISA synthesis of closely-related PLMA–PBzMA diblock copolymer nano-objects indicated that pure worms could only be prepared at copolymer concentrations of at least 17.5% w/w.23,28,29 However, the PSMA13–PBzMAx PISA formulation reported herein provides access to a pure worm phase at copolymer concentrations as low as 5.0% w/w.27 Worm gels are formed in all cases, as indicated by the minimal angular frequency dependence of the storage modulus (G′) and loss modulus (G′′) exhibited by each dispersion (see Fig. S2, ESI†). Fielding and co-workers found23 that a 20% w/w PLMA16-PBzMA37 worm gel in n-dodecane was transformed on heating into a free-flowing fluid via a worm-to-sphere transition. Both SAXS and rheological studies indicated good thermoreversibility for such worm dispersions at (or above) 5.0% w/w solids.23 Similarly, PSMA13–PBzMAx worm gels also display thermoresponsive behavior: degelation occurs on heating up to 90 °C as judged by oscillatory rheology studies (Fig. 3). | ||
| Fig. 3 Variation of storage modulus (G′) with temperature for PSMA13–PBzMAx worm gels (cone and plate geometry; 1.0% applied strain at an angular frequency of 10 rad s−1; data recorded at 5 °C intervals, with 5 min being allowed for thermal equilibration before each measurement). The critical gelation temperature (CGT) for each worm gel is indicated by a star symbol and associated arrow for each data set. Full data sets containing G′ and G′′ are shown in Fig. S3 (ESI†). The inset graph shows G′ measured at 20 °C for each worm gel vs. copolymer concentration. | ||
Formally, degelation occurs when the loss modulus, G′′, exceeds the storage modulus, G′.2,3 Thus, the critical gelation temperature (CGT) can be defined as the temperature above which G′ < G′′.
On lowering the copolymer concentration from 20 to 5.0% w/w, the CGT is reduced from 65 to 57 °C. Moreover, a monotonic reduction in G′ is observed for worm gels synthesized at progressively lower concentration. More specifically, the G′ value determined at 20 °C is reduced from ∼104 at 20% w/w solids to ∼102 Pa at 5.0% w/w solids. This is because macroscopic gelation involves multiple inter-worm contacts, which results in the formation of a 3D percolating network.8,50,51 Reducing the copolymer concentration from 20 to 5.0% w/w leads to significantly fewer inter-worm contacts and hence correspondingly weaker gels. This also accounts for the subtle reduction in CGT observed on lowering the copolymer concentration. Thus, fewer inter-worm contacts enables degelation to occur earlier during the worm-to-sphere transformation (i.e. at lower temperatures). However, other parameters such as worm length, worm core diameter (Dwc) and worm stiffness also affect the physical properties of worm gels.7,8
The G′ value observed at 90 °C is much greater than that expected for a free-flowing dispersion of purely spherical nanoparticles. This suggests that the worm-to-sphere transition is not complete at 90 °C, particularly at higher copolymer concentrations. In fact, a dispersion of spherical nanoparticles should have a comparable viscosity to that of the solvent alone.46,52 Indeed, Fielding and co-workers reported that the morphological transformation from worms to spheres was only complete after heating PLMA16–PBzMA37 worms in n-dodecane up to 160 °C.10 In this case, Lw was reduced from ∼600 nm at 20 °C to ∼350 nm at 90 °C, so the corresponding worm aspect ratio (Lw/Dwc) was only reduced by ∼50% at this lower temperature.
For a 20% w/w dispersion of such PLMA16–PBzMA37 worms, the CGT was determined to be ∼47 °C. The CGTs observed for PSMA13–PBzMAx worm gels (Fig. 3) are significantly higher than those reported for the PLMA16–PBzMA37 worm gel. One plausible explanation is that the PSMA13–PBzMAx worms are longer than the PLMA16–PBzMA37 worms. If this is correct, then a greater reduction in Lw would be required to lower the number of inter-worm contacts sufficiently to induce degelation. However, this hypothesis could not be examined owing to the limited q range available for the SAXS experiments. Alternatively, surface plasticization of the core-forming PBzMA block may simply occur at higher temperatures for PSMA–PBzMA worms in mineral oil compared to PLMA–PBzMA worms in n-dodecane.
The CGC for the 20% w/w PSMA13–PBzMA64 worm gel was determined via serial dilution using the tube inversion method.23 The CGC can be defined as the lowest concentration at which a free-standing gel can be obtained at any given temperature.3 For PSMA13–PBzMA64 worms prepared at 20% w/w, the CGC was determined to be ∼4.0% w/w at 20 °C (Fig. 4), which is significantly lower than that reported for various PLMA–PBzMA worms in n-dodecane, mineral oil or a poly(α-olefin).23,29 Again, the most likely explanation for this observation is that the PSMA13–PBzMA64 worms are longer than the PLMA–PBzMA worms, which would lead to a larger number of inter-worm contacts and thus afford free-standing gels at a lower copolymer concentration.8 Although these studies were conducted on the 20% w/w dispersion of PSMA13–PBzMA64 worms, similar CGC values are expected for the PSMA13–PBzMAx worms prepared at lower copolymer concentrations in mineral oil because (i) the copolymer chains are of comparable molecular weight as judged by GPC analysis (see Fig. S1, ESI†), and (ii) SAXS analysis indicates the formation of comparable worm-like nanoparticles (Fig. 2). Moreover, the PSMA13–PBzMA64 worms prepared at 5.0% w/w formed a free-standing gel, which indicates that the CGC for this gel must be below this copolymer concentration.
 | ||
| Fig. 4 Digital images of PSMA13–PBzMA64 worm dispersions in mineral oil. Serial dilution of the as-synthesized 20% w/w copolymer dispersion enables the critical gelation concentration (CGC) to be estimated using the tube inversion method. | ||
Shear-induced alignment of PSMA13–PBzMAx worms
To investigate the shear-induced alignment of PSMA13–PBzMAx worms, the 20% w/w PSMA13–PBzMA64 worm gel was analyzed using SIPLI.36,53 The apparent turbidity of this worm gel (see Fig. 4) is accentuated by the relatively long path length (∼12 mm) of the sample vial. In practice, such dispersions are sufficiently transparent to enable SIPLI studies (for which the effective path length is 2 mm). For all variable temperature measurements, this sample was loaded onto the fused quartz plate pre-heated to 150 °C in order to erase the thermal history of the gel: the worms are transformed into spheres at this temperature and the latter are unable to form a 3D network.23 The effective viscosity of the dispersion (η) was measured during a 150–20–150 °C thermal cycle conducted at a cooling/heating rate of 2 °C min−1 and using a maximum (sample edge) shear rate of 1, 10 or 100 s−1. Reference data for mineral oil alone are provided in Fig. S5 (ESI†). At 1 s−1 (Fig. 5), no birefringence was detected at 150 °C because only isotropic spherical micelles exist at this temperature. This is consistent with effective solution viscosities of ∼0.01 Pa s, which are characteristic of a free-flowing Newtonian fluid.52 On cooling, the dispersion became gradually more viscous, indicating the formation of anisotropic worms from the stochastic fusion of multiple spheres.23 The dispersion viscosity rapidly increased on cooling from 120 °C to 90 °C, suggesting that progressively longer worms were being formed. The first distinct Maltese cross motif was observed at 100 °C, confirming that the worms present at this temperature are sufficiently aligned to cause birefringence at a shear rate at or below 1 s−1. Interestingly, birefringence was observed across the entire sample, suggesting a very low critical shear rate for the worm alignment. Such shear-induced alignment is observed on cooling from 100 °C to 60 °C as indicated by the persistence of the Maltese cross over this temperature range. The dispersion viscosity continues to increase between 90 °C and 65 °C (albeit more slowly than in the 90–120 °C interval), suggesting higher worm aspect ratios over this temperature range. Between 65 °C and 35 °C, the relatively rapid increase in dispersion viscosity suggests further evolution in the copolymer morphology and/or inter-worm contacts, which ultimately generate the 3D gel network. For example, the presence of branched worms should increase the probability of inter-worm contacts, leading to more viscous dispersions. Interestingly, the Maltese cross pattern almost completely disappears on cooling to 55 °C. This observation is consistent with the formation of branched worms (or a transient worm network) since such structures are likely to oppose the alignment and, as a result, prevent material birefringence. Nevertheless, a modest degree of residual alignment remains visible in the PLIs recorded between 55 °C and 20 °C (Fig. 5). Given the relatively high viscosity of the 20% w/w dispersion over this temperature range, it is possible that partially-aligned structures may become ‘trapped’ at the shear geometry centre, thus preventing the fully disordered state of the branched worm network. In this case, it is likely that branched worms form an infinite network that becomes stretched at low shear rates (i.e. towards the centre of the rotating plate). Such partial alignment leads to birefringence owing to stress along the flow direction. However, network degradation most likely occurs at higher shear rates (i.e. towards the edge of the rotating plate). Thus, the branched worms are able to rotate, which reduces the stress and thus leads to weaker birefringence. In this context, we note that no stress birefringence (orientation) was observed for related aqueous branched worm systems.22 Immediately after the 150 °C to 20 °C temperature ramp shown in Fig. 5, the same 20% w/w dispersion was heated from 20 °C to 150 °C at 2 °C min−1 using a constant maximum shear rate of 1 s−1 (see Fig. 5). On initial inspection, the dispersion viscosity-temperature profiles obtained for the cooling and heating ramps appear to mirror one another. However, there are subtle differences in the characteristic temperatures at which changes in viscosity occur. The first major difference is the viscosity maximum observed at ∼45 °C during the heating step. This feature occurs well before any evidence for shear alignment at 55 °C, suggesting that it may correspond to the transition from branched worms to linear worms. The PLIs obtained on heating from 20 °C to 150 °C shown in Fig. 5 indicate that shear alignment occurs between 55 °C and 90 °C, which differs from the 60–100 °C range for which shear alignment is observed during the cooling ramp. Similar hysteresis has been reported for PLMA16–PBzMA37 worms in n-dodecane.23 On the other hand, the pronounced shoulder in the viscosity profile occurs at ∼100 °C in both cases. This is presumably because the number of spheres increases to the point where this population dictates the dispersion viscosity. The final effective viscosity of the 20% w/w dispersion at 150 °C (η ∼ 0.01 Pa s) is the same as that observed at the beginning of the variable-temperature study. This indicates good thermoreversibility for the sphere-to-worm-to-sphere transition on the timescale of these SIPLI experiments. | ||
| Fig. 5 Dispersion viscosity-temperature profile and corresponding polarized light images (PLIs) obtained for a 20% w/w dispersion of PSMA13–PBzMA64 nano-objects on cooling from 150 °C to 20 °C (blue data, left of the dashed vertical line) and on heating from 20 °C to 150 °C (red data, right of the dashed vertical line) at a rate of 2 °C min−1 when employing a constant maximum (sample edge) shear rate of 1 s−1. Selected PLIs represent the sample birefringence observed at various temperatures (see Fig. S4 for additional PLIs, ESI†). Arrows show the planes of polarization for the polarizer (P) and the analyzer (A), crossed at 90°. A Maltese cross motif indicates shear-induced alignment of anisotropic objects, whereas its absence indicates either no alignment or no anisotropic objects. All PLIs recorded during the cooling cycle are shown in Fig. S1 (ESI†). The diameter of the sample is 25 mm. | ||
Further investigation of the shear alignment and dispersion viscosity of PSMA13–PBzMAx nano-objects in mineral oil involved varying the maximum shear rate employed during these SIPLI experiments. The same 20% w/w dispersion of PSMA13–PBzMA64 nano-objects was used to conduct comparable variable-temperature SIPLI experiments at maximum shear rates of 10 and 100 s−1, and these data were compared to that obtained at 1 s−1 (Fig. 6). As expected, higher shear rates yielded less viscous dispersions owing to their shear-thinning nature.54,55 This is particularly evident in Fig. 6, where the dispersion viscosity, η ∼ 345 Pa s at 20 °C at a maximum shear rate of 1 s−1, whereas η ∼ 40 Pa s at 10 s−1 and η ∼ 4 Pa s at 100 s−1 at the same temperature. However, when free-flowing dispersions of spheres are formed at 150 °C, the dispersion viscosity becomes essentially independent of shear rate, indicating Newtonian behavior.52,56 Interestingly, there are subtle differences for the temperature ranges over which shear alignment is observed for the same dispersion at maximum shear rates of 1, 10 and 100 s−1. Nevertheless, birefringence is consistently observed between 95 °C and 75 °C during the cooling ramp and between 65 °C and 90 °C during the heating ramp.
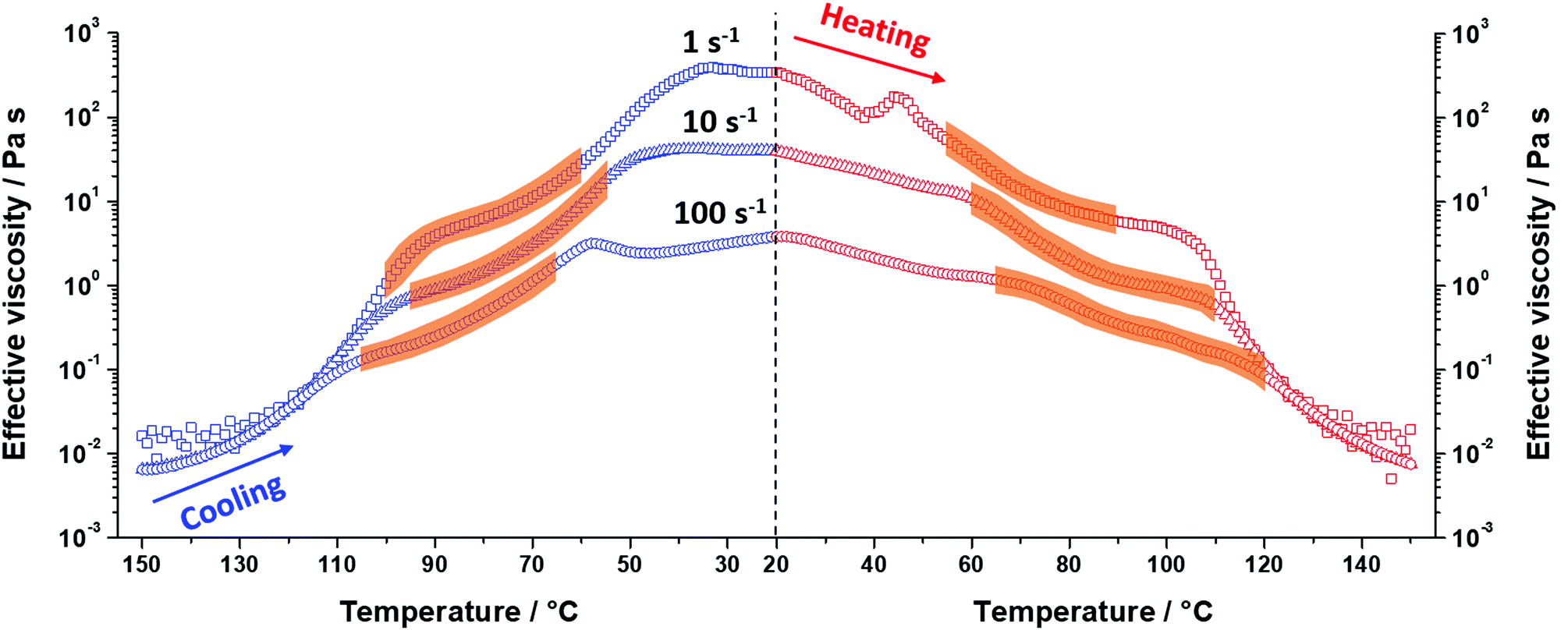 | ||
| Fig. 6 Combination of the dispersion viscosity-temperature profiles recorded for the cooling (blue data, left of dashed vertical line) and heating (red data, right of dashed vertical line) temperature ramps (cooling/heating rate = 2 °C min−1) for a 20% w/w dispersion of PSMA13–PBzMA64 nano-objects at the maximum (sample edge) shear rate of 1 s−1 (squares), 10 s−1 (triangles) or 100 s−1 (circles). The orange-shaded regions represent the temperature ranges over which sample birefringence was observed. | ||
Thus far, only PSMA13–PBzMA64 worms synthesized at 20% w/w have been analyzed using the SIPLI technique. Hence PSMA13–PBzMAx worm gels prepared at 10 and 15% w/w were compared to this 20% w/w dispersion. The same thermal cycle was used in these experiments and the maximum shear rate was 100 s−1 for all measurements (Fig. 7). As expected, the more concentrated dispersions are more viscous at 20 °C. The same features can be identified in the viscosity-temperature profiles recorded for the 15 and 10% w/w dispersions as for those observed for the 20% w/w dispersion. An initial low-viscosity fluid (η ∼ 0.01 Pa s) at 150 °C became progressively more viscous on cooling, indicating the formation of anisotropic worms. This interpretation is consistent with the observation of sample birefringence, as indicated by the characteristic Maltese cross motif. A further increase in viscosity (η ∼ 1–3 Pa s) suggests the formation of branched worms, which explains the subsequent loss of sample birefringence. There is significant overlap for the temperature ranges over which sample birefringence is observed for these dispersions, but there are subtle differences in the onset temperatures. Specifically, sample birefringence is observed earlier (i.e. at a higher onset temperature) when cooling the 20% w/w dispersion from 150 °C to 20 °C. This suggests more efficient sphere-sphere fusion at this higher copolymer concentration, as previously reported for the regeneration of worms from their constituent spheres in closely-related PISA formulations.23
 | ||
| Fig. 7 Combination of the dispersion viscosity-temperature profile recorded for the cooling (blue data) and heating (red data) temperature ramps (cooling/heating rate = 2 °C min−1) for a 20% w/w (squares), 15% w/w (triangles) and 10% w/w (circles) dispersion of PSMA13–PBzMAx nano-objects at the maximum (sample edge) shear rate = 100 s−1. The highlighted regions represent the temperature ranges over which sample birefringence is observed. | ||
Conclusions
In summary, PSMA13–PBzMA65 worms prepared via PISA at 5.0 to 20% w/w solids in mineral oil form free-standing gels at 20 °C. A pure worm morphology was confirmed in each case by a combination of TEM and SAXS studies performed on dilute dispersions. Moreover, SAXS analysis indicated essentially the same mean worm core diameter of 11.9–13.1 nm at each copolymer concentration. SAXS results, supported by SIPLI measurements, also provided some evidence for worm branching, but the degree of branching could not be determined. Each copolymer dispersion exhibited thermoreversible behavior: the initial free-standing worm gel at 20 °C is converted into a free-flowing fluid comprising spheres on heating up to 150 °C. The critical gelation temperature (CGT) varied between 57 °C and 65 °C for PSMA13–PBzMA62–64 worm gels prepared at 5.0 to 20% w/w solids. The critical gelation concentration (CGC) was determined to be around 4.0% w/w as judged by the tube inversion method after serial dilution of a 20% w/w PSMA13–PBzMA64 worm gel. These worm gels exhibit shear-thinning behavior at 20 °C, and shear-induced alignment was observed between 80 °C and 110 °C for all worm gels using the SIPLI technique. On cooling from 150 °C, isotropic spherical nanoparticles formed shear-aligned anisotropic worms. Further cooling resulted in the loss of birefringence. This suggests the formation of either branched worms or worm clusters. Lower dispersion viscosities were observed at 20 °C for less concentrated worm gels (10% w/w vs. 20% w/w) or after exposure to higher shear rates. However, the dispersion viscosity for the spherical nanoparticles formed at 150 °C was more or less independent of the shear rate, indicating that such free-flowing dispersions behave as Newtonian fluids.Author contributions
M. J. D. performed all synthesis and analysis. M. J. D., O. O. M. and S. P. A. contributed to the writing, reviewing and editing of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Lubrizol Corporation is thanked for funding a PhD studentship for M. J. D. and for permission to publish this work. The Leverhulme Trust is also thanked for post-doctoral funding of M. J. D. (RPG-2016-330). S. P. A. acknowledges an EPSRC Established Career Fellowship (EP/R003009/1). Dr Svetomir Tzokov at the University of Sheffield Biomedical Science Electron Microscopy Suite is thanked for TEM assistance. All staff on the I22 beamline at Diamond Light Source are thanked for help and advice during the synchrotron SAXS studies (experiment no. SM10237).Notes and references
- Y.-Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960 CrossRef CAS PubMed.
- S. Jain and F. S. Bates, Science, 2003, 300, 460 CrossRef CAS PubMed.
- N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- Y.-Y. Won, K. Paso, H. T. Davis and F. S. Bates, J. Phys. Chem. B, 2001, 105, 8302–8311 CrossRef CAS.
- M. J. Rymaruk, C. T. O’Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 6849–6860 CrossRef CAS.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- K. A. Simon, N. J. Warren, B. Mosadegh, M. R. Mohammady, G. M. Whitesides and S. P. Armes, Biomacromolecules, 2015, 16, 3952–3958 CrossRef CAS PubMed.
- R. Spelat, F. Ferro, P. Contessotto, N. J. Warren, G. Marsico, S. P. Armes and A. Pandit, Mater. Today Bio, 2020, 5, 100040 CrossRef PubMed.
- A. L. A. Binch, L. P. D. Ratcliffe, A. H. Milani, B. R. Saunders, S. P. Armes and J. A. Hoyland, Biomacromolecules, 2021, 22, 837–845 CrossRef CAS PubMed.
- D. E. Mitchell, J. R. Lovett, S. P. Armes and M. I. Gibson, Angew. Chem., Int. Ed., 2016, 55, 2801–2804 CrossRef CAS PubMed.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS PubMed.
- Y. Geng and D. E. Discher, J. Am. Chem. Soc., 2005, 127, 12780–12781 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- S. M. Loverde, M. L. Klein and D. E. Discher, Adv. Mater., 2012, 24, 3823–3830 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- A. B. Lowe, Polymer, 2016, 106, 161–181 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. D. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Macromolecules, 2018, 51, 8357–8371 CrossRef CAS PubMed.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad and E. Rizzardo, Polym. Int., 2020, 69, 658–661 CrossRef CAS.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS PubMed.
- I. R. Dorsman, M. J. Derry, V. J. Cunningham, S. L. Brown, C. N. Williams and S. P. Armes, Polym. Chem., 2021, 12, 1224–1235 RSC.
- M. Y. Lin, H. J. M. Hanley, G. C. Straty, D. G. Peiffer, M. W. Kim and S. K. Sinha, Int. J. Thermophys., 1994, 15, 1169–1178 CrossRef CAS.
- V. Castelletto and I. W. Hamley, Polym. Adv. Technol., 2006, 17, 137–144 CrossRef CAS.
- V. Castelletto, P. Parras, I. W. Hamley, P. Bäverbäck, J. S. Pedersen and P. Panine, Langmuir, 2007, 23, 6896–6902 CrossRef CAS PubMed.
- B. Lonetti, J. Kohlbrecher, L. Willner, J. K. G. Dhont and M. P. Lettinga, J. Phys.: Condens. Matter, 2008, 20, 404207 CrossRef.
- O. O. Mykhaylyk, Soft Matter, 2010, 6, 4430–4440 RSC.
- O. O. Mykhaylyk, P. Chambon, C. Impradice, J. P. A. Fairclough, N. J. Terrill and A. J. Ryan, Macromolecules, 2010, 43, 2389–2405 CrossRef CAS.
- C. Holland, F. Vollrath, A. J. Ryan and O. O. Mykhaylyk, Adv. Mater., 2012, 24, 105–109 CrossRef CAS PubMed.
- O. O. Mykhaylyk, A. J. Parnell, A. Pryke and J. P. A. Fairclough, Macromolecules, 2012, 45, 5260–5272 CrossRef CAS.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- E. R. Draper, O. O. Mykhaylyk and D. J. Adams, Chem. Commun., 2016, 52, 6934–6937 RSC.
- J. S. Trent, Macromolecules, 1984, 17, 2930–2931 CrossRef CAS.
- J. Filik, A. W. Ashton, P. C. Y. Chang, P. A. Chater, S. J. Day, M. Drakopoulos, M. W. Gerring, M. L. Hart, O. V. Magdysyuk, S. Michalik, A. Smith, C. C. Tang, N. J. Terrill, M. T. Wharmby and H. Wilhelm, J. Appl. Crystallogr., 2017, 50, 959–966 CrossRef CAS PubMed.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- O. Glatter and O. Kratky, Small-angle X-ray Scattering, Academic Press, London, 1982 Search PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- K. Vogtt, G. Beaucage, M. Weaver and H. Jiang, Langmuir, 2015, 31, 8228–8234 CrossRef CAS PubMed.
- J. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk, J. A. Lane, M. J. Derry and S. P. Armes, Chem. Sci., 2015, 6, 4207–4214 RSC.
- A. P. Chatterjee, J. Chem. Phys., 2010, 132, 224905 CrossRef PubMed.
- R. H. J. Otten and P. V. D. Schoot, J. Chem. Phys., 2011, 134, 094902 CrossRef PubMed.
- M. J. Derry, T. Smith, P. S. O’Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS PubMed.
- O. O. Mykhaylyk, N. J. Warren, A. J. Parnell, G. Pfeifer and J. Laeuger, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2151–2170 CrossRef CAS.
- S. R. Raghavan and E. W. Kaler, Langmuir, 2001, 17, 300–306 CrossRef CAS.
- V. Croce, T. Cosgrove, G. Maitland, T. Hughes and G. Karlsson, Langmuir, 2003, 19, 8536–8541 CrossRef CAS.
- G. M. Kavanagh and S. B. Ross-Murphy, Prog. Polym. Sci., 1998, 23, 533–562 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: GPC data, additional rheology data and PLIs, viscosity-temperature profile for mineral oil, structural model details for SAXS analysis. See DOI: 10.1039/d1sm01011e |
| ‡ Present address: Aston Institute of Materials Research, Aston University, Aston Triangle, Birmingham, B4 7ET, UK. |
| This journal is © The Royal Society of Chemistry 2021 |